

Abstract
Thiamin plays a critical role in cellular energy metabolism. Mammalian cells obtain the vitamin from their surroundings, converted it to thiamin pyrophosphate (TPP) in the cytoplasm, followed by uptake of TPP by mitochondria via a carrier-mediated process that involves the MTPPT (product of the SLC25A19 gene). Previous studies have characterized different physiological/biological aspects of the human MTPPT (hMTPPT), but less is known about structural features that are important for its function. Here, we used a protein-docking model (“Phyre2” and “DockingServer”) to predict residues that may be important for function (substrate recognition) of the hMTPPT; we also examined the role of conserved positively-charged residues predicted (“PRALINE”) to be in the trans-membrane domains (TMDs) in uptake of the negatively-charged TPP. Among the six residues predicted by the docking model (i.e., Thr29, Arg30, Ile33, Ser34, Asp37 and Phe298), only Ile33, Ser34 and Asp37 were found to be critical for function. While no change in translational efficiency/protein stability of the Ser34 mutant was observed, both the Ile33 and Asp37 mutants showed a decrease in this parameter(s); there was also a decrease in the expression of the latter two mutants in mitochondria. A need for a polar residue at position 34 of the hMTPPT was evident. Our findings with the positively-charged residues (i.e., His82, His137, Lys231 and Lys291) predicted in the TMD showed that His137 and Lys291 are important for function (via a role in proper delivery of the protein to mitochondria). These investigations provide important information about the structure-function relationship of the hMTPPT
Keywords: Thiamin pyrophosphate transporter, mitochondria, site-directed mutagenesis, protein-docking modeling, multiple sequence alignment, uptake
Graphical abstract
Role of selective residues in the hMTPPT protein in TPP uptake by mitochondria

1. Introduction
Thiamin (vitamin B1) is essential for normal cellular functions due to its involvement (as a co-factor, mainly in the form of thiamin pyrophosphate, TPP) in critical cellular metabolic reactions related to oxidative energy metabolism, ATP production, and reduction of cellular oxidative stress [1–6]. The vitamin also plays a critical role in maintaining normal structure and function of the mitochondria [7]. All mammalian cells cannot synthesize thiamin endogenously; rather they obtain the vitamin from their surroundings via transport across the cell membrane. The latter process is carrier-mediated and involves the well-characterized thiamin transporter-1 & -2 (THTR-1 and THTR-2; products of SLC19A2 and SLC19A3 genes, respectively) [8–10]. Internalized thiamin is then converted enzymatically to the di- (pyro) phosphate form (TPP) in the cytoplasm [5, 11, 12], followed by uptake of the majority of the TPP by the mitochondria [13–16]. The latter process is carrier-mediated and involves the mitochondrial thiamin pyrophosphate transporter (MTPPT; product of the SLC25A19 gene [17]). Mutations in the SLC25A19 gene lead to a drastic depletion in mitochondrial TPP level and to the development of Amish congenital lethal microcephaly (an autosomal recessive disorder) [17, 18], as well as to the development of neuropathy and bilateral striatal necrosis [19].
The MTPPT is a 35 kDa protein of 320 amino acid residues, and like all other members of the mitochondrial carrier family (MCF) of proteins, consists of three tandem repeats (i.e., three modules) of approx. 100 amino acids each, and is predicted to have six trans-membrane domains (TMDs). Members of the MCF show high level of specificity for their substrate(s), a capability facilitated by the existence of specific sites within their structure that recognize the particular substrate(s). There is little currently known about the sites in the MTPPT protein that are important for substrate recognition/function. Our aim in this study was to shed light into this issue. Thus, we used programs that can predict potential substrate recognition/interaction sites in membrane transporters to identify potential sites in the MTPPT protein that may be involved in TPP recognition/interaction. For this, we first predicted the structure of MTPPT [by mean of homology modeling (“Phyre2”) using the “mitochondrial ADP/ATP carrier” (PDB ID: 1OKC) as template; 20–22], then submitted the MTPPT structure to the program “DockingServer” (http://www.dockingserver.com/web) [23, 24] using TPP as the substrate. We also examined (using “PRALINE”) possible role of conserved (among mammals) positively-charged residues predicted by homology modeling to be located at potentially strategic sites in the TMD of the MTPPT polypeptide [21] on transport function of the negatively charged TPP, since previous studies with other membrane transporters have shown a role for such sites in transporting negatively charged substrates [25–28]. Our results showed an important role for Ile33, Ser34, Asp37, His137 and Lys291 residues in function of the hMTPPT and provided insight into how their roles are played.
2. Materials and Methods
2.1. Materials
A living color GFP monoclonal antibody was from Clontech (Mountain view, CA), and the pyruvate dehydrogenase (PDH) monoclonal antibody was from Abcam (Cambridge, MA). HepG2 cells were purchased from ATCC (Manassas, VA). Mutated oligonucleotides were bought from Sigma Genosys (Woodlands, TX). 3H-thiamin pyrophosphate (TPP) (specific activity ~1.3Ci/mmol) was obtained from Moravek Biochemicals (Brea, CA) and all other chemicals used in this study were either of analytical or of molecular biology grades.
2.2. Generation of hMTPPT mutants: Site-directed mutagenesis
All the mutations were introduced in the open reading frame (ORF) of hMTPPT (SLC25A19 gene; i.e, in a phMTPPT-EGFP) using the Quick change™ II site-directed mutagenesis kit (Santa Clara, CA) and primers (Table-1) as described previously [29]. All mutations were made to alanine (unless otherwise stated) since such a substitution is predicted to have the minimal effect on secondary structure of proteins [30–32]. All the mutated constructs were verified by DNA sequencing (Laragen, Los Angeles, CA).
