

Abstract
The canonical transient receptor potential channel 4 (TRPC4) comprises an endothelial store–operated Ca2+ entry channel, and TRPC4 inactivation confers a survival benefit in pulmonary arterial hypertension (PAH). Endothelial Ca2+ signals mediated by TRPC4 enhance vascular permeability in vitro, but the contribution of TRPC4-dependent Ca2+ signals to the regulation of endothelial permeability in PAH is poorly understood. We tested the hypothesis that TRPC4 increases vascular permeability and alters the frequency of endothelial Ca2+ transients in PAH. We measured permeability in isolated lungs, and found that TRPC4 exaggerated permeability responses to thapsigargin in Sugen/hypoxia-treated PAH rats. We compared endothelial Ca2+ activity of wild-type with TRPC4-knockout rats using confocal microscopy, and evaluated how Ca2+ signals were influenced in response to thapsigargin and sequential treatment with acetylcholine. We found that thapsigargin-stimulated Ca2+ signals were increased in PAH, and recovered by TRPC4 inactivation. Store depletion revealed bimodal Ca2+ responses to acetylcholine, with both short- and long-duration populations. Our results show that TRPC4 underlies an exaggerated endothelial permeability response in PAH. Furthermore, TRPC4 increased the frequency of endothelial Ca2+ transients in severe PAH, suggesting that TRPC4 provides a Ca2+ source associated with endothelial dysfunction in the pathophysiology of PAH. This phenomenon represents a new facet of the etiology of PAH, and may contribute to PAH vasculopathy by enabling inflammatory mediator flux across the endothelial barrier.
The current model of the endothelium extends beyond the concept of a homogeneous cell monolayer in contact with the blood.1, 2, 3, 4, 5, 6 Recent advances in high-resolution microscopy have enabled the measurement of individual cellular behavior rather than tissue-wide averaged responses, and we are now beginning to understand the heterogeneous nature of local endothelia and their major impact on physiology and disease.7, 8, 9, 10 Endothelial dysfunction underlies the pathogenesis of pulmonary arterial hypertension (PAH), a deadly disease of high pulmonary artery pressure culminating in right-sided heart failure. Endothelial dysfunction in PAH is characterized by the aberrant production of endothelial-dependent vasoconstrictors and vasodilators within pulmonary arterioles, leading to sustained elevation of pulmonary artery pressure, and the initiation of endothelial hyperproliferation, leading to occlusive lesion formation within the microcirculation.11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22
Although spatially heterogeneous, vascular dysfunction in PAH is linked through the nexus of cell signaling mediated by intracellular Ca2+, where diverse spatially and temporally encoded endothelial and smooth muscle calcium signals are upstream regulators of vasoconstrictor and vasodilator production,23, 24, 25 and elevation of cytosolic calcium via canonical transient receptor potential proteins stimulates cell proliferation associated with vascular remodeling in pulmonary arterioles.15, 26 Activation of endothelial store–operated Ca2+ entry increases vascular permeability in extra-alveolar pulmonary arteries,6, 15, 27 although its contribution to the pathophysiology of PAH is controversial.28
Recently, our laboratory found that genetic knockout of the canonical transient receptor potential channel 4 (TRPC4), an ion channel that is involved in the formation of a store-operated Ca2+ entry complex, confers a substantial survival benefit in severe PAH.29 This phenomenon is associated with a reduction of both vascular lesion density and number,29 and maintenance of cardiac output.29 We identified ongoing endothelial Ca2+ transients in wild-type and TRPC4-deficient rats, and found that TRPC4 potentiates tissue-wide Ca2+ responses to acetylcholine (ACh). Furthermore, we found that PAH impairs endothelial-dependent vasodilation in response to ACh. However, the precise role of TRPC4 as a subunit of a functional store-operated Ca2+ entry complex involved in regulating localized endothelial Ca2+ signals and its impact on lung vascular permeability in PAH remain unknown. Therefore, we tested the hypothesis that TRPC4 underlies a permeability defect and encodes localized, high-frequency endothelial Ca2+ signaling events in severe PAH.
Materials and Methods
Animals
All experimental procedures were performed in accordance with and approved by the Institutional Animal Care and Use Committee of the University of South Alabama (Mobile, AL). Severe occlusive PAH in rats was induced by a single s.c. 20 mg/kg injection of the vascular endothelial growth factor receptor type II inhibitor SU5416 (Cayman Chemical, Ann Arbor, MI) at 7 weeks after birth, followed by exposure to 3 weeks of normobaric hypoxia (10% O2) and then reexposure to normoxia (21% O2) for 2 to 3 additional weeks (PAH). TRPC4-knockout Fischer rats were generated by Transposagen Biopharmaceuticals (Lexington, KY), as part of the Knockout Rat Consortium (Trpc4tm1Bni, targeted mutation 1, Bernd Nilius), and were bred and genotyped both at Transposagen Biopharmaceuticals and at the University of South Alabama. Control rats were exposed to normoxia for 6 to 8 weeks. All rats were male and aged 12 to 16 weeks.
Genotyping of the TRPC4-Knockout Rats
Rat tail snips were collected according to the guidelines of the University of South Alabama Animal Care and Use Committee. DNA was extracted from tail snips, as described previously,29 and 2 μL of the resulting DNA solution was subjected to PCR analysis using three primers (primer A, 5′-GTGTTGGTCTCCATTACTTCAGCT-3′; primer B, 5′-ATTCTTCCCTTTGAGCCCACT-3′; and transposon primer, 5′-CTGACCTAAGACAGGGAATT-3′) in a total volume of 20 μL containing 1× GoTaq Green PCR master mix (Promega, Madison, WI) and 1 μmol/L of each primer. The cycling parameters were denaturation at 94°C for 5 minutes; then 30 cycles of 94°C for 30 seconds, 54°C for 30 seconds, and 72°C for 1 minute; and extension at 72°C for 7 minutes.
