

Abstract
Purpose
This study was undertaken to identify causal mutations responsible for autosomal recessive retinitis pigmentosa (arRP) in consanguineous families.
Methods
Large consanguineous families were ascertained from the Punjab province of Pakistan. An ophthalmic examination consisting of a fundus evaluation and electroretinography (ERG) was completed, and small aliquots of blood were collected from all participating individuals. Genomic DNA was extracted from white blood cells, and a genome-wide linkage or a locus-specific exclusion analysis was completed with polymorphic short tandem repeats (STRs). Two-point logarithm of odds (LOD) scores were calculated, and all coding exons and exon–intron boundaries of RP1 were sequenced to identify the causal mutation.
Results
The ophthalmic examination showed that affected individuals in all families manifest cardinal symptoms of RP. Genome-wide scans localized the disease phenotype to chromosome 8q, a region harboring RP1, a gene previously implicated in the pathogenesis of RP. Sanger sequencing identified a homozygous single base deletion in exon 4: c.3697delT (p.S1233Pfs22*), a single base substitution in intron 3: c.787+1G>A (p.I263Nfs8*), a 2 bp duplication in exon 2: c.551_552dupTA (p.Q185Yfs4*) and an 11,117 bp deletion that removes all three coding exons of RP1. These variations segregated with the disease phenotype within the respective families and were not present in ethnically matched control samples.
Conclusions
These results strongly suggest that these mutations in RP1 are responsible for the retinal phenotype in affected individuals of all four consanguineous families.
Introduction
Retinitis pigmentosa (RP) is the most common inherited retinal dystrophy, affecting approximately 1 in 5,000 individuals worldwide [1,2]. RP primarily affects the rod photoreceptors, while the cone cells are compromised as the disease progresses [3]. Affected individuals exhibit night blindness in the initial stages of the disease followed by a progressive reduction in the visual field [3]. Ocular findings include atrophic changes in the photoreceptors and the RPE followed by the appearance of melanin-containing structures in the retinal vascular layer [3]. The fundus changes include a pale optic nerve, attenuation of the retinal vessels, and bone spicule-like pigmentation in the mid-peripheral retina [3]. Electroretinography (ERG) recordings show severely diminished or completely extinguished rod response while the cone response is somewhat normal in early stages but is undetectable as the disease progresses [3].
RP is a genetically heterogeneous disorder that manifests as an autosomal dominant, autosomal recessive, and X-linked trait. To date, 73 genes have been implicated in the pathogenesis of RP. Of these genes, 27 have been associated with autosomal dominant RP (adRP) [4-30] while mutations in 50 genes have been identified in patients with autosomal recessive RP (arRP; RetNet) [31-77]. Interestingly, mutations in RHO (Gene ID: 6010; OMIM: 180380), RP1 (Gene ID: 6101; OMIM: 603937), NRL (Gene ID: 4901; OMIM: 162080), RPE65 (Gene ID: 6121; OMIM: 180069), BEST1 (Gene ID: 7439; OMIM: 607854), NR2E3 (Gene ID: 10002; OMIM: 604485), IMPDH1 (Gene ID: 3614; OMIM: 146690) have been identified in familial cases of both adRP and arRP. Likewise, causal mutations in OFD1 (Gene ID: 8481; OMIM: 300170), RP2 (Gene ID: 6102; OMIM: 300757), and RPGR (Gene ID: 6103; OMIM: 312610) have been identified in RP cases with an X-linked inheritance pattern [78-80].
RP1 was localized to chromosome 8q and consists of four exons that encode for a 2,156 amino acid protein [81]. Pierce and colleagues first identified mutations in RP1 responsible for adRP, and subsequently, they estimated that the nonsense mutation at codon 677 (p.R677*) is present in approximately 3% of the dominant RP cases in North America [81]. The RP1 protein localizes to the connecting cilia of the rod and cone cells in the ocular retina and is required for correct stacking of the outer segment disc [81,82].
Here, we report four consanguineous familial cases with multiple members who manifest cardinal symptoms of RP. Genome-wide linkage analyses localized the disease phenotype to chromosome 8q, harboring RP1, while bidirectional Sanger sequencing identified causal mutations in RP1 that segregated with the disease phenotype in their respective families and were absent in the ethnically matched controls and the genome-variant databases.
Methods
Clinical ascertainment
More than 300 consanguineous Pakistani families with non-syndromic retinal dystrophies were recruited to identify new disease loci responsible for inherited visual diseases. The institutional review boards (IRBs) of the National Centre of Excellence in Molecular Biology (Lahore, Pakistan), the National Eye Institute (Bethesda, MD) and the Johns Hopkins University (Baltimore, MD) approved the study. All participating family members provided informed written consent that was endorsed by the respective IRBs and is consistent with the tenets of the Declaration of Helsinki.
A detailed clinical and medical history was obtained from the individual families. Funduscopy was performed at Layton Rehmatulla Benevolent Trust (LRBT) Hospital (Lahore, Pakistan). ERG measurements were recorded by using equipment manufactured by LKC (Gaithersburg, MD). Dark-adapted rod responses were determined through incident flash attenuated by −25 dB, whereas rod–cone responses were measured at 0 dB. The 30 Hz flicker responses were recorded at 0 dB to a background illumination of 17 to 34 cd/m2.
All participating members voluntarily provided an approximately 10 ml blood sample that was stored in 50 ml Sterilin® Falcon (Sarstedt, Inc. Newton, NC) tubes containing 400 μl of 0.5 M EDTA. Blood samples were stored at −20 °C for long-term storage.
Genomic DNA extraction
Genomic DNA was extracted from white blood cells using a non-organic modified procedure as described previously [83]. The concentration of the extracted genomic DNA was estimated with a SmartSpec plus Bio-Rad Spectrophotometer (Bio-Rad, Hercules, CA).
Genome-wide scan and exclusion analysis
The Applied Biosystems MD-10 linkage mapping panels (Applied Biosystems, Foster City, CA) were used to complete a genome-wide scan for family PKRP117. PCR was completed in a 5 μl reaction volume containing 40 ng of genomic DNA, various combinations of 10 μM fluorescently labeled primer pairs, 10X PCR buffer (100 mM Tris HCl pH 8.4, 400 mM NaCl, 15 mM MgCl2, 2.5 mM Spermidine), 2 mM deoxynucleotide triphosphate (dNTP) mix, and 0.2 U OneTaq DNA polymerase (New England BioLabs Inc, Ipswich, MA). Initial denaturation was performed for 5 minutes (min) at 95 °C, followed by 10 cycles consisting of denaturation at 94 °C for 15 s, annealing at 55 °C for 15 s and elongating at 72 °C for 30 s, and then 20 cycles consisting of denaturation at 89 °C for 15 s, annealing at 55 °C for 15 s, and elongation at 72 °C for 30 s. The final extension was performed for 10 min at 72 °C, followed by a final hold at 16 °C. PCR products were mixed with a loading cocktail containing HD-400 size standards (Applied Biosystems) and resolved in an Applied Biosystems 3100 DNA Analyzer. Genotypes were assigned using the Gene Mapper software from Applied Biosystems. Exclusion analyses were completed for PKRP262, PKRP344, and PKRP358 using closely spaced short tandem repeat (STR) markers.
Linkage analysis
Linkage analysis was performed with alleles of PKRP117 obtained through the genome-wide scan and the alleles of PKRP262, PKRP344, and PKRP358 obtained through exclusion analysis using the FASTLINK version of MLINK from the LINKAGE Program Package [84,85]. Maximum LOD scores were calculated using ILINK from the LINKAGE Program Package. Autosomal recessive retinitis pigmentosa was investigated as a fully penetrant disorder that has an affected allele frequency of 0.001.
Mutation screening
The sequences of primer pairs used to amplify RP1 exons were designed using the Primer3 software. The sequences of the primer pairs used for sequencing are shown in Appendix 1. PCR reactions were completed in 10 μl volume containing 20 ng of genomic DNA. PCR amplification consisted of a denaturation step at 95 °C for 5 min followed by a two-step touchdown procedure. The first step of ten cycles consisted of denaturation at 95 °C for 30 s, followed by a primer set-specific annealing for 30 s (annealing temperature decreases by 1 °C per cycle) and elongation at 72 °C for 45 s. The second step of 30 cycles consisted of denaturation at 95 °C for 30 s followed by annealing (annealing temperature −10 °C) for 30 s and elongation at 72 °C for 45 s, followed by a final elongation at 72 °C for 5 min.
The PCR primers for each exon were used for bidirectional sequencing using the BigDye Terminator Ready reaction mix (Applied Biosystems, Foster City, CA), according to the manufacturer’s instructions. The sequencing products were resolved on an ABI PRISM 3100 DNA analyzer (Applied Biosystems), and results were analyzed with Applied Biosystems SeqScape software.
In silico analysis
The degree of evolutionary conservation of the splice donor site (c.787+1G) in other RP1 orthologs was examined using the UCSC Genome Browser (Genome). The effect of the c.787+1G>A mutation on RP1 mRNA splicing was predicted with an online bioinformatics tool, the Human Splicing Finder 2.4.1 (HSF).
Results
In an ongoing effort to investigate the genetic load of retinal dystrophies in the Pakistani population, we recruited a large cohort of familial cases with multiple members in these families manifesting cardinal symptoms of early onset RP. Among these families, PKRP117, PKRP262, PKRP344, and PKRP358 were recruited from the Punjab province of Pakistan. In PKRP117, we were able to enroll a total of 19 family members, including nine affected individuals (Figure 1A). A detailed medical history, including the onset and progression of the ocular disease, was obtained by interviewing family elders, especially the parents of the affected individuals, which revealed that all affected individuals complained of night blindness during the early years of their life. Exclusion analysis with closely spaced STR markers spanning known RP loci suggested linkage to chromosome 8q harboring RP1. Maximum two-point LOD scores of 3.21, 6.95, and 5.95 at θ = 0 were obtained with markers D8S1737, D8S509, and D8S2332, respectively (Table 1).