Table 1.
Primers used to generate the specified mutation sites in hMTPPT
| Mutation Sites | Forward and Reverse Primer sequences |
|---|---|
| Thr29Ala | 5′-GGTCTGTGTCTGGACTTGTTGCTCGGGCGCTG-3′; 5′-CAGCGCCCGAGCAACAAGTCCAGACACAGACC-3′ |
| Arg30Ala | 5′-GGTCTGTGTCTGGACTTGTTACTGCGGCGCTGATCAG-3′; 5′-CTGATCAGCGCCGCAGTAACAAGTCCAGACACAGACC-3′ |
| Ile33Ala | 5′-GTTACTCGGGCGCTGGCCAGTCCCTTCGACGT-3′; 5′-ACGTCGAAGGGACTGGCCAGCGCCCGAGTAAC-3′ |
| Ser34Ala | 5′-CTCGGGCGCTGATCGCTCCCTTCGACGTCA-3′; 5′-TGACGTCGAAGGGAGCGATCAGCGCCCGAG-3′ |
| Asp37Ala | 5′-CTGATCAGTCCCTTCGCCGTCATCAAGATCCGT-3′; 5′-ACGGATCTTGATGACGGCGAAGGGACTGATCAG-3′ |
| Phe298Ala | 5′-CTGCCCTCTCCACAGGCGCCATGTTCTTCTCGTATG-3′; 5′-CATACGAGAAGAACATGGCGCCTGTGGAGAGGGCAG-3′ |
| His82Ala | 5′-CGACAGCTTTCTGGAAAGGAGCCGTCCCAGCTC-3′; 5′-GAGCTGGGACGGCTCCTTTCCAGAAAGCTGTCG-3′ |
| His137Ala | 5′-CCACCCTCACTGTGGCCCCCGTGGATGTTC-3′; 5′-GAACATCCACGGGGGCCACAGTGAGGGTGG-3′ |
| Lys231Ala | 5′-GAGCTGGTGTCATCAGCGCGACCCTGACATATCCGC-3′; 5′-GCGGATATGTCAGGGTCGCGCTGATGACACCAGCTC-3′ |
| Lys291Ala | 5′-CCCCAGCTTGCTGGCGGCTGCCCTCTCC-3′; 5′-GGAGAGGGCAGCCGCCAGCAAGCTGGGG-3′ |
| Ser34Val | 5′-TACTCGGGCGCTGATCGTTCCCTTCGACGTCATC-3′; 5′-GATGACGTCGAAGGGAACGATCAGCGCCCGAGTA-3′ |
| Ser34Thr | 5′-CTCGGGCGCTGATCACTCCCTTCGAC-3′; 5′-GTCGAAGGGAGTGATCAGCGCCCGAG-3′ |
| His137Arg | 5′-CACCCTCACTGTGCGCCCCGTGGATGTTC-3′; 5′-GAACATCCACGGGGCGCACAGTGAGGGTG-3′ |
Mutated sites of each primer are shown in bold italic text.
2.3. Cell culture and transfection
HepG2 cells, grown in T75 flasks (Corning, NY) in Dulbecco’s modified Eagle’s medium supplemented with 10% (v/v) fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin were transiently transfected with 10 μg of wild-type (WT) hMTPPT and mutated plasmid construct at 80–90% confluency using 10 μl of Lipofectamine2000 (Invitrogen, CA). For stable selection, HepG2 cells were selected using G418 (0.5 mg/ml; Invitrogen) for 6–8 weeks and the over expression of the WT hMTPPT and mutant constructs were confirmed by Western blot analysis.
2.4. Isolation of mitochondria and uptake measurement
Mitochondria were isolated from HepG2 cells stably expressing WT hMTPPT and mutant constructs using mitochondria isolation kit (Thermo Scientific, Rockford, IL). The 3H-TPP uptake on isolated mitochondria was performed as previously described [29]. Briefly, the freshly prepared mitochondrial fraction was suspended in uptake buffer [140mM KCl, 0.3mM EDTA, 5mM MgCl2, 10mM MES, 10mM HEPES and 10mM succinate (to maintain the function of mitochondria), pH 7.4] and uptake was performed as described before [29] using rapid filtration method [33]. The mitochondrial suspension (20 μl; containing ~15–20 μg/μl of protein) was then added to 80 μl of uptake buffer containing 0.38 μM of 3H-TPP and incubated at 37°C for 5 min. The uptake was stoped by the addition of 1 ml of ice-cold stop buffer [100mM KCl, 100mM mannitol, and 10mM KH2PO4, pH 7.4] followed by rapid filtration. After washing the filter with ice-cold stop buffer, the radioactivity was measured using Beckman liquid scintillation counter. The mitochondrial suspension (20 μl) was used to determine the protein concentration and uptake normalized with this protein concentration.
2.5. Western Blot
Mitochondria were isolated from HepG2 stable cells and the total protein was isolated by using RIPA buffer (Sigma) followed by sonication. The total protein and mitochondrial protein (~60 μg) were resolved in NuPAGE 4–12% Bis-Tris gradient minigels (Invitrogen) followed by transfer onto PVDF membrane (Fisher Scientific), and subsequently membranes were incubated with anti-GFP monoclonal antibody (Clontech, CA) and anti-pyruvate dehydrogenase (PDH) monoclonal antibodies (Abcam, MA). The detection of specific protein bands were performed by incubating with anti-mouse IRDye-800 and anti-mouse IRDye-680 secondary antibodies (LI-COR Bioscience, Lincoln, NE) at 1:30,000 dilutions sequentially. Finally, the bands were visualized using the Odyssey infrared imaging system (LI-COR Bioscience) [30]. The density of individual band was determined by using odyssey application software (version 3.0).
