

Abstract
Pulmonary-artery smooth-muscle-cell (PA-SMC) proliferation in pulmonary hypertension (PH) may be linked to dysregulated mammalian target of rapamycin (mTOR) signaling. The mTOR pathway involves two independent complexes, mTORC1 and mTORC2, which phosphorylate S6K and Akt, respectively, and differ in their sensitivity to rapamycin. Here, we evaluated rapamycin-sensitive mTOR substrates and PA-SMC proliferation in rats with monocrotaline-induced PH (MCT-PH). Compared to cells from control rats, cultured PA-SMCs from MCT-PH rats exhibited increased growth responses to PDGF, serotonin (5-HT), IL-1β, IGF-1, or fetal calf serum (FCS), with increases in phosphorylated (Ser-473)Akt, (Thr-308)Akt, GSK3, and S6K reflecting activated mTORC1 and mTORC2 signaling. Treatment with rapamycin (0.5 μM) or the Akt inhibitor A-443654 (0.5 μM) reduced FCS-stimulated growth of PA-SMCs from MCT-PH rats to the level in control rats while inhibiting Akt, GSK3, and S6K activation. Neither the tyrosine-kinase inhibitor imatinib (0.1 μM) nor the 5-HT transporter inhibitor fluoxetine (5 μM) normalized the increased PA-SMC growth response to FCS. Rapamycin treatment (5 mg/Kg/day) of MCT-PH rats from day 21 to day 28 markedly reduced P-Akt, P-GSK3, and P-S6K in pulmonary arteries and normalized growth of derived PA-SMCs. This effect was not observed after 1 one week of imatinib (100 mg/Kg/d) or fluoxetine (20 mg/Kg/d). Rapamycin given preventively (days 1 to 21) or curatively (days 21 to 42) inhibited MCT-PH to a greater extent than did imatinib or fluoxetine. Experimental PH in rats is associated with a sustained proliferative PA-SMC phenotype linked to activation of both mTORC1 and mTORC2 signaling and suppressed by rapamycin treatment.
INTRODUCTION
Hyperplasia of pulmonary-artery smooth muscle cells (PA-SMCs) is a hallmark pathological feature of all forms of pulmonary hypertension (PH) that leads to structural remodeling and occlusion of the pulmonary vessels(1). The intracellular signaling pathway involving serine/threonine kinase (Akt) and mammalian target of rapamycin (mTOR) is now recognized as a critical player in cell proliferation and cancer (2). In PA-SMCs, Akt and mTOR signaling can be activated by numerous growth factors (3–5), as well as physical stimuli such as shear stress and hypoxia (6). Thus, the Akt/mTOR signaling pathway is shared by various physical and biological stimuli that act on PA-SMCs and can induce PH. Consequently, treatments targeting this pathway may hold promise in PH.
One major molecular target for antiproliferative therapies directed to the Akt pathway is the mTOR protein, which plays a central role in controlling cell growth, proliferation, and survival and is regulated by mitogenic and nutrient signals (7–9). In the cell, mTOR is found in two distinct protein complexes with specific binding partners, raptor in mTOR complex 1 (mTORC1) and rictor in mTORC2 (7–9). The mTORC1 substrates include S6 kinases (S6K), while mTORC2 phosphorylates the hydrophobic motif of Akt family members at Ser473, leading to subsequent phosphorylation of downstream effectors such as GSK3. Activation of mTORC1 exerts a negative feed-back effect on Akt. Consequently, rapamycin, which binds only to mTORC1, inhibits the mTORC1 substrate S6K but can simultaneously activate the Akt-GSK3 pathway (10). In contrast, mTORC2 inhibition is associated with variable inactivation of Akt and downstream Akt effectors such as GSK3. Long-term rapamycin treatment can also affect mTORC2 activity (11, 12). The effects of rapamycin may therefore differ according to cell types and treatment conditions.
Studies of rapamycin in animal models of PH showed contradictory results according to the rapamycin dose, with no relationship to Akt/mTOR signaling (13–16). The hypothesis that dysregulated mTOR signaling is involved in PA-SMC hyperplasia during PH progression rests mainly on recent results from our laboratory and others showing increased mTORC1 and mTORC2 substrate phosphorylation in pulmonary-vascular smooth muscle from rats with monocrotaline (MCT)- or hypoxia-induced PH, as well as increased P-GSK3 in remodeled vessels from patients with PH (17, 18). Of note, a recently published case-report describes a dramatic improvement in PH in a patient given rapamycin for a pancreatic tumor (19). The potential usefulness of rapamycin derivatives in PH is still under investigation.
Here, we investigated whether PA-SMCs from rats with MCT-induced PH exhibited an abnormal proliferative phenotype similar to that previously described in patients with PH. We found an increased PA-SMC growth response to a variety of growth factors and we therefore investigated whether this sustained proliferative phenotype was related to alteration of the mTOR signaling pathway. Finally we determined whether rapamycin treatment normalized PA-SMC growth when added in vitro to cell cultures or given in vivo to rats and whether rapamycin treatment was effective in preventing or reversing PH in rats with MCT-induced PH.
METHODS
Animal model and experimental design
All experiments were performed according to the NIH Guide for the Care and Use of Laboratory Animals. Male Wistar rats (200–250 g) were studied after a single subcutaneous MCT injection (60 mg/Kg; Sigma, Saint-Quentin-Fallavier, France). Rats were assigned at random (8–10/group to fluoxetine (20 mg/Kg/day), imatinib (100 mg/Kg/day), rapamycin (5 mg/Kg/day), or vehicle only, given once daily by gavage.
Studies on cultured rat PA-SMCs, assessment of PA-SMC growth and apoptosis
PA-SMCs from rat pulmonary arteries were cultured and characterized as previously described (17). After 48 hours’ incubation in DMEM, the cells were treated with FCS (15%), platelet-derived growth factor (PDGF)-BB (20 ng/mL), 5-hydroxytryptophan (5-HT, 200 ng/mL), interleukin-1β (IL-1β, 50 ng/mL), or insulin-like growth factor (IGF, 50 ng/mL). After 48 hours, MTT (0.2 mg/mL) was added and tetrazolium salt reduction to formazan was quantified by spectrophotometry. Rapamycin (0.01–0.5 μM) or vehicle was added before and 24 hours after growth factor exposure; and fluoxetine (5–10 μM) or imatinib (0.1–1 μM) or vehicle was added in serum-free medium for 24 hours prior to growth factor exposure. The apoptotic cell count was determined using annexin V flow cytometry.