Figure 1.

Pedigree drawings with a haplotype formed from the alleles of chromosome 8q microsatellite markers. A: PKRP117. B: PKRP262. C: PKRP344. D: PKRP358. Alleles forming the risk haplotype are black, heterozygous alleles cosegregating with the phenotype are gray, and alleles not cosegregating with RP are shown in white. Square = male; circle = female; filled symbol = affected individual; the double line between individuals = consanguineous marriage; the diagonal line through a symbol = deceased family member.
Table 1. Two-point LOD scores for microsatellite markers used for linkage analysis of families PKRP117, PKRP262, PKRP344, and PKRP358.
| Marker | cM | Mb | Family | 0 | 0.01 | 0.05 | 0.1 | 0.2 | 0.3 | Zmax | θmax |
|---|---|---|---|---|---|---|---|---|---|---|---|
| D8S1110 |
67.27 |
52.26 |
PKRP117 |
-∞ |
-1.03 |
0.73 |
1.2 |
1.21 |
0.81 |
1.21 |
0.2 |
| |
|
|
PKRP262 |
-∞ |
-1.82 |
-0.63 |
-0.2 |
0.08 |
0.11 |
0.11 |
0.3 |
| |
|
|
PKRP344 |
-∞ |
0.03 |
0.6 |
0.73 |
0.66 |
0.43 |
0.73 |
0.1 |
| |
|
|
PKRP358 |
-∞ |
-0.61 |
-0.02 |
0.14 |
0.18 |
0.13 |
0.18 |
0.2 |
| D8S1737 |
67.27 |
53.87 |
PKRP117 |
3.22 |
3.14 |
2.86 |
2.48 |
1.68 |
0.85 |
3.22 |
0 |
| |
|
|
PKRP262 |
2.54 |
2.47 |
2.2 |
1.91 |
1.24 |
0.57 |
2.54 |
0 |
| |
|
|
PKRP344 |
1.62 |
1.58 |
1.43 |
1.24 |
0.85 |
0.48 |
1.62 |
0 |
| |
|
|
PKRP358 |
1.42 |
1.39 |
1.25 |
1.09 |
0.77 |
0.47 |
1.42 |
0 |
| D8S509 |
69.4 |
54.68 |
PKRP117 |
6.95 |
6.83 |
6.35 |
5.71 |
4.35 |
2.86 |
6.95 |
0 |
| |
|
|
PKRP262 |
2.51 |
2.44 |
2.18 |
1.86 |
1.2 |
0.53 |
2.51 |
0 |
| |
|
|
PKRP344 |
1.15 |
1.13 |
1.04 |
0.93 |
0.67 |
0.4 |
1.15 |
0 |
| |
|
|
PKRP358 |
2.13 |
2.08 |
1.85 |
1.57 |
1.03 |
0.54 |
2.13 |
0 |
| D8S2332 |
69.4 |
55.21 |
PKRP117 |
5.95 |
5.83 |
5.38 |
4.79 |
3.55 |
2.24 |
5.95 |
0 |
| |
|
|
PKRP262 |
2.51 |
2.44 |
2.18 |
1.86 |
1.2 |
0.53 |
2.51 |
0 |
| |
|
|
PKRP344 |
-∞ |
0.03 |
0.6 |
0.73 |
0.66 |
0.43 |
0.73 |
0.1 |
| PKRP358 | 2.52 | 2.45 | 2.2 | 1.88 | 1.27 | 0.7 | 2.52 | 0 |
In parallel, we enrolled three affected and nine unaffected members of PKRP262, four affected and three unaffected members of PKRP344, and four affected and three unaffected members of PKRP358 (Figure 1B-D). Fundus photographs of the affected individuals revealed typical symptoms of RP, including attenuated retinal arteries, waxy, pale optic disc, and bone spicule-like pigment deposits in the lateral and mid-periphery of the retina (Figure 2). Likewise, scotopic ERG recordings measured at −25 dB and photopic responses at 0 dB (30 Hz flicker) were undetectable in the affected individuals, suggestive of compromised rod photoreceptor and cone cells, while unaffected individuals exhibited rod and cone responses in the normal range (Figure 3).
Figure 2.
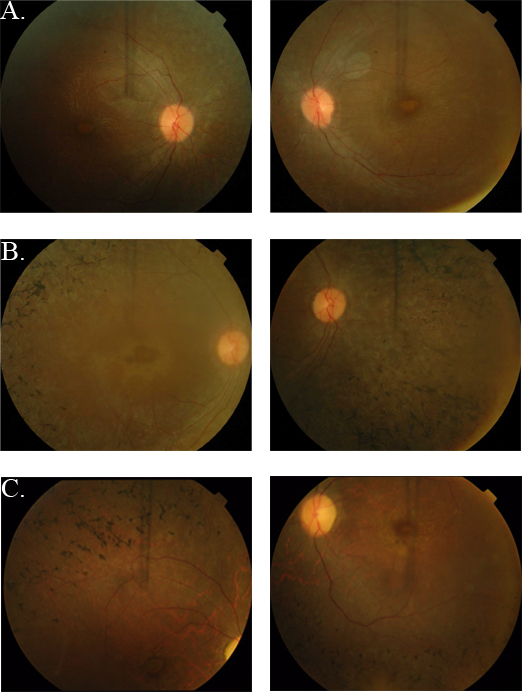
Fundus photographs of individuals with retinal dystrophy. A: OD and OS of individual 8 (affected, 25 years) of PKRP262. B: OD and OS of individual 12 (affected, 10 years) of PKRP262. C: OD and OS of individual 16 (affected, 12 years) of PKRP358. Fundus photographs of affected individuals show bone spicule-like pigmentation in the mid-periphery of the retina, attenuated retinal arteriole, severe maculopathy, and disc pallor. OD = oculus dexter; OS = oculus sinister.
Figure 3.

Electroretinography responses of individuals with retinal dystrophy. Stimulus conditions: scotopic 0 dB bright flashes elicit rod responses (left column of each pair) and photopic 0 dB, 30 Hz flicker elicits cone responses (right column of each pair). Responses are of A: OD and B: OS: individual 8 (affected, 25 years); C: OD and D: OS: individual 12 (affected, 10 years); E: OD and F: OS: individual 7 (unaffected, 60 years) of PKRP262. Affected individuals exhibit non-detectable electroretinography responses whereas the unaffected individual exhibits normal a and b waves suggestive of normal rod and cone functions. OD = oculus dexter; OS = oculus sinister.
Exclusion analysis localized all three familial cases (PKRP262, PKRP344, and PKRP358) to a region of chromosome 8q harboring RP1. Alleles for markers D8S1737, D8S509, and D8S2332 yielded 2-point LOD scores of 2.54, 2.51, and 2.51 at θ = 0 and 1.42, 2.13, and 2.52 at θ = 0 for families PKRP262 and PKRP358, respectively, while the alleles for markers D8S1737 and D8S509 yielded 2-point LOD scores of 1.62 and 1.15 at θ = 0 for PKRP344 (Table 1). Although these LOD scores are less than a 2-point LOD score of 3.0, which is traditionally considered sufficient for linkage, they are the maximum two-point LOD scores attainable by PKRP262, PKRP344, and PKRP358 and were considered worthy of further investigation because the known RP locus, RP1, was included in the region.
To identify the causal mutation responsible for the RP phenotype in these four families, we sequenced all coding exons and the exon–intron boundaries of RP1. In PKRP117, we identified a 1-bp homozygous deletion in exon 4, c.3697delT, that is predicted to result in a frameshift p.S1233Pfs22* (Figure 4A-C). Likewise, we identified a homozygous variation in intron 3, c.787+1G>A (p.I263Nfs8*), in PKRP262 that affects the conserved splice donor site (Figure 4D-F) and a 2 bp duplication in exon 2, c.551_552dupTA (p.Q185Yfs4*), in PKRP344 (Figure 4G-I).
Figure 4.

Sequence chromatograms of RP1 variations identified in families PKRP117, PKRP262, and PKRP344. A: Unaffected individual 7 homozygous for the wild-type. B: Unaffected individual 9 heterozygous. C: Affected individual 10 of PKRP117 homozygous for a single base pair deletion: c.3697delT in exon 4 of RP1. D: Unaffected individual 14 homozygous for the wild-type. E: Unaffected individual 11 heterozygous carrier. F: Affected individual 12 of PKRP262 homozygous for G to A transition in intron 3: c.787+1G >A. G: Control homozygous for the wild-type allele. H: Unaffected individual 9 heterozygous carrier. I: Affected individual 10 of PKRP344 homozygous for 2 bp duplication in exon 2: c.551_552dupTA.
In PKRP358, PCR of all three coding exons of RP1 did not yield any amplification products for the affected individuals while the genomic DNA of the unaffected individuals produced amplified products of the appropriate size. One plausible explanation is that the affected individuals of PKRP358 harbor a homozygous deletion that removes the coding region of RP1. We designed six primer pairs between exons 1 and 2 of RP1 and another six primer pairs downstream of RP1 using Primer3 software. The sequences are available upon request. Briefly, each primer pair was PCR-amplified in a 10 μl reaction volume containing 20 ng of genomic DNA, 1 μl of 10X PCR buffer, 2 mM dNTP mix, 1 μl of 10 μM forward and reverse primer, 500 mM Betaine, 700 mM Dimethyl sulfoxide (DMSO), and 0.2 U OneTaq DNA polymerase. The initial denaturation step was at 95 °C for 5 min followed by a two-step touchdown procedure. The first step of 10 cycles consisted of denaturation at 95 °C for 30 s, followed by a 66 °C annealing for 30 s (annealing temperature decreased by 1 °C after every cycle) and elongation at 72 °C for 45 s. The second step of 25 cycles consisted of denaturation at 95 °C for 30 s followed by 56 °C annealing for 30 s and elongation at 72 °C for 45 s, followed by a final elongation at 72 °C for 5 min. The amplification pattern of these 12 primer pairs helped us identify a 11,117 bp deletion (chr8:55,531,690-55, 542, 807) that removes all three coding exons of RP1 (Figure 5A-C).