2.6. Real-Time PCR
Total RNA isolated from the HepG2 cells stably expressing the different hMTPPT constructs using TRIzol reagent was treated with DNase I and subjected to reverse transcription using iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). The mRNA expression level was quantified in a CFX96 real-time PCR system (Bio-Rad), using iQ SYBR Green Super mix (Bio-Rad) and primers specific hMTPPT (forward: 5′-AGCATGAGCGCCTGTCGC-3′ and reverse: 5′-TGAGCTGGGACTGTCCTTTCCA-3′) and human β-actin (forward: 5′-CATCCTGCGTCTGGACCT-3′ and reverse: 5′-TAATGTCACGCACGATTTCC-3′). Real-time PCR conditions were used as previously described [34]. Data were normalized to β-actin and calculated using a relative relationship method [35] supplied by the iCycler manufacturer (Bio-Rad).
2.7. Comparative protein modeling and computational analysis
We subjected the hMTPPT polypeptide sequence to Phyre2 fold recognition program (http://www.sbg.bio.ic.ac.uk/phyre2) and obtained several homology models as described before [20]. Among these models, we validated and chose only one by comparing the score of the energy value to the experimentally determined energy value of native protein of similar length in “ProSA-Web” (https://prosa.services.came.sbg.ac.at/prosa.php) [22]. The selected model was designed based on “Mitochondrial ADP/ATP carrier” (PDB ID: 1OKC) as the most suitable template [21]. The template gives 93% coverage for hMTPPT with 100% confidence. The generated three dimensional (3D) structure of hMTPPT was visualized by “RasMol” (www.rasmol.org) and docking calculations were carried out on hMTPPT model using “DockingServer” (http://www.dockingserver.com) [23, 24, 36, 37].
The topology of hMTPPT determined from secondary structure prediction by “TMpred” program (http://www.ch.embnet.org/software/TMPRED_form.html) [38] matches the relative orientation of amino acids as determined by comparative 3D modeling. Conserved amino acids residues were identified by multiple sequence alignment using “PRALINE” software (www.ibi.vu.nl/programs/pralinewww/) [39].
2.8. Statistical Analysis
All the data are presented as mean ± SE of at least 3 independent determinations. The uptake was expressed in terms of femtomoles per milligram of protein per 5 min. Statistical analysis of each experiment was performed by Student’s t-test and significance was set at P < 0.05.
3. Results
3.1.1. The hMTPPT-TPP docking model and prediction of the TPP recognition/interaction site(s)
The hMTPPT is a 35 kDa protein (320 amino acids) and is predicted to have six TMDs with both the amino and carboxy terminals oriented towards the cytosolic compartment. From the available hMTPPT amino acid sequence, we generated a comparative homology model (“Methods”), which was then docked with TPP as the substrate (using “DockingServer” program [23, 24, 36, 37]) to predict the substrate recognition/interaction site(s). This in silico docking model predicted residues Thr29, Arg30, Ile33, Ser34, Asp37, and Phe298 of the hMTPPT (Fig. 1A) as having a potential role(s) in substrate (TPP) recognition/interacting. Of these residues, Thr29, Arg30, Ile33, Ser34 and Asp37 appear to be located in the 1st TMD, while Phe298 is located in the 6th TMD, as predicted by “TMpred” program [38]. Also, all the predicted residues were found to be conserved among the mammals (human, sumatran orangutan, chimpanzee, crab-eating macaque, bovine, sheep, wild boar, mouse and rat; data not shown). Figure 1B shows a schematic diagram of the secondary structures of hMTPPT with location of the above-mentioned potential recognition/interaction residues.
Figure 1. Amino acid residues in the hMTPPT protein predicted to have potential role in recognition/function.
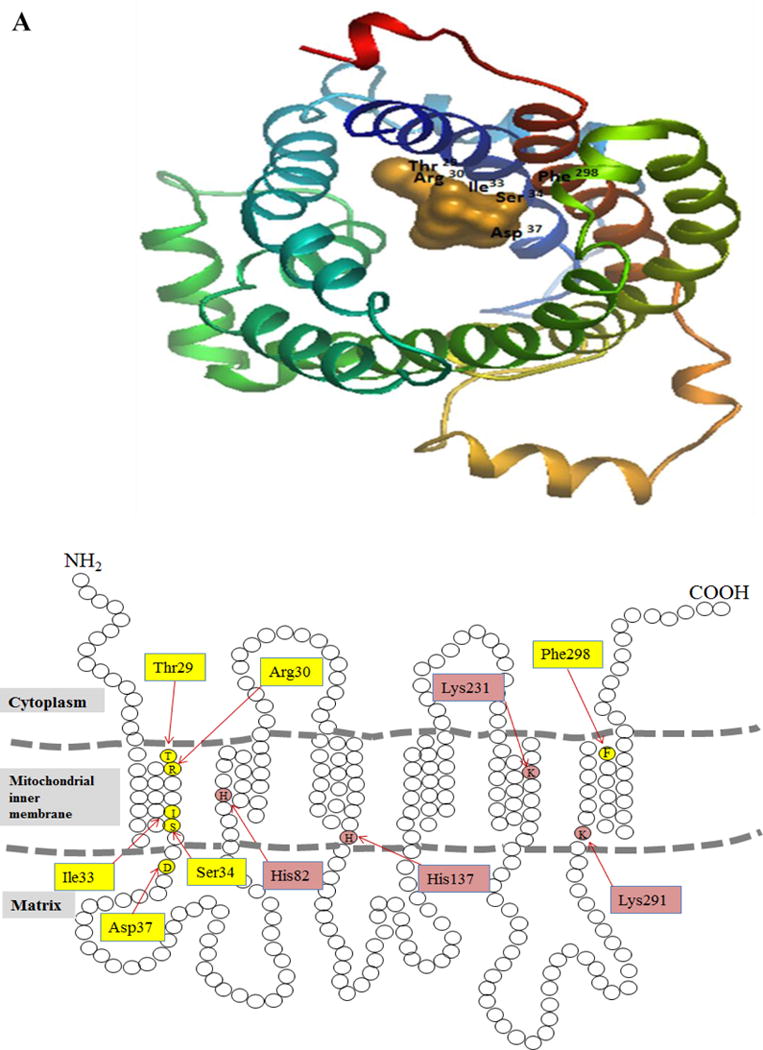