Isolated Lung and Assessment of Endothelial Permeability
Animals were anesthetized using 65 mg pentobarbital (Nembutal; Sigma-Aldrich, St. Louis, MO)/kg body weight. Once a surgical plane was achieved, defined by the absence of a withdrawal reflex after toe and tail pinch, animals were intubated and ventilated, a sternotomy was performed, and pulmonary artery and left ventricular catheters were inserted. Blood was taken by heart puncture from the right ventricle. Heart and lungs were removed en bloc and suspended in a humidified chamber where mechanical ventilation and blood flow was established. Rat lungs were perfused at constant flow (40 mL/minute per kg body weight) with buffer (in mmol/L: 119.0 NaCl, 4.7 KCl, 1.17 MgSO4, 1.0 Na2HPO4, 1.18 KH2PO4, 2.2 NaHCO3, and 5.5 glucose) containing 4% bovine serum albumin/6% autologous blood and physiological (2.2 mmol/L) CaCl2 at pH 7.30 to 7.40 at 38°C, and mean pulmonary artery pressure measurements were recorded.
Filtration coefficient (Kf) values were measured as previously described, using zone 3 conditions.30 Baseline Kf was calculated as the rate of weight gain obtained 13 to 15 minutes after a 10 cm H2O increase in pulmonary venous pressure, normalized per 100 g predicted wet lung weight. A dose of 75 nmol/L thapsigargin was added to the perfusate reservoir and circulated for 10 minutes, and a second Kf was determined after the treatment in both the wild-type and TRPC4-knockout groups under normotensive and pulmonary arterial hypertensive conditions.
Calcium Imaging
Intrapulmonary branches of the left pulmonary artery were dissected from the lung in cold HEPES/bicarbonate-buffered physiological saline solution. Pulmonary arteries (approximately 5 mm in length) were dissected from surrounding connective tissue, cut longitudinally, and mounted in a custom viewing chamber. Tissue preparations were loaded with 1 mL of physiological saline solution (in mmol/L: 2 CaCl, 134 NaCl, 6 KCl, 1 MgCl, 10 HEPES, and 10 glucose) containing 10 μmol/L Fluo-4 fluorescent Ca2+ indicator dye and 0.03% Pluronic for 35 minutes at 25°C. After a 5-minute wash and a 10-minute equilibration period, tissues were mounted in a custom chamber with 1 mL of buffer and viewed on a Perkin Elmer RS-3 spinning disk inverted confocal microscope (Perkin Elmer, Waltham, MA). Excitation and emission wavelengths were 488 and 510 nm, respectively. Fluorescence intensity recordings were captured at ×20 magnification. Each recording began with an unstimulated 3-minute interval, followed by stimulation with 1 μmol/L thapsigargin, and a 3-minute treatment sampling interval, then 1 μmol/L ACh and a final 3-minute treatment sampling interval. Stimulation was performed with volume swaps using 0.5 mL of drug solution at 2 μmol/L for either ACh or thapsigargin, swapped with 0.5 mL of viewing buffer for an effective concentration of 1 μmol/L. Image sequences were acquired with Perkin Elmer Ultraview software version 1.0.0.9 at 9.8 frames per second. Ca2+ events were analyzed offline with LC_pro custom analysis software version 12.18.11 (linear base) as a plugin for ImageJ software version 1.49v (NIH, Bethesda, MD) and statistical processing using custom analysis scripts with R software version 3.1.2 (“Pumpkin Helmet”; http://www.r-project.org).31, 32
Image Registration
Treatment with thapsigargin and ACh caused significant movement of the endothelial cell plane, because of the vasodilation effect of both drugs. We therefore used StackReg version 07.07.11, ImageJ-based image stack registration software, to compensate for this motion. Image sequences of endothelial Ca2+ activity en face were registered by selecting the anchor frame to be the last frame before drug treatment, and the stackreg plugin was set to the translation setting for continuous, unidirectional linear motion compensation throughout each sequence.
Data Analysis
All data were analyzed using the R statistical processing program. For Fulton index and pulmonary artery pressure measurements, data were analyzed using a two-tailed t-test. Filtration coefficient, cell number, modal Ca2+ event number, and event duration measurements were analyzed using a two-way analysis of variance with Tukey's post hoc test for pairwise comparisons.
Results
Store Depletion Enhances Endothelial Permeability in Pulmonary Arterial Hypertension
To determine the effect of pulmonary arterial hypertension on endothelial barrier responses to store depletion, we induced severe pulmonary hypertension in wild-type and TRPC4-knockout littermates with Sugen/hypoxia treatment, and measured whole lung permeability. Consistent with previous reports, both wild-type and TRPC4-knockout animals developed severe pulmonary arterial hypertension, with right ventricular hypertrophy.29, 33 Fulton index values computed as the weight of the right ventricle divided by the weight of the left ventricle and septum were 0.76 ± 0.04 for wild-type hypertensive and 0.71 ± 0.04 for TRPC4-knockout hypertensive groups, compared with 0.26 ± 0.02 for wild-type normotensive animals (P < 0.01) (Figure 1A). Isolated perfused lungs were prepared as previously described and equilibrated so that a baseline isogravimetric period was established, where lung pressures and weight remained stable.30 Under these conditions, mean pulmonary artery pressure was 9 ± 1 and 48 ± 6.23 cm H2O in normotensive and pulmonary arterial hypertensive wild-type lungs, respectively (P < 0.01). Mean pulmonary artery pressures were comparable between hypertensive wild-type (48 ± 6.23) and TRPC4-knockout rats (43 ± 4.69) (Figure 1B). Next, baseline permeability was measured. Baseline filtration coefficients were not significantly different for hypertensive animals (wild-type: 0.21 ± 0.04; TRPC4-knockout: 0.32 ± 0.06) compared with normotensive animals (wild-type: 0.17 ± 0.05; TRPC4-knockout: 0.24 ± 0.03) (Figure 1C).