Figure 5.

Identification of a splice donor site mutation in RP1. A: Illustration of the deletion breakpoints that remove all three coding exons of RP1. B: Forward sequence chromatograms of bases flanking the homozygous deletion. C: Reverse sequence chromatograms of bases flanking the homozygous deletion. D: In silico analysis of the splice donor site mutation in RP1. The HSF2 algorithm predicted a consensus value (CV) of 80.96 for the wild-type splice donor site (c.787+1G) and 48.29 (c.787+1A) for the mutant. The consensus value deviation of −32.67 suggests that the loss of the wild-type splice site will result in retention of intron 3 of RP1 resulting in a frame shift and eventually a premature stop codon (p.I263Nfs8*).
All four mutations segregated with the disease phenotype in their respective families. All affected individuals were homozygous for the mutant allele while unaffected individuals were either heterozygous carriers or homozygous for the wild-type allele. These mutations were absent in ethnically matched control chromosomes and were not found in the 1000 Genomes, the NHLBI Exome Sequencing Project, and the dbSNP databases.
The mutation identified in PKRP117, c.3697delT (p.S1233Pfs22*), is expected to produce a truncated protein lacking 903 amino acids of the C-terminal, while the transcript harboring the mutation identified in PKRP344, c.551_552dupTA (p.Q185Yfs4*), is expected to degrade through nonsense-mediated decay. Moreover, affected individuals in PKRP358 are believed to have no expression of RP1 due to the large deletion that removes all coding exons of RP1. Therefore, the respective mutations in PKRP117, PKRP344, and PKRP358 are likely to have caused the RP phenotype in these families. However, we sought additional evidence to strengthen the candidacy of the splice donor variation identified in PKRP262.
First, we examined the evolutionary conservation of c.787+1G and found that the +1G of the splice donor site is completely conserved in RP1 orthologs in general and mammals in particular (Appendix 2). Second, we evaluated the effect of the c.787+1G>A variation on RP1 mRNA splicing using Human Splice Finder 2 (HSF2). The HSF2 generated consensus values of 80.96 and 48.29 for the wild-type (c.787+1G) and mutant (c.787+1A) nucleotides, respectively. The predicted consensus value deviation of −32.67 for c.787+1G>A suggests that the loss of the wild-type splice site will result in the retention of intron 3 of RP1 (Figure 5D), resulting in a frame shift and eventually a premature stop codon (p.I263Nfs8*).
Discussion
We previously reported homozygous mutations responsible for arRP in three consanguineous familial cases that implicated RP1 in the pathogenesis of arRP [48]. Here, we report four consanguineous families recruited from the Punjab province of Pakistan with multiple members who manifest cardinal symptoms of RP. Linkage analyses with closely spaced STR markers localized the linkage interval in all four families to chromosome 8q12.1, harboring RP1, while Sanger sequencing of RP1 identified mutations that segregate with the disease phenotype in their respective families and are predicted to produce truncated RP1 proteins.
A total of 55 mutations have been identified in RP1 associated with RP in multiple ethnic populations [7,48,86-112]. 37 result in adRP while 17 mutations are responsible for arRP and one mutation responsible for sporadic case. The majority of the mutations identified in RP1 are either nonsense codons or lead to premature termination of RP1. Nonsense mutations in mammalian genes generally lead to unstable mRNAs that are degraded by nonsense-mediated decay [113]. However, nonsense-mediated decay does not occur if the mutation is present in the last exon, and therefore, most of the reported mutations are expected to produce stable transcripts translated into truncated proteins.
The precise mechanism that dictates whether a single allele would be sufficient or whether a homozygous mutation would be required for manifestation of the disease phenotype is not yet completely understood and probably varies among different examples. Nonetheless, it is conceivable that variations resulting in a mutant protein with a deleterious effect would manifest as an autosomal dominant trait while functionally null alleles would manifest as an autosomal recessive disease. We previously speculated that disruption of RP1 within or immediately after the bifocal gene product (BIF) domain might result in a protein with a deleterious effect whereas truncation of RP1 before the BIF motif or within the terminal portion of the protein would result in a loss of RP1 function [48].
More than 50 causal mutations have been identified in patients with adRP and arRP within the past decade, including our earlier observation (Table 2). As shown in Figure 6, pathogenic mutations in the heterozygous state in RP1 responsible for adRP seem to reside between amino acid residues 617–1551. In contrast, the homozygous mutations responsible for arRP cluster in two regions: first, between amino acids 193–736 and, second, between amino acids 1243–1890 (Figure 6). Taken together, these data support our earlier speculation [48]: Truncation mutations especially in the central region (800–1,200) of RP1 that is structurally not well defined, produce mutant proteins with a deleterious effect while mutations close to the N- and C-terminals of RP1 (>600 or <1,600) produce loss of function of the mutant proteins.
Table 2. List of pathogenic mutations identified in RP1.
| No. | Nucleotide change | Amino Acid Change | Coding Exon | Inheritance | Reference |
|---|---|---|---|---|---|
| 1 |
c.2029C>T |
p.Arg677X |
4 |
adRP |
[7,86,87] |
| 2 |
c.2232T>A |
p.Cys744X |
4 |
adRP |
[7] |
| 3 |
c.2303delC |
p.Lys769ArgfsX6 |
4 |
adRP |
[7] |
| 4 |
c.2287_2290del |
p.Asn763LeufsX11 |
4 |
adRP |
[90] |
| 5 |
c.2035C>T in cis with c.5377C>T |
p.Gln679X in cis with p.Pro1793Ser |
4 |
adRP |
[90] |
| 6 |
c.2280_2284del |
p.Leu762TyrfsX17 |
4 |
adRP |
[7] |
| 7 |
c.1498_1499insGT |
p.Met500SerfsX33 |
4 |
adRP |
[88] |
| 8 |
c.2171_2186del |
p.Gly724GlufsX9 |
4 |
adRP |
[88] |
| 9 |
c.2594_2596del |
p.Thr865_Leu866delinsIle |
4 |
adRP |
[88] |
| 10 |
c.2613dupA |
p.Arg872ThrfsX2 |
4 |
adRP |
[88] |
| 11 |
c.2284_2289del |
p.Leu762_Asn763del |
4 |
adRP |
[88] |
| 12 |
c.2206_2207insT |
p.Thr736IlefsX4 |
4 |
adRP |
[88] |
| 13 |
c.2239delA |
p.Ser747ValfsX16 |
4 |
adRP |
[87] |
| 14 |
c. 3157delT |
p.Tyr1053ThrfsX4 |
4 |
adRP |
[87] |
| 15 |
c.2185delG |
p.Glu729LysfsX9 |
4 |
adRP |
[90] |
| 16 |
c.2167G>T |
p.Gly723X |
4 |
adRP |
[89,90] |
| 17 |
c.2332A>T |
p.Lys778X incomplete penetrance |
4 |
adRP |
[91] |
| 18 |
c.1118C>T |
p.Thr373Ile |
4 |
arRP |
[90,93] |
| 19 |
c.2336_2337delCT |
p.Ser779X |
4 |
adRP |
[92] |
| 20 |
c.1606insTGAA |
p.Glu488X |
4 |
arRP |
[48] |
| 21 |
c.4703delA |
p.Arg1519fsX1521 |
4 |
arRP |
[48] |
| 22 |
c.5400delA |
p.Asn1751fsX1754 |
4 |
arRP |
[48] |
| 23 |
c.2005G>A |
p.Ala669Thr |
4 |
Sporadic case |
[93] |
| 24 |
c.2056C>T |
p.Gln686X |
4 |
adRP |
[95] |
| 25 |
c.2115delA |
p.Gly706ValfsX7 |
4 |
adRP |
[95] |
| 26 |
c.2164_2165delinsG |
p.Lys722GlufsX16 |
4 |
adRP |
[95] |
| 27 |
c.2590_2599del |
p.Ile864LysfsX11 incomplete penetrance |
4 |
adRP |
[96] |
| 28 |
c.2951A>G |
p.Asp984Gly |
4 |
adRP |
[94,97,102] |
| 29 |
c.2732C>A |
p.Ser911X incomplete penetrance |
4 |
adRP |
[96] |
| 30 |
c.2342C>G |
p.Ser781* |
4 |
adRP |
[111] |
| 31 |
c.606C>A |
p.Asp202Glu |
2 |
arRP |
[98] |
| 32 |
c.662delC |
p.Ala221GlyfsX43 |
3 |
arRP |
[98] |
| 33 |
c.2847delT |
p.Asn949LysfsX32 |
4 |
arRP |
[99] |
| 34 |
c.5_6delGT |
p.Ser2ArgfsX16 |
2 |
adRP |
[101] |
| 35 |
c.4108A>G |
p.Lys1370Glu |
4 |
adRP |
[100] |
| 36 |
c.4941dupT |
p.Pro1648SerfsX13 |
4 |
adRP |
[101] |
| 37 |
c.4955G>T |
p.Arg1652Leu |
4 |
adRP |
[100] |
| 38 |
c.2025delA |
p.Lys675AsnfsX7 |
4 |
adRP |
[112] |
| 39 |
c.2169delA |
p.Ile725TyrfsX13 |
4 |
adRP |
[112] |
| 40 |
c.2275A>T |
p.Arg759X |
4 |
adRP |
[112] |
| 41 |
c.1625C>G |
p.Ser542X |
4 |
arRP |
[105,107] |
| 42 |
c.33396G>A |
p.Trp1131X |
4 |
arRP |
[104] |
| 43 |
c.3418delGG |
p.Gly1140LysfsX4 |
4 |
arRP |
[109] |
| 44 |
c.3428delA |
p.Asn1143IlefsX25 |
4 |
arRP |
[104] |
| 45 |
c.3677_3678dupA |
p.Glu1227MetfsX29 |
4 |
arRP |
[104] |
| 46 |
c.4552A>T |
p.Lys1518X |
4 |
arRP |
[104] |
| 47 |
c.1012C>T |
p.Arg338X |
4 |
arRP |
[103] |
| 48 |
c.2180_2181delinsAA |
p.Cys727X |
4 |
adRP |
[110] |
| 49 |
c.2181T>A |
p.Cys727X |
4 |
adRP |
[110] |
| 50 |
c.2194C>T |
p.Gln732X |
4 |
adRP |
[110] |
| 51 |
c.5173C>T |
p.Gln1725X |
4 |
arRP |
[107] |
| 52 |
c.4327C>T |
p.Arg1443Trp |
4 |
adRP |
[106] |
| 53 |
c.4804C>T |
p.Gln1602X |
4 |
arRP |
[107] |
| 54 |
c.2585C>G |
p.Ser862X |
4 |
adRP |
[110] |
| 55 | c.1186C>T | p.Arg396X | 4 | arRP | [109] |
adRP: autosomal dominant RP; arRP: autosomal recessive RP.
