

Abstract
Müller glia can be stimulated to de-differentiate, proliferate and form Müller glia-derived progenitor cells (MGPCs) that are capable of producing retinal neurons. The signaling pathways that influence the de-differentiation of mature Müller glia and proliferation of MGPCs may include the Wnt-pathway. The purpose of this study was to investigate how Wnt-signaling influences the formation of MGPCs in the chick retina in vivo. In NMDA-damaged retinas where MGPCs are known to form, we find dynamic changes in retinal levels of potential readouts of Wnt-signaling, including dkk1, dkk3, axin2, c-myc, tcf-1 and cd44. We find accumulations of nuclear β-catenin in MGPCs that peaks at 3 days and rapidly declines by 5 days after NMDA-treatment. Inhibition of Wnt-signaling with XAV939 in damaged retinas suppressed the formation of MGPCs, increased expression of ascl1a and decreased hes5, but had no effect upon the differentiation of progeny produced by MGPCs. Activation of Wnt-signaling, with GSK3β-inhibitors, in the absence of retinal damage, failed to stimulate the formation of MGPCs, whereas activation of Wnt-signaling in damaged retinas stimulated the formation of MGPCs. In the absence of retinal damage, FGF2/MAPK-signaling stimulated the formation of MGPCs by activating a signaling network that includes Wnt/β-catenin. In FGF2-treated retinas, inhibition of Wnt-signaling reduced numbers of proliferating MGPCs, whereas activation of Wnt-signaling failed to influence the formation of proliferating MGPCs. Our findings indicate that Wnt-signaling is part of a network initiated by FGF2/MAPK or retinal damage, and activation of canonical Wnt-signaling is required for the formation of proliferating MGPCs.
Keywords: retina, Müller glia, progenitor, proliferation, Wnt, β-catenin
INTRODUCTION
Müller glia have the potential to become progenitor cells in the retinas of fish, birds and mammals (reviewed by Fischer and Bongini, 2010; Gallina et al., 2014a; Goldman, 2014; Lenkowski and Raymond, 2014). Different signaling pathways have been shown to influence the formation of Müller glia-derived progenitor cells (MGPCs) and neuronal regeneration in the retina. In different model systems similar cell-signaling pathways influence the formation of MGPCs. For example, EGF, HB-EGF, FGF2 and IGF1 signal through the MAPK pathway to stimulate Müller glia to de-differentiate, proliferate and become MGPCs in fish (Wan et al., 2012; Wan et al., 2014), birds (Fischer et al., 2009a; Fischer et al., 2009b) and mammals (Karl et al., 2008). By comparison, Notch-signaling in retinas of mammals and chick stimulates the proliferation of MGPCs (Das et al., 2006; Ghai et al., 2010; Hayes et al., 2007), whereas Notch-signaling in fish inhibits the proliferation of MGPCs (Wan et al., 2012). In rodent and chick retinas, CNTF/Jak/Stat-signaling may stimulate glial reactivity, instead of stimulating the formation of proliferating MGPCs as in the zebrafish model system (reviewed by Gallina et al., 2014a). Recently, we have shown that Glucocortiocoid Receptor (GCR) -signaling inhibits the formation of proliferating MGPCs and inhibits the neuronal differentiation of MGPC-progeny by interfering with MAPK signaling (Gallina et al., 2014b).
Wnt/β-catenin-signaling may be one of the key pathways that drive MGPCs to regenerate retina in the fish. During development of the nervous system, Wnt-signaling is involved in many different important functions including cell proliferation, migration, cell fate specification, axis patterning, axon guidance and dendrite and synapse formation (reviewed by Ciani and Salinas, 2005; Moon et al., 2004). In the retina, Müller glia are known to express high levels of Dkk’s which are secreted to sequester Wnt-ligands and intracellular components of the Wnt-signaling (Roesch et al., 2008; Roesch et al., 2012). In the rodent model in vitro, Wnt-signaling stimulates the proliferation of MGPCs (Das et al., 2006; Osakada et al., 2007). However, in vivo studies have shown a modest albeit significant increase in proliferating MGPCs in damaged mouse retina with activation of the Wnt-pathway (Das et al., 2006). Further, in axin2−/− mice with elevated Wnt-signaling, the retina contains a subpopulation of Müller glia that are Wnt-responsive and proliferate at elevated levels in response to damage (Liu et al., 2012). In normal and damaged zebrafish retinas, activation of the Wnt-signaling stimulates the formation of the MGPCs and neuronal regeneration (Meyers et al., 2012; Ramachandran et al., 2011). During retinal development in chick, Wnt2b maintains the proliferative and undifferentiated state of neural stem cells in peripheral regions of the retina (Kubo et al., 2005). Further, Wnt2b/β-catenin-signaling suppresses neurogenesis in peripheral regions of the developing optic cup to promote specification of ciliary body and iris in the chick eye (Cho and Cepko, 2006). During embryonic retinal development in the chick, β-catenin must be down-regulated to permit retinal regeneration from prospective ciliary marginal tissues at the peripheral edge of the developing neural retina (Zhu et al., 2014). In this study we investigate whether Wnt/β-catenin-signaling influences Müller glia and the formation of MGPCs in normal and damaged chick retina in vivo.
METHODS
Animals
The use of animals in these experiments was in accordance with the guidelines established by the National Institutes of Health and the Ohio State University. Fertilized eggs and newly hatched wild type leghorn chickens (Gallus gallus domesticus) were obtained from Meyer Hatchery (Polk, Ohio). The stage of the chick embryos was determined according to the guidelines established by Hamburger and Hamilton in 1951 (Hamburger and Hamilton, 1992). Postnatal chicks were kept on a cycle of 12 hours light, 12 hours dark (lights on at 8:00 AM). Chicks were housed in a stainless steel brooder at about 25°C and received water and Purina™ chick starter ad libitum.
Intraocular injections
Chickens were anesthetized via inhalation of 2.5% isoflurane in oxygen and intraocular injections performed as described previously (Fischer et al., 1998; Fischer et al., 1999; Fischer et al., 1999). Injection paradigms were started on P6 or P7; there is no appreciable difference in experimental outcomes if injections paradigms begin at P6 through P28 (Fischer et al., 2014a; Todd and Fischer 2015). For all experiments, the right eyes of chicks were injected with the “test” compound and the contra-lateral left eyes were injected with vehicle as a control. Compounds were injected in 20 μl sterile saline with 0.05 mg/ml bovine serum albumin added as a carrier. Compounds used in these studies included NMDA (38.5 or 154 μg/dose), FGF2 (200 ng/dose), IGF1 (400 ng/dose; R&D Systems), smoothened agonist (SAG; 500 ng/dose; EMD Millipore; CAS 364590-63-6), XAV939 (200ng/dose; Selleck), Wnt-c59 (200ng or 1000ng/dose; Selleck), a cocktail of 3 different GSK3β inhibitors (ABC) which included 1-Azakenpaullone (500 ng/dose; Selleck Chemicals), BIO (500ng/dose; R&D Systems), CHIR 99021 (500ng/dose; R&D Systems). Assuming a volume of liquid vitreous of 200 μl, the estimated initial maximum concentrations of factors would be; FGF2 – 46 nM, IGF1 – 176 nM, SAG – 4.2 μM, XAV939 – 3.2 μM, Wnt-c59 – 3.5 μM or 17.3 μM, 1-Azakenpaullone 7.6 μM, BIO – 7.0 μM and CHIR 99021 – 5.4 μM. XAV939 and the GSK3β cocktail were used in 25% DMSO. Two μg of BrdU or 1 μg EdU were injected to label proliferating cells. Injection paradigms are included in each figure.
Quantitative Reverse transcriptase PCR
Individual retinas were placed in 1.5 ml of Trizol Reagent (Invitrogen) and total RNA was isolated according to the Trizol protocol and resuspended in 50 μl RNAse free water. Genomic DNA was removed by using the DNA FREE kit provided by Ambion. cDNA was synthesized from mRNA by using Superscript™ III First Strand Synthesis System (Invitrogen) and oligo dT primers according to the manufacturer’s protocol. Control reactions were performed using all components with the exception of the reverse transcriptase to exclude the possibility that primers were amplifying genomic DNA.
PCR primers were designed by using the Primer-BLAST primer design tool at NCBI (http://www.ncbi.nlm.nih.gov/tools/primer-blast/). Primer sequences and predicted product sizes are listed in table 1. PCR reactions were performed by using standard protocols, Platinum™ Taq (Invitrogen) and an Eppendorf thermal cycler. PCR products were run on an agarose gel to verify the predicted product sizes.
Table 1.
PCR Primer sequences and predicted product sizes
| Gene | Forward primer 5′-3′ | Reverse primer 5′-3′ | Product size (bp) |
|---|---|---|---|
| tcf1 | CGA CCT CAAG TCC TCG TTG G | TCC GGG AGC TTC TCC TGA TA | 123 |
| cd44 | ACG AGG AGC AAA GCA TGT GA | TTG TAA CGT GAG CCG TCC TC | 101 |
| dkk3 | CCA GCT TTG TGT TTG GGG TG | GTA ACG GAG TGC ACA CAG GA | 143 |
| dkk1 | GCG ACT GAT TGC AGT ACG TT | CTG GAA ACT CAG CGC GTA CC | 127 |
| cMyc | ACA CAA CTA CGC TGC TCC TC | TTC GCC TCT TGT CGT TCT CC | 155 |
| axin2 | GCTTCAGGCAGATGGACCTT | CTTGAGCTGCTTGGAGACGA | 104 |
| cNotch1 | GGC TGG TTA TCA TGG AGT TA | CAT CCA CAT TGA TCT CAC AG | 154 |
| cHes5 | GGA GAA GGA GTT CCA GAG AC | AAT TGC AGA GCT TCT TTG AG | 143 |
| ascl1a | AGG GAA CCA CGT TTA TGC AG | TTA TAC AGG GCC TGG TGA GC | 187 |
| gapdh | CAT CCA AGG AGT GAG CCA AG | TGG AGG AAG AAA TTG GAG GA | 161 |
Fixation, sectioning and immunocytochemistry
Tissues were fixed, sectioned and immunolabeled as described previously (Fischer et al., 2008; Fischer et al., 2009b). Working dilutions and sources of antibodies used in this study included the following: (1) mouse anti-nuclear β-catenin was used at 1:40 (PY654; Developmental Studies Hybridoma Bank (DSHB), University of Iowa): (2) rabbit anti-Sox9 was used at 1:2000 (AB5535; Chemicon); (3) mouse anti-BrdU was used at 1:80 (G3B4; DSHB); (4) rat anti-BrdU was used at 1:200 (OBT00030S; Serrotec); (5) mouse anti-Nkx2.2 was used at 1:50 (74.5A5; DSHB); (5) mouse anti-neurofilament was used at 1:80 (RT-97; DSHB); (6) goat anti-Sox2 was used at 1:1000 (Y-17; Santa Cruz Immunochemicals); (7) rabbit anti-phospho-histone H3 (pHisH3) was used at 1:600 (06570; Upstate); (8) mouse anti-Pax6 was used at 1:50 (PAX6; DSHB); (9) mouse anti-CD45 was used at 1:200 (HIS-C7; Cedi Diagnostic); (10)rabbit anti-p38 MAPK was used at 1:400 (12F8; Cell Signaling Technologies); (11) goat anti-Egr1 was used at 1:1,000 (AF2818; R&D Systems); (12) rabbit pERK1/2 was used at 1:200 (137F5; Cell Signaling Technologies); (13) rabbit anti-pCREB was used at 1:600 (87G3; Cell Signaling Technologies); (14) rabbit anti-cFos was used at 1:400 (K-25; Santa Cruz Immunochemicals).