A. Docking model of hMTPPT with TPP created by the docking server and the protein (hMTPPT) was shown as a multi-color cartoon representation and substrate (TPP) as orange colored. The homology model of hMTPPT was generated based on the crystal structure of the mitochondrial ADP/ATP carrier in complex with carboxyatractyloside (PDB ID: 1OKC) [21]. B. Predicted topology model of hMTPPT showing six TMDs with both NH2 and COOH- terminal oriented towards the cytosol. The location of predicted substrate recognition site and conserved positively-charged residues are shown by yellow and pink colored boxes, respectively. C. MTPPT protein sequence from human (NP_001119594), sumatran orangutan (NP_001127123), chimpanzee (NP_001233547), crab-eating macaque (NP_001306520), bovine (NP_001039352), sheep (NP_001136362), wild boar (NP_001157986), mouse (NP_001239313) and rat (NP_001007675) were aligned by using PRALINE software. Conserved and unconserved residues are indicated by red to blue color, respectively and identities of conserved positively-charged residues (which are located in potentially strategic position based on homology model) in TMD of hMTPPT are shown in boxes.
3.1.2. Role of the predicted putative recognition/interaction sites in hMTPPT function
Using the approach of site-directed mutagenesis, we mutated the individual putative recognition/interaction site Thr29, Arg30, Ile33, Ser34, Asp37 and Phe298 and examined the effect of each mutation on function of the hMTPPT in TPP transport. For this, WT and mutated hMTPPT constructs were stably expressed in HepG2 cells followed by isolation of mitochondria and assaying for 3H-TPP uptake (“Methods”). The results showed that mutating Ile33, Ser34 and Asp37 to lead to a significant (P < 0.01 for all) decrease in 3H-TPP uptake, while mutating Thr29, Arg30 and Phe298 to have no effect (Fig. 2 A).
Figure 2. Effect of mutating residues in the hMTPPT protein predicted to play a role in substrate recognition/interaction on 3H-TPP uptake by mitochondria and on the level of expression of hMTPPT mRNA and protein.

A. Uptake of 3H-TPP (0.38 μM; 37 °C) by mitochondria isolated from HepG2 cells stably expressing WT hMTPPT and the mutants Thr29, Arg30, Ile33, Ser34, Asp37 and Phe298. Uptake was measured after 5 min incubation (buffer, pH 7.4, 37°C) and calculated as described in “Method”. Data are mean ± SE of at least three independent experiments, **P < 0.01. B. The level of mRNA expression of the WT hMTPPT and mutants (Ile33, Ser34 and Asp37) determined by real-time PCR (RT-PCR) using total RNA isolated from HepG2 cells stably expressing these constructs. Data are presented as mean ± SE of three to four independent experiments after normalization with β-actin. (C) & (D) show Western blot analysis performed using equal amount of whole HepG2 cell homogenate protein and purified isolated mitochondria, respectively (“Methods”). The blots were probed with anti-GFP monoclonal antibody and anti-PDH monoclonal antibody; data was normalized relative to the mitochondrial internal control, PDH. Representative blots are shown in inset. Data are mean ± SE from at least three different samples from three different batches of cells. *P < 0.05 and ** P < 0.01.
To determine whether the reduction in TPP uptake by the Ile33, Ser34 and Asp37 hMTPPT mutants was due to an alteration in mRNA level of the different constructs, we examined (by mean of RT-PCR) mRNA levels of these mutants in HepG2 cells stably expressing these constructs compared the finding to level of mRNA expression of the WT hMTPPT. The results showed no effect of these mutations on expression of the hMTPPT mRNA (Fig. 2B). We then examined whether the Ile33, Ser34 and Asp37 mutations lead to changes in translation efficiency/stability of the hMTPPT protein. For that, we performed Western blotting using whole cell homogenate from HepG2 cells expressing these mutants and the WT hMTPPT. The results showed that while the level of expression of the Ser34 hMTPPT mutant was similar to that of the WT, the level of expression of the Ile33 and Asp37 mutants was significantly (P < 0.05 for both) lower than that of WT (Fig. 2C). The latter findings suggest that the Ile33 and Asp37 mutations affect translational efficiency/protein stability of hMTPPT.