Assessment of pulmonary hypertension
At various times after MCT administration, rats were anesthetized with ketamine and xylazine. The following were measured as previously described (17) : pulmonary arterial pressure (Pap), right ventricular hypertrophy index (right ventricular free wall weight over sum of septum plus left ventricular free wall weights, RV/LV+S), and pulmonary vessel muscularization.
Evaluation of in situ PA-SMC death and proliferation
Ki67 and caspase 3 immunostainings were performed using an anti-Ki67 rabbit antibody (1:500, Abcam, Cambridge, MA, USA) and anti-caspase 3 active rabbit antibody (1:200, R&D Systems, Minneapolis, MN, USA)(17).
Immunoblotting of Akt, S6K, and GSK3 proteins and immunofluorescence studies
Akt, S6K, and GSK3 proteins were measured in lung tissues and isolated PA-SMCs using Western blotting with the following antibodies: anti-p-Akt ser 473, anti-p-Akt Thr 308, anti-Akt, anti-p-GSK3, anti-GSK3, anti-p-S6K, and anti-S6K (Cell Signaling Technology, Boston, MA, USA); and anti–β-actin antibody (Sigma). Lung cryosections were incubated with antibodies against anti-p-Akt ser 473 or anti-p-S6K (Cell Signaling Technology) and alpha-SMA (Sigma) then with Alexa 488 anti-mouse and 555 anti–rabbit secondary antibodies (Cell Signaling Technology). Nuclei were stained with Hoechst 333342 (1 μg/mL, Cell Signaling Technology).
Drugs
Rapamycin (Sirolimus®, Pfizer, city, NY, USA), fluoxetine (Arrows, Lyon France), and imatinib (Gleevec®, Novartis, Basel, Switzerland) used for animal studies were from the pharmacy of the Henri Mondor Hospital (Créteil, France). Fluoxetine and rapamycin for cell experiments were from Sigma. Imatinib solid caplets were ground into a fine powder, dissolved in water or phosphate-buffered saline (PBS) then used to prepare a concentrated stock solution. The Akt inhibitor (A-443654) was a gift from Abbott Laboratories (Chicago, IL, USA) and was supplied as a powder to be dissolved in PBS or water(20).
Statistical analyses
The data are described as mean±SEM. Parametric tests were used after verification that the variables were normally distributed in each group. The effect on PA-SMC growth of FCS, PDGF, 5-HT, IL-1β, and IFG-1 at various times after monocrotaline injection was evaluated by two-way ANOVA with repeated measures, testing for treatment and time effects. Comparisons of treatments with A-443654, rapamycin, fluoxetine, imatinib, and vehicle on growth of PA-SMCs from control and MCT-treated rats were performed with two-way ANOVA. The effects of chronic treatment of MCT-treated rats with rapamycin, fluoxetine, imatinib, or vehicle on physiological or biological parameters were assessed using one-way ANOVA. When a significant difference was found, group means were compared by post-test analysis. P values lower than 0.05 were considered significant.
RESULTS
Hyperplasia of cultured PA-SMCs from rats with monocrotaline (MCT)-induced pulmonary hypertension; effects of rapamycin, imatinib, and fluoxetine
Rats studied 3 and 4 weeks after MCT administration (3wk- and 4wk-MCT rats, respectively) had severe PH with marked increases in Pap and pulmonary vessel muscularization compared to control rats injected with vehicle. PH was more severe in 4wk-MCT than in 3 wk-MCT rats (Figure 1A). Cells from pulmonary arteries of MCT-treated rats grew faster than those from vehicle-treated rats when stimulated with varying doses of FCS, PDGF, 5-HT, IL-1β, or IGF-1 (Figures 1B, 1C). The stimulated PA-SMC growth paralleled the severity of pulmonary vascular remodeling, with a higher growth rate of cells from 4wk-MCT than from 3 wk-MCT rats. With rapamycin (0.5 μM) or the selective Akt inhibitor A-443654 (0.5 μM), the growth-stimulating effect of FCS was markedly reduced and PA-SMC growth was not different between MCT-treated and vehicle-treated rats (Figure 2A). In contrast, cell pretreatment with imatinib (0.1 μM) or fluoxetine (5 μM) failed to normalize FCS-induced growth of PA-SMCs from MCT-treated rats (Figure 2A). This increased growth responsiveness of PA-SMCs from MCT-treated rats was associated with increased cell levels of (Ser-473)P-Akt, (Thr-308)P-Akt, P-GSK-3β, and P-S6K proteins at baseline or after stimulation with FCS, compared to cells from control rats (Figure 2B). Treatment of PA-SMCs with 0.5 μM rapamycin markedly inhibited (Ser-473)P-Akt, (Thr-308)P-Akt, P-GSK-3β, and P-S6K in cells from both control and MCT-treated rats, abolishing the differences between these two groups. PA-SMC treatment with the Akt inhibitor A-443654 also markedly decreased P-GSK3 and P-S6K in cells from both control and MCT-treated rats and was associated with increased Akt phosphorylation, as expected based on the reported mechanism of action of this drug (20). In contrast, imatinib and fluoxetine were less efficient than rapamycin in reducing (Ser-473) p-Akt, (Thr-308) p-Akt, and P-GSK-3β levels and failed to alter p-S6K levels in either group (Figure 2B). Interestingly, rapamycin normalized FCS-induced growth of PA-SMCs from MCT-treated rats only in concentrations greater than 20 nM (Figure 3A). Thus, rapamycin 20 nM failed to normalize FCS-induced growth of PA-SMCs from MCT-treated rats but markedly reduced p-S6K levels without affecting (Ser-473) p-Akt or P-GSK-3β levels (Figure 3B). These results were consistent with selective mTORC1 inhibition by rapamycin 20 nM and with dual mTORC1 and mTORC2 inhibition by rapamycin 500 nM.
Figure 1.