None of the observed labeling was due to non-specific labeling of secondary antibodies or autofluorescence because sections labeled with secondary antibodies alone were devoid of fluorescence. Secondary antibodies included donkey-anti-goat-Alexa488/568, goat-anti-rabbit-Alexa488/568/647, goat-anti-mouse-Alexa488/568/647, goat anti-rat-Alexa488 and goat-anti-mouse-IgM-Alexa568 (Invitrogen) diluted to 1:1000 in PBS plus 0.2% Triton X-100. Some sections were stained with Draq5 (Life Technologies) which was diluted to 2.5 nM in the secondary antibody solution.
Labeling for EdU
Immunolabeled Sections were fixed in 4% formaldehyde in PBS for 5 minutes at room temperature, washed for 5 minutes with PBS, permeabilized with 0.5% Triton X-100 in PBS for 1 minute at room temperature, and washed twice for 5 minutes in PBS. Sections were incubated for 30 minutes at room temperature in 2M Tris, 50 mM CuSO4, Alexa Fluor 568 Azide (Thermo Fisher Scientific), and 0.5M ascorbic acid in dH2O. Sections were washed with PBS for 5 minutes and further processed for immunofluorescence as required.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)
To identify dying cells that contained fragmented DNA the TUNEL method was used. We used an In Situ Cell Death Kit (TMR red; Roche Applied Science), as per the manufacturer’s instructions.
Photography, measurements, cell counts and statistics
Photomicrographs were obtained using a Leica DM5000B microscope equipped with epifluorescence and Leica DC500 digital camera. Confocal images were obtained using a Zeiss LSM 510 imaging system at the Hunt-Curtis Imaging Facility at the Ohio State University. Images were optimized for color, brightness and contrast, multiple channels overlaid and figures constructed by using Adobe Photoshop. Cell counts were performed on representative images. To avoid the possibility of region-specific differences within the retina, cell counts were consistently made from the same region of retina for each data set.
The identity of BrdU-labeled cells was determined based on previous findings that 100% of the proliferating cells in the chick retina are comprised of Sox2/9+ Müller glia in the INL/ONL, Sox2/9/Nkx2.2+ NIRG cells in the IPL, GCL and NFL (the NIRG cells do not migrate distally into the retina), and CD45+ (Sox2−/9−) microglia (Fischer et al., 2010; Zelinka et al., 2012). Sox2+ nuclei in the INL were identified as Müller glia based on their large size and fusiform shape which was distinctly different from the Sox2+ nuclei of cholinergic amacrine cells which are small and round (Fischer et al., 2010).
Similar to previous reports (Fischer et al., 2009a; Fischer et al., 2009b; Fischer et al., 2010; Ghai et al., 2009), immunofluorescence was quantified by using ImagePro 6.2 (Media Cybernetics, Bethesda, MD, USA). Identical illumination, microscope, and camera settings were used to obtain images for quantification. Retinal areas were sampled from 5.4 MP digital images. Areas were sampled over the inner nuclear layer (INL) and outer nuclear layer (ONL) of treated and control tissues from one individual which were embedded and cut from one block, and placed consecutively on glass slides to ensure equal exposure to reagents. Measurement for content in the nuclei of Müller glia/MGPCs were made by selecting the total area of pixel values ≥70 for Sox2 or Sox9 immunofluorescence (in the red channel), and copying nuclear β-catenin or Pax6 (in the green channel). This copied data was pasted into a separate file for quantification or onto 70% grayscale background for figures. Measurements were made for pixels with intensity values of ≥70 (0 = black and 255 = saturated). The total area was calculated for clusters (≥50) of pixels with intensities > 70. The density sum was calculated as the sum of pixel values in areas above threshold for each field of view. The average pixel intensity was calculated for all pixels within threshold regions. These calculations were determined for retinal regions sampled from at least six different retinas for each experimental condition.
Where significance of difference was determined between two treatment groups accounting for inter-individual variability (means of treated-control values) we performed a two-tailed, paired t-test. Where significance of difference was determined between two treatment groups we performed a two-tailed, unpaired t-test.
RESULTS
Wnt-signaling in NMDA-damaged retinas
To determine whether Wnt-signaling might be involved in the formation of MGPCs in the chick retina, we used qRT-PCR to probe for changes in gene expression in NMDA-damaged retinas where MGPCs are known to form (Fischer and Reh, 2001). We probed for expression of dkk1 and dkk3, secreted proteins which are known to sequester Wnt-ligands, and other potential read-outs of Wnt-signaling including c-myc and cd44 (reviewed by Clevers and Nusse, 2012) and axin2 (Lustig et al., 2002). We found that levels of dkk1 were reduced at 1 day after NMDA-treatment, but were not significantly different at 4hrs, 2 days and 3 days after treatment (Fig. 1a). By comparison, dkk3 was decreased only at 3 days after treatment (Fig. 1b). Levels of axin2 were significantly reduced by 1 day after treatment and remained reduced through 3 days after treatment (Fig. 1c). Retinal levels of tcf1 were significantly decreased at 2 days after treatment and returned to control levels by 3 days after treatment (Fig. 1d). Retinal levels of c-myc and cd44 were elevated at 4 hrs after NMDA-treatment and remained elevated through day 3 (Figs. 1e,f). These findings suggest that Wnt-signaling is dynamically regulated following NMDA-treatment, and this pathway could be involved in the formation of MPGCs in the chick retina.
Figure 1.
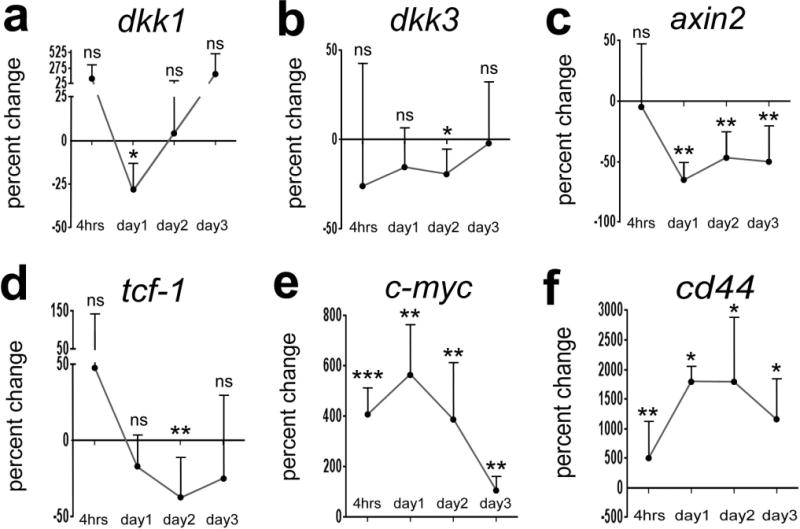
Components of the Wnt-pathway are dynamically regulated in NMDA-damaged retinas. qRT-PCR was used to detect retinal mRNA for dkk1 (a), dkk3 (b), axin2 (c), c-myc (d), PparD (e), tcf-1 (f) and cd44 (f). cDNA was generated from retinas (n≥4) that were treated with saline (control) or NMDA (treated), and harvested at 4hrs, 1 day, 2 days, 3 days or 5 days later. Significance of difference (*p<0.05) was determined by using a Mann-Whitney U test.
To determine whether Wnt-signaling is active in Müller glia and/or MGPCs in NMDA damaged retinas we probed for nuclear β-catenin as a cell-level read-out of signaling. β-catenin is the key mediator of the canonical Wnt-signaling. Nuclear β-catenin binds to TCF/Lef1 to induce transcriptional activity, which is a hallmark of Wnt/β-catenin signaling activation (Behrens et al., 1996; Rhee et al., 2007). We found that the Sox9+ nuclei of Müller glia in normal retinas contained no nuclear β-catenin (Fig. 2a). By contrast, we observed significant levels of nuclear β-catenin in INL neurons; amacrine cells in the inner INL and bipolar cells in the outer INL (Fig. 2a). This observation agrees with the findings that Wnt-signaling is active in mature neurons and is needed for synapse function and maintenance (reviewed by Inestrosa and Arenas, 2010). At 4hrs after NMDA-treatment we observed some accumulation of nuclear β-catenin in Müller glia, and the levels of nuclear β-catenin appeared to increase in these cells from 1 to 2 days after NMDA-treatment (Fig. 2a). Levels of nuclear β-catenin in Müller glia/MGPCs appeared maximal at 3 days after treatment, but were reduced by 5 days after treatment (Fig. 2a). Conversely, levels of nuclear β-catenin in neurons in the ONL and INL were decreased at 1, 2 and 3 days after treatment (Fig. 2a). We determined the percentage of Sox9-positive cells in the INL and ONL that were positive for nuclear β-catenin. Consistent with the qualitative studies, we found that the percentage of β-catenin+/Sox9+ rapidly increased after NMDA-treatment, was maximal at 2–3 days after treatment, and rapidly decreased by 5 days treatment (Fig. 2b).
Figure 2.
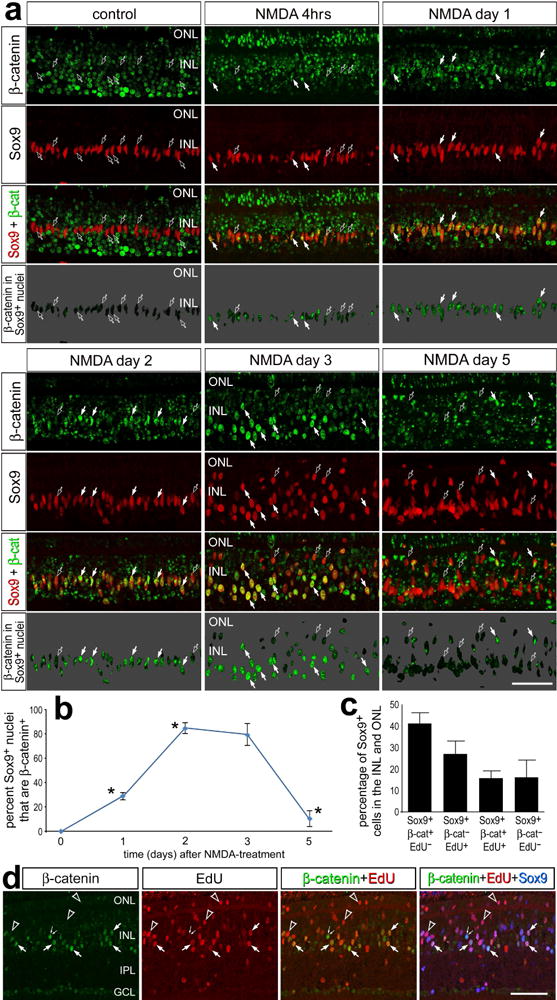
Nuclear β-catenin accumulates in MGPCs following NMDA-treatment. (a) Sections of the retina were labeled with antibodies to nuclear β-catenin (green) and Sox9 (red). Arrows indicate nuclei of Müller glia/MGPCs that are positive for Sox9 and nuclear β-catenin, and hollow arrows indicate nuclei of Müller glia/MGPCs that are positive for Sox9 and negative for nuclear β-catenin. The panels with a 70% grayscale background display β-catenin isolated in the Sox9+ nuclei of Müller glia. The plot in b illustrates the percentage of the Sox9+ nuclei in the INL and ONL that are β-catenin+ in untreated retinas and in retinas treated with NMDA at 1, 2, 3 and 5 days after treatment. Two-way ANOVA was to determine significance of difference (*p<0.01) between consecutive time points. The histogram in c illustrates the mean (±SD; n=5) percentages of Sox9+ Müller glia/MGPCs in the INL and ONL that are positive for EdU, β-catenin, or EdU and β-catenin in NMDA-damaged retinas at 3 days after treatment. (d) Sections of the retina were labeled for EdU (red) and antibodies to nuclear β-catenin (green) and Sox9 (blue). Hollow arrow-head indicate Sox9+ nuclei labeled for EdU, but not β-catenin; small double arrows indicate Sox9+ nuclei labeled for β-catenin, but not EdU; and arrows indicate Sox9+ nuclei labeled for β-catenin and EdU. The scale bar (50 μm) in the bottom right corner of panel a applies to a alone, and the bar in d applies to d alone. Abbreviations: ONL – outer nuclear layer, INL – inner nuclear layer.