Figure 1.

Pulmonary arterial hypertension (PAH) attenuates thapsigargin (thaps)–dependent increases in lung vascular permeability in TRPC4-knockout rats. A: After thoracotomy of anesthetized hypertensive wild-type (wt PAH) and TRPC4-knockout (ko PAH) rats, hearts were dissected, cut at the septum, and weighed. Fulton indexes for both groups were calculated as the right ventricular weight (RV), divided by the left ventricular weight including the septum (LV + S). Values were not significantly different between groups. B: Pulmonary arterial pressure was measured (PPA) for both wt PAH and ko PAH groups. There is no significant difference between PPA for either group. C: Lung filtration coefficients (Kf) were measured before (bl) and after stimulation with 75 μmol/L thaps. Thapsigargin induces a significant increase in lung vascular permeability in wild-type hypertensive rats that is abolished in TRPC4-deficient rats. n = 6 to 11 for all groups. ∗∗P < 0.01.
To bypass upstream signal cascades and deplete intracellular Ca2+ stores directly, thapsigargin (75 nmol/L) was added to the recirculating bath for 10 minutes, and a second filtration coefficient measurement was obtained (Figure 1C). Thapsigargin treatment did not significantly increase filtration coefficient absolute values in normotensive wild-type animals (0.36 ± 0.12), although Kf values expressed as fold-change over baseline were significantly greater than the predicted value of 1 (2 ± 0.13; P < 0.01), suggesting a comparable sensitivity to endothelial permeability evoked by activation of store-operated Ca2+ entry between Fischer and Sprague-Dawley rats.27 Thapsigargin did not increase the filtration coefficient in normotensive lungs from TRPC4-knockout (0.3 ± 0.03) rats at baseline. However, thapsigargin significantly increased the filtration coefficient in wild-type animals with pulmonary arterial hypertension (0.57 ± 0.08) (P < 0.01 versus baseline). Thapsigargin-dependent increases in filtration coefficient values were abolished in TRPC4-knockout hypertensive rats (0.36 ± 0.06). Thus, TRPC4 underlies thapsigargin-induced increases in permeability in the hypertensive pulmonary circulation.
Pulmonary Arterial Hypertension Alters Endothelial Morphology, and Increases Both Basal and Thapsigargin-Evoked Cytosolic Ca2+ Transients
Next, endothelial cell morphology and Ca2+ signaling in excised pulmonary artery segments were examined en face using confocal microscopy and the Ca2+ indicator dye, Fluo-4. Notably, arteries from PAH rats were markedly thicker on gross observation en face compared with normotensive controls. Opened pulmonary artery segments loaded with Fluo-4 revealed an atypical polygonal appearance of the pulmonary endothelium of hypertensive animals compared with normotensive wild-type and TRPC4-knockout controls (Figure 2A). However, endothelial morphology was preserved in hypertensive animals harboring a TRPC4 deletion. There was no significant difference in endothelial cell number per 133 × 133-μm confocal viewing field in hypertensive wild-type and TRPC4-knockout animals (141 ± 42 and 154 ± 47, respectively) compared with normotensive wild-type (173 ± 20) and TRPC4-knockout (205 ± 2) controls (Figure 2B).
Figure 2.

Endothelial cell morphology and number measurements from excised vessel segments. A: Time-lapse fluorescent intensity images show relative Ca2+ fluorescence in fields of intact endothelium. Endothelial cell morphology is revealed through preparing pulmonary artery segments en face, loading with the fluorescent Ca2+ indicator dye, Fluo-4, and measuring basal cell Ca2+ dynamics for 3 minutes. Wild-type (wt), normotensive endothelial cells are elliptical, in contrast to severely hypertensive [pulmonary arterial hypertension (PAH)] wild-type rats, who exhibit a polygonal appearance. Normotensive rats genetically lacking TRPC4 (ko) exhibit endothelium, a slightly round appearance compared with wt, and endothelial cells of ko rats with severe hypertension are markedly larger and more elliptical than cells in wt PAH rats. B: Endothelial cells in each 133 × 133-μm field were counted for each replicate. There is no significant difference in endothelial cell number for PAH or TRPC4-knockout groups. n = 4 for all groups (B).
We next examined whether adaptation to pulmonary arterial hypertension altered endothelial Ca2+ signaling. To determine whether endothelial Ca2+ event frequency and duration were affected by PAH, baseline Ca2+ was analyzed for 3 minutes and then 1 μmol/L thapsigargin was applied for an additional 3 minutes (Supplemental Movie S1). Transient increases in Ca2+ (events) were detected and counted by automated region-of-interest analysis using LC_Pro, a plugin for ImageJ (Figure 3). Event duration was defined by the difference in time between half-maximal peak values on either side of a detected signal peak. Histograms of the number of events versus event duration during baseline measurement periods (basal) for wild-type, TRPC4-knockout, wild-type hypertensive, and TRPC4-knockout hypertensive groups are shown in Figure 3A. Figure 3B shows histograms for the corresponding measurement period after 1 μmol/L thapsigargin stimulation.
Figure 3.

Pulmonary arterial hypertension (PAH) increases basal and thapsigargin (thaps)–evoked Ca2+ signals. A: Histograms of baseline Ca2+ event distributions with respect to event duration show distinct, right-shifted distributions for wild-type (wt), TRPC4-knockout (ko), hypertensive wild-type (wt PAH), hypertensive TRPC4-knockout (ko PAH) groups. B: Histograms of event distributions versus event duration after stimulation with thapsigargin reveal similarly right-shifted distributions compared with baseline, with an expanded maximum event number within the wild-type hypertensive group. C: Intracellular Ca2+ event numbers are quantified as bar charts. Basal (bl) responses (open bars) show a significant increase in the number of events in hypertension (wt PAH) relative to control (wt). TRPC4 inactivation (ko PAH) recovers this response. Thapsigargin-stimulated cell responses (closed bars) are also significantly increased in PAH groups, and partially abolished after TRPC4 inactivation. D: Signal half maximum duration measurements are not significantly different between all groups basally (open bars), but PAH cell response duration after thapsigargin stimulation is significantly reduced relative to normotensive control. n = 3 to 4 for all groups (C and D). ∗P < 0.05, ∗∗P < 0.01.