Figure 6.

A schematic of the distribution of causal mutations reported in RP1 responsible for RP. The red and green bars indicate parts of RP1 where mutations responsible for autosomal dominant retinitis pigmentosa (RP) and autosomal recessive RP, respectively, have been identified. Asterisks are the mutations identified in this study. Note: The deletion identified in PKRP358 removes all three coding exons of RP1.
Moreover, the regions that include amino acid residues 600–750 and 1,250–1,550 include mutations that have been associated with both adRP and arRP, although no single mutation has been associated with both adRP and arRP. Thus, it is tempting to speculate that “zones” exist in RP1 in which the nature of the mutation instead of its location in the polypeptide dictates the inheritance pattern. Additional functional investigations are required to define these zones and elucidate mechanistic details of mutations leading to the particular genetic trait.
Acknowledgments
We are thankful to all family members for their participation in this study. This study was supported in part by Higher Education Commission, Islamabad, Pakistan, and by the National Eye Institute Grant R01EY021237 (RA and SAR).
Appendix 1. The primer sequences for the amplification of RP1.
To access the data, click or select the words “Appendix 1.”
Appendix 2. Sequence alignment of the exon-intron junction illustrating conservations of splice-donor site of intron 3 of RP1. The c.787+1G shown in red are fully conserved in RP1 orthologs.
To access the data, click or select the words “Appendix 2.”
References
- 1.Berson EL. Retinitis pigmentosa. The Friedenwald Lecture. Invest Ophthalmol Vis Sci. 1993;34:1659–76. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=8473105&dopt=Abstract [PubMed] [Google Scholar]
- 2.Bird AC. Retinal photoreceptor dystrophies LI. Edward Jackson Memorial Lecture. Am J Ophthalmol. 1995;119:543–62. doi: 10.1016/s0002-9394(14)70212-0. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=7733180&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 3.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–809. doi: 10.1016/S0140-6736(06)69740-7. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=17113430&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 4.Farrar GJ, Kenna P, Redmond R, McWilliam P, Bradley DG, Humphries MM, Sharp EM, Inglehearn CF, Bashir R, Jay M. Autosomal dominant retinitis pigmentosa: absence of the rhodopsin proline----histidine substitution (codon 23) in pedigrees from Europe. Am J Hum Genet. 1990;47:941–5. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=2239971&dopt=Abstract [PMC free article] [PubMed] [Google Scholar]
- 5.Kajiwara K, Hahn LB, Mukai S, Travis GH, Berson EL, Dryja TP. Mutations in the human retinal degeneration slow gene in autosomal dominant retinitis pigmentosa. Nature. 1991;354:480–3. doi: 10.1038/354480a0. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=1684223&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 6.Dryja TP, Hahn LB, Kajiwara K, Berson EL. Dominant and digenic mutations in the peripherin/RDS and ROM1 genes in retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1997;38:1972–82. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=9331261&dopt=Abstract [PubMed] [Google Scholar]
- 7.Bowne SJ, Daiger SP, Hims MM, Sohocki MM, Malone KA, McKie AB, Heckenlively JR, Birch DG, Inglehearn CF, Bhattacharya SS, Bird A, Sullivan LS. Mutations in the RP1 gene causing autosomal dominant retinitis pigmentosa. Hum Mol Genet. 1999;8:2121–8. doi: 10.1093/hmg/8.11.2121. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=10484783&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bessant DA, Payne AM, Mitton KP, Wang QL, Swain PK, Plant C, Bird AC, Zack DJ, Swaroop A, Bhattacharya SS. A mutation in NRL is associated with autosomal dominant retinitis pigmentosa. Nat Genet. 1999;21:355–6. doi: 10.1038/7678. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=10192380&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 9.Payne AM, Downes SM, Bessant DA, Plant C, Moore T, Bird AC, Bhattacharya SS. Genetic analysis of the guanylate cyclase activator 1B (GUCA1B) gene in patients with autosomal dominant retinal dystrophies. J Med Genet. 1999;36:691–3. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=10507726&dopt=Abstract [PMC free article] [PubMed] [Google Scholar]
- 10.Vithana EN, Abu-Safieh L, Allen MJ, Carey A, Papaioannou M, Chakarova C, Al-Maghtheh M, Ebenezer ND, Willis C, Moore AT, Bird AC, Hunt DM, Bhattacharya SS. A human homolog of yeast pre-mRNA splicing gene, PRP31, underlies autosomal dominant retinitis pigmentosa on chromosome 19q13.4 (RP11). Mol Cell. 2001;8:375–81. doi: 10.1016/s1097-2765(01)00305-7. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=11545739&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 11.McKie AB, McHale JC, Keen TJ, Tarttelin EE, Goliath R, van Lith-Verhoeven JJ, Greenberg J, Ramesar RS, Hoyng CB, Cremers FP, Mackey DA, Bhattacharya SS, Bird AC, Markham AF, Inglehearn CF. Mutations in the pre-mRNA splicing factor gene PRPC8 in autosomal dominant retinitis pigmentosa (RP13). Hum Mol Genet. 2001;10:1555–62. doi: 10.1093/hmg/10.15.1555. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=11468273&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 12.Wada Y, Abe T, Takeshita T, Sato H, Yanashima K, Tamai M. Mutation of human retinal fascin gene (FSCN2) causes autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2001;42:2395–400. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=11527955&dopt=Abstract [PubMed] [Google Scholar]
- 13.Chakarova CF, Hims MM, Bolz H, Abu-Safieh L, Patel RJ, Papaioannou MG, Inglehearn CF, Keen TJ, Willis C, Moore AT, Rosenberg T, Webster AR, Bird AC, Gal A, Hunt D, Vithana EN, Bhattacharya SS. Mutations in HPRP3, a third member of pre-mRNA splicing factor genes, implicated in autosomal dominant retinitis pigmentosa. Hum Mol Genet. 2002;11:87–92. doi: 10.1093/hmg/11.1.87. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=11773002&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 14.Bowne SJ, Sullivan LS, Blanton SH, Cepko CL, Blackshaw S, Birch DG, Hughbanks-Wheaton D, Heckenlively JR, Daiger SP. Mutations in the inosine monophosphate dehydrogenase 1 gene (IMPDH1) cause the RP10 form of autosomal dominant retinitis pigmentosa. Hum Mol Genet. 2002;11:559–68. doi: 10.1093/hmg/11.5.559. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=11875050&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maita H, Kitaura H, Keen TJ, Inglehearn CF, Ariga H, Iguchi-Ariga SM. PAP-1, the mutated gene underlying the RP9 form of dominant retinitis pigmentosa, is a splicing factor. Exp Cell Res. 2004;300:283–96. doi: 10.1016/j.yexcr.2004.07.029. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=15474994&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 16.Rebello G, Ramesar R, Vorster A, Roberts L, Ehrenreich L, Oppon E, Gama D, Bardien S, Greenberg J, Bonapace G, Waheed A, Shah GN, Sly WS. Apoptosis-inducing signal sequence mutation in carbonic anhydrase IV identified in patients with the RP17 form of retinitis pigmentosa. Proc Natl Acad Sci USA. 2004;101:6617–22. doi: 10.1073/pnas.0401529101. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=15090652&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abid A, Ismail M, Mehdi SQ, Khaliq S. Identification of novel mutations in the SEMA4A gene associated with retinal degenerative diseases. J Med Genet. 2006;43:378–81. doi: 10.1136/jmg.2005.035055. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=16199541&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coppieters F, Leroy BP, Beysen D, Hellemans J, De BK, Haegeman G, Robberecht K, Wuyts W, Coucke PJ, De BE. Recurrent mutation in the first zinc finger of the orphan nuclear receptor NR2E3 causes autosomal dominant retinitis pigmentosa. Am J Hum Genet. 2007;81:147–57. doi: 10.1086/518426. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=17564971&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chakarova CF, Papaioannou MG, Khanna H, Lopez I, Waseem N, Shah A, Theis T, Friedman J, Maubaret C, Bujakowska K, Veraitch B, Abd El-Aziz MM. Prescott dQ, Parapuram SK, Bickmore WA, Munro PM, Gal A, Hamel CP, Marigo V, Ponting CP, Wissinger B, Zrenner E, Matter K, Swaroop A, Koenekoop RK, Bhattacharya SS. Mutations in TOPORS cause autosomal dominant retinitis pigmentosa with perivascular retinal pigment epithelium atrophy. Am J Hum Genet. 2007;81:1098–103. doi: 10.1086/521953. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=17924349&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fingert JH, Oh K, Chung M, Scheetz TE, Andorf JL, Johnson RM, Sheffield VC, Stone EM. Association of a novel mutation in the retinol dehydrogenase 12 (RDH12) gene with autosomal dominant retinitis pigmentosa. Arch Ophthalmol. 2008;126:1301–7. doi: 10.1001/archopht.126.9.1301. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=18779497&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 21.Davidson AE, Millar ID, Urquhart JE, Burgess-Mullan R, Shweikh Y, Parry N, O’Sullivan J, Maher GJ, McKibbin M, Downes SM, Lotery AJ, Jacobson SG, Brown PD, Black GC, Manson FD. Missense mutations in a retinal pigment epithelium protein, bestrophin-1, cause retinitis pigmentosa. Am J Hum Genet. 