We performed triple-labeling for nuclear β-catenin, Sox9 and EdU to examine whether there was a correlation between proliferation and Wnt/β-catenin-signaling in MGPCs at 3 days after NMDA-treatment. We have found that about 40% of Sox9+ nuclei were positive for β-catenin, but negative for EdU (Figs. 2c,d). By comparison, about 25% of the Sox9+ nuclei were positive for EdU, but negative for β-catenin, and only about 15% Sox9+ nuclei were positive for β-catenin and EdU (Figs. 2c,d). About 15% of the Sox9+ nuclei in the INL and ONL were negative for both EdU and β-catenin (Figs. 2c,d). Collectively, these data indicate that the accumulation of nuclear β-catenin is not sufficient to drive the proliferation of MGPCs and that some of the proliferation of MGPCs may occur independent of Wnt/β-catenin-signaling.
Wnt-signaling influences the formation of MGPCs in damaged retinas
To examine whether Wnt-signaling influences the formation of proliferating MGPCs, we interfered with signaling by making intraocular injections of the small molecule inhibitor XAV939. XAV939 antagonizes the Wnt/β-catenin pathway by inhibiting tankyrase 1/2 which inhibits participation of axin2 in the β-catenin destruction complex, thus promoting axin2 stabilization, degradation β-catenin, and inhibition of Wnt-signaling (Huang et al., 2009). Alternatively, to prevent processing and release of Wnt-ligands, we applied a small molecule inhibitor (Wnt-c59) to Porcupine; an enzyme required for the processing and secretion of Wnt-ligands (Kadowaki et al., 1996). In normal retinas, XAV939 reduced levels of nuclear β-catenin in retinal neurons across all cell layers (Figs. 3a,b). Similarly, Wnt-c59 reduced levels of nuclear β-catenin in retinal neurons (data not shown).
Figure 3.
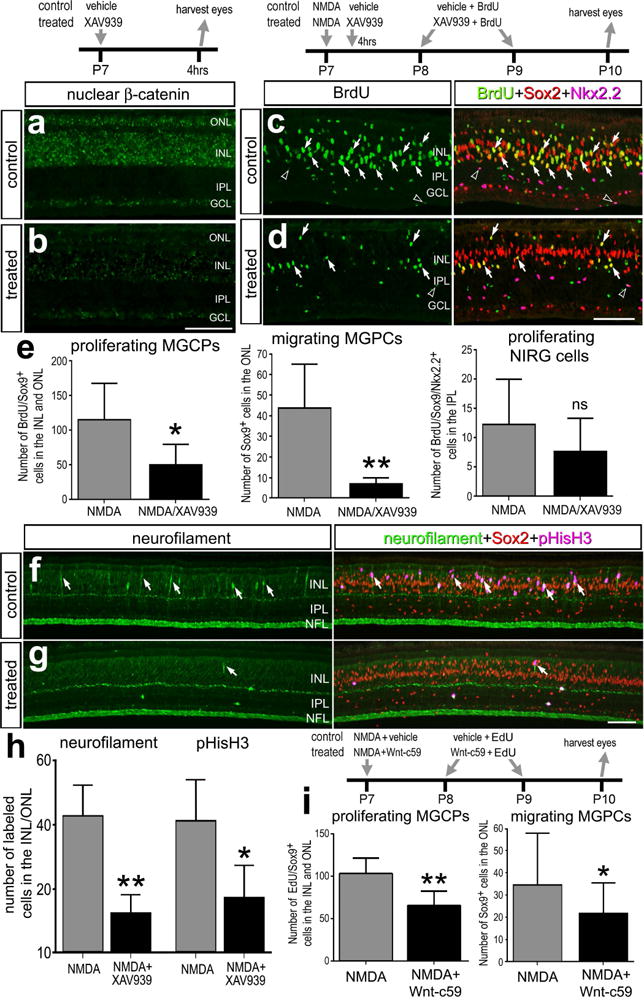
Inhibition of Wnt-signaling with XAV939 or Wnt-c59 reduces numbers of proliferating MGPCs in damaged retinas. Eyes were injected with NMDA at P7, vehicle or XAV939 4hrs later, BrdU + vehicle or XAV939 at P8 and P9, and eyes harvested at P10. Retinal sections were labeled with antibodies to nuclear β-catenin (a and b), BrdU (green), Sox2 (red) and Nkx2.2 (magenta; c and d), neurofilament (green), and Sox2 (red) and pHisH3 (magenta; f and g). Arrows indicate Müller glia and/or MGPCs. The histograms in e illustrate the mean (± SD; n=8) number of BrdU-positive Müller glia, Sox9-positive nuclei in the ONL, or BrdU-positive NIRG cells per field of view (14,400 μm2) in retinas treated with NMDA or NMDA+XAV939. The histogram in h illustrates the mean (± SD; n=8) number of MGPCs labeled for neurofilament or pHisH3 in retinas treated with NMDA or NMDA+XAV939. i; Eyes were injected with NMDA and vehicle or Wnt-c59 at P7, EdU + vehicle or EdU + Wnt-c59 at P8 and P9, and eyes harvested at P10. The histograms in i the mean (± SD; n=8) number of EdU-positive Müller glia and Sox9-positive nuclei in the ONL. Significance of difference (*p<0.05; **P<0.01) between control and treated groups was determined by using a two-tailed, paired t test. The scale bar (50 μm) in panel b applies to a and b, the bar d applies to c and d, and the bar in g applies to f and g. Abbreviations: ONL – outer nuclear layer, INL – inner nuclear layer, IPL – inner plexiform layer, GCL – ganglion cell layer.
To determine whether the effects of Wnt/β-catenin-signaling are specific to MGPCs or other types of retinal glia, we determined whether agonists and antagonists to Wnt/β-catenin influenced the proliferation of NIRG cells. NIRG (non-astrocytic inner retinal glial) cells are a unique type of retinal glia (Fischer et al., 2010) that are derived from optic nerve progenitors and migrate into retina during late stages of embryonic development (Rompani and Cepko, 2010), and when microglia and NIRG cells are ablated the formation of MGPCs is inhibited (Fischer et al., 2014a). Application of XAV939 after NMDA-treatment significantly reduced the numbers of BrdU-labeled MGPCs, and significantly reduced numbers of Sox9+ nuclei (Müller glia and/or MGPCs) that migrated into the ONL, whereas numbers of BrdU-labeled NIRG cells were unaffected (Figs. 3c–e). To further examine the formation of MGPCs, we probed for the expression of neurofilament and phospho-histone H3 (pHisH3) which are known to be transiently expressed by proliferating MGPCs (Fischer and Reh, 2001). Treatment of NMDA-damaged retinas with XAV939 significantly reduced numbers of MGPCs that were positive for neurofilament and pHisH3 (Figs. 3f–h).
Similar to the effects of XAV939, we found that Wnt-c59 inhibited the formation of MGPCs in NMDA-damaged retinas (Fig. 3i), whereas the proliferation of NIRG cells was unaffected (not shown). Although lower doses (200ng) of Wnt-c59 had no effect upon the proliferation of MGPCs (not shown), higher doses (1 μg) significantly reduced numbers of EdU+/Sox9+ MGPCs in the INL and ONL compared to numbers seen in control retinas (Fig. 3i). Similar to XAV939, Wnt-c59 suppressed the nuclear migration of Müller glia/MGPCs; we found significantly fewer Sox9+ nuclei in the ONL of NMDA-damaged retinas treated with Wnt-c59 compared to numbers of Sox9+ nuclei in the ONL of control retinas (Fig. 3i).
To examine whether inhibition of Wnt-signaling influences the de-differentiation of Müller glia we probed for the expression of Pax6. Müller glia are known to up-regulate Pax6 during the transition to a progenitor-like phenotype (Fischer et al., 2002b; Fischer and Reh, 2001) and Pax6 is known to be required for MGPC-mediated regeneration of zebrafish retina (Thummel et al., 2010). We found reduced levels of Pax6 in Müller glia in retinas treated with XAV939 compared to levels of Pax6 seen in Müller glia in retinas treated with NMDA and vehicle (Figs. 4a,b). This effect was highly significant when the density sum was divided by total number of Sox9+ nuclei per field of view to account for XAV939-mediated decreases in proliferation (Fig. 4a). To examine whether XAV939 effectively inhibited Wnt-signaling in NMDA-damaged retinas we probed for the accumulation of nuclear β-catenin. We found that XAV939 significantly reduced levels of nuclear β-catenin, particularly in the Müller glia/MGPCs and when accounting for XAV939-mediated decreases in proliferation (Figs. 4c,d). We have recently shown that reactive microglia are required for the formation of proliferating MGPCs (Fischer et al., 2014b). Thus, we examined whether microglial reactivity was influenced by XAV939. There was no change in the accumulation of reactive CD45+ microglia in NMDA-damaged retinas treated with XAV939 (Fig. 4e).
Figure 4.