We quantified the number of events at the most frequent (modal) value of signal duration for both unstimulated and thapsigargin-stimulated treatment intervals (Figure 3C). In normotensive wild-type and TRPC4-knockout pulmonary arteries, 1 ± 0.58 and 3.5 ± 1.25 Ca2+ events occurred at the modal signal duration, whereas this number was significantly increased in wild-type PAH pulmonary arteries, 19 ± 10.3 (P < 0.05). In PAH, TRPC4 inactivation partially restored the number of Ca2+ events at the modal signal duration (10 ± 3.8) (Figure 3C). Thus, basal Ca2+ events were more prominent in pulmonary endothelium from PAH arteries.
After treatment with 1 μmol/L thapsigargin, the number of Ca2+ events in wild-type normotensive pulmonary arteries expanded to 10.3 ± 9.3, whereas a significant expansion (55 ± 14.6) occurred in wild-type PAH pulmonary arteries (Figure 3C). TRPC4-knockout cell responses in normotensive (12.5 ± 3.7) and PAH (25.7 ± 3.5) conditions were not significantly different from wild-type controls.
In oscillating cells, we measured the duration of Ca2+ events (eg, half-maximal time of an increase and corresponding decrease in Ca2+). Event duration values were not significantly different among groups at baseline (Figure 3D), indicating that the only parameter changed by PAH at baseline was the number of Ca2+ signal transient events (Figure 3C). However, PAH significantly reduced the duration of Ca2+ events in response to 1 μmol/L thapsigargin (13.8 ± 9.9 seconds) relative to normotensive control (41.8 ± 34.7 seconds) (Figure 3D). Thus, the endothelium of PAH pulmonary arteries responds to thapsigargin uniquely, with an increased frequency of events occurring at an attenuated duration. These data suggest that TRPC4 regulates the number of short-duration endothelial Ca2+ signals, and its activation is associated with increased vascular permeability.
Pulmonary Arterial Hypertension Attenuates ACh-Induced Endothelial Cell Ca2+ Signals after Store Depletion
To determine the effect of TRPC4 in regulating endothelial Ca2+ entry after physiological stimuli, we next examined the Ca2+ signals initiated by ACh subsequent to store depletion with thapsigargin. Histograms of the number of detected ACh-induced Ca2+ events versus event duration reveal bimodal population distributions for wild-type, TRPC4-knockout, wild-type hypertensive, and TRPC4-knockout hypertensive groups (Figure 4A). ACh induced both long-duration (>30 seconds) and short-duration (<30 seconds) responses in normotensive and PAH pulmonary artery endothelium. Figure 4B illustrates representative short- and long-duration ACh-induced Ca2+ signals, which were characterized by a rapid increase in Ca2+. Long-duration signals exhibited a sustained increase and gradual decrease in Ca2+ levels.
Figure 4.

Bimodal distribution of temporal acetylcholine (ACh) responses in rat pulmonary artery endothelium after store depletion. A: All groups display a robust increase in the number of Ca2+ events in responses to ACh relative to baseline. Counts of event duration per group [wild-type (wt), TRPC4-knockout (ko), wt pulmonary arterial hypertension (PAH), and TRPC4-knockout PAH] plotted as histograms reveal the existence of both long (gray bars) and short (black bars) duration modes of response. B: Signal envelopes of selected short- and long-duration cellular response were characterized by quick onset to peak fluorescence. Short responses exhibit rapid return to basal Ca2+ levels, whereas long responses show sustained and gradual decreases to baseline. C: The number of events of both short mode (left) and long mode (right) duration responses to ACh is not significantly affected by animal genotype or PAH; however, long-duration responses are completely abolished in both normotensive ko and wt PAH groups. D: In addition, the duration of short responses is significantly greater in normotensive ko and ko PAH groups compared with wt. The duration of long responses is attenuated in normotensive knockout animals, and significantly increased in PAH. n = 4 for all groups (C and D). ∗∗P < 0.01.
ACh stimulation increased the number of short- and long-duration Ca2+ events differentially after store depletion (Figure 4C). The wild-type PAH group displayed a decreased number of long-duration events (3.02 ± 1.6) relative to wild-type controls (44.6 ± 38) (Figure 4C). Furthermore, TRPC4-knockout PAH animals exhibited a restoration of the number of long-duration responses (52.1 ± 42) to control levels. The number of short-duration events was not significantly changed with respect to animal group (Figure 4C). TRPC4-knockout significantly increased the duration of short ACh responses in both normotensive and hypertensive conditions (Figure 4D). Notably, TRPC4 inactivation significantly decreased the duration of normotensive long-duration responses (50.5 ± 11.04) relative to wild-type (87.8 ± 16.73) (Figure 4D). Together, these data reveal that ACh stimulation after store depletion with thapsigargin elicits two populations of Ca2+ responses with respect to response duration. Only the long-duration responses are abolished in PAH, suggesting that PAH inhibits TRPC4-dependent potentiation of endothelial Ca2+ responses.