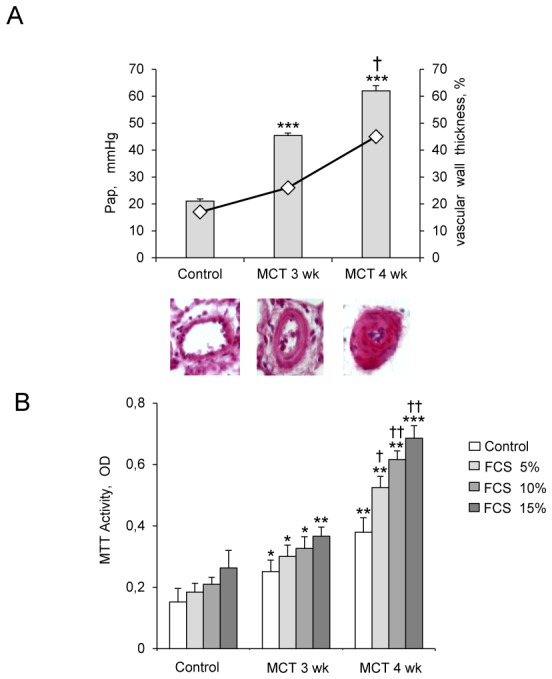
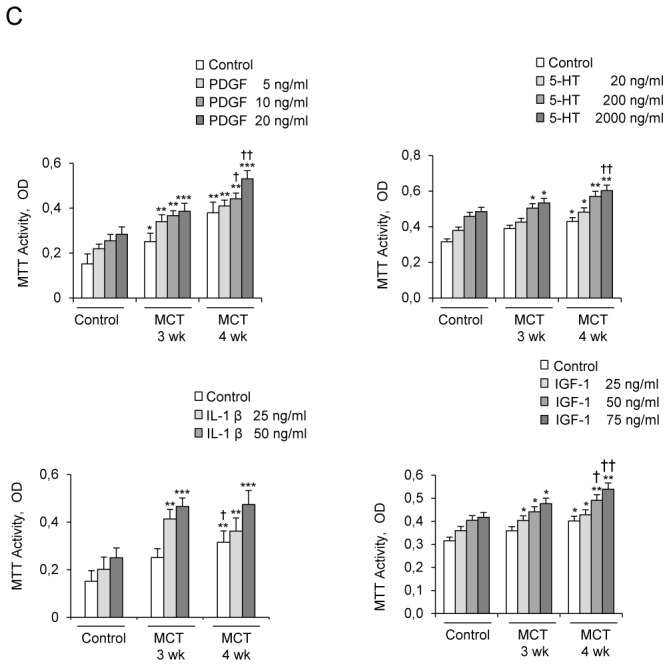
(A) Development of pulmonary hypertension 3 and 4 weeks following monocrotaline (MCT) injection in rats, as assessed by the increases in mean pulmonary arterial pressure (Pap, bar graphs), and muscularization of distal pulmonary vessels as assessed by pulmonary arterial wall thickness (diamonds). Representative photomicrographs of pulmonary vessels and pulmonary artery wall thickness from rats on days 21 and 28 after MCT or saline (controls). (B and C) Proliferation of pulmonary-artery smooth muscle cells (PA-SMCs) derived from control rats and MCT-treated rats after 3 weeks (3wk-MCT) or 4 weeks (4wk-MCT). The cells were starved of FCS for 48 hours then exposed to vehicle or increasing concentrations of FCS, PDGF, 5-HT, IL-1β or IGF-1. Each point is the mean±SE of at least five determinations 21 and 28 days following MCT administration (mean±SE, n=30 at each time point). *P<0.05, **P<0.01, **P<0.001 compared with values in untreated control rats (day 0); †P<0.05 and ††P<0.01 compared with respective values in 3wk-MCT rats.
Figure 2.
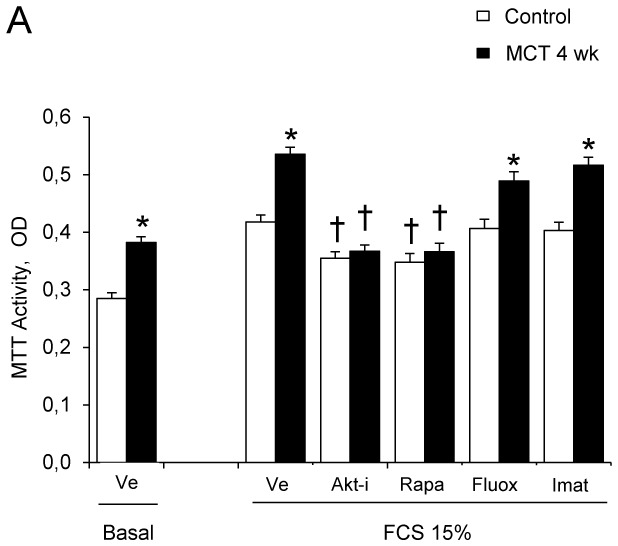
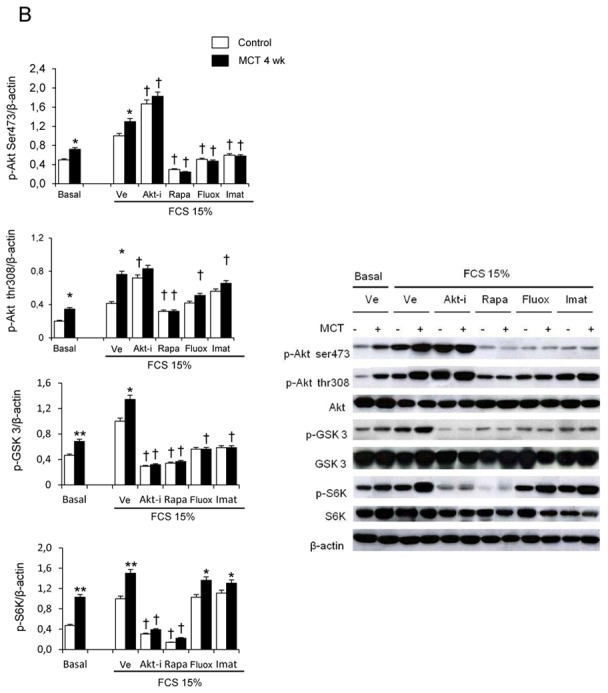
(A). Effects of the Akt inhibitor A-443654 (0.5 μM), rapamycin (0.5 μM), fluoxetine (5 μM), or imatinib (0.1 μM) on proliferation of PA-SMCs from control rats or rats 4 weeks after monocrotaline injection (MCT-4wk), under basal conditions and after stimulation with 15% fetal calf serum (FCS). The cells were starved of FCS for 48 hours then exposed to the drugs according to different protocols. Cell proliferation was measured with the tetrazolium assay 48 hours after stimulation with 15% FCS or vehicle. Each point is the mean±SE of at least five determinations (mean±SE, n=30 at each time point). (B) Effects of treatment with the Akt inhibitor A-443654(0.5 μM), rapamycin (0.5 μM), fluoxetine (5 μM), or imatinib (0.1 μM) on Ser 473, p-Akt, p-GSK3 and p-S6K protein levels in cultured PA-SMCs stimulated with 15% FCS for 24 hours. Values are mean±SEM of 30 values from five independent experiments. *P<0.05, **P<0.01 compared with values in control rats not given MCT; †P<0.05 compared with respective values in control or MCT-4wk rats treated with vehicle.