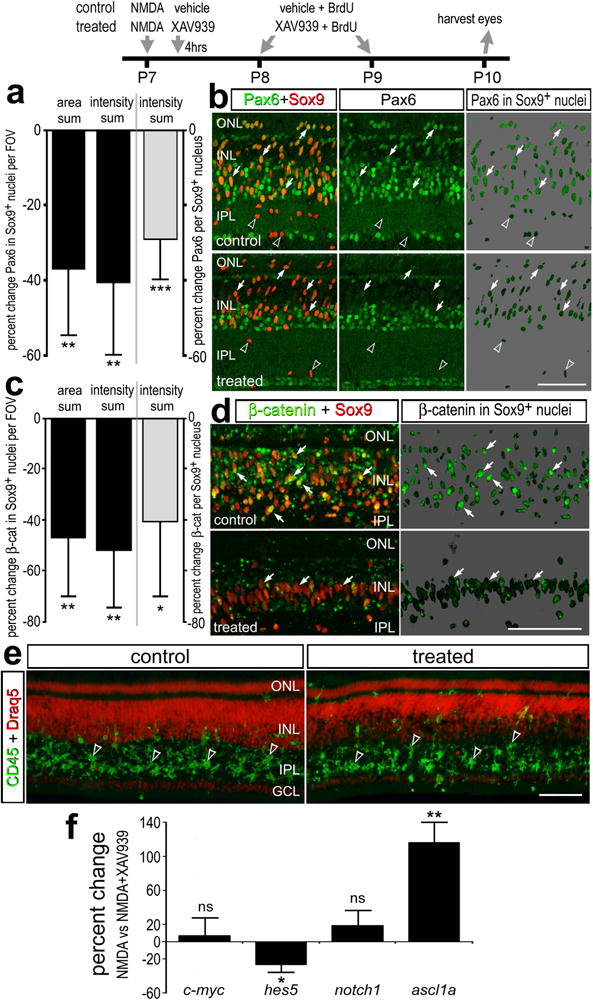
Changes in Pax6, nuclear β-catenin and gene expression in retinas treated with a Wnt-signaling inhibitor in NMDA-damaged retinas. Eyes were injected with NMDA at P7, vehicle or XAV939 4hrs later, BrdU + vehicle or XAV939 at P8 and P9, and eyes harvested at P10. The histograms in a and c illustrate the mean (± SD; n=6) percent change in area sum, intensity sum per field of view, or intensity sum divided by the total number of Sox9+ nuclei per field view for levels of Pax6 (a) or nuclear β-catenin (c) in the Sox9+ nuclei of Müller glia/MGPCs. Significance of difference (*p<0.05, **p<0.01, ***p<0.001) was determined by using a Mann-Whitney U test. Retinal sections were labeled with antibodies to Pax6 (green) and Sox9 (red; b), nuclear β-catenin (green) and Sox9 (red; d), or Draq5 (red) and CD45 (green; e). b,d; The panels with the 70% grayscale background show β-catenin in the nuclei of Sox9+ Müller glia/MGPCs. Arrows indicate Müller glia and/or MGPCs and arrow-heads indicate CD45-positive microglia in the IPL. The scale bar (50 μm) in panel d applies to b and d, and the bar in e applies to e alone. Abbreviations: ONL – outer nuclear layer, INL – inner nuclear layer, IPL – inner plexiform layer, GCL – ganglion cell layer, FOV – field of view. f; qRT-PCR was used to measure genes that are changed in response to Wnt-inhibitor in damaged-retinas. These genes included c-myc, hes5, notch1, and ascl1a. Significance of difference (*p<0.05; **p<0.001) was determined by using a Mann-Whitney U test. ns – not significant.
We performed qRT-PCR on retinas treated with NMDA ± XAV939 to better understand how Wnt-signaling might influence the formation of MGPCs. We probed for changes in the expression of c-myc, hes5, notch1 and ascl1a; genes known to be expressed by MGPCs and genes that could be regulated by Wnt/β-catenin (reviewed by Gallina et al., 2014; Goldman, 2014). There was no significant change in levels of c-myc or notch1, whereas levels of hes5 were significantly reduced and levels of ascl1a were significantly increased (Fig. 4f). These findings suggest that neurogenesis may be increased while gliogenesis is decreased, even though the total numbers of MGPCs are reduced by XAV939. In support of this hypothesis, decreased Wnt-signaling and decreased nuclear β-catenin are known to be required for retinal neurogenesis during development (Cho and Cepko, 2006; Liu et al., 2007; Zhu et al., 2014). Further, increased ascl1a is needed for the formation of MGPCs in the zebrafish retina (Fausett et al., 2008) and forced-expression of ascl1a in mammalian Müller glia enhances neurogenic potential (Pollak et al., 2013). Therefore, we tested whether there was increased neural differentiation of the progeny produced by MGPCs. When XAV939 was applied at 1 and 2 days after NMDA, and retinas were harvested at 5 days later, there was no effect upon the percentage of cells that differentiated as neurons or glia (not shown). When XAV939 was applied at 3, 4 and 5 days after NMDA-treatment, after the proliferation of MGPCs, there was no effect upon the percentage of cells that differentiated as neurons or glia (not shown).
To test whether activation of Wnt-signaling facilitates the formation of MGPCs in damaged retinas, we applied a cocktail (ABC) of GSK3β-inhibitors. These inhibitors are expected to suppress GSK3β-mediated phosphorylation of β-catenin, thereby inhibiting degradation and promoting nuclear translocation of β-catenin. We found that application of ABC following a low dose of NMDA resulted in a 4-fold increase in the number of BrdU-labeled MGPCs compared to controls (Figs. 5a,b). In response to treatment with GSK3β-inhibitors, the percentage of Sox9+ Müller glia/MGPCs was increased more than 4-fold, from 5% to more than 20% (Fig. 5c). By comparison, inhibition of GSK3β had no effect upon numbers of proliferating NIRG cells (Figs. 5a,d). We found significant increases in levels of nuclear β-catenin in the Sox9+ nuclei of MGPCs in retinas treated with GSK3β-inhibitors (Figs. 5e,f), indicating that Wnt-signaling was elevated during the reprogramming of Müller glia. In retinas treated with GSK3β-inhibitors, the percentage of Sox9+ Müller glia/MGPCs that were positive for nuclear β-catenin was increased nearly 3-fold, from 30% to more than 85% (Fig. 5f). We found that GSK3β-inhibitors had no effect upon numbers of dying cells; there was no significant change in the number of TUNEL-positive cells in retinas treated with NMDA compared to NMDA/ABC (NMDA – 15.8±10.9 vs NMDA/ABC – 17.0±11.7). In addition, activation of Wnt-signaling with GSK3β-inhibitors did not influence the neuronal and glial differentiation of the progeny of MGPCs (data not shown).
Figure 5.
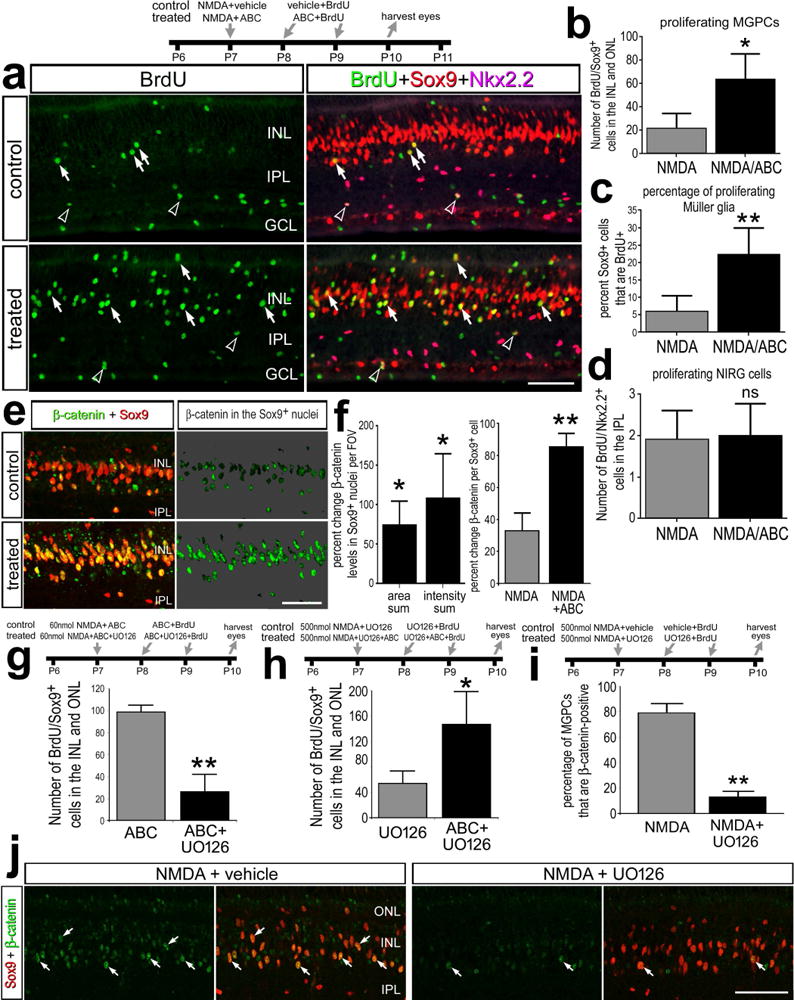
Activation of Wnt-signaling with GSK3β-inhibitors increases numbers of proliferating MGPCs and stimulates the accumulation of nuclear β-catenin in MGPCs in NMDA-damaged retinas. Eyes were injected with a relatively low dose (60 nmol) or high dose (500 nmol) of NMDA ± GSK3β-inhibitors ± MEK-inhibitor (UO126) at P7, GSK3β-inhibitors ± MEK inhibitor and BrdU at P8 and P9, and retinas were harvest at 3 days after NMDA-treatment at P10. Sections of the retina were labeled with antibodies to BrdU (green), Sox9 (red) and Nkx2.2 (magenta; a), or nuclear β-catenin (green) and Sox9 (red; e and j). The histograms in b, g and h illustrate the mean (± SD; n=9) number of proliferating Müller glia/MPGCs per field of view (14,400 μm2). The histogram in c illustrates the mean (± SD; n=6) percentage of Sox9-positive nuclei in the INL and ONL that are BrdU-positive. The histograms in d illustrates the mean (± SD; n=9) number of proliferating NIRG cells per field of view (14,400 μm2). e; β-catenin in the nuclei of Sox9+ Müller glia/MGPCs on a 70% grayscale background. Arrows indicate the nuclei of Müller glia and or MGPCs, and hollow arrow-heads indicate nuclei of proliferating NIRG cells. Histograms f illustrates the mean (± SD; n=6) percent change in area sum and intensity sum per field of view for levels of nuclear β-catenin in the Sox9+ nuclei of Müller glia/MGPCs, and (f and i) the mean percentage of Sox9+ nuclei that are β-catenin+. Significance of difference (*p<0.05, **p<0.01) between control and treated groups was determined by using a paired, two-tailed t test. The scale bars in a, e and j represent 50 μm. Abbreviations: ONL – outer nuclear layer, INL – inner nuclear layer, IPL – inner plexiform layer, GCL – ganglion cell layer.
To investigate whether there is cross-talk between MAPK- and Wnt/β-catenin-signaling in damaged retinas, we applied the MEK inhibitor UO126, which is known to inhibit the formation of proliferating MGPCs in NMDA-damaged chick retinas (Fischer et al., 2009b). In retinas damaged by a low dose of NMDA and treated with GSK3β-inhibitors, UO126 suppressed the proliferation of MGPCs (Fig. 5g). UO126 has been shown to suppress the proliferation of MGPCs in NMDA-damaged retinas (Fig. 5h; Fischer et al., 2009b). In retinas damaged by a high dose of NMDA and treated with UO126, GSK3β-inhibitors stimulated the proliferation of MGPCs (Fig. 5h). In retinas damaged by a high dose of NMDA, UO126 significantly reduced the percentage of Sox9+ Müller glia/MGPCs that are positive for nuclear β-catenin (Figs. 5i,j). Collectively, these findings suggest that Wnt/β-catenin-signaling is activated downstream of MAPK-signaling in damaged retinas, and activation of β-catenin-signaling can compensate for inhibition of MEK to facilitate the formation of proliferating MGPCs.