In another study, we examined the effect of mutating Ile33, Ser34 and Asp37 in the hMTPPT protein on expression of the mutant proteins in mitochondria, i.e., determine whether these mutations affect targeting/delivery of the protein to mitochondria. This was again done by mean of Western blotting, where an identical amount of purified mitochondria (~60 μg) isolated from HepG2 cells stably expressing the different mutants and the WT hMTPPT was used. Data was normalized relative to the internal control PDH, a mitochondrial protein. The results showed a significantly lower level of expression of the Ile33 (P < 0.01) and Asp37 (P < 0.05) mutants in mitochondria compared to the WT hMTPPT; mitochondrial expression of the Ser34 mutant was similar to that of the WT protein (Fig. 2D). Focusing on the Ser34 hMTPPT mutant (which did not show changes in mRNA synthesis and in translational efficiency/protein stability), we sought to determine whether the inhibition in functionality was due to a loss of a polar residue at that position. For this we examined the effect of replacing the polar Ser at position 34 with another polar residue (threonine; Thr) or with a non-polar residue (valine; Val) on transport function of hMTPPT. The results showed that substituting Ser with Thr resulted in normal hMTPPT transport function, while replacing it with Val led to an impairment in functionality (Fig. 3A). There was no change in level of mRNA (Fig. 3B) or protein (Fig. 3C & D) expression of the different mutants.
Figure 3. Effect of the hMTPPT Ser34Ala, Ser34Thr and Ser34Val mutants on 3H-TPP uptake and on the level of expression of hMTPPT mRNA and protein.

A. Uptake of 3H-TPP (0.38 μM) by mitochondria of HepG2 cells stably expressing WT hMTPPT and the Ser34Ala, Ser34Val and Ser34Thr mutants. Uptake was measured after 5 min incubation (buffer, pH 7.4, 37 °C) and calculated as described in “Method”. Data are mean ± SE of at least three independent experiments. **P < 0.01 B. The level of mRNA expression of Ser34Ala, Ser34Thr and Ser34Val mutants compared to WT hMTPPT. The RT-PCR was performed using total RNA isolated from HepG2 cells expressing the WT hMTPPT and mutants. Data are mean ± SE of three to four independent experiments after normalization with β-actin expression. Western blot analyses were performed with an equal amount of whole cell homogenate (C), and purified mitochondria (D) from HepG2 cells expressing WT hMTPPT or mutants (“Methods”). The blot was probed with anti-GFP monoclonal antibody and anti-PDH monoclonal antibody; data was then normalized relative to the mitochondrial internal control, PDH. Representative blots are shown in inset. I and II are indicates the Ser34Ala, Ser34Val, Ser34Thr and His137Ala, His137Arg, respectively. Data are mean ± SE from at least three different samples from three different batches of cells.
3.2.1. Prediction of conserved positively-charged residues in the TMD of the hMTPPT protein that may play a role in transport function
The hMTPPT protein is predicted (by the multiple sequence alignment “PRALINE” prediction software) to have four positively-charged residues in its TMDs (His82, His137, Lys231 and Lys291) that are conserved among mammals (human, sumatran orangutan, chimpanzee, crab-eating macaque, bovine, sheep, wild boar, mouse and rat) (Fig. 1C). Of those, His82 and His137 are predicted (“TMpred” program) to be located at the 2nd and 3rd TMD respectively, while Lys231 and Lys291 are predicted to be located at the 5th and 6th TMD, respectively.
3.2.2. Role of His82, His137, Lys231 and Lys291 in transport function of hMTPPT
We used site-directed mutagenesis to introduce individual mutations in the predicted conserved positively-charged residues i.e, His82, His137, Lys231 and Lys291, then examined the consequence of that mutation on functionality of hMTPPT in transporting the negatively charged TPP. For this, 3H-TPP uptake assay was performed using purified mitochondria isolated from HepG2 cells stably expressing the WT and the mutated constructs (“Methods”). The results showed that mutating His137 and Lys291 to lead to a significant (P < 0.01 for both) decrease in 3H-TPP uptake, while mutating His82 and Lys231 was without an effect (Fig. 4A).
Figure 4. Effect of mutating conserved positively-charged residues in hMTPPT TMDs on 3H-TPP uptake by mitochondria and on the level of mRNA and protein expression of the transporter.

A. Uptake of 3H-TPP (0.38μM) by mitochondria of HepG2 cells stably expressing WT hMTPPT and the mutants His82, His137, Lys231 and Lys 291. Freshly isolated mitochondria were incubated in uptake buffer, pH 7.4 at 37°C and 3H-TPP (0.38μM) was added to the incubation buffer to start the uptake assay. Uptake was measured after 5 min incubation and calculated as described in “Method”. Data are mean ± SE of at least three independent experiments. **P < 0.01. B. The level of mRNA expression of the WT hMTPPT and mutants (His137 and Lys291) determined by real-time PCR (RT-PCR) using total RNA isolated from HepG2 cells stably expressing these constructs. Data are presented as mean ± SE of three to four independent experiments after normalization with β-actin. (C) & (D) show Western blot analysis performed using equal amount of whole HepG2 cell homogenate protein and purified isolated mitochondria, respectively (“Methods”). The blots were probed with anti-GFP monoclonal antibody and anti-PDH monoclonal antibody; data was normalized relative to the mitochondrial internal control, PDH. Representative blots are shown in inset. Data are mean ± SE of at least three different samples from three different batches of cells. ** P < 0.01.