Figure 3.
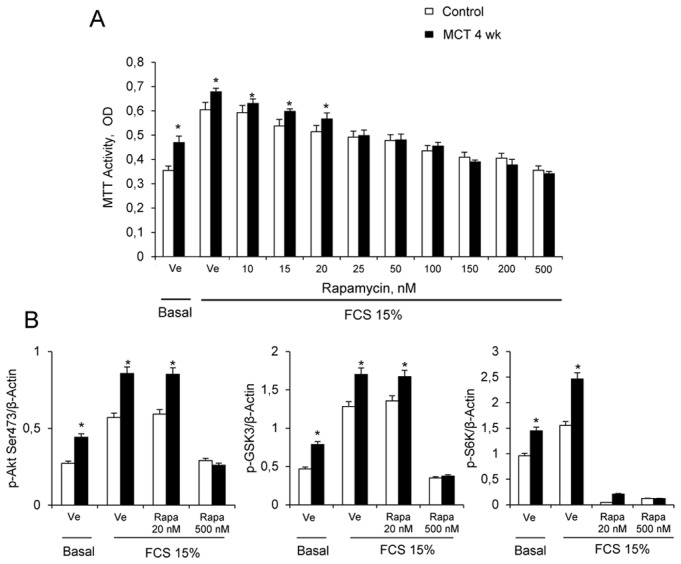
(A). Effects of increasing concentrations of rapamycin on proliferation of PA-SMCs from control rats or rats 4 weeks after monocrotaline injection (MCT-4wk) under basal conditions and after stimulation with 15% fetal calf serum (FCS); (mean±SE, n=12 at each time point). (B) Effects of treatment with 20 nM and 100 nM rapamycin on Ser 473, p-Akt, p-GSK3, and p-S6K protein levels in cultured PA-SMCs stimulated with 15% FCS for 24 hours. Values are mean±SEM of 12 values. *P<0.05, **P<0.01 compared with values in control rats not given MCT.
Treatment of rats with established monocrotaline (MCT)-induced pulmonary hypertension by rapamycin abrogated the proliferative phenotype of derived cultured PA-SMCs and inhibited mTORC1 and mTORC2 activation
In rats studied 4 weeks after MCT administration, the development of PH (Figure 4A) was associated with large increases in (Ser-473) p-Akt, (Thr-308) p-Akt, p-GSK3, and p-S6K protein levels in proximal and distal pulmonary arteries (Figure 4B and 4C); with no change in total Akt, GSK3, or S6K (Figure 4B). Activation of p-Akt and p-S6K was mainly found in the media of pulmonary arteries, with immunofluorescence studies showing prominent p-Akt immunostaining in distal remodeled pulmonary vessels (Figure 4C).
Figure 4.
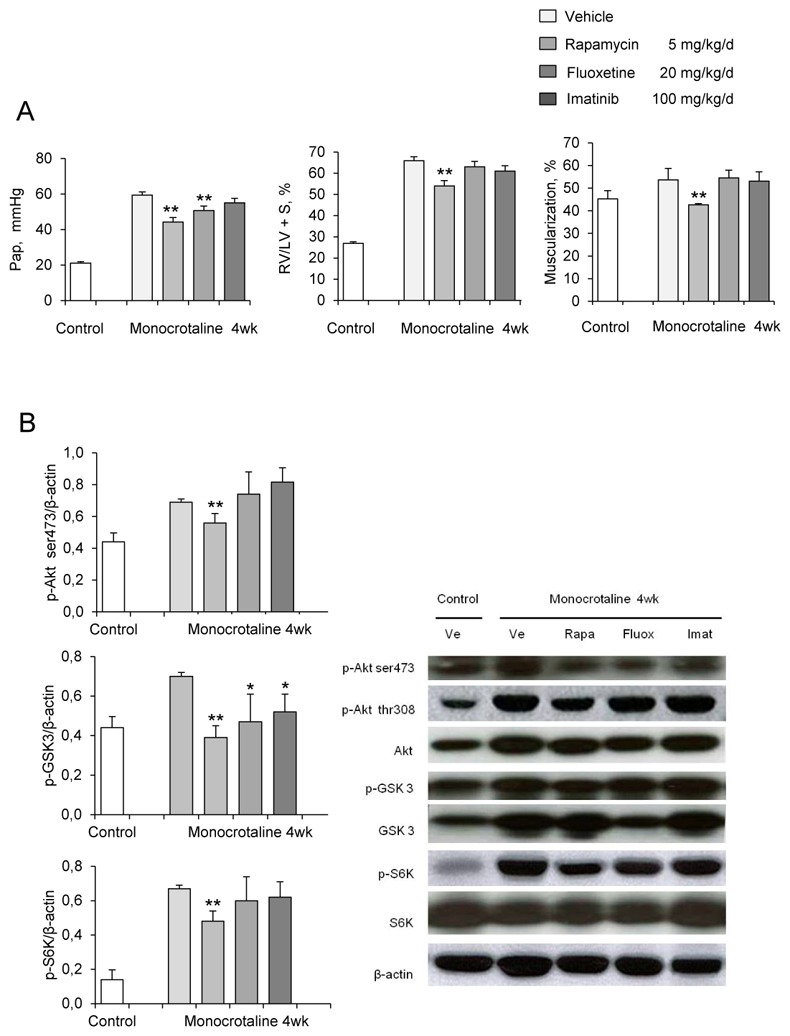
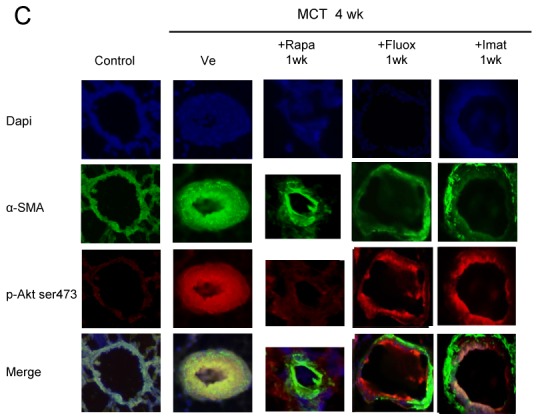
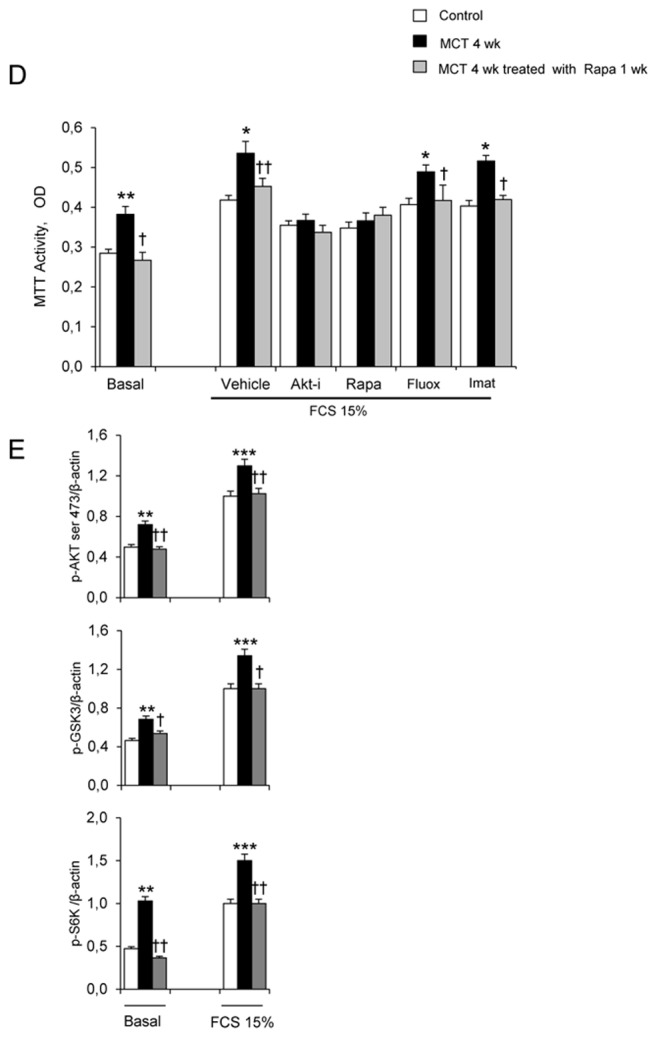
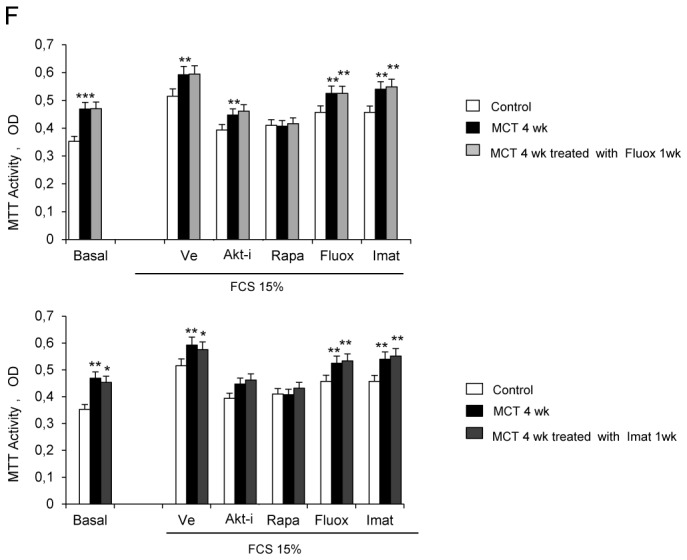
(A) Pulmonary arterial pressure (Pap), right ventricular hypertrophy index [RV/(LV+S) weight ratio], and pulmonary vessel muscularization (percentage of partially and fully muscularized pulmonary vessels) on day 28 after monocrotaline (MCT) administration in the groups treated from day 21 to day 28 with rapamycin (5 mg/Kg/d), fluoxetine (20 mg/Kg/d), or imatinib (100 mg/Kg/d). Control rats were kept in the same conditions after saline administration (n=10 in each group). *P<0.05 and **P<0.01 vs. values in MCT-4wk rats treated with vehicle. (B) Changes in Akt, GSK3, and S6K phosphorylation in proximal pulmonary arteries after MCT injection. Protein levels were evaluated by Western blot in pulmonary artery extracts 28 days after MCT administration. Rapamycin, fluoxetine, imatinib, or vehicle was given from day 21 to day 28 (n=30 in