FGF2/MAPK-signaling activates Wnt-signaling in undamaged retinas
We examined whether activation of Wnt-signaling acted synergistically with FGF2/MAPK to enhance the formation of proliferating MGPCs. We have recently reported that 4 consecutive daily intraocular injections of FGF2 stimulate the formation of proliferating MGPCs in the absence of retinal damage (Fischer et al., 2014a). Thus, we tested whether activation of Wnt-signaling is down-stream of FGF2/MAPK in undamaged retinas. Two days after the last dose of FGF2, we found that levels of β-catenin were increased in the nuclei of Müller glia in FGF2-treated retinas compared to those seen in control retinas (Figs. 6a,b). These findings indicate that Wnt-signaling is downstream of MAPK-signaling, and becomes activated by FGF2/MAPK-signaling in Müller glia/MGPCs in the absence of retinal damage. We next examined whether inhibition of Wnt-signaling influences the formation of MGPCs in FGF2-treated retinas. We found that co-application of XAV939 with FGF2 suppressed the proliferation of MGPCs and migration of Sox9+ nuclei into the ONL (Figs. 6c–f), whereas the proliferation of NIRG cells was unaffected (not shown). To test whether activation of Wnt/β-catenin-signaling acts synergistically with FGF2-treatment, we tested whether 3 consecutive daily intraocular injections of FGF2 combined with GSK3 β-inhibitors to stimulate the formation of MGPCs. Three consecutive daily intraocular injections of FGF2 are expected to “prime” the Müller glia for reprogramming, without driving the proliferation of large numbers of MGPCs (Fischer et al., 2009a; Fischer et al., 2014b). We found that the formation of proliferating MGPCs was not affected by FGF2 and GSK3β-inhibitors (Figs. 6g–i).
Figure 6.
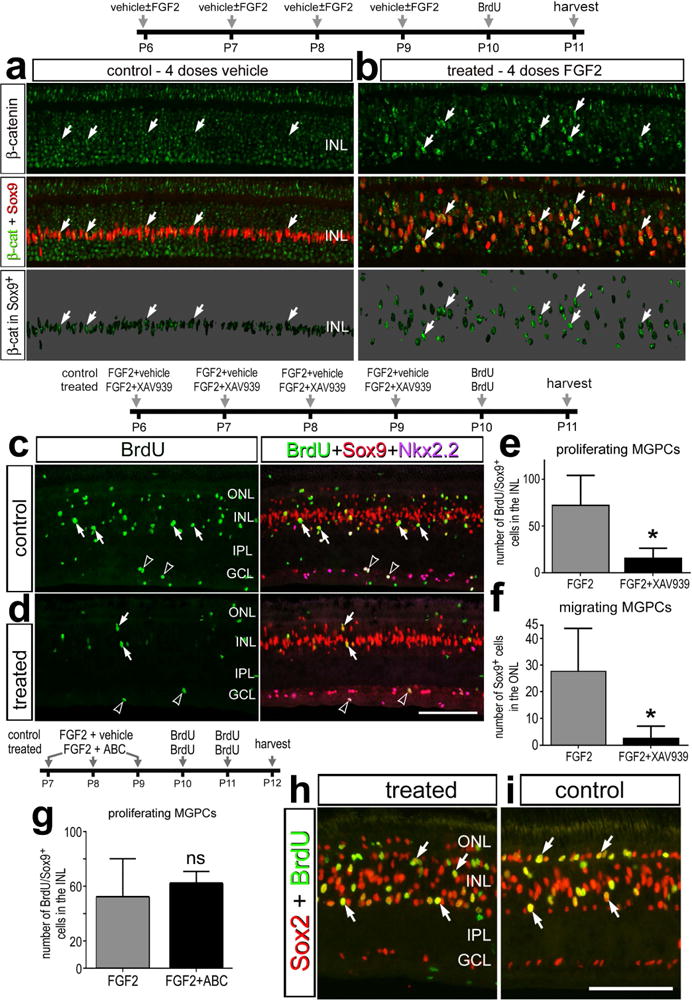
FGF2-mediated formation of proliferating MGPCs recruits and requires the activation of Wnt-signaling. Eyes were injected with 4 consecutive daily injections of FGF2 ± XAV939, BrdU 24 hrs after the last injection of FGF2, and retinas harvested 48 hrs (a–f) or 24 hrs later. Alternatively, eyes were injected with FGF2 alone (control) or FGF2+GSK3β-inhibitors (treated) at P7, P8 and P9, BrdU at P10 and P11, and retinas harvested at P12 (g–i). Retinal sections were labeled with antibodies to nuclear β-catenin (green) and Sox9 (red; a,b), BrdU (green), Sox9 (red), Nkx2.2 (magenta; c,d), or BrdU (green) and Sox2 (red; h,i). a,b; β-catenin in the nuclei of Sox9+ Müller glia/MGPCs on 70% grayscale background. Arrows indicate the nuclei of Müller glia and/or MGPCs, and hollow arrow head indicate the nuclei of proliferating NIRG cells. The histograms in e and g illustrate the mean (± SD; n=8) number of proliferating Müller glia per field of view (14,400 μm2 for e, 28,800 μm2 for g). The histogram in f illustrates the mean (± SD; n=8) number of Sox9-positive nuclei in the ONL per field of view (14,400 μm2). Significance of difference (*p<0.001; ns – not significant) between control and treated groups was determined by using a two-tailed t test. The scale bar (50 μm) in panel d applies to e and d, and the bar in i applies to a,b,h and i. Abbreviations: ONL – outer nuclear layer, INL – inner nuclear layer, IPL – inner plexiform layer, GCL – ganglion cell layer.
To investigate the specificity of the XAV939 we probed for the accumulation of nuclear β-catenin in the Müller glia/MGPCs. One day after the last dose of FGF2, we found that XAV939 significantly reduced levels of β-catenin in the Sox9+ nuclei of Müller glia/MGPCs (Figs. 7a,b). This effect was highly significant when the density sum was divided by total number of Sox9+ nuclei per field of view to account for XAV939-mediated decreases in proliferation (Fig. 7b). To better understand how XAV939 might influence the formation of MGPCs we examined whether FGF2/MAPK-signaling was influenced by probing for readouts of MAPK-signaling. Intraocular injections of FGF2 are known to selectively up-regulate pERK1/2, pCREB, p38 MAPK, cFos and Egr1 in Müller glia (Fischer et al., 2009a). We found that levels of p38 MAPK were significantly increased in Müller glia treated with FGF2 and XAV939 or Wnt-c59, compared to levels seen in glia treated with FGF2 alone (Figs. 7c,d). By comparison, levels of cFos (Fig. 7e), Egr1 and pERK1/2 (not shown) in Müller glia were unaffected by XAV939 or Wnt-c59 in FGF2-treated retinas.
Figure 7.
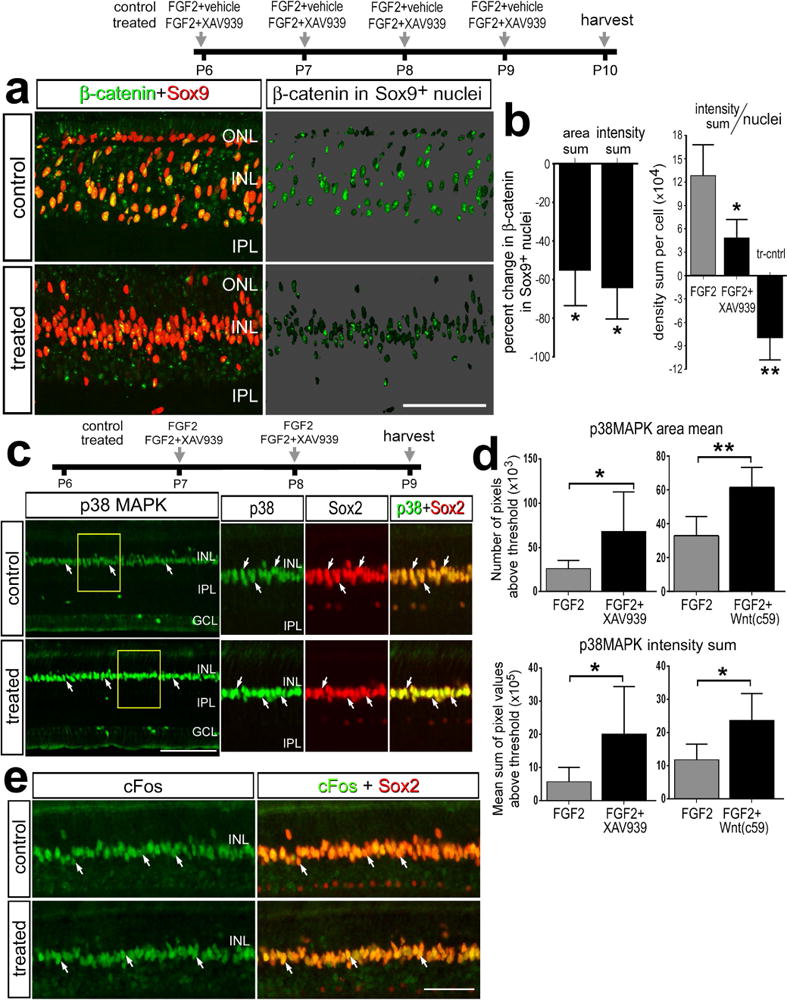
Application of XAV939 or Wnt-c59 with FGF2 blocks the accumulation of nuclear β-catenin in MGPCs, and increases levels of p38MAPK in Müller glia, whereas levels of cFos are unaffected. a,b; Eyes were injected with 4 consecutive daily injections of FGF2 ± XAV939 or Wnt-c59, BrdU 24 hrs after the last injection of FGF2, and retinas harvested 24 hrs later. c–e; Eyes were injected with FGF2 alone (control) or FGF2+XAV939 or Wnt-c59 (treated) at P7 and P8, and tissues harvested at P9. Sections of the retina were labeled for nuclear β-catenin (green) and Sox9 (red; a), Sox2 (red) and p38 MAPK (green) (c), or Sox2 (red) and cFos (green; e). a; β-catenin in the nuclei of Sox9+ Müller glia/MGPCs on 70% grayscale background. Areas indicated by yellow boxes in c are enlarged 2-fold in the panels to the right. Arrows indicate the nuclei of Müller glia and/or MGPCs. The histograms in b illustrates the mean (± SD; n=6) percent change in the area sum, intensity sum per field of view, and intensity sum divided by the total number of Sox9+ nuclei per field of view for nuclear β-catenin in the Sox9+ nuclei of Müller glia/MGPCs. The histograms in d illustrate the mean (± SD; n=6) percent change in the area sum and intensity sum per field of view for p38 MAPK in the INL. Significance of difference (*p<0.01) was determined by using a Mann-Whitney U test.