2009;85:581–92. doi: 10.1016/j.ajhg.2009.09.015. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=19853238&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Friedman JS, Ray JW, Waseem N, Johnson K, Brooks MJ, Hugosson T, Breuer D, Branham KE, Krauth DS, Bowne SJ, Sullivan LS, Ponjavic V, Granse L, Khanna R, Trager EH, Gieser LM, Hughbanks-Wheaton D, Cojocaru RI, Ghiasvand NM, Chakarova CF, Abrahamson M, Goring HH, Webster AR, Birch DG, Abecasis GR, Fann Y, Bhattacharya SS, Daiger SP, Heckenlively JR, Andreasson S, Swaroop A. Mutations in a BTB-Kelch protein, KLHL7, cause autosomal-dominant retinitis pigmentosa. Am J Hum Genet. 2009;84:792–800. doi: 10.1016/j.ajhg.2009.05.007. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=19520207&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao C, Bellur DL, Lu S, Zhao F, Grassi MA, Bowne SJ, Sullivan LS, Daiger SP, Chen LJ, Pang CP, Zhao K, Staley JP, Larsson C. Autosomal-dominant retinitis pigmentosa caused by a mutation in SNRNP200, a gene required for unwinding of U4/U6 snRNAs. Am J Hum Genet. 2009;85:617–27. doi: 10.1016/j.ajhg.2009.09.020. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=19878916&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Menotti-Raymond M, Deckman KH, David V, Myrkalo J, O’Brien SJ, Narfstrom K. Mutation discovered in a feline model of human congenital retinal blinding disease. Invest Ophthalmol Vis Sci. 2010;51:2852–9. doi: 10.1167/iovs.09-4261. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=20053974&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tanackovic G, Ransijn A, Ayuso C, Harper S, Berson EL, Rivolta C. A missense mutation in PRPF6 causes impairment of pre-mRNA splicing and autosomal-dominant retinitis pigmentosa. Am J Hum Genet. 2011;88:643–9. doi: 10.1016/j.ajhg.2011.04.008. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=21549338&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bowne SJ, Humphries MM, Sullivan LS, Kenna PF, Tam LC, Kiang AS, Campbell M, Weinstock GM, Koboldt DC, Ding L, Fulton RS, Sodergren EJ, Allman D, Millington-Ward S, Palfi A, McKee A, Blanton SH, Slifer S, Konidari I, Farrar GJ, Daiger SP, Humphries P. A dominant mutation in RPE65 identified by whole-exome sequencing causes retinitis pigmentosa with choroidal involvement. Eur J Hum Genet. 2011;19:1074–81. doi: 10.1038/ejhg.2011.86. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=21654732&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen X, Liu Y, Sheng X, Tam PO, Zhao K, Chen X, Rong W, Liu Y, Liu X, Pan X, Chen LJ, Zhao Q, Vollrath D, Pang CP, Zhao C. PRPF4 mutations cause autosomal dominant retinitis pigmentosa. Hum Mol Genet. 2014;23:2926–39. doi: 10.1093/hmg/ddu005. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=24419317&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 28.Sullivan LS, Koboldt DC, Bowne SJ, Lang S, Blanton SH, Cadena E, Avery CE, Lewis RA, Webb-Jones K, Wheaton DH, Birch DG, Coussa R, Ren H, Lopez I, Chakarova C, Koenekoop RK, Garcia CA, Fulton RS, Wilson RK, Weinstock GM, Daiger SP. A dominant mutation in hexokinase 1 (HK1) causes retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2014;55:7147–58. doi: 10.1167/iovs.14-15419. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=25190649&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Y, Chen X, Xu Q, Gao X, Tam PO, Zhao K, Zhang X, Chen LJ, Jia W, Zhao Q, Vollrath D, Pang CP, Zhao C. SPP2 Mutations Cause Autosomal Dominant Retinitis Pigmentosa. Sci Rep. 2015;5:14867. doi: 10.1038/srep14867. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=26459573&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma X, Guan L, Wu W, Zhang Y, Zheng W, Gao YT, Long J, Wu N, Wu L, Xiang Y, Xu B, Shen M, Chen Y, Wang Y, Yin Y, Li Y, Xu H, Xu X, Li Y. Whole-exome sequencing identifies OR2W3 mutation as a cause of autosomal dominant retinitis pigmentosa. Sci Rep. 2015;5:9236. doi: 10.1038/srep09236. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=25783483&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosenfeld PJ, Cowley GS, McGee TL, Sandberg MA, Berson EL, Dryja TP. A null mutation in the rhodopsin gene causes rod photoreceptor dysfunction and autosomal recessive retinitis pigmentosa. Nat Genet. 1992;1:209–13. doi: 10.1038/ng0692-209. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=1303237&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 32.Bayes M, Giordano M, Balcells S, Grinberg D, Vilageliu L, Martinez I, Ayuso C, Benitez J, Ramos-Arroyo MA, Chivelet P. Homozygous tandem duplication within the gene encoding the beta-subunit of rod phosphodiesterase as a cause for autosomal recessive retinitis pigmentosa. Hum Mutat. 1995;5:228–34. doi: 10.1002/humu.1380050307. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=7599633&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 33.Huang SH, Pittler SJ, Huang X, Oliveira L, Berson EL, Dryja TP. Autosomal recessive retinitis pigmentosa caused by mutations in the alpha subunit of rod cGMP phosphodiesterase. Nat Genet. 1995;11:468–71. doi: 10.1038/ng1295-468. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=7493036&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 34.Dryja TP, Finn JT, Peng YW, McGee TL, Berson EL, Yau KW. Mutations in the gene encoding the alpha subunit of the rod cGMP-gated channel in autosomal recessive retinitis pigmentosa. Proc Natl Acad Sci USA. 1995;92:10177–81. doi: 10.1073/pnas.92.22.10177. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=7479749&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gu SM, Thompson DA, Srikumari CR, Lorenz B, Finckh U, Nicoletti A, Murthy KR, Rathmann M, Kumaramanickavel G, Denton MJ, Gal A. Mutations in RPE65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nat Genet. 1997;17:194–7. doi: 10.1038/ng1097-194. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=9326941&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 36.Maw MA, Kennedy B, Knight A, Bridges R, Roth KE, Mani EJ, Mukkadan JK, Nancarrow D, Crabb JW, Denton MJ. Mutation of the gene encoding cellular retinaldehyde-binding protein in autosomal recessive retinitis pigmentosa. Nat Genet. 1997;17:198–200. doi: 10.1038/ng1097-198. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=9326942&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 37.Banerjee P, Kleyn PW, Knowles JA, Lewis CA, Ross BM, Parano E, Kovats SG, Lee JJ, Penchaszadeh GK, Ott J, Jacobson SG, Gilliam TC. TULP1 mutation in two extended Dominican kindreds with autosomal recessive retinitis pigmentosa. Nat Genet. 1998;18:177–9. doi: 10.1038/ng0298-177. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=9462751&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 38.Cremers FP, van De Pol DJ. van DM, den Hollander AI, van Haren FJ, Knoers NV, Tijmes N, Bergen AA, Rohrschneider K, Blankenagel A, Pinckers AJ, Deutman AF, Hoyng CB. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum Mol Genet. 1998;7:355–62. doi: 10.1093/hmg/7.3.355. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=9466990&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 39.Nakazawa M, Wada Y, Tamai M. Arrestin gene mutations in autosomal recessive retinitis pigmentosa. Arch Ophthalmol. 1998;116:498–501. doi: 10.1001/archopht.116.4.498. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=9565049&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 40.Morimura H, Saindelle-Ribeaudeau F, Berson EL, Dryja TP. Mutations in RGR, encoding a light-sensitive opsin homologue, in patients with retinitis pigmentosa. Nat Genet. 1999;23:393–4. doi: 10.1038/70496. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=10581022&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 41.den Hollander AI, ten Brink JB, de Kok YJ. van SS, van den Born LI, van Driel MA, van De Pol DJ, Payne AM, Bhattacharya SS, Kellner U, Hoyng CB, Westerveld A, Brunner HG, Bleeker-Wagemakers EM, Deutman AF, Heckenlively JR, Cremers FP, Bergen AA. Mutations in a human homologue of Drosophila crumbs cause retinitis pigmentosa (RP12). Nat Genet. 1999;23:217–21. doi: 10.1038/13848. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=10508521&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 42.Gal A, Li Y, Thompson DA, Weir J, Orth U, Jacobson SG. pfelstedt-Sylla E, Vollrath D. Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat Genet. 2000;26:270–1. doi: 10.1038/81555. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=11062461&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 43.Rivolta C, Sweklo EA, Berson EL, Dryja TP. Missense mutation in the USH2A gene: association with recessive retinitis pigmentosa without hearing loss. Am J Hum Genet. 2000;66:1975–8. doi: 10.1086/302926. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=10775529&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bareil C, Hamel CP, Delague V, Arnaud B, Demaille J, Claustres M. Segregation of a mutation in CNGB1 encoding the beta-subunit of the rod cGMP-gated channel in a family with autosomal recessive retinitis pigmentosa. Hum Genet. 2001;108:328–34. doi: 10.1007/s004390100496. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=11379879&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 45.Ruiz A, Kuehn MH, Andorf JL, Stone E, Hageman GS, Bok D. Genomic organization and mutation analysis of the gene encoding lecithin retinol acyltransferase in human retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2001;42:31–7. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=11133845&dopt=Abstract [PubMed] [Google Scholar]