Discussion
Herein, we used isolated lung studies and confocal microscopy of endothelial Ca2+ activity to determine lung vascular permeability and the frequency of TRPC4-dependent pulmonary artery endothelial Ca2+ transients in PAH. We used a model of severe hypertension in Fischer rats that uniquely recapitulates key features of the human PAH, including occlusive vascular lesions and early death because of right-sided heart failure.29, 33 Previously, we found that rats lacking TRPC4 exhibit a substantial survival benefit in PAH, associated with a reduction of both lesion number and density.29 However, the role of TRPC4 as a functional store-operated Ca2+ entry channel constituent in the regulation of arteriolar endothelial barrier function in PAH has remained unstudied. Herein, we identified a defect in lung vascular permeability in PAH mediated by TRPC4, and discovered specific endothelial Ca2+ signals associated with this phenomenon.
It is well known that vascular remodeling leads to increased pulmonary vascular resistance and reduced vascular compliance in severe pulmonary hypertension, and endothelial dysfunction is an initiating event in the pathogenesis of PAH.11, 16, 20, 34, 35 However, endothelial barrier disruption and increased vascular permeability subsequent to endothelial dysfunction in PAH is a controversial proposition. In rat models of hypoxic pulmonary hypertension, vascular permeability has been shown to be decreased.28 Furthermore, in an aortocaval fistula model of heart failure with right ventricular hypertrophy, thapsigargin-dependent increases in vascular permeability are ablated, suggesting that pulmonary vascular permeability as a result of endothelial dysfunction is not a determinant of the pathology of pulmonary hypertension secondary to heart failure.28 These findings are contradicted by studies addressing the role of bone morphogenetic protein receptor type 2 (BMPR2) mutations in familial pulmonary arterial hypertension, where silencing of BMPR2 in human endothelial cells increases permeability,36 and mice transfected with mutated BMPR2 exhibit substantial vascular leak in vivo.37 Herein, we demonstrated a TRPC4-dependent increase in the permeability of isolated lungs from PAH Fischer rats after store depletion with thapsigargin. Despite the disparities in the field, our data support a role for endothelial permeability in PAH. It is not likely that this is merely an artifact of the model system because multiple studies of BMPR2 mutations in idiopathic and familial PAH recapitulate this phenomenon. Furthermore, BMPR2 silencing and Sugen models of PAH exhibit interaction with respect to permeability, indicating that vascular endothelial growth factor receptor and BMPR pathways are linked in regulating endothelial barrier function.36 We suggest that increased vascular permeability may be a distinguishing feature of a subset of PAH patients, and therefore warrants further study.
Vascular remodeling is a hallmark of PAH, and an underlying mechanism of increasing pulmonary vascular resistance.11, 13, 17, 18, 20, 21, 26 Vascular remodeling of the vessel media and intima occurs heterogeneously along arteries and arterioles in this disease.13, 34 Notably, medial thickening is seen along arteries and arterioles with distal extension of smooth muscle into typically nonmuscularized segments in PAH.13 Medial remodeling may be reversible in PAH, as seen in some animal models of hypoxic hypertension.13 Herein, we observed gross thickening of the media in larger pulmonary arterioles that was not recovered by TRPC4 inactivation, suggesting that this process may be independent of regulation by this store-operated Ca2+ entry channel. Measurements of Ca2+ activity in this study were confined to the vascular endothelium. Given the role of medial remodeling in the pathogenesis of PAH, and that store-operated Ca2+ entry mediated by TRP channels underlies proliferative phenotypic switching of vascular smooth muscle,38 a follow-up investigation of smooth muscle Ca2+ activity and smooth muscle/endothelial signal interaction in PAH is appropriate.
Human PAH is characterized by endothelial cell remodeling of the small arterioles and endothelial hyperproliferation resulting in plexiform lesion formation.4, 11, 16, 18, 22 Of the various models of PAH and hypoxic hypertension, only the Sugen/hypoxia model of severe pulmonary hypertension reproduces the characteristic endothelial cell remodeling of small arterioles and mortality seen in human PAH.29 Within the lung, endothelial cells from conduit arteries and microvessels exhibit distinct phenotypic heterogeneity and embryonic origin.1, 5 This heterogeneity is likely to extend to the distinct regulation of endothelial barrier integrity throughout the pulmonary vasculature.39, 40 Indeed, we demonstrated a permeability defect in severe PAH associated with increased Ca2+ activity within conduit arterial endothelium.
The paradigm of Ca2+ signaling is now known to comprise a wide array of spatially and temporally regulated signal transients, ranging from elementary store release events, to Ca2+-induced propagation of traveling intercellular Ca2+ waves.41, 42 These signaling signatures lead to diverse and disparate functional effects, such as proliferation, migration, apoptosis, and secretion, in virtually every animal cell.41 We now appreciate such diversity within the context of endothelial physiology, where subtly differentiable signals underlie continuous maintenance and regulation of vascular tone.42 Thus, it is necessary and relevant to unravel the complex nature of Ca2+ signals with respect to their specific functional effects in the pathogenesis of human disease, which has only recently been enabled by robust, high-throughput signal analysis algorithms.32, 42 Herein, we used these algorithms to determine the functional effects of pulmonary artery endothelial Ca2+ signatures in PAH. We identified specific high-frequency, low-duration Ca2+ signal transients that encode endothelial permeability in PAH in response to store depletion. This finding supports the role of specific spatially and temporally delimited Ca2+ signals in modulating distinct physiological functions, and represents a step toward unraveling the complex network of overlapping messages into its functional constituents.