Nuclear β–catenin in ciliary marginal zone (CMZ) progenitors and NPE of the ciliary body
Wnt-signaling has been reported to influence the embryonic retinal development and regeneration of peripheral regions of embryonic retina (Cho and Cepko, 2006; Kubo et al., 2005; Liu et al., 2007; Zhu et al., 2014). We failed to detect significant levels of nuclear β-catenin in the neural retina, including maturing Müller glia during later stages of retinal development (not shown). Consistent with a previous report of early stages of retinal development (Zhu et al., 2014), we observed nuclear β-catenin in the developing non-pigmented epithelium (NPE) of the ciliary body in E8 eyes (Fig. 8a), and this pattern of expression was maintained through the time of hatching (E21) and for at least the first 3 weeks of postnatal development (Figs. 8b,d). Thus, we examined whether Wnt/β-catenin-signaling was influenced under conditions where proliferation and neurogenesis are activated in the CMZ and the NPE of the ciliary body. Intraocular injections of IGF1 and FGF2 in the post-hatch chick eye are known to stimulate the proliferation of CMZ progenitors (Fischer et al., 2002a; Fischer and Reh, 2000), proliferation of NPE cells in the ciliary body, and differentiation of neurons in the NPE (Fischer and Reh, 2003a). We found that treatment with IGF1 and FGF2 did not induce nuclear β-catenin in Sox9-positive CMZ progenitors (Figs. 8b,c). By comparison, levels of nuclear β-catenin were elevated in control NPE cells, but were greatly reduced by treatment with IGF1 and FGF2 (Figs. 8d,e), where proliferation and neurogenesis are known to be induced (Fischer and Reh, 2003b).
Figure 8.
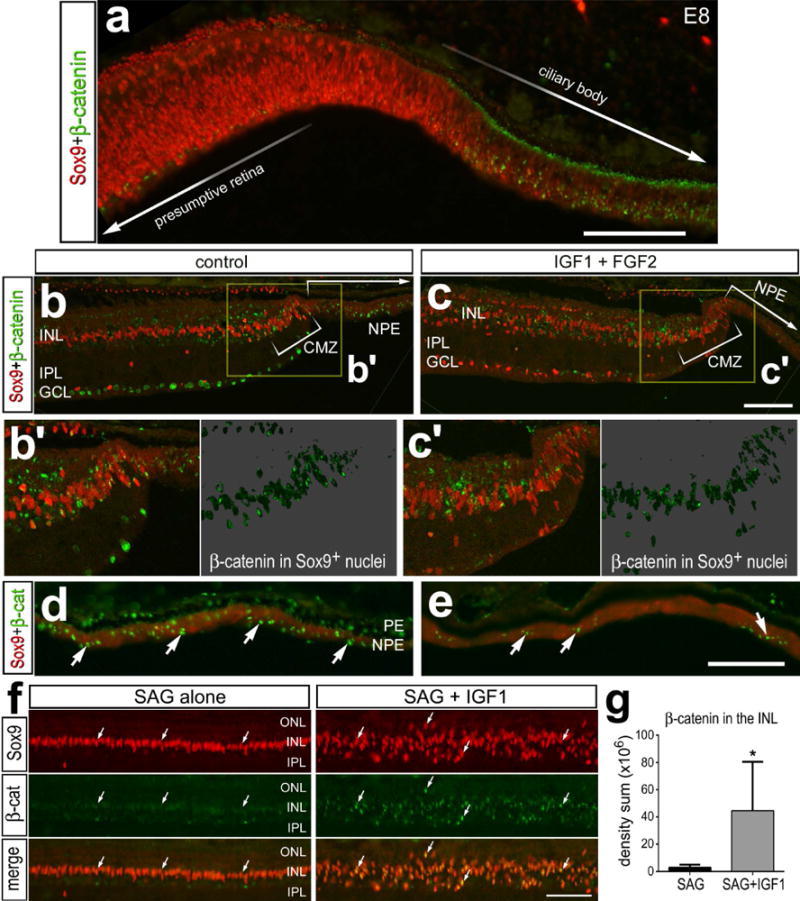
The combination of IGF1 and FGF2 decreases nuclear β-catenin in the non-pigmented epithelium of the ciliary body, whereas IGF1 and Hh-agonist (SAG) increase nuclear β-catenin in Müller glia. Tissues were obtained from: (1) E8 chick embryos (a), (2) P11 eyes that received 4 consecutive daily intraocular injections of IGF1 alone or IGF1+FGF2 (b–e), or (3) P11 eyes that received 4 consecutive daily intraocular injections of IGF1 alone or IGF1+SAG (f,g). Sections of the peripheral retina (a,f) and CMZ (b,c), and epithelia of the ciliary body (d,e) were labeled with antibodies to Sox9 (red) and (green). The areas boxed-out in yellow in b and c are enlarged 2-fold in the lower panels (b’ and c’). Arrows indicate the nuclei labeled for β-catenin. b’ and c’; β-catenin in the nuclei of Sox9+ Müller glia/MGPCs on 70% grayscale background. The scale bar (50 μm) in panel a applies to a alone, the bar c applies to b and c, the bar in e applies to d and e, and the bar in f applies to f alone. Abbreviations: ONL – outer nuclear layer, INL – inner nuclear layer, IPL – inner plexiform layer, GCL – ganglion cell layer, CMZ – ciliary marginal zone, PE – pigmented epithelium.
We have recently reported that Hedgehog (Hh)-signaling is part of the network of signaling pathways that drives the formation of MGPCs (Todd and Fischer, 2015). Müller glia do not normally respond to Hh-agonists, but become responsive in damaged and undamaged retinas downstream of FGF/MAPK-signaling (Todd and Fischer, 2015). The combination of IGF1 and Hh-agonist (SAG) was shown to stimulate the nuclear migration of Müller glia, a symptom of glial reprogramming, but failed to drive the proliferation of MGPCs (Todd and Fischer, 2015). We examined whether IGF1 and SAG failed to drive the formation of proliferating MGPCs because of a failure to recruit and activate Wnt-signaling. Surprisingly, we found that the combination of IGF1 and SAG induced a significant increase in nuclear β-catenin in Sox9+ Müller glia (Fig. 8f,g), suggesting that accumulations of nuclear β-catenin and Wnt-signaling in Müller glia may not be sufficient to drive the transition to proliferating MGPCs.
DISCUSSION
Our findings indicate that Wnt-signaling and nuclear β-catenin are key players in the cell-signaling network that drives the formation of MGPCs in the chick retina. These findings are consistent with those recently reported from the zebrafish model system (Meyers et al., 2012; Ramachandran et al., 2011; Wan et al., 2014). Similarly, in explant cultures of the rodent retina, the formation of MGPCs is stimulated by Wnt-ligands (Osakada et al., 2007). We find that Wnt-signaling is activated in Müller glia and/or MGPCs down-stream of FGF2/MAPK and in response to excitotoxic retinal damage. Similarly, in the zebrafish model there is convergence and cross-talk between MAPK, PI3K, β-catenin and pStat3 signaling during the formation of MGPCs (Wan et al., 2014). In the zebrafish model, the cell-signaling network that drives the formation of MGPCs can be initiated, in the absence of damage, by many different factors including Wnt-ligands, HB-EGF, TNFα, CNTF/cytokines or the combination IGF1/insulin and FGF2 (reviewed by Goldman 2014). By comparison, in the chick model, the signaling network that drives the formation of MGPCs can be initiated, in the absence of damage, by only FGF2, but not other factors (unpublished observations and (Fischer et al., 2014a; Gallina et al., 2014b; Ghai et al., 2010). In the mammalian model, there currently are no known means by which to activate the cell-signaling network in Müller glia to stimulate the formation of MGPCs without first damaging the retina.
In normal healthy retinas, we find that Wnt-signaling and nuclear β-catenin are not active in Müller glia. There was a distinct absence of nuclear β-catenin in normal Müller glia and intraocular injections of GSK3β-inhibitors failed to stimulate any detectable activation of cell-signaling in Müller glia, and thus failed to stimulate the formation of proliferating MGPCs. However, in NMDA-damaged retinas, but not FGF2-treated retinas, inhibition of GSK3β resulted in the accumulation of nuclear β-catenin (activation of Wnt-signaling) and proliferation of MGPCs. Both retinal damage and FGF2-treatment are known to activate MAPK-signaling in the Müller glia, and the activation of this pathway is required for the formation of proliferating MGPCs (Fischer and Reh, 2001; Fischer et al., 2009a; Fischer et al., 2009b). Collectively, these findings suggest that FGF2/MAPK-signaling is necessary for activation Wnt-signaling in Müller glia, and that Wnt-signaling contributes to the network of signaling pathways that drives the formation of MGPCs. It remains uncertain whether MAPK-signaling is soley sufficient to activate Wnt/β-catenin-signaling. Similarly, recent reports using the zebrafish model indicate that the MGPC-signaling network can be initiated by insulin/PI3K and FGF2/MAPK and involves the recruitment of Jak/Stat- and Wnt-signaling (Wan et al., 2014; Zhao et al., 2014).
Axin2 and tcf-1 are targets of Wnt-signaling and are known to be up-regulated by Wnt-signaling (reviewed by Clevers and Nusse, 2012). We detected retina-wide decreases in levels of axin2 and tcf-1 following NMDA-treatment. These decreases in Wnt/β-catenin-signaling likely reflect the net changes in neurons, whereas any changes in axin2 and tcf-1 in Müller glia would be masked; both of these cell-types undergo fluctuations of nuclear β-catenin. Indeed, we observed a decrease in nuclear β-catenin in retinal neurons after NMDA-treatment and after treatment with Wnt-inhibitor. By comparison, c-myc was increased in NMDA-treated retinas, and, in principle, should be expressed only in proliferating cells such as the MGPCs, not in post-mitotic neurons. Thus, the assays that distinguish discrete changes in β-catenin in the nuclei of Müller glia/MGPCs provide the most accurate read-out of changes in Wnt-signaling during reprogramming.
The neurogenic capacity of MGPCs may not be influenced by Wnt-signaling. Wnt/β-catenin-signaling must be down-regulated to permit retinal neurogenesis during development (Cho and Cepko, 2006; Liu et al., 2007). During embryonic retinal development in the chick, β-catenin must be down-regulated to permit retinal regeneration from prospective ciliary marginal tissues at the peripheral edge of the developing neural retina (Zhu et al., 2014). In the zebrafish retina, Wnt/β-catenin-signaling is necessary and sufficient for the formation of multipotent proliferating MGPCs without biasing cell fate specification (Ramachandran et al., 2011). Further, Wnt/β-catenin-signaling is known to prevent differentiation and maintains retinal progenitors in a proliferative state (Meyers et al., 2012). In the chick retina, we found that nuclear β-catenin is rapidly down-regulated in MGPCs following the cessation of treatment with FGF2 and between 3 and 5 days after NMDA-treatment. Interestingly, when Wnt-signaling was inhibited following NMDA-treatment, numbers of proliferating MGPCs were reduced and levels of the pro-neural bHLH transcription factor ascl1a were increased and the pro-glial bHLH factor hes5 were decreased. When Wnt-signaling was inhibited after NMDA-treatment, despite decreases hes5 and increases in ascl1a, we found that cell fate specification was unaffected. This was unexpected because Hes5 is known to promote gliogenesis (Hojo et al., 2000), and Ascl1a is known to promote neurogenesis in the developing retina (Tomita et al., 1996; Verma-Kurvari et al., 1996). During development, Notch-signaling is known to maintain neural progenitors in an undifferentiated, multipotent state (Bao and Cepko, 1997; Furukawa et al., 2000). In regenerating zebrafish retina, Notch-signaling suppresses the proliferation of MGPCs (Conner et al., 2014; Wan et al., 2012). By comparison, Notch-signaling promotes the proliferation of MGPCs and suppresses the neuronal differentiation of MGPC-derived progeny in the chick retina (Ghai et al., 2010; Hayes et al., 2007). In zebrafish, ascl1a is known to be downstream of Wnt-signaling during the re-programming of Müller glia into neurogenic MGPCs (Ramachandran et al., 2011). ascl1a is expressed by Müller glia in the fish retina within 4 hours after damage (Fausett et al., 2008). By comparison, ascl1a is not up-regulated after injury in the rodent retina (Karl et al., 2008), and ascl1a is up-regulated between 2–4 days after damage in the chick retina (Fischer and Reh, 2001; Hayes et al., 2007). These findings indicate fundamental differences between mammals, birds and fish in how Wnt-signaling might regulate the formation of MGPCs. Thus, independent of XAV939-mediated inhibition of Wnt/β-catenin-signaling there must be factors or signaling pathways that override changes in Hes5 and Ascl1a to prevent neuronal differentiation. The pathways that inhibit neuronal differentiation may include Notch- (Hayes et al., 2007), Jak/Stat- and BMP/smad-signaling (unpublished observations); signaling pathways which are known to promote gliogenesis and inhibit neurogenesis during development (reviewed by Guillemot, 2007). Collectively, our findings suggest that Wnt-signaling acts to stimulate proliferation during the formation of MGPCs in the chick retina, but does not influence the differentiation of the progeny produced by MGPCs.