To define whether the reduction of TPP uptake by the His137 and Lys291 hMTPPT mutants was due to reduction in mRNA level, we examined (by mean of RT-PCR) the mRNA expression of the mutants stably expressed in HepG2 cells. The results showed similar level of expression of the two mutants to that of the WT hMTPPT (Fig. 4B). We then examined whether the His137 and Lys291 mutations led to changes in translational efficiency/stability of the hMTPPT protein. For that we performed Western blotting using whole cell homogenate of HepG2 stably expressing these mutants. The results showed that the protein levels of the His137 and Lys291 mutants were similar to that of the WT hMTPPT (Fig. 4C). We further tested if there was a change in the level of expression of the His137 and Lys291 mutant hMTPPT proteins in mitochondria. Again Western blotting was employed and equal amount of purified mitochondria isolated from HepG2 cells expressing these mutants and the WT hMTPPT were used; results were normalized relative to the internal control, PDH, a mitochondrial protein. The results showed that the His137 and Lys291 mutations to be associated with a significant (P < 0.01 for both) decrease in expression of the hMTPPT protein in mitochondria (Fig. 4D). It is interesting to mention here that replacing the positively charged His at position 137 with the positively charged arginine (Arg) was found to be sufficient to maintain normal hMTPPT function (TPP uptake of 422 ± 33 and 539 ± 20 fmol/mg protein/5min for WT and His137Arg, respectively). Whether the latter is an indication for the importance of having a positively charged residue at position 137 for normal hMTPPT function is not clear and needs of further investigations.
4. Discussion
The hMTPPT is both physiologically and clinically important transporter as it is responsible for uptake of the metabolically important TPP into mitochondria (there is no synthesis of TPP inside this cellular organelle), and that mutations in this transporter cause Amish congenital lethal microcephaly [17, 18, 40] and neuropathy and bilateral striatal necrosis [19]. Previous studies have delineated different physiological and biological aspects of the MTPPT system [29, 41], but little is known about the structure-function relationship of this mitochondrial transporter. Thus, our aim in this study was to address this issue, and for that we employed a comparative protein-docking modeling [36, 37] approach as well as a multiple sequence alignment [39] approach to predict amino acid residues that may be important for function of this transporter. We then experimentally tested the relevance of these amino acid residues in hMTPPT functionality using site-directed mutagenesis and other physiological/biological means.
Our investigations with the protein-docking model have predicted Thr29, Arg30, Ile33, Ser34, Asp37 and Phe298 residues in the hMTPPT as being putative substrate recognition/interaction sites. Thus, we mutated each of these residues and examined the effect of such mutations on TPP uptake by purified mitochondria isolated from HepG2 cells stably expressing these mutants. The results showed an important role for Ile33, Ser34 and Asp37 (but not Thr29, Arg30 and Phe298) in hMTPPT function, as indicated by the significant inhibition in TPP uptake observed with these mutants compared to the WT hMTPPT. This inhibition in TPP uptake was not due to alteration in mRNA level of the mutated constructs as no change was observed in mRNA expression compared to the WT hMTPPT. There was, however, a reduction in the level of protein expression of the Ile33 and Asp37 hMTPPT mutants in whole cellular homogenates, suggesting a likely change(s) in translational efficiency/protein stability. We also observed a decrease in the level of expression of the Ile33 and Asp37 hMTPPT mutant proteins in mitochondria, suggesting a role for these two residues in proper delivery of hMTPPT to the mitochondrial compartment.
The inhibition observed in TPP uptake as a result of mutating Ser34 of the hMTPPT protein was the severest among the mutations predicted by the protein-docking model. Since this did not appear to be due to changes in translation efficiency/trafficking to mitochondria of the hMTPPT protein, we sought to further examine whether it was due to a requirement for a polar residue at position 34. To test this possibility, we investigated the effect of replacing that polar Ser at position 34 with another polar residue, Thr, or with the non-polar residue Val on hMTPPT function. Substitution with Thr was found to be sufficient in maintaining normal function for hMTPPT, while substitution with Val impaired functionality. There was no change in transcriptional/translational efficiency of all the latter hMTPPT mutants or in their level of expression in mitochondria. These findings suggest that a polar residue at the position 34 of the hMTPPT protein is necessary for normal function of the carrier protein.
Previous studies have reported that positively charged residues in TMD of membrane transporters of negatively charged substrates may play a role in functionality of these carriers [25–28]. For this reason we examined potential role of conserved positively charged residues in the hMTPPT TMD in transport function of this carrier; four such residues (His82, His137, Lys231 and Lys291) were predicted in TMD of hMTPPT [21]. The results, however, showed that only His137 and Lys291 (but not His82 and Lys231) are important for function as indicated by the significant inhibition in TPP uptake by mitochondria from HepG2 cells stably expressing these mutants compared to WT hMTPPT. This inhibition in TPP uptake was not due to alterations in level of mRNA expression of the mutated constructs; it was also not due to changes in translational efficiency/stability of the hMTPPT protein as indicated by lack of change in hMTPPT protein level in whole cell homogenates compared to WT hMTPPT. There was, however, a reduction in the level of protein expression of the His137 and Lys291 hMTPPT mutants in mitochondria, suggesting a role for these residues in proper delivery of the synthesized hMTPPT to that cellular organelle.
In summary, results of the current investigations provide valuable information regarding structure-function relationship of the hMTPPT, a transporter that is important for normal cell physiology and metabolism.
Highlights.
The Ile33, Ser34 and Asp37 residues of hMTPPT are critical for functionality.
The Ile33 and Asp37 are involved in hMTPPT stability and delivery to mitochondria.
A need for polar residue at position 34 of hMTPPT was evident.
The His137 and Lys291 in TMD of hMTPPT are also important for hMTPPT function.
Requirement for positively-charged residue at position 137 of hMTPPT is revealed.
Acknowledgments
This study was supported by grants from the National Institutes of Health (AA018071, DK56061, DK58057 and DK107474) and the Department of Veterans Affairs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
No conflicts of interest, financial or otherwise, are declared by the authors.