Endothelial Ca2+ signals are initiators of cell proliferation associated with disorganized growth in PAH.15 Several Ca2+-permeable transient receptor potential proteins, including TRPC1, TRPC3, and TRPC6, are involved in vascular smooth muscle cell proliferation in response to chronic hypoxia-induced pulmonary hypertension,15, 26 and there is evidence that both TRPC1 and TRPC4 may regulate pulmonary artery endothelial cell contraction via store-operated Ca2+ influx.39, 40, 43, 44 In addition, elevation of endothelial Ca2+ via Ca2+ influx pathways increases endothelial permeability in many in vitro models of vascular injury.43, 44 There is substantial evidence that TRPC4 is a functional component of an endothelial store-operated Ca2+ entry channel complex, and that activation of store-operated Ca2+ entry induces endothelial cell gap formation.39, 40 Thus, it is likely that the increased TRPC4-dependent endothelial cell Ca2+ activity in severe PAH is an underlying mechanism of the permeability defect observed in this study. Furthermore, store depletion and activation of Ca2+ entry via TRPC1 and TRPC4 preferentially disrupts the barrier of extra-alveolar vessels.27 Consistent with these findings, we observed a robust TRPC4-dependent increase in vascular permeability and associated endothelial Ca2+ activity after store depletion with thapsigargin. However, a key limitation of this work is the missing link between information encoded by endothelial Ca2+ activity and the regulation of TRPC4-dependent permeability. We plan to elucidate the cell signaling mechanisms involved in this phenomenon, but we first require the ability to rescue TRPC4 expression in TRPC4-deficient animals. We are currently developing conditional genetic approaches for modulating TRPC4 expression specifically within the vascular endothelium to address this limitation.
In summary, PAH accompanies endothelial disorganization, which is reduced by TRPC4 inactivation without affecting cell number. Severe hypertension significantly increases a specific high-frequency, low-duration Ca2+ signal after store depletion, which is linked to vascular permeability. Loss of TRPC4 recovers normotensive permeability and Ca2+ signal frequency and duration, demonstrating that TRPC4-dependent Ca2+ activity underlies increased permeability in PAH. Agonist stimulation after endothelial store depletion reveals two distinct classes of responses, which are differentially altered in PAH, suggesting that the coupling between store release and Ca2+ influx signaling may be lost in PAH, contributing to endothelial dysfunction.
Acknowledgments
We thank Drs. Mark Taylor, Masahiko Oka, and Ivan McMurtry for providing laboratory space and Drs. Thomas C. Rich and Silas J. Leavesley for edits to the manuscript.
Footnotes
Supported by NIH grants HL60024 (M.F., N.X., C.Z., and T.S.) and HL66299 (M.F. and T.S.) and American Heart Association grant 14PRE18470024 (N.X.).
Disclosures: None declared.
Supplemental material for this article can be found at http://dx.doi.org/10.1016/j.ajpath.2016.02.002.
Supplemental Data
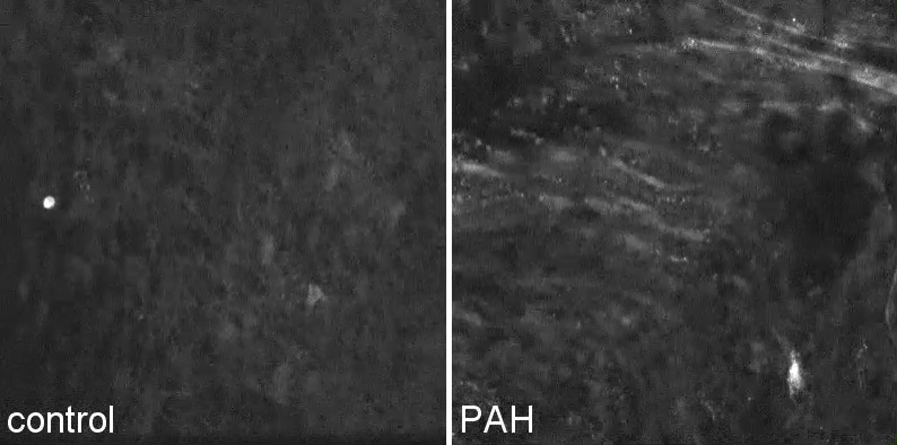
Representative image sequences of basal and thapsigargin-stimulated Ca2+ activity in pulmonary artery endothelium. Recordings of basal endothelial Ca2+ activity show an increased frequency of intracellular Ca2+ transients in severely hypertensive [pulmonary arterial hypertension (PAH)] pulmonary artery endothelium relative to normotensive (control) over a 3-minute unstimulated sampling interval compressed into 4 seconds (1 to 4 seconds). Thapsigargin stimulation over a 3-minute sampling interval (+thaps, 4 to 8 seconds) elicits synchronized increases in endothelial Ca2+, which is elevated in frequency in hypertensive (PAH) groups relative to normotensive controls (control). High-frequency smooth muscle Ca2+ activity was filtered from endothelial focal plane measurements in hypertensive arteries (PAH).