each group). The bar graph shows the mean±SEM values in pulmonary arteries (n=30 in each group). *P<0.05, **P<0.01 vs. values from MCT-4wk rats treated with vehicle. (C) Representative photographs of immunofluorescence staining for P-Akt in control rats and MCT-4wk rats treated with rapamycin, fluoxetine, imatinib, or vehicle from day 21 to 28. P-Akt was predominantly expressed in pulmonary vessels and co-localized with α-SMA in PA-SMCs. No immunoreactivity was detected in sections incubated with secondary anti-rabbit and anti-mouse antibody but no primary antibody. (D) Proliferation of PA-SMCs derived from control and MCT-4wk rats treated with vehicle or rapamycin from day 21 to day 28. The cells were starved of fetal calf serum (FCS) for 48 hours then exposed to vehicle or 15% FCS with the Akt inhibitor (0.5 μM), rapamycin (0.5 μM), fluoxetine (5 μM), or imatinib (0.1 μM). Cell proliferation was measured with the tetrazolium assay. Each point is the mean±SE of at least five determinations 21 and 28 days following MCT administration (mean±SE, n=30 at each time point). *P<0.05 compared with values in control rats not given monocrotaline; †P<0.05 compared with values in MCT-4wk rats treated with vehicle instead of rapamycin. (E) Ser 473, p-Akt, andp-GSK3 protein levels in cultured PA-SMCs from control and MCT-4wk rats treated from day 21 to day 28 with rapamycin or vehicle. Cells were studied after treatment with either 15% FCS or vehicle for 48 hours. Values are mean±SE of 30 values from five independent experiments.*P<0.05 compared with values in control rats not given monocrotaline; †P<0.05 compared with values in MCT-4wk rats treated with vehicle instead of rapamycin. (F) Proliferation of PA-SMCs derived from control and MCT-4wk rats treated with vehicle, fluoxetine, or imatinib from day 21 to day 28. The cells were starved of FCS for 48 hours then exposed to vehicle or 15% FCS with the Akt inhibitor (0.5 μM), rapamycin (0.5 μM), fluoxetine (5 μM), or imatinib (0.1 μM). Cell proliferation was measured with the tetrazolium assay. Each point is the mean±SE of 30 determinations 21 and 28 days following MCT administration (mean±SE). *P<0.05 compared with values in control rats not given monocrotaline; †P<0.05 compared with values in MCT-4wk rats treated with vehicle instead of rapamycin.