- 46.Nishiguchi KM, Friedman JS, Sandberg MA, Swaroop A, Berson EL, Dryja TP. Recessive NRL mutations in patients with clumped pigmentary retinal degeneration and relative preservation of blue cone function. Proc Natl Acad Sci USA. 2004;101:17819–24. doi: 10.1073/pnas.0408183101. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=15591106&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tuson M, Marfany G, Gonzalez-Duarte R. Mutation of CERKL, a novel human ceramide kinase gene, causes autosomal recessive retinitis pigmentosa (RP26). Am J Hum Genet. 2004;74:128–38. doi: 10.1086/381055. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=14681825&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Riazuddin SA, Zulfiqar F, Zhang Q, Sergeev YV, Qazi ZA, Husnain T, Caruso R, Riazuddin S, Sieving PA, Hejtmancik JF. Autosomal recessive retinitis pigmentosa is associated with mutations in RP1 in three consanguineous Pakistani families. Invest Ophthalmol Vis Sci. 2005;46:2264–70. doi: 10.1167/iovs.04-1280. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=15980210&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 49.Zangerl B, Goldstein O, Philp AR, Lindauer SJ, Pearce-Kelling SE, Mullins RF, Graphodatsky AS, Ripoll D, Felix JS, Stone EM, Acland GM, Aguirre GD. Identical mutation in a novel retinal gene causes progressive rod-cone degeneration in dogs and retinitis pigmentosa in humans. Genomics. 2006;88:551–63. doi: 10.1016/j.ygeno.2006.07.007. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=16938425&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abd El-Aziz MM, El-Ashry MF, Chan WM, Chong KL, Barragan I, Antinolo G, Pang CP, Bhattacharya SS. A novel genetic study of Chinese families with autosomal recessive retinitis pigmentosa. Ann Hum Genet. 2007;71:281–94. doi: 10.1111/j.1469-1809.2006.00333.x. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=17156103&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 51.Zhang Q, Zulfiqar F, Xiao X, Riazuddin SA, Ahmad Z, Caruso R, MacDonald I, Sieving P, Riazuddin S, Hejtmancik JF. Severe retinitis pigmentosa mapped to 4p15 and associated with a novel mutation in the PROM1 gene. Hum Genet. 2007;122:293–9. doi: 10.1007/s00439-007-0395-2. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=17605048&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 52.Hartong DT, Dange M, McGee TL, Berson EL, Dryja TP, Colman RF. Insights from retinitis pigmentosa into the roles of isocitrate dehydrogenases in the Krebs cycle. Nat Genet. 2008;40:1230–4. doi: 10.1038/ng.223. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=18806796&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.den Hollander AI, McGee TL, Ziviello C, Banfi S, Dryja TP, Gonzalez-Fernandez F, Ghosh D, Berson EL. A homozygous missense mutation in the IRBP gene (RBP3) associated with autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2009;50:1864–72. doi: 10.1167/iovs.08-2497. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=19074801&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang H, den Hollander AI, Moayedi Y, Abulimiti A, Li Y, Collin RW, Hoyng CB, Lopez I, Abboud EB, Al-Rajhi AA, Bray M, Lewis RA, Lupski JR, Mardon G, Koenekoop RK, Chen R. Mutations in SPATA7 cause Leber congenital amaurosis and juvenile retinitis pigmentosa. Am J Hum Genet. 2009;84:380–7. doi: 10.1016/j.ajhg.2009.02.005. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=19268277&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Escher P, Gouras P, Roduit R, Tiab L, Bolay S, Delarive T, Chen S, Tsai CC, Hayashi M, Zernant J, Merriam JE, Mermod N, Allikmets R, Munier FL, Schorderet DF. Mutations in NR2E3 can cause dominant or recessive retinal degenerations in the same family. Hum Mutat. 2009;30:342–51. doi: 10.1002/humu.20858. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=19006237&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Riazuddin SA, Iqbal M, Wang Y, Masuda T, Chen Y, Bowne S, Sullivan LS, Waseem NH, Bhattacharya S, Daiger SP, Zhang K, Khan SN, Riazuddin S, Hejtmancik JF, Sieving PA, Zack DJ, Katsanis N. A splice-site mutation in a retina-specific exon of BBS8 causes nonsyndromic retinitis pigmentosa. Am J Hum Genet. 2010;86:805–12. doi: 10.1016/j.ajhg.2010.04.001. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=20451172&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bandah-Rozenfeld D, Collin RW, Banin E, van den Born LI, Coene KL, Siemiatkowska AM, Zelinger L, Khan MI, Lefeber DJ, Erdinest I, Testa F, Simonelli F, Voesenek K, Blokland EA, Strom TM, Klaver CC, Qamar R, Banfi S, Cremers FP, Sharon D, den Hollander AI. Mutations in IMPG2, encoding interphotoreceptor matrix proteoglycan 2, cause autosomal-recessive retinitis pigmentosa. Am J Hum Genet. 2010;87:199–208. doi: 10.1016/j.ajhg.2010.07.004. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=20673862&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Collin RW, Safieh C, Littink KW, Shalev SA, Garzozi HJ, Rizel L, Abbasi AH, Cremers FP, den Hollander AI, Klevering BJ, Ben-Yosef T. Mutations in C2ORF71 cause autosomal-recessive retinitis pigmentosa. Am J Hum Genet. 2010;86:783–8. doi: 10.1016/j.ajhg.2010.03.016. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=20398884&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dvir L, Srour G, Abu-Ras R, Miller B, Shalev SA, Ben-Yosef T. Autosomal-recessive early-onset retinitis pigmentosa caused by a mutation in PDE6G, the gene encoding the gamma subunit of rod cGMP phosphodiesterase. Am J Hum Genet. 2010;87:258–64. doi: 10.1016/j.ajhg.2010.06.016. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=20655036&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Langmann T, Di Gioia SA, Rau I, Stohr H, Maksimovic NS, Corbo JC, Renner AB, Zrenner E, Kumaramanickavel G, Karlstetter M, Arsenijevic Y, Weber BH, Gal A, Rivolta C. Nonsense mutations in FAM161A cause RP28-associated recessive retinitis pigmentosa. Am J Hum Genet. 2010;87:376–81. doi: 10.1016/j.ajhg.2010.07.018. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=20705278&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li L, Nakaya N, Chavali VR, Ma Z, Jiao X, Sieving PA, Riazuddin S, Tomarev SI, Ayyagari R, Riazuddin SA, Hejtmancik JF. A mutation in ZNF513, a putative regulator of photoreceptor development, causes autosomal-recessive retinitis pigmentosa. Am J Hum Genet. 2010;87:400–9. doi: 10.1016/j.ajhg.2010.08.003. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=20797688&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Khan MI, Kersten FF, Azam M, Collin RW, Hussain A, Shah ST, Keunen JE, Kremer H, Cremers FP, Qamar R, den Hollander AI. CLRN1 mutations cause nonsyndromic retinitis pigmentosa. Ophthalmology. 2011;118:1444–8. doi: 10.1016/j.ophtha.2010.10.047. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=21310491&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 63.Stone EM, Luo X, Heon E, Lam BL, Weleber RG, Halder JA, Affatigato LM, Goldberg JB, Sumaroka A, Schwartz SB, Cideciyan AV, Jacobson SG. Autosomal recessive retinitis pigmentosa caused by mutations in the MAK gene. Invest Ophthalmol Vis Sci. 2011;52:9665–73. doi: 10.1167/iovs.11-8527. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=22110072&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zelinger L, Banin E, Obolensky A, Mizrahi-Meissonnier L, Beryozkin A, Bandah-Rozenfeld D, Frenkel S, Ben-Yosef T, Merin S, Schwartz SB, Cideciyan AV, Jacobson SG, Sharon D. A missense mutation in DHDDS, encoding dehydrodolichyl diphosphate synthase, is associated with autosomal-recessive retinitis pigmentosa in Ashkenazi Jews. Am J Hum Genet. 2011;88:207–15. doi: 10.1016/j.ajhg.2011.01.002. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=21295282&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Estrada-Cuzcano A, Neveling K, Kohl S, Banin E, Rotenstreich Y, Sharon D, Falik-Zaccai TC, Hipp S, Roepman R, Wissinger B, Letteboer SJ, Mans DA, Blokland EA, Kwint MP, Gijsen SJ, van Huet RA, Collin RW, Scheffer H, Veltman JA, Zrenner E, den Hollander AI, Klevering BJ, Cremers FP. Mutations in C8orf37, encoding a ciliary protein, are associated with autosomal-recessive retinal dystrophies with early macular involvement. Am J Hum Genet. 2012;90:102–9. doi: 10.1016/j.ajhg.2011.11.015. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=22177090&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Abu-Safieh L, Alrashed M, Anazi S, Alkuraya H, Khan AO, Al-Owain M, Al-Zahrani J, Al-Abdi L, Hashem M, Al-Tarimi S, Sebai MA, Shamia A, Ray-Zack MD, Nassan M, Al-Hassnan ZN, Rahbeeni Z, Waheeb S, Alkharashi A, Abboud E, Al-Hazzaa SA, Alkuraya FS. Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res. 2013;23:236–47. doi: 10.1101/gr.144105.112. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=23105016&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Davidson AE, Schwarz N, Zelinger L, Stern-Schneider G, Shoemark A, Spitzbarth B, Gross M, Laxer U, Sosna J, Sergouniotis PI, Waseem NH, Wilson R, Kahn RA, Plagnol V, Wolfrum U, Banin E, Hardcastle AJ, Cheetham ME, Sharon D, Webster AR. Mutations in ARL2BP, encoding ADP-ribosylation-factor-like 2 binding protein, cause autosomal-recessive retinitis pigmentosa. Am J Hum Genet. 2013;93:321–9. doi: 10.1016/j.ajhg.2013.06.003. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=23849777&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Davidson AE, Sergouniotis PI, Mackay DS, Wright GA, Waseem NH, Michaelides M, Holder GE, Robson AG, Moore AT, Plagnol V, Webster AR. RP1L1 variants are associated with a spectrum of inherited retinal diseases including retinitis pigmentosa and occult macular dystrophy. Hum Mutat. 2013;34:506–14. doi: 10.1002/humu.22264. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=23281133&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 69.Nishiguchi KM, Tearle RG, Liu YP, Oh EC, Miyake N, Benaglio P, Harper S, Koskiniemi-Kuendig H, Venturini G, Sharon D, Koenekoop RK, Nakamura M, Kondo M, Ueno S, Yasuma TR, Beckmann JS, Ikegawa S, Matsumoto N, Terasaki H, Berson EL, Katsanis N, Rivolta C. Whole genome sequencing in patients with retinitis pigmentosa reveals pathogenic DNA structural changes and NEK2 as a new disease gene. Proc Natl Acad Sci USA. 2013;110:16139–44. doi: 10.1073/pnas.1308243110. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=24043777&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Siemiatkowska AM, van den Born LI, van Hagen PM, Stoffels M, Neveling K, Henkes A, Kipping-Geertsema M, Hoefsloot LH, Hoyng CB, Simon A, den Hollander AI, Cremers FP, Collin RW. Mutations in the mevalonate kinase (MVK) gene cause nonsyndromic retinitis pigmentosa. Ophthalmology. 2013;120:2697–705. doi: 10.1016/j.ophtha.2013.07.052. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=24084495&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 71.Ajmal M, Khan MI, Neveling K, Khan YM, Azam M, Waheed NK, Hamel CP, Ben-Yosef T, De BE, Koenekoop RK, Collin RW, Qamar R, Cremers FP. A missense mutation in the splicing factor gene DHX38 is associated with early-onset retinitis pigmentosa with macular coloboma. J Med Genet. 2014;51:444–8. doi: 10.1136/jmedgenet-2014-102316. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=24737827&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 72.El SS, Neuille M, Terray A, Orhan E, Condroyer C, Demontant V, Michiels C, Antonio A, Boyard F, Lancelot ME, Letexier M, Saraiva JP, Leveillard T, Mohand-Said S, Goureau O, Sahel JA, Zeitz C, Audo I. Whole-exome sequencing identifies KIZ as a ciliary gene associated with autosomal-recessive rod-cone dystrophy. Am J Hum Genet. 2014;94:625–33. doi: 10.1016/j.ajhg.2014.03.005. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=24680887&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jin ZB, Huang XF, Lv JN, Xiang L, Li DQ, Chen J, Huang C, Wu J, Lu F, Qu J. SLC7A14 linked to autosomal recessive retinitis pigmentosa. Nat Commun. 2014;5:3517. doi: 10.1038/ncomms4517. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=24670872&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ali S, Khan SY, Naeem MA, Khan SN, Husnain T, Riazuddin S, Ayyagari R, Riazuddin S, Hejtmancik JF, Riazuddin SA. Phenotypic variability associated with the D226N allele of IMPDH1. Ophthalmology. 2015;122:429–31. doi: 10.1016/j.ophtha.2014.07.057. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=25439607&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 75.Wang F, Li H, Xu M, Li H, Zhao L, Yang L, Zaneveld JE, Wang K, Li Y, Sui R, Chen R. A homozygous missense mutation in NEUROD1 is associated with nonsyndromic autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2015;56:150–5. doi: 10.1167/iovs.14-15382. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=25477324&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Haer-Wigman L, Newman H, Leibu R, Bax NM, Baris HN, Rizel L, Banin E, Massarweh A, Roosing S, Lefeber DJ, Zonneveld-Vrieling MN, Isakov O, Shomron N, Sharon D, den Hollander AI, Hoyng CB, Cremers FP, Ben-Yosef T. Non-syndromic retinitis pigmentosa due to mutations in the mucopolysaccharidosis type IIIC gene, heparan-alpha-glucosaminide N-acetyltransferase (HGSNAT). Hum Mol Genet. 2015;24:3742–51. doi: 10.1093/hmg/ddv118. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=25859010&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Avila-Fernandez A, Perez-Carro R, Corton M, Lopez-Molina MI, Campello L, Garanto A, Fernandez-Sanchez L, Duijkers L, Lopez-Martinez MA, Riveiro-Alvarez R, Da Silva LR, Sanchez-Alcudia R, Martin-Garrido E, Reyes N, Garcia-Garcia F, Dopazo J, Garcia-Sandoval B, Collin RW, Cuenca N, Ayuso C. Whole-exome sequencing reveals ZNF408 as a new gene associated with autosomal recessive retinitis pigmentosa with vitreal alterations. Hum Mol Genet. 2015;24:4037–48. doi: 10.1093/hmg/ddv140. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=25882705&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 78.Buraczynska M, Wu W, Fujita R, Buraczynska K, Phelps E, Andreasson S, Bennett J, Birch DG, Fishman GA, Hoffman DR, Inana G, Jacobson SG, Musarella MA, Sieving PA, Swaroop A. Spectrum of mutations in the RPGR gene that are identified in 20% of families with X-linked retinitis pigmentosa. Am J Hum Genet. 1997;61:1287–92. doi: 10.1086/301646. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=9399904&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mears AJ, Gieser L, Yan D, Chen C, Fahrner S, Hiriyanna S, Fujita R, Jacobson SG, Sieving PA, Swaroop A. Protein-truncation mutations in the RP2 gene in a North American cohort of families with X-linked retinitis pigmentosa. Am J Hum Genet. 1999;64:897–900. doi: 10.1086/302298. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=10053026&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Coene KL, Roepman R, Doherty D, Afroze B, Kroes HY, Letteboer SJ, Ngu LH, Budny B. van WE, Gorden NT, Azhimi M, Thauvin-Robinet C, Veltman JA, Boink M, Kleefstra T, Cremers FP, van BH, de Brouwer AP. OFD1 is mutated in X-linked Joubert syndrome and interacts with LCA5-encoded lebercilin. Am J Hum Genet. 2009;85:465–81. doi: 10.1016/j.ajhg.2009.09.002. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=19800048&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pierce EA, Quinn T, Meehan T, McGee TL, Berson EL, Dryja TP. Mutations in a gene encoding a new oxygen-regulated photoreceptor protein cause dominant retinitis pigmentosa. Nat Genet. 1999;22:248–54. doi: 10.1038/10305. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=10391211&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 82.Liu Q, Zhou J, Daiger SP, Farber DB, Heckenlively JR, Smith JE, Sullivan LS, Zuo J, Milam AH, Pierce EA. Identification and subcellular localization of the RP1 protein in human and mouse photoreceptors. Invest Ophthalmol Vis Sci. 2002;43:22–32. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=11773008&dopt=Abstract [PMC free article] [PubMed] [Google Scholar]
- 83.Kaul H, Riazuddin SA, Shahid M, Kousar S, Butt NH, Zafar AU, Khan SN, Husnain T, Akram J, Hejtmancik JF, Riazuddin S. Autosomal recessive congenital cataract linked to EPHA2 in a consanguineous Pakistani family. Mol Vis. 2010;16:511–7. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=20361013&dopt=Abstract [PMC free article] [PubMed] [Google Scholar]
- 84.Lathrop GM, Lalouel JM. Easy calculations of lod scores and genetic risks on small computers. Am J Hum Genet. 1984;36:460–5. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=6585139&dopt=Abstract [PMC free article] [PubMed] [Google Scholar]
- 85.Schaffer AA, Gupta SK, Shriram K, Cottingham RW., Jr Avoiding recomputation in linkage analysis. Hum Hered. 1994;44:225–37. doi: 10.1159/000154222. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=8056435&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 86.Guillonneau X, Piriev NI, Danciger M, Kozak CA, Cideciyan AV, Jacobson SG, Farber DB. A nonsense mutation in a novel gene is associated with retinitis pigmentosa in a family linked to the RP1 locus. Hum Mol Genet. 1999;8:1541–6. doi: 10.1093/hmg/8.8.1541. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=10401003&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 87.Jacobson SG, Cideciyan AV, Iannaccone A, Weleber RG, Fishman GA, Maguire AM, Affatigato LM, Bennett J, Pierce EA, Danciger M, Farber DB, Stone EM. Disease expression of RP1 mutations causing autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2000;41:1898–908. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=10845615&dopt=Abstract [PubMed] [Google Scholar]