{kind=link}
References
- 1.Aird W.C. Phenotypic heterogeneity of the endothelium, I: structure, function, and mechanisms. Circ Res. 2007;100:158–173. doi: 10.1161/01.RES.0000255691.76142.4a. [DOI] [PubMed] [Google Scholar]
- 2.Aird W.C. Endothelial cell heterogeneity. Cold Spring Harb Perspect Med. 2012;2:a006429. doi: 10.1101/cshperspect.a006429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ochoa C.D., Wu S., Stevens T. New developments in lung endothelial heterogeneity: Von Willebrand factor, P-selectin, and the Weibel-Palade body. Semin Thromb Hemost. 2010;36:301–308. doi: 10.1055/s-0030-1253452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stevens T. Molecular and cellular determinants of lung endothelial cell heterogeneity. Chest. 2005;128:558S–564S. doi: 10.1378/chest.128.6_suppl.558S. [DOI] [PubMed] [Google Scholar]
- 5.Stevens T. Functional and molecular heterogeneity of pulmonary endothelial cells. Proc Am Thorac Soc. 2011;8:453–457. doi: 10.1513/pats.201101-004MW. [DOI] [PubMed] [Google Scholar]
- 6.Stevens T., Phan S., Frid M.G., Alvarez D., Herzog E., Stenmark K.R. Lung vascular cell heterogeneity: endothelium, smooth muscle, and fibroblasts. Proc Am Thorac Soc. 2008;5:783–791. doi: 10.1513/pats.200803-027HR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aird W.C. Mechanisms of endothelial cell heterogeneity in health and disease. Circ Res. 2006;98:159–162. doi: 10.1161/01.RES.0000204553.32549.a7. [DOI] [PubMed] [Google Scholar]
- 8.Aird W.C. Endothelial cell heterogeneity and atherosclerosis. Curr Atheroscler Rep. 2006;8:69–75. doi: 10.1007/s11883-006-0067-z. [DOI] [PubMed] [Google Scholar]
- 9.Aird W.C. Endothelium in health and disease. Pharmacol Rep. 2008;60:139–143. [PubMed] [Google Scholar]
- 10.Mullen M.J., Kharbanda R.K., Cross J., Donald A.E., Taylor M., Vallance P., Deanfield J.E., MacAllister R.J. Heterogenous nature of flow-mediated dilatation in human conduit arteries in vivo: relevance to endothelial dysfunction in hypercholesterolemia. Circ Res. 2001;88:145–151. doi: 10.1161/01.res.88.2.145. [DOI] [PubMed] [Google Scholar]
- 11.Vaillancourt M., Ruffenach G., Meloche J., Bonnet S. Adaptation and remodelling of the pulmonary circulation in pulmonary hypertension. Can J Cardiol. 2015;31:407–415. doi: 10.1016/j.cjca.2014.10.023. [DOI] [PubMed] [Google Scholar]
- 12.Farber H.W., Loscalzo J. Pulmonary arterial hypertension. N Engl J Med. 2004;351:1655–1665. doi: 10.1056/NEJMra035488. [DOI] [PubMed] [Google Scholar]
- 13.Sakao S., Tatsumi K., Voelkel N.F. Reversible or irreversible remodeling in pulmonary arterial hypertension. Am J Respir Cell Mol Biol. 2010;43:629–634. doi: 10.1165/rcmb.2009-0389TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galiè N., Corris P.A., Frost A., Girgis R.E., Granton J., Jing Z.C., Klepetko W., McGoon M.D., McLaughlin V.V., Preston I.R., Rubin L.J., Sandoval J., Seeger W., Keogh A. Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62:D60–D72. doi: 10.1016/j.jacc.2013.10.031. [DOI] [PubMed] [Google Scholar]
- 15.Firth A.L., Remillard C.V., Yuan J.X. TRP channels in hypertension. Biochim Biophys Acta. 2007;1772:895–906. doi: 10.1016/j.bbadis.2007.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Humbert M., Morrell N.W., Archer S.L., Stenmark K.R., MacLean M.R., Lang I.M., Christman B.W., Weir E.K., Eickelberg O., Voelkel N.F., Rabinovitch M. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43:13S–24S. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 17.Sakao S., Tatsumi K. Vascular remodeling in pulmonary arterial hypertension: multiple cancer-like pathways and possible treatment modalities. Int J Cardiol. 2011;147:4–12. doi: 10.1016/j.ijcard.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 18.Firth A.L., Mandel J., Yuan J.X. Idiopathic pulmonary arterial hypertension. Dis Model Mech. 2010;3:268–273. doi: 10.1242/dmm.003616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Machado R.D. The molecular genetics and cellular mechanisms underlying pulmonary arterial hypertension. Scientifica. 2012;2012:106576. doi: 10.6064/2012/106576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morrell N.W., Adnot S., Archer S.L., Dupuis J., Jones P.L., MacLean M.R., McMurtry I.F., Stenmark K.R., Thistlethwaite P.A., Weissmann N., Yuan J.X., Weir E.K. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54:S20–S31. doi: 10.1016/j.jacc.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Voelkel N.F., Gomez-Arroyo J., Abbate A., Bogaard H.J., Nicolls M.R. Pathobiology of pulmonary arterial hypertension and right ventricular failure. Eur Respir J. 2012;40:1555–1565. doi: 10.1183/09031936.00046612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jonigk D., Golpon H., Bockmeyer C.L., Maegel L., Hoeper M.M., Gottlieb J., Nickel N., Hussein K., Maus U., Lehmann U., Janciauskiene S., Welte T., Haverich A., Rische J., Kreipe H., Laenger F. Plexiform lesions in pulmonary arterial hypertension composition, architecture, and microenvironment. Am J Pathol. 2011;179:167–179. doi: 10.1016/j.ajpath.2011.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aromolaran A.A., Blatter L.A. Modulation of intracellular Ca2+ release and capacitative Ca2+ entry by CaMKII inhibitors in bovine vascular endothelial cells. Am J Physiol Cell Physiol. 2005;289:C1426–C1436. doi: 10.1152/ajpcell.00262.2005. [DOI] [PubMed] [Google Scholar]
- 24.Lowry J.L., Brovkovych V., Zhang Y., Skidgel R.A. Endothelial nitric-oxide synthase activation generates an inducible nitric-oxide synthase-like output of nitric oxide in inflamed endothelium. J Biol Chem. 2013;288:4174–4193. doi: 10.1074/jbc.M112.436022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pries A.R., Kuebler W.M. Normal endothelium. Handb Exp Pharmacol. 2006;76(Pt 1):1–40. doi: 10.1007/3-540-32967-6_1. [DOI] [PubMed] [Google Scholar]