References
- 1.Berdanier CD. Advanced Nutrition-Micronutrients. CRC Press; New York: 1998. pp. 80–88. [Google Scholar]
- 2.Singleton CK, Martin PR. Molecular mechanisms of thiamin utilization. Curr Mol Med. 2001;1:197–207. doi: 10.2174/1566524013363870. [DOI] [PubMed] [Google Scholar]
- 3.Calingasan N, Chun W, Park L, Uchida K, Gibson GE. Oxidative stress is associated with region-specific neuronal death during thiamine deficiency. J Neuropathol Exp Neurol. 1999;58:946–958. doi: 10.1097/00005072-199909000-00005. [DOI] [PubMed] [Google Scholar]
- 4.Portari GV, Marchini JS, Vannucchi H, Jordao AA. Antioxidant effect of thiamine on acutely alcoholized rats and lack of efficacy using thiamine or glucose to reduce blood alcohol content. Basic Clin Pharmacol Toxicol. 2008;103:482–486. doi: 10.1111/j.1742-7843.2008.00311.x. [DOI] [PubMed] [Google Scholar]
- 5.Gangolf M, Czerniecki J, Radermecker M, Detry O, Nisolle M, Jouan C, Martin D, Chantraine F, Lakaye B, Wins P, Grisar T, Bettendorff L. Thiamine status in humans and content of phosphorylated thiamine derivatives in biopsies and cultured cells. PLoS One. 2010;5:e13616. doi: 10.1371/journal.pone.0013616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frederikse PH, Farnsworth P, Zigler JS., Jr Thiamin deficiency in vivo produces fiber cell degeneration in mouse lenses. Biochem Biophys Res Commun. 1999;258:703–707. doi: 10.1006/bbrc.1999.0560. [DOI] [PubMed] [Google Scholar]
- 7.Bettendorff L, Goessens G, Sluse F, Wins P, Bureau M, Laschet J, Grisar T. Thiamin deficiency in cultured neuroblastoma cells: effect on mitochondrial function and peripheral benzodiazepine receptors. J Neurochem. 1995;64:2013–2021. doi: 10.1046/j.1471-4159.1995.64052013.x. [DOI] [PubMed] [Google Scholar]
- 8.Diaz GA, Banikazemi M, Oishi K, Desnick RJ, Gelb BD. Mutations in a new gene encoding a thiamin transporter cause thiamin-responsive megaloblastic anaemia syndrome. Nat Genet. 1999;22:309–312. doi: 10.1038/10385. [DOI] [PubMed] [Google Scholar]
- 9.Fleming JC, Tartaglini E, Steinkamp MP, Schorderet DF, Cohen N, Neufeld EJ. The gene mutated in thiamine-responsive anaemia with diabetes and deafness (TRMA) encodes a functional thiamine transporter. Nat Genet. 1999;22:305–308. doi: 10.1038/10379. [DOI] [PubMed] [Google Scholar]
- 10.Rajagopal A, Edmondnson A, Goldman ID, Zhao R. SLC19A3 encodes a second thiamine transporter ThTr2. Biochim Biophys Acta. 2001;1537:175–178. doi: 10.1016/s0925-4439(01)00073-4. [DOI] [PubMed] [Google Scholar]
- 11.Deus D, Blum H. Subcellular distribution of thiamin pyrophosphokinase activity in rat liver and erythrocytes. Biochim Biophys Acta. 1970;219:489–492. doi: 10.1016/0005-2736(70)90229-4. [DOI] [PubMed] [Google Scholar]
- 12.Cusaro G, Rindi G, Sciorelli G. Subcellular distribution on thiamin pyrophospho kinase and thiamin-pyrophosphatase activities in rat isolated enterocytes. Int J Vit Nutr Res. 1997;47:99–106. [PubMed] [Google Scholar]
- 13.Bettendorff L. Thiamin homeostasis in neuroblastoma cells. Neurochem Int. 1995;26:295–303. doi: 10.1016/0197-0186(94)00123-c. [DOI] [PubMed] [Google Scholar]
- 14.Bettendorff L, Wins P, Lesourd M. Subcellular localization and compartmentation of thiamin derivatives in rat brain. Biochim Biophys Acta. 1994;1222:1–6. doi: 10.1016/0167-4889(94)90018-3. [DOI] [PubMed] [Google Scholar]
- 15.Bettendorff L. The compartmentation of phosphorylated thiamin derivatives in cultured neuroblastoma cells. Biochim Biophys Acta. 1994;1222:7–14. doi: 10.1016/0167-4889(94)90019-1. [DOI] [PubMed] [Google Scholar]
- 16.Barile M, Passarella S, Quangliariello E. Thiamin pyrophosphate uptake into isolated rat liver mitochondria. Arch Biochem Biophys. 1990;280:352–357. doi: 10.1016/0003-9861(90)90341-u. [DOI] [PubMed] [Google Scholar]
- 17.Lindhurst MJ, Fiermonte G, Song S, Struys E, De Leonardis F, Schwartzberg PL, Chen A, Castegna A, Verhoeven N, Mathews CK, Palmieri F, Biesecker LG. Knockout of Slc25a19 causes mitochondrial thiamine pyrophosphate depletion, embryonic lethality, CNS malformations, and anemia. Proc Natl Acad Sci U S A. 2006;103:15927–15932. doi: 10.1073/pnas.0607661103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Siu VM, Ratko S, Prasad AN, Prasad C, Rupar CA. Amish microcephaly: long-term survival and biochemical characterization. Am J Med Genet. 2010;152 A:1747–1751. doi: 10.1002/ajmg.a.33373. [DOI] [PubMed] [Google Scholar]
- 19.Spiegel R, Shaag A, Edvardson S, Mandel H, Stepensky P, Shalev SA, Horovitz Y, Pines O, Elpeleg O. SLC25A19 mutation as a cause of neuropathy and bilateral striatal necrosis. Ann Neurol. 2009;66:419–424. doi: 10.1002/ana.21752. [DOI] [PubMed] [Google Scholar]