To investigate whether brief in vivo rapamycin treatment altered the proliferative phenotype of derived cultured PA-SMCs from4wk-MCT rats, cells were collected after rapamycin treatment from day 21 to day 28 following MCT injection. Rapamycin treatment completely abolished the increased PA-SMC growth seen in cells from vehicle-treated 4wk-MCT rats, so that MTT activity in PA-SMCs from rapamycin- and MCT-treated rats was similar to that in control rats not given MCT (Figure 4D). Direct exposure of cultured PA-SMCs to rapamycin produced only a slight additional inhibitory effect not seen with in vitro imatinib or fluoxetine treatment. Rapamycin treatment for 1 week was associated with slight but significant reductions in Pap, RV/LV+S, and pulmonary-artery muscularization (Figure 4A) and with large decreases in pulmonary-artery levels of P-Akt, P-GSK3, and P-S6K (Figures 4B, and 4C). More importantly, PA-SMCs from 4wk-MCT rats given rapamycin for 1 week exhibited the same levels of p-Akt, p-GSK3 and p-S6K as did PA-SMCs from control rats not given MCT, both at baseline and after FCS stimulation (Figure 4E).
In vivo treatment of MCT-treated rats with either fluoxetine or imatinib for 1 week did not noticeably affect PH parameters, although fluoxetine slightly reduced Pap without affecting RV hypertrophy or pulmonary vessel muscularization (Figure 4A). Fluoxetine or imatinib for 1 week failed to suppress the proliferative phenotype of derived cultured PA-SMCs (Figure 4F). Both fluoxetine and imatinib slightly decreased P-GSK3 levels in pulmonary arteries without affecting P-Akt or P-S6K levels (Figures 4B and 4C).
Preventive and curative effects of rapamycin on MCT-induced PH; comparison with fluoxetine and imatinib
Chronic treatment with rapamycin (5 mg/Kg/day), fluoxetine (20 mg/Kg/day), or imatinib (100 mg/Kg/day) from day 0 to day 21 after MCT administration attenuated PH severity. Reductions in Pap and pulmonary artery muscularization on day 21 were larger with rapamycin than with imatinib or fluoxetine (Figure 5A). Rapamycin treatment was associated with reductions in pulmonary-artery levels of p-Akt, P-GSK3, and p-S6K, which did not differ from those induced by imatinib or fluoxetine (Figure 5B).
Figure 5.

(A) Pulmonary arterial pressure; right ventricular hypertrophy index (RV/LV+S weights); and percentages of partially muscularized and fully muscularized vessels on day 21 after saline (controls) or monocrotaline (MCT) administration combined with rapamycin (Rapa), fluoxetine (Fluox), imatinib (Imat), or vehicle (Ve) from day 0 to day 21 (n=10 in each group). *P<0.01 compared with values in control rats. (B) Changes in Akt, GSK3, and S6K phosphorylation in proximal pulmonary arteries after MCT injection. Protein expression and phosphorylation were evaluated by Western blot in pulmonary artery extracts 21 days after MCT administration. Treatment with rapamycin, fluoxetine, imatinib, or vehicle was given from day 0 to day 21 (n=10 in each group). The bar graph shows the mean±SEM values in pulmonary arteries (n=10 in each group). *P<0.05 and **P<0.01 vs. values in MCT-4wk rats treated with vehicle.
Rapamycin treatment from day 21 to day 42 after MCT injection was more potent in reducing Pap and RV/LV+S, compared to imatinib or fluoxetine (Figure 6A). Rapamycin had more pronounced effects than did imatinib on pulmonary vessel muscularization on day 42. Rapamycin treatment for 3 weeks consistently and markedly reduced lung p-Akt, p-GSK3, and pS6K levels; whereas fluoxetine and imatinib induced smaller decreases in p-Akt and p-GSK3 levels and had no effects on P-S6K (Figures 6B, and 6C).
Figure 6.

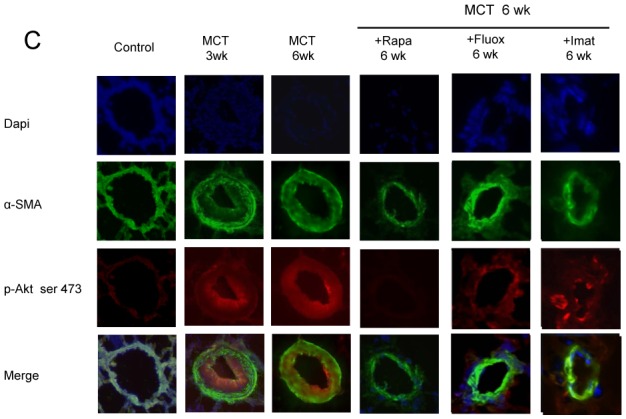
(A) Pulmonary arterial pressure; right ventricular hypertrophy index (RV/LV+S weights); and percentage of partially muscularized and fully muscularized vessels on day 42 after saline (controls) or monocrotaline (MCT) administration combined with either rapamycin (Rapa), fluoxetine (Fluox), imatinib (Imat), or vehicle (Ve) from day 21 to day 42 (n=10 in each group). *P<0.01 compared with values in control rats. (B) Changes in Akt, GSK3, and S6K phosphorylation in proximal pulmonary arteries after MCT. Protein expression and phosphorylation were evaluated by Western blot in pulmonary artery extracts 42 days after MCT administration. Treatment with rapamycin, fluoxetine, imatinib, or vehicle was given from day 21 to day 42 The bar graph shows the mean±SEM values in pulmonary arteries (n=10 in each group). *P<0.05 and **P<0.01 vs. values in MCT-4wk rats treated with vehicle. (C) Representative photographs of immunofluorescence staining for P-Akt in control rats and MCT-6wk rats treated with rapamycin, fluoxetine, imatinib, or vehicle from day 21 to day 42. No immunoreactivity was detected in sections incubated with secondary anti-rabbit and anti-mouse antibody but no primary antibody.