- 88.Payne A, Vithana E, Khaliq S, Hameed A, Deller J, Abu-Safieh L, Kermani S, Leroy BP, Mehdi SQ, Moore AT, Bird AC, Bhattacharya SS. RP1 protein truncating mutations predominate at the RP1 adRP locus. Invest Ophthalmol Vis Sci. 2000;41:4069–73. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=11095597&dopt=Abstract [PubMed] [Google Scholar]
- 89.Sohocki MM, Daiger SP, Bowne SJ, Rodriquez JA, Northrup H, Heckenlively JR, Birch DG, Mintz-Hittner H, Ruiz RS, Lewis RA, Saperstein DA, Sullivan LS. Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum Mutat. 2001;17:42–51. doi: 10.1002/1098-1004(2001)17:1<42::AID-HUMU5>3.0.CO;2-K. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=11139241&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Berson EL, Grimsby JL, Adams SM, McGee TL, Sweklo E, Pierce EA, Sandberg MA, Dryja TP. Clinical features and mutations in patients with dominant retinitis pigmentosa-1 (RP1). Invest Ophthalmol Vis Sci. 2001;42:2217–24. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=11527933&dopt=Abstract [PubMed] [Google Scholar]
- 91.Dietrich K, Jacobi FK, Tippmann S, Schmid R, Zrenner E, Wissinger B, Apfelstedt-Sylla E. A novel mutation of the RP1 gene (Lys778ter) associated with autosomal dominant retinitis pigmentosa. Br J Ophthalmol. 2002;86:328–32. doi: 10.1136/bjo.86.3.328. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=11864893&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kawamura M, Wada Y, Noda Y, Itabashi T, Ogawa S, Sato H, Tanaka K, Ishibashi T, Tamai M. Novel 2336-2337delCT mutation in RP1 gene in a Japanese family with autosomal dominant retinitis pigmentosa. Am J Ophthalmol. 2004;137:1137–9. doi: 10.1016/j.ajo.2003.12.037. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=15183808&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 93.Khaliq S, Abid A, Ismail M, Hameed A, Mohyuddin A, Lall P, Aziz A, Anwar K, Mehdi SQ. Novel association of RP1 gene mutations with autosomal recessive retinitis pigmentosa. J Med Genet. 2005;42:436–8. doi: 10.1136/jmg.2004.024281. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=15863674&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang DY, Fan BJ, Chan WM, Tam OS, Chiang WY, Lam SC, Pang CP. Digenic association of RHO and RP1 genes with retinitis pigmentosa among Chinese population in Hong Kong. Zhonghua Yi Xue Za Zhi. 2005;85:1613–7. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=16185528&dopt=Abstract [PubMed] [Google Scholar]
- 95.Gamundi MJ, Hernan I, Martinez-Gimeno M, Maseras M, Garcia-Sandoval B, Ayuso C, Antinolo G, Baiget M, Carballo M. Three novel and the common Arg677Ter RP1 protein truncating mutations causing autosomal dominant retinitis pigmentosa in a Spanish population. BMC Med Genet. 2006;7:35. doi: 10.1186/1471-2350-7-35. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=16597330&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Roberts L, Bartmann L, Ramesar R, Greenberg J. Novel variants in the hotspot region of RP1 in South African patients with retinitis pigmentosa. Mol Vis. 2006;12:177–83. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=16568030&dopt=Abstract [PubMed] [Google Scholar]
- 97.Chiang SW, Wang DY, Chan WM, Tam PO, Chong KK, Lam DS, Pang CP. A novel missense RP1 mutation in retinitis pigmentosa. Eye (Lond) 2006;20:602–5. doi: 10.1038/sj.eye.6701944. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=15933747&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 98.Aldahmesh MA, Safieh LA, Alkuraya H, Al-Rajhi A, Shamseldin H, Hashem M, Alzahrani F, Khan AO, Alqahtani F, Rahbeeni Z, Alowain M, Khalak H, Al-Hazzaa S, Meyer BF, Alkuraya FS. Molecular characterization of retinitis pigmentosa in Saudi Arabia. Mol Vis. 2009;15:2464–9. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=19956407&dopt=Abstract [PMC free article] [PubMed] [Google Scholar]
- 99.Singh HP, Jalali S, Narayanan R, Kannabiran C. Genetic analysis of Indian families with autosomal recessive retinitis pigmentosa by homozygosity screening. Invest Ophthalmol Vis Sci. 2009;50:4065–71. doi: 10.1167/iovs.09-3479. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=19339744&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang X, Chen LJ, Law JP, Lai TY, Chiang SW, Tam PO, Chu KY, Wang N, Zhang M, Pang CP. Differential pattern of RP1 mutations in retinitis pigmentosa. Mol Vis. 2010;16:1353–60. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=20664799&dopt=Abstract [PMC free article] [PubMed] [Google Scholar]
- 101.Chen LJ, Lai TY, Tam PO, Chiang SW, Zhang X, Lam S, Lai RY, Lam DS, Pang CP. Compound heterozygosity of two novel truncation mutations in RP1 causing autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2010;51:2236–42. doi: 10.1167/iovs.09-4437. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=19933189&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 102.Kim C, Kim KJ, Bok J, Lee EJ, Kim DJ, Oh JH, Park SP, Shin JY, Lee JY, Yu HG. Microarray-based mutation detection and phenotypic characterization in Korean patients with retinitis pigmentosa. Mol Vis. 2012;18:2398–410. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=23049240&dopt=Abstract [PMC free article] [PubMed] [Google Scholar]
- 103.Siemiatkowska AM, Astuti GD, Arimadyo K, den Hollander AI, Faradz SM, Cremers FP, Collin RW. Identification of a novel nonsense mutation in RP1 that causes autosomal recessive retinitis pigmentosa in an Indonesian family. Mol Vis. 2012;18:2411–9. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=23077400&dopt=Abstract [PMC free article] [PubMed] [Google Scholar]
- 104.Al-Rashed M, Abu SL, Alkuraya H, Aldahmesh MA, Alzahrani J, Diya M, Hashem M, Hardcastle AJ, Al-Hazzaa SA, Alkuraya FS. RP1 and retinitis pigmentosa: report of novel mutations and insight into mutational mechanism. Br J Ophthalmol. 2012;96:1018–22. doi: 10.1136/bjophthalmol-2011-301134. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=22317909&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 105.Avila-Fernandez A, Corton M, Nishiguchi KM, Munoz-Sanz N, Benavides-Mori B, Blanco-Kelly F, Riveiro-Alvarez R, Garcia-Sandoval B, Rivolta C, Ayuso C. Identification of an RP1 prevalent founder mutation and related phenotype in Spanish patients with early-onset autosomal recessive retinitis. Ophthalmology. 2012;119:2616–21. doi: 10.1016/j.ophtha.2012.06.033. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=22917891&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 106.Ma L, Sheng XL, Li HP, Zhang FX, Liu YN, Rong WN, Zhang JL. Identification of a novel p.R1443W mutation in RP1 gene associated with retinitis pigmentosa sine pigmento. Int J Ophthalmol. 2013;6:430–5. doi: 10.3980/j.issn.2222-3959.2013.04.04. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=23991373&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Corton M, Nishiguchi KM, Avila-Fernandez A, Nikopoulos K, Riveiro-Alvarez R, Tatu SD, Ayuso C, Rivolta C. Exome sequencing of index patients with retinal dystrophies as a tool for molecular diagnosis. PLoS One. 2013;8:e65574. doi: 10.1371/journal.pone.0065574. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=23940504&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.de Sousa DM, Hernan I, Pascual B, Borras E, Mane B, Gamundi MJ, Carballo M. Detection of novel mutations that cause autosomal dominant retinitis pigmentosa in candidate genes by long-range PCR amplification and next-generation sequencing. Mol Vis. 2013;19:654–64. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=23559859&dopt=Abstract [PMC free article] [PubMed] [Google Scholar]
- 109.Bocquet B, Marzouka NA, Hebrard M, Manes G, Senechal A, Meunier I, Hamel CP. Homozygosity mapping in autosomal recessive retinitis pigmentosa families detects novel mutations. Mol Vis. 2013;19:2487–500. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=24339724&dopt=Abstract [PMC free article] [PubMed] [Google Scholar]
- 110.Sullivan LS, Bowne SJ, Reeves MJ, Blain D, Goetz K, Ndifor V, Vitez S, Wang X, Tumminia SJ, Daiger SP. Prevalence of mutations in eyeGENE probands with a diagnosis of autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2013;54:6255–61. doi: 10.1167/iovs.13-12605. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=23950152&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pan X, Chen X, Liu X, Gao X, Kang X, Xu Q, Chen X, Zhao K, Zhang X, Chu Q, Wang X, Zhao C. Mutation analysis of pre-mRNA splicing genes in Chinese families with retinitis pigmentosa. Mol Vis. 2014;20:770–9. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=24940031&dopt=Abstract [PMC free article] [PubMed] [Google Scholar]
- 112.Audo I, Mohand-Said S, Dhaenens CM, Germain A, Orhan E, Antonio A, Hamel C, Sahel JA, Bhattacharya SS, Zeitz C. RP1 and autosomal dominant rod-cone dystrophy: novel mutations, a review of published variants, and genotype-phenotype correlation. Hum Mutat. 2012;33:73–80. doi: 10.1002/humu.21640. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=22052604&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 113.Hentze MW, Kulozik AE. A perfect message: RNA surveillance and nonsense-mediated decay. Cell. 1999;96:307–10. doi: 10.1016/s0092-8674(00)80542-5. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=10025395&dopt=Abstract [DOI] [PubMed] [Google Scholar]