Inhibition of GSK3β combined with FGF2-treatment fails to enhance the formation of proliferating MGPCs. These findings suggest that FGF2/MAPK- and Wnt/β-catenin-signaling do not act synergistically to stimulate the formation of MGPCs. However, FGF2 alone resulted in a significant and selective accumulation of nuclear β-catenin in Müller glia, indicating that FGF2/MAPK-signaling recruits the Wnt-pathway into the signaling network that drives the formation of proliferating MGPCs. It is likely that inhibition of GSK3β combined with activation of FGF2/MAPK fails to enhance the formation of MGPCs because the Wnt/β-catenin-pathway is saturated by FGF-treatment. Since activation and inhibition of Wnt-signaling had no apparent effect upon normal Müller glia in undamaged retinas, we propose that FGF2-treatment renders Müller glia responsive to canonical Wnt-signaling. By comparison, the combination of IGF1 and Hh-agonist, which induces nuclear migration but fails to drive proliferation (Todd and Fischer, 2015), induced a selective activation of Wnt-signaling in Müller glia. In damaged retinas, we did not find a correlation between the accumulation of nuclear β-catenin and proliferation of MGPCs. Collectively, these findings suggest that the Wnt/β-catenin-signaling is a necessary component of the network of pathways that drive the formation of proliferating MGPCs. However, Wnt/β-catenin-signaling is not sufficient to drive the reprogramming of Müller glia into proliferating MGPCs.
Wnt-signaling has been shown to promote neuronal survival in degenerating retinas. However, we found that Wnt-signaling had no influence on the survival of neurons in NMDA-treated retinas. By comparison, Wnt/β-catenin-signaling in Muller glia has been shown to protect photoreceptors in a mouse model of retinal degeneration (Patel et al., 2014). Similarly, Wnt/β-catenin-signaling in Müller glia mediates the neuroprotective effects of Norrin on retinal ganglion cells (Seitz et al., 2010). It is possible that the damage caused by NMDA was too severe and/or too acute for Wnt-signaling to influence the neuroprotective properties of the Müller glia. By comparison, we have reported previously that activation of cell-signaling on Müller glia through CNTF (Fischer et al., 2004) or FGF2/MAPK (Fischer et al., 2009a) potently protects neurons against NMDA-induced retinal damage.
The formation of MGPCs in FGF2-treated retinas was suppressed by XAV939, and resulted in increased levels of p38 MAPK in the Müller glia. Similarly, we have reported previously that p38 MAPK is significantly up-regulated in Müller glia in FGF2-treated retinas when the microglia were ablated and the formation of proliferating MGPCs is suppressed (Fischer et al., 2014a). Activation of p38 MAPK is known to suppress pERK1/2- and Notch-mediated proliferation and self-renewal of stem cells, including those that drive neurogenesis (Yang et al., 2006), hematopoiesis (Schraml et al., 2009), and angiogenesis (Matsumoto et al., 2002). p38 MAPK-signaling in retinal glia is typically associated with ocular inflammation, ischemic stress or infection (Roth et al., 2003; Shamsuddin and Kumar, 2011; Takeda et al., 2002). Collectively, these findings suggest that increased p38 MAPK-signaling enhances glial reactivity and inflammatory responses, while suppressing the progenitor-like potential of the Müller glia.
Conclusions
In the chick retina, Müller glia do not respond to Wnt-signaling in normal, undamaged retina. However, Müller glia can be made responsive to Wnt-signaling by retinal damage or treatment with FGF2. Wnt-signaling is part of the signaling network that is activated in Müller glia by FGF2-treatment in the absence of damage or by damage to retinal neurons. In damaged retinas, inhibition of MAPK-signaling fails to override the proliferation-stimulating effects of increased Wnt-signaling (GSK3β–inhibition). Although activation of Wnt-signaling is required for the de-differentiation and proliferation of MGPCs, our findings indicate that the neurogenic potential of MGPCs is not affected by Wnt/β-catenin-signaling.
Acknowledgments
The antibodies to Nkx2.2, Pax6, neurofilament, and BrdU developed by Drs. T.M. Jessell/S. Brenner-Morton, A. Kawakami, J. Wood and S.J. Kaufman, respectively, were obtained from the Developmental Studies Hybridoma Bank, which was developed under the auspices of the NICHD and is maintained by the University of Iowa, Department of Biological Sciences, Iowa City, IA 52242. This work was supported by a grant (EY022030-03) from the National Institutes of Health, National Eye Institute.
References
- Bao ZZ, Cepko CL. The expression and function of Notch pathway genes in the developing rat eye. J Neurosci. 1997;17:1425–34. doi: 10.1523/JNEUROSCI.17-04-01425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382:638–42. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- Cho SH, Cepko CL. Wnt2b/beta-catenin-mediated canonical Wnt signaling determines the peripheral fates of the chick eye. Development. 2006;133:3167–77. doi: 10.1242/dev.02474. [DOI] [PubMed] [Google Scholar]
- Ciani L, Salinas PC. WNTs in the vertebrate nervous system: from patterning to neuronal connectivity. Nat Rev Neurosci. 2005;6:351–62. doi: 10.1038/nrn1665. [DOI] [PubMed] [Google Scholar]
- Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149:1192–205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- Conner C, Ackerman KM, Lahne M, Hobgood JS, Hyde DR. Repressing Notch Signaling and Expressing TNFalpha Are Sufficient to Mimic Retinal Regeneration by Inducing Muller Glial Proliferation to Generate Committed Progenitor Cells. J Neurosci. 2014;34:14403–19. doi: 10.1523/JNEUROSCI.0498-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das AV, Mallya KB, Zhao X, Ahmad F, Bhattacharya S, Thoreson WB, Hegde GV, Ahmad I. Neural stem cell properties of Muller glia in the mammalian retina: regulation by Notch and Wnt signaling. Dev Biol. 2006;299:283–302. doi: 10.1016/j.ydbio.2006.07.029. [DOI] [PubMed] [Google Scholar]
- Fausett BV, Gumerson JD, Goldman D. The proneural basic helix-loop-helix gene ascl1a is required for retina regeneration. J Neurosci. 2008;28:1109–17. doi: 10.1523/JNEUROSCI.4853-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer AJ, Bongini R. Turning Muller glia into neural progenitors in the retina. Mol Neurobiol. 2010;42:199–209. doi: 10.1007/s12035-010-8152-2. [DOI] [PubMed] [Google Scholar]
- Fischer AJ, Dierks BD, Reh TA. Exogenous growth factors induce the production of ganglion cells at the retinal margin. Development. 2002a;129:2283–91. doi: 10.1242/dev.129.9.2283. [DOI] [PubMed] [Google Scholar]
- Fischer AJ, McGuire CR, Dierks BD, Reh TA. Insulin and fibroblast growth factor 2 activate a neurogenic program in Muller glia of the chicken retina. J Neurosci. 2002b;22:9387–98. doi: 10.1523/JNEUROSCI.22-21-09387.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer AJ, Reh TA. Identification of a proliferating marginal zone of retinal progenitors in postnatal chickens. Dev Biol. 2000;220:197–210. doi: 10.1006/dbio.2000.9640. [DOI] [PubMed] [Google Scholar]
- Fischer AJ, Reh TA. Muller glia are a potential source of neural regeneration in the postnatal chicken retina. Nat Neurosci. 2001;4:247–52. doi: 10.1038/85090. [DOI] [PubMed] [Google Scholar]
- Fischer AJ, Reh TA. Growth factors induce neurogenesis in the ciliary body. Dev Biol. 2003a;259:225–40. doi: 10.1016/s0012-1606(03)00178-7. [DOI] [PubMed] [Google Scholar]
- Fischer AJ, Reh TA. Potential of Muller glia to become neurogenic retinal progenitor cells. Glia. 2003b;43:70–6. doi: 10.1002/glia.10218. [DOI] [PubMed] [Google Scholar]
- Fischer AJ, Ritchey ER, Scott MA, Wynne A. Bullwhip neurons in the retina regulate the size and shape of the eye. Dev Biol. 2008;317:196–212. doi: 10.1016/j.ydbio.2008.02.023. [DOI] [PubMed] [Google Scholar]
- Fischer AJ, Schmidt M, Omar G, Reh TA. BMP4 and CNTF are neuroprotective and suppress damage-induced proliferation of Muller glia in the retina. Mol Cell Neurosci. 2004;27:531–42. doi: 10.1016/j.mcn.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Fischer AJ, Scott MA, Ritchey ER, Sherwood P. Mitogen-activated protein kinase-signaling regulates the ability of Müller glia to proliferate and protect retinal neurons against excitotoxicity. Glia. 2009a;57:1538–1552. doi: 10.1002/glia.20868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer AJ, Scott MA, Tuten W. Mitogen-activated protein kinase-signaling stimulates Muller glia to proliferate in acutely damaged chicken retina. Glia. 2009b;57:166–81. doi: 10.1002/glia.20743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer AJ, Scott MA, Zelinka C, Sherwood P. A novel type of glial cell in the retina is stimulated by insulin-like growth factor 1 and may exacerbate damage to neurons and Muller glia. Glia. 2010;58:633–49. doi: 10.1002/glia.20950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer AJ, Zelinka C, Gallina D, Scott MA, Todd L. Reactive microglia and macrophage facilitate the formation of Muller glia-derived retinal progenitors. Glia. 2014a;62:1608–1628. doi: 10.1002/glia.22703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer AJ, Zelinka C, Gallina D, Scott MA, Todd L. Reactive microglia and macrophage facilitate the formation of Muller glia-derived retinal progenitors. Glia. 2014b;62:1608–28. doi: 10.1002/glia.22703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa T, Mukherjee S, Bao ZZ, Morrow EM, Cepko CL. rax, Hes1, and notch1 promote the formation of Muller glia by postnatal retinal progenitor cells. Neuron. 2000;26:383–94. doi: 10.1016/s0896-6273(00)81171-x. [DOI] [PubMed] [Google Scholar]