DISCUSSION
A major finding from the present study is that cultured PA-SMCs from rats with MCT-induced PH, when exposed to various growth factors, exhibit an abnormal proliferative phenotype linked to sustained activation of the intracellular mTOR signaling pathway. This proliferative phenotype was abolished by rapamycin added in vitro to cell cultures or given in vivo to rats, suggesting that it was mediated by mTOR signaling. Rapamycin inhibited both mTORC1 and mTORC2 substrates, whereas imatinib and fluoxetine less efficiently inhibited mTORC2 substrates and failed to inhibit the mTORC1 substrate S6K or to normalize PA-SMC growth. We also found that low doses of rapamycin, which selectively blocked mTORC1, were unable to normalize PA-SMC growth. Taken together, these results are consistent with a major role for mTORC1 and mTORC2 in mediating the abnormal PA-SMC proliferative phenotype during PH progression. Also, they suggest that rapamycin derivatives may hold promise for treating PH.
That PA-SMCs from remodeled pulmonary vessels from patients with various types of PH maintain an abnormal proliferative phenotype in vitro has been demonstrated in several studies using various growth factors including 5-HT, PDGF, and bFGF (21–23). This abnormality was initially considered an intrinsic cell alteration specific of human PH, in analogy with the abnormal behavior of tumor cells (24). The present results obtained with PA-SMCs from rats developing PH after MCT injection indicate that the proliferative phenotype can be acquired during PH progression. Faster growth of cultured PA-SMCs from rats with experimental PH than from control rats was shown recently in the hypoxia-induced PH model (18). The increased PA-SMC growth in both MCT- and hypoxia-induced PH suggests independence of the abnormal proliferative phenotype from the cause of PH. Moreover, the increased growth capacity was not specific of a given growth factor but was observed instead in response to PDGF, 5-HT, IL-1β, IGF-1, and the growth factor mixture in serum, indicating signaling pathway dysregulation downstream of the action of these effectors.
In the present study, both the mTORC1 and the mTORC2 signaling pathways were activated in cells from MCT-PH rats compared to cells from control rats, as shown by increased phosphorylation of the mTORC1 substrate S6K and of the mTORC2 substrate Akt(Ser-473). Akt phosphorylation at Thr-308 was also increased in PA-SMCs from MCT-treated rats, consistent with activation of the PI3K/PDK signaling cascade and of the Akt downstream effector GSK3. Interestingly, rapamycin in the concentration of 0.5 μM inhibited mTORC1-dependent S6K phosphorylation and mTORC2-dependent Akt phosphorylation at Ser473, as well as GSK3 phosphorylation. Rapamycin is known to bind mTORC1 and can also bind and inhibit mTORC2 during long-term treatment(11, 12). Our results therefore indicate that rapamycin in the dose used in our experiments inhibits not only mTORC1 in PA-SMCs, but also mTORC2.
Rapamycin treatment markedly inhibited the PA-SMC growth-promoting effect of FCS, abolishing the difference between cells from MCT-PH and control rats. Similarly, the Akt inhibitor A-443654, which inhibited mTORC2 and mTORC1 downstream effectors, normalized the growth of cells from MCT-PH rats. These results indicate that the differing proliferation rates of cells from MCT-PH and control rats were related to different activities of the mTORC1/mTORC2 signaling pathways and that dysregulation of these pathways persisted in cultured cells separated from their in vivo environment. Therefore, both mTORC1 and mTORC2 activation contributed to the increased PA-SMC growth in MCT-induced PH. Neither fluoxetine nor imatinib affected mTORC1-dependent S6K phosphorylation; both drugs decreased mTORC2-dependent Ser-473 Akt and GSK3 phosphorylation to a lesser extent than rapamycin and failed to normalize the growth of PA-SMCs from MCT-treated rats. To better delineate the roles for mTORC1 and mTORC2, we used various doses of rapamycin. We found that low-dose rapamycin failed to fully normalize the growth of PA-SMCs from MCT-treated rats and effectively inhibited mTORC1-dependent S6K phosphorylation without affecting mTORC2 substrates. Thus, selective mTORC1 inhibition by low-dose rapamycin is not sufficient to suppress the sustained growth capacity of cells from MCT-PH rats. Higher doses that simultaneously inhibit mTORC1 and mTORC2 signaling are required to normalize the growth of PA-SMCs from MCT-treated rats. These results are consistent with those obtained in chronically hypoxic rats and indicate that both mTORC1 and mTORC2 are required for the increased PA-SMC proliferation associated with PH development (18).
An important goal of this study was to investigate whether in vivo rapamycin treatment altered the mTOR signaling pathway in pulmonary vessels and normalized the growth of derived cultured PA-SMCs. We therefore studied rats with established PH after only 1 week of rapamycin, imatinib, or fluoxetine, when only minor changes in pulmonary-artery muscularization had occurred. The decreases in p-Akt, p-GSK3, and pS6K in pulmonary arteries at this time point, and notably in smooth-muscle cells, are consistent with a primary rapamycin effect on the Akt signaling pathway followed by secondary inhibition of PA-SMC proliferation. At this time point, fluoxetine or imatinib had no effect on pS6K or pAkt phosphorylation in the lung or pulmonary arteries. Rapamycin given in vivo to rats therefore inhibited the mTOR signaling pathway in a manner very similar to that seen with direct cell exposure, inhibiting both mTORC1 and mTORC2 substrates in pulmonary arteries, whereas imatinib and fluoxetine effects predominated on mTORC2 substrates.
Cultured PA-SMCs collected after 1 week of rapamycin treatment from rats with established MCT-induced PH exhibited the same growth rate as cells from control rats without PH. Also, no reduction in PA-SMC growth was noted in cells from rats given imatinib or fluoxetine according to the same protocol. These results show clearly that short-term in vivo rapamycin treatment can reverse the abnormal PA-SMC proliferative phenotype and normalize PA-SMC growth during PH progression, an effect not shared by fluoxetine or imatinib.
We then evaluated the curative and preventive effects of rapamycin treatment on the development of MCT-induced PH. Daily treatment started 3 weeks after MCT injection produced large decreases in Pap, right ventricular hypertrophy, and number of muscularized pulmonary vessels compared with vehicle-treated rats. Similar reductions in these parameters were obtained when rapamycin was given preventively. In both protocols, rapamycin appeared to have more potent effects than either fluoxetine or imatinib.