- 26.Yu Y., Fantozzi I., Remillard C.V., Landsberg J.W., Kunichika N., Platoshyn O., Tigno D.D., Thistlethwaite P.A., Rubin L.J., Yuan J.X. Enhanced expression of transient receptor potential channels in idiopathic pulmonary arterial hypertension. Proc Natl Acad Sci U S A. 2004;101:13861–13866. doi: 10.1073/pnas.0405908101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chetham P.M., Babál P., Bridges J.P., Moore T.M., Stevens T. Segmental regulation of pulmonary vascular permeability by store-operated Ca2+ entry. Am J Physiol. 1999;276:L41–L50. doi: 10.1152/ajplung.1999.276.1.L41. [DOI] [PubMed] [Google Scholar]
- 28.Townsley M.I., King J.A., Alvarez D.F. Ca2+ channels and pulmonary endothelial permeability: insights from study of intact lung and chronic pulmonary hypertension. Microcirculation. 2006;13:725–739. doi: 10.1080/10739680600930362. [DOI] [PubMed] [Google Scholar]
- 29.Alzoubi A., Almalouf P., Toba M., O'Neill K., Qian X., Francis M., Taylor M.S., Alexeyev M., McMurtry I.F., Oka M., Stevens T. TRPC4 inactivation confers a survival benefit in severe pulmonary arterial hypertension. Am J Pathol. 2013;183:1779–1788. doi: 10.1016/j.ajpath.2013.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alvarez D.F., Gjerde E.A., Townsley M.I. Role of EETs in regulation of endothelial permeability in rat lung. Am J Physiol Lung Cell Mol Physiol. 2004;286:L445–L451. doi: 10.1152/ajplung.00150.2003. [DOI] [PubMed] [Google Scholar]
- 31.Abramoff M.D., Magalhaes P.J., Ram S.J. Image processing with ImageJ. Biophotonics Int. 2004;11:36–42. [Google Scholar]
- 32.Francis M., Qian X., Charbel C., Ledoux J., Parker J.C., Taylor M.S. Automated region of interest analysis of dynamic Ca2+ signals in image sequences. Am J Physiol Cell Physiol. 2012;303:C236–C243. doi: 10.1152/ajpcell.00016.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang B., Deng Y., Suen C., Taha M., Chaudhary K.R., Courtman D.W., Stewart D.J. Marked strain-specific differences in the SU5416 rat model of severe pulmonary arterial hypertension. Am J Respir Cell Mol Biol. 2016;54:461–468. doi: 10.1165/rcmb.2014-0488OC. [DOI] [PubMed] [Google Scholar]
- 34.Tuder R.M., Abman S.H., Braun T., Capron F., Stevens T., Thistlethwaite P.A., Haworth S.G. Development and pathology of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S3–S9. doi: 10.1016/j.jacc.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 35.Fishman A.P. Etiology and pathogenesis of primary pulmonary hypertension: a perspective. Chest. 1998;114:242S–247S. doi: 10.1378/chest.114.3_supplement.242s. [DOI] [PubMed] [Google Scholar]
- 36.Burton V.J., Ciuclan L.I., Holmes A.M., Rodman D.M., Walker C., Budd D.C. Bone morphogenetic protein receptor II regulates pulmonary artery endothelial cell barrier function. Blood. 2011;117:333–341. doi: 10.1182/blood-2010-05-285973. [DOI] [PubMed] [Google Scholar]
- 37.Johnson J.A., Hemnes A.R., Perrien D.S., Schuster M., Robinson L.J., Gladson S., Loibner H., Bai S., Blackwell T.R., Tada Y., Harral J.W., Talati M., Lane K.B., Fagan K.A., West J. Cytoskeletal defects in BMPR2-associated pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2012;302:L474–L484. doi: 10.1152/ajplung.00202.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fernandez R.A., Wan J., Song S., Smith K.A., Gu Y., Tauseef M., Tang H., Makino A., Mehta D., Yuan J.X. Upregulated expression of STIM2, TRPC6, and Orai2 contributes to the transition of pulmonary arterial smooth muscle cells from a contractile to proliferative phenotype. Am J Physiol Cell Physiol. 2015;308:C581–C593. doi: 10.1152/ajpcell.00202.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cioffi D.L., Lowe K., Alvarez D.F., Barry C., Stevens T. TRPing on the lung endothelium: calcium channels that regulate barrier function. Antioxid Redox Signal. 2009;11:765–776. doi: 10.1089/ars.2008.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cioffi D.L., Stevens T. Regulation of endothelial cell barrier function by store-operated calcium entry. Microcirculation. 2006;13:709–723. doi: 10.1080/10739680600930354. [DOI] [PubMed] [Google Scholar]
- 41.Berridge M.J. Inositol trisphosphate and calcium oscillations. Biochem Soc Symp. 2007;74:1–7. doi: 10.1042/BSS0740001. [DOI] [PubMed] [Google Scholar]
- 42.Taylor M.S., Francis M., Qian X., Solodushko V. Dynamic Ca2+ signal modalities in the vascular endothelium. Microcirculation. 2012;19:423–429. doi: 10.1111/j.1549-8719.2012.00180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tiruppathi C., Ahmmed G.U., Vogel S.M., Malik A.B. Ca2+ signaling, TRP channels, and endothelial permeability. Microcirculation. 2006;13:693–708. doi: 10.1080/10739680600930347. [DOI] [PubMed] [Google Scholar]
- 44.Tiruppathi C., Freichel M., Vogel S.M., Paria B.C., Mehta D., Flockerzi V., Malik A.B. Impairment of store-operated Ca2+ entry in TRPC4(-/-) mice interferes with increase in lung microvascular permeability. Circ Res. 2002;91:70–76. doi: 10.1161/01.res.0000023391.40106.a8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representative image sequences of basal and thapsigargin-stimulated Ca2+ activity in pulmonary artery endothelium. Recordings of basal endothelial Ca2+ activity show an increased frequency of intracellular Ca2+ transients in severely hypertensive [pulmonary arterial hypertension (PAH)] pulmonary artery endothelium relative to normotensive (control) over a 3-minute unstimulated sampling interval compressed into 4 seconds (1 to 4 seconds). Thapsigargin stimulation over a 3-minute sampling interval (+thaps, 4 to 8 seconds) elicits synchronized increases in endothelial Ca2+, which is elevated in frequency in hypertensive (PAH) groups relative to normotensive controls (control). High-frequency smooth muscle Ca2+ activity was filtered from endothelial focal plane measurements in hypertensive arteries (PAH).