- Gallina D, Todd L, Fischer AJ. A comparative analysis of Muller glia-mediated regeneration in the vertebrate retina. Exp Eye Res. 2014a;123:121–130. doi: 10.1016/j.exer.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallina D, Zelinka C, Fischer AJ. Glucocorticoid receptors in the retina, Müller glia and the formation of Müller glia-derived progenitors. Development. 2014b;141:3340–3351. doi: 10.1242/dev.109835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghai K, Zelinka C, Fischer AJ. Serotonin released from amacrine neurons is scavenged and degraded in bipolar neurons in the retina. J Neurochem. 2009;111:1–14. doi: 10.1111/j.1471-4159.2009.06270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghai K, Zelinka C, Fischer AJ. Notch signaling influences neuroprotective and proliferative properties of mature Muller glia. J Neurosci. 2010;30:3101–12. doi: 10.1523/JNEUROSCI.4919-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman D. Muller glial cell reprogramming and retina regeneration. Nat Rev Neurosci. 2014 doi: 10.1038/nrn3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemot F. Cell fate specification in the mammalian telencephalon. Prog Neurobiol. 2007;83:37–52. doi: 10.1016/j.pneurobio.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Hamburger V, Hamilton HL. A series of normal stages in the development of the chick embryo. 1951. Dev Dyn. 1992;195:231–72. doi: 10.1002/aja.1001950404. [DOI] [PubMed] [Google Scholar]
- Hayes S, Nelson BR, Buckingham B, Reh TA. Notch signaling regulates regeneration in the avian retina. Dev Biol. 2007;312:300–11. doi: 10.1016/j.ydbio.2007.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hojo M, Ohtsuka T, Hashimoto N, Gradwohl G, Guillemot F, Kageyama R. Glial cell fate specification modulated by the bHLH gene Hes5 in mouse retina. Development. 2000;127:2515–22. doi: 10.1242/dev.127.12.2515. [DOI] [PubMed] [Google Scholar]
- Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner S, Hild M, Shi X, Wilson CJ, Mickanin C, Myer V, Fazal A, Tomlinson R, Serluca F, Shao W, Cheng H, Shultz M, Rau C, Schirle M, Schlegl J, Ghidelli S, Fawell S, Lu C, Curtis D, Kirschner MW, Lengauer C, Finan PM, Tallarico JA, Bouwmeester T, Porter JA, Bauer A, Cong F. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461:614–20. doi: 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- Inestrosa NC, Arenas E. Emerging roles of Wnts in the adult nervous system. Nat Rev Neurosci. 2010;11:77–86. doi: 10.1038/nrn2755. [DOI] [PubMed] [Google Scholar]
- Kadowaki T, Wilder E, Klingensmith J, Zachary K, Perrimon N. The segment polarity gene porcupine encodes a putative multitransmembrane protein involved in Wingless processing. Genes Dev. 1996;10:3116–28. doi: 10.1101/gad.10.24.3116. [DOI] [PubMed] [Google Scholar]
- Karl MO, Hayes S, Nelson BR, Tan K, Buckingham B, Reh TA. Stimulation of neural regeneration in the mouse retina. Proc Natl Acad Sci U S A. 2008;105:19508–13. doi: 10.1073/pnas.0807453105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo F, Takeichi M, Nakagawa S. Wnt2b inhibits differentiation of retinal progenitor cells in the absence of Notch activity by downregulating the expression of proneural genes. Development. 2005 doi: 10.1242/dev.01856. [DOI] [PubMed] [Google Scholar]
- Lenkowski JR, Raymond PA. Muller glia: Stem cells for generation and regeneration of retinal neurons in teleost fish. Prog Retin Eye Res. 2014 doi: 10.1016/j.preteyeres.2013.12.007. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Hunter DJ, Rooker S, Chan A, Paulus YM, Leucht P, Nusse Y, Nomoto H, Helms JA. Wnt signaling promotes muller cell proliferation and survival after injury. Invest Ophthalmol Vis Sci. 2012;54:444–53. doi: 10.1167/iovs.12-10774. [DOI] [PubMed] [Google Scholar]
- Liu H, Xu S, Wang Y, Mazerolle C, Thurig S, Coles BL, Ren JC, Taketo MM, van der Kooy D, Wallace VA. Ciliary margin transdifferentiation from neural retina is controlled by canonical Wnt signaling. Dev Biol. 2007;308:54–67. doi: 10.1016/j.ydbio.2007.04.052. [DOI] [PubMed] [Google Scholar]
- Lustig B, Jerchow B, Sachs M, Weiler S, Pietsch T, Karsten U, van de Wetering M, Clevers H, Schlag PM, Birchmeier W, Behrens J. Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol Cell Biol. 2002;22:1184–93. doi: 10.1128/MCB.22.4.1184-1193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto T, Turesson I, Book M, Gerwins P, Claesson-Welsh L. p38 MAP kinase negatively regulates endothelial cell survival, proliferation, and differentiation in FGF-2-stimulated angiogenesis. J Cell Biol. 2002;156:149–60. doi: 10.1083/jcb.200103096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers JR, Hu L, Moses A, Kaboli K, Papandrea A, Raymond PA. beta-catenin/Wnt signaling controls progenitor fate in the developing and regenerating zebrafish retina. Neural Dev. 2012;7:30. doi: 10.1186/1749-8104-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon RT, Kohn AD, De Ferrari GV, Kaykas A. WNT and beta-catenin signalling: diseases and therapies. Nat Rev Genet. 2004;5:691–701. doi: 10.1038/nrg1427. [DOI] [PubMed] [Google Scholar]
- Osakada F, Ooto S, Akagi T, Mandai M, Akaike A, Takahashi M. Wnt signaling promotes regeneration in the retina of adult mammals. J Neurosci. 2007;27:4210–9. doi: 10.1523/JNEUROSCI.4193-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AK, Surapaneni K, Yi H, Nakamura RE, Karli SZ, Syeda S, Lee T, Hackam AS. Activation of Wnt/beta-catenin signaling in Muller glia protects photoreceptors in a mouse model of inherited retinal degeneration. Neuropharmacology. 2014;91:1–12. doi: 10.1016/j.neuropharm.2014.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak J, Wilken MS, Ueki Y, Cox KE, Sullivan JM, Taylor RJ, Levine EM, Reh TA. Ascl1 reprograms mouse Muller glia into neurogenic retinal progenitors. Development. 2013;140:2619–2631. doi: 10.1242/dev.091355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Zhao XF, Goldman D. Ascl1a/Dkk/beta-catenin signaling pathway is necessary and glycogen synthase kinase-3beta inhibition is sufficient for zebrafish retina regeneration. Proc Natl Acad Sci U S A. 2011;108:15858–63. doi: 10.1073/pnas.1107220108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee J, Buchan T, Zukerberg L, Lilien J, Balsamo J. Cables links Robo-bound Abl kinase to N-cadherin-bound beta-catenin to mediate Slit-induced modulation of adhesion and transcription. Nat Cell Biol. 2007;9:883–92. doi: 10.1038/ncb1614. [DOI] [PubMed] [Google Scholar]
- Roesch K, Jadhav AP, Trimarchi JM, Stadler MB, Roska B, Sun BB, Cepko CL. The transcriptome of retinal Muller glial cells. J Comp Neurol. 2008;509:225–38. doi: 10.1002/cne.21730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesch K, Stadler MB, Cepko CL. Gene expression changes within Muller glial cells in retinitis pigmentosa. Mol Vis. 2012;18:1197–214. [PMC free article] [PubMed] [Google Scholar]
- Rompani SB, Cepko CL. A common progenitor for retinal astrocytes and oligodendrocytes. J Neurosci. 2010;30:4970–80. doi: 10.1523/JNEUROSCI.3456-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schraml E, Fuchs R, Kotzbeck P, Grillari J, Schauenstein K. Acute adrenergic stress inhibits proliferation of murine hematopoietic progenitor cells via p38/MAPK signaling. Stem Cells Dev. 2009;18:215–27. doi: 10.1089/scd.2008.0072. [DOI] [PubMed] [Google Scholar]
- Seitz R, Hackl S, Seibuchner T, Tamm ER, Ohlmann A. Norrin mediates neuroprotective effects on retinal ganglion cells via activation of the Wnt/beta-catenin signaling pathway and the induction of neuroprotective growth factors in Muller cells. J Neurosci. 2010;30:5998–6010. doi: 10.1523/JNEUROSCI.0730-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thummel R, Enright JM, Kassen SC, Montgomery JE, Bailey TJ, Hyde DR. Pax6a and Pax6b are required at different points in neuronal progenitor cell proliferation during zebrafish photoreceptor regeneration. Exp Eye Res. 2010;90:572–82. doi: 10.1016/j.exer.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd L, Fischer AJ. Hedgehog-signaling stimulates the formation of proliferating Müller glia-derived progenitor cells in the retina. Development. 2015;142:2610–2622. doi: 10.1242/dev.121616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita K, Nakanishi S, Guillemot F, Kageyama R. Mash1 promotes neuronal differentiation in the retina. Genes Cells. 1996;1:765–74. doi: 10.1111/j.1365-2443.1996.tb00016.x. [DOI] [PubMed] [Google Scholar]
- Verma-Kurvari S, Savage T, Gowan K, Johnson JE. Lineage-specific regulation of the neural differentiation gene MASH1. Dev Biol. 1996;180:605–17. doi: 10.1006/dbio.1996.0332. [DOI] [PubMed] [Google Scholar]
- Wan J, Ramachandran R, Goldman D. HB-EGF is necessary and sufficient for Muller glia dedifferentiation and retina regeneration. Dev Cell. 2012;22:334–47. doi: 10.1016/j.devcel.2011.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan J, Zhao XF, Vojtek A, Goldman D. Retinal Injury, Growth Factors, and Cytokines Converge on beta-Catenin and pStat3 Signaling to Stimulate Retina Regeneration. Cell Rep. 2014;9:285–97. doi: 10.1016/j.celrep.2014.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SR, Kim SJ, Byun KH, Hutchinson B, Lee BH, Michikawa M, Lee YS, Kang KS. NPC1 gene deficiency leads to lack of neural stem cell self-renewal and abnormal differentiation through activation of p38 mitogen-activated protein kinase signaling. Stem Cells. 2006;24:292–8. doi: 10.1634/stemcells.2005-0221. [DOI] [PubMed] [Google Scholar]
- Zelinka CP, Scott MA, Volkov L, Fischer AJ. The Reactivity, Distribution and Abundance of Non-Astrocytic Inner Retinal Glial (NIRG) Cells Are Regulated by Microglia, Acute Damage, and IGF1. PLoS One. 2012;7:e44477. doi: 10.1371/journal.pone.0044477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao XF, Wan J, Powell C, Ramachandran R, Myers MG, Jr, Goldman D. Leptin and IL-6 family cytokines synergize to stimulate muller glia reprogramming and retina regeneration. Cell Rep. 2014;9:272–84. doi: 10.1016/j.celrep.2014.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Luz-Madrigal A, Haynes T, Zavada J, Burke AK, Del Rio-Tsonis K. beta-Catenin inactivation is a pre-requisite for chick retina regeneration. PLoS One. 2014;9:e101748. doi: 10.1371/journal.pone.0101748. [DOI] [PMC free article] [PubMed] [Google Scholar]