- 20.Kelley LA, Sternberg MJE. Protein Structure prediction on the web: a case study using the Phyre server. Nat Prot. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 21.Pebay-Peyroula E, Dahout-Gonzalez C, Kahn R, Trezeguet V, Lauquin GJM, Brandolin G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature. 2003;426:39–44. doi: 10.1038/nature02056. [DOI] [PubMed] [Google Scholar]
- 22.Widerstein M, Sipple MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucl Acids Res. 2007;35:W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hazai E, Kovacs S, Demko L, Bikadi Z. Docking Server (www.dockingserver.com) Virtual drug Ltd; Budapest, Hungary: 2009. [Google Scholar]
- 24.Bikadi Z, Demko L, Hazai E. Functional and structural characterization of protein based analysis of its hydrogen bonding networks by hydrogen bonding plot. Arch Biochem Biophys. 2007;461:225–234. doi: 10.1016/j.abb.2007.02.020. [DOI] [PubMed] [Google Scholar]
- 25.Unal ES, Zhao R, Chang MH, Fiser A, Romero MF, Goldman ID. The functional roles of the His247 and His281 residues in folate and proton translocation mediated by the human proton-coupled folate transporter SLC46A1. J Biol Chem. 2009;284:17846–57. doi: 10.1074/jbc.M109.008060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou F, Pan Z, Ma J, You G. Mutational analysis of histidine residues in human organic anion transporter 4 (hOAT4) Biochem J. 2004;384:87–92. doi: 10.1042/BJ20040751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Said HM, Mohammadkhani R. Involvement of histidine residues and sulflaydryl groups in the function of the biotin transport carrier of rabbit intestinal brush-border membrane. Biochim Biophys Acta. 1992;1107:238–244. doi: 10.1016/0005-2736(92)90410-n. [DOI] [PubMed] [Google Scholar]
- 28.Dridi L, Haimeur A, Ouellette M. Structure-function analysis of the highly conserved charged residues of the membrane protein FT1, the main folic acid transporter of the protozoan parasite Leishmania. Biochem Pharmacol. 2010;79:30–8. doi: 10.1016/j.bcp.2009.07.019. [DOI] [PubMed] [Google Scholar]
- 29.Subramanian VS, Nabokina SM, Lin-Moshier Y, Marchant JS, Said HM. Mitochondrial Uptake of Thiamin Pyrophosphate: Physiological and Cell Biological Aspects. PLoS One. 2013;8(8):e73503. doi: 10.1371/journal.pone.0073503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morrison KL, Weiss GA. Combinatorial alanine-scanning. Curr Opin Chem Bio. 2001;5:302–307. doi: 10.1016/s1367-5931(00)00206-4. [DOI] [PubMed] [Google Scholar]
- 31.Peterson BZ, Johnson BD, Hockerman GH, Acheson M, Scheuer T, Catterall WA. Analysis of the Dihydropyridine Receptor Site of L-type Calcium Channels by Alanine-scanning Mutagenesis. J Biol Chem. 1997;272:18752–18758. doi: 10.1074/jbc.272.30.18752. [DOI] [PubMed] [Google Scholar]
- 32.Blaber M, Zhang XJ, Matthews BW. Structural basis of amino acid alpha helix propensity. Science. 1993;260:1637–1640. doi: 10.1126/science.8503008. [DOI] [PubMed] [Google Scholar]
- 33.Hopfer U, Nelson K, Perrotto J, Isselbacher KJ. Glucose transport inisolated brush border membrane from rat small intestine. J Biol Chem. 1973;248:25–32. [PubMed] [Google Scholar]
- 34.Subramanian VS, Subramanya SB, Said HM. Relative contribution of THTR-1 and THTR-2 in thiamin uptake by pancreatic acinar cells: studies utilizing Slc19a2 and Slc19a3 knockout mouse models. Am J Physiol Gastro intest Physiol. 2011;302:G572–578. doi: 10.1152/ajpgi.00484.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C[T]) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 36.Bikadi Z, Hazai E. Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. J Cheminf. 2009;1:15. doi: 10.1186/1758-2946-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huey R, Morris GM, Olson AJ, Goodsell DS. A Semiempirical Free Energy Force Field with Charge-Based Desolvation. J Comput Chem. 2007;28:1145–1152. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- 38.Hofmann K, Stoffel W. TMbase - A database of membrane spanning proteins segments. Biol Chem Hoppe-Seyler. 1993;374:166. [Google Scholar]
- 39.Bawono P, Heringa J. PRALINE: a versatile multiple sequence alignment toolkit. Methods Mol Biol. 2014;1079:245–62. doi: 10.1007/978-1-62703-646-7_16. [DOI] [PubMed] [Google Scholar]
- 40.Rosenberg MJ, Agarwala R, Bouffard G, Davis J, Fiermonte G, Hilliard MS, Koch T, Kalikin LM, Makalowska I, Morton DH, Petty EM, Weber JL, Palmieri F, Kelley RI, Schäffer AA, Biesecker LG. Mutant deoxynucleotide carrier is associated with congenital microcephaly. Nat Genet. 2002;32:175–179. doi: 10.1038/ng948. [DOI] [PubMed] [Google Scholar]
- 41.Nabokina SM, Valle JE, Said HM. Characterization of the human mitochondrial thiamine pyrophosphate transporter SLC25A19 minimal promoter: a role for NF-Y in regulating basal transcription. Gene. 2013;528(2):248–55. doi: 10.1016/j.gene.2013.06.073. [DOI] [PMC free article] [PubMed] [Google Scholar]