Taken together, these results emphasize the importance for treating PH of targeting intracellular signaling pathways common to various growth factors, instead of selectively targeting one particular extracellular effector acting on PA-SMC growth. Also, they emphasize the importance of the mTOR signaling pathway in mediating PA-SMC growth during PH progression, regardless of the cause of PH. These data provide further support for studies aimed at identifying the best molecular targets in the mTOR signaling pathway for antiproliferative therapies directed against PH. Given the ability of rapamycin to block either mTORC1 or both mTORC1 and mTORC2 depending on the dose, clinical studies will be needed to determine whether rapamycin derivatives may provide clinical benefits in patients with PH when used in doses devoid of systemic toxicity. The dramatic improvement of PH in a patient given rapamycin for a pancreatic tumor, recently described in a case-report, is consistent with therapeutic efficacy in the doses used in clinical practice (19). Treatment with rapamycin derivatives has already been approved for immune suppression after organ transplantation; prevention of vessel restenosis after angioplasty; and treatment of renal cell carcinoma, T-cell lymphoma, and tuberous sclerosis (10, 25). The potential usefulness of rapamycin derivatives in human PH therefore deserves further investigation.
Acknowledgments
Funding: This research was supported by grants from INSERM, the Ministère de la Recherche, the Delegation a la Recherche Clinique de l’AP-HP, and the Fondation de France.
We are grateful to the pharmaceutical company Abbott for generously donating the A-443654 drug used in this study.
References
- 1.Morrell NW, Adnot S, Archer SL, Dupuis J, Jones PL, MacLean MR, McMurtry IF, Stenmark KR, Thistlethwaite PA, Weissmann N, et al. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54:S20–31. doi: 10.1016/j.jacc.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase akt pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 3.Garat CV, Fankell D, Erickson PF, Reusch JE, Bauer NN, McMurtry IF, Klemm DJ. Platelet-derived growth factor bb induces nuclear export and proteasomal degradation of creb via phosphatidylinositol 3-kinase/akt signaling in pulmonary artery smooth muscle cells. Mol Cell Biol. 2006;26:4934–4948. doi: 10.1128/MCB.02477-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iwasaki H, Eguchi S, Ueno H, Marumo F, Hirata Y. Endothelin-mediated vascular growth requires p42/p44 mitogen-activated protein kinase and p70 s6 kinase cascades via transactivation of epidermal growth factor receptor. Endocrinology. 1999;140:4659–4668. doi: 10.1210/endo.140.10.7023. [DOI] [PubMed] [Google Scholar]
- 5.Liu Y, Fanburg BL. Serotonin-induced growth of pulmonary artery smooth muscle requires activation of phosphatidylinositol 3-kinase/serine-threonine protein kinase b/mammalian target of rapamycin/p70 ribosomal s6 kinase 1. Am J Respir Cell Mol Biol. 2006;34:182–191. doi: 10.1165/rcmb.2005-0163OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Humar R, Kiefer FN, Berns H, Resink TJ, Battegay EJ. Hypoxia enhances vascular cell proliferation and angiogenesis in vitro via rapamycin (mtor)-dependent signaling. FASEB J. 2002;16:771–780. doi: 10.1096/fj.01-0658com. [DOI] [PubMed] [Google Scholar]
- 7.Wullschleger S, Loewith R, Hall MN. Tor signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 8.Foster KG, Fingar DC. Mammalian target of rapamycin (mtor): Conducting the cellular signaling symphony. J Biol Chem. 2010;285:14071–14077. doi: 10.1074/jbc.R109.094003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laplante M, Sabatini DM. Mtor signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guertin DA, Sabatini DM. The pharmacology of mtor inhibition. Sci Signal. 2009;2:pe24. doi: 10.1126/scisignal.267pe24. [DOI] [PubMed] [Google Scholar]
- 11.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mtorc2 assembly and akt/pkb. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 12.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of akt/pkb by the rictor-mtor complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 13.McMurtry MS, Bonnet S, Michelakis ED, Haromy A, Archer SL. Statin therapy, alone or with rapamycin, does not reverse monocrotaline pulmonary arterial hypertension: The rapamcyin-atorvastatin-simvastatin study. Am J Physiol Lung Cell Mol Physiol. 2007;293:L933–940. doi: 10.1152/ajplung.00310.2006. [DOI] [PubMed] [Google Scholar]
- 14.Paddenberg R, Stieger P, von Lilien AL, Faulhammer P, Goldenberg A, Tillmanns HH, Kummer W, Braun-Dullaeus RC. Rapamycin attenuates hypoxia-induced pulmonary vascular remodeling and right ventricular hypertrophy in mice. Respir Res. 2007;8:15. doi: 10.1186/1465-9921-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou H, Liu H, Porvasnik SL, Terada N, Agarwal A, Cheng Y, Visner GA. Heme oxygenase-1 mediates the protective effects of rapamycin in monocrotaline-induced pulmonary hypertension. Lab Invest. 2006;86:62–71. doi: 10.1038/labinvest.3700361. [DOI] [PubMed] [Google Scholar]
- 16.Nishimura T, Faul JL, Berry GJ, Veve I, Pearl RG, Kao PN. 40-o-(2-hydroxyethyl)-rapamycin attenuates pulmonary arterial hypertension and neointimal formation in rats. Am J Respir Crit Care Med. 2001;163:498–502. doi: 10.1164/ajrccm.163.2.2006093. [DOI] [PubMed] [Google Scholar]
- 17.Gary-Bobo G, Houssaini A, Amsellem V, Rideau D, Pacaud P, Perrin A, Bregeon J, Marcos E, Dubois-Rande JL, Sitbon O, et al. Effects of hiv protease inhibitors on progression of monocrotaline- and hypoxia-induced pulmonary hypertension in rats. Circulation. 2010;122:1937–1947. doi: 10.1161/CIRCULATIONAHA.110.973750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krymskaya VP, Snow J, Cesarone G, Khavin I, Goncharov DA, Lim PN, Veasey SC, Ihida-Stansbury K, Jones PL, Goncharova EA. Mtor is required for pulmonary arterial vascular smooth muscle cell proliferation under chronic hypoxia. FASEB J. 2011;25:1922–1933. doi: 10.1096/fj.10-175018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wessler JD, Steingart RM, Schwartz GK, Harvey BG, Schaffer W. Dramatic improvement in pulmonary hypertension with rapamycin. Chest. 2010;138:991–993. doi: 10.1378/chest.09-2435. [DOI] [PubMed] [Google Scholar]
- 20.Han EK, Leverson JD, McGonigal T, Shah OJ, Woods KW, Hunter T, Giranda VL, Luo Y. Akt inhibitor a-443654 induces rapid akt ser-473 phosphorylation independent of mtorc1 inhibition. Oncogene. 2007;26:5655–5661. doi: 10.1038/sj.onc.1210343. [DOI] [PubMed] [Google Scholar]
- 21.Eddahibi S, Humbert M, Fadel E, Raffestin B, Darmon M, Capron F, Simonneau G, Dartevelle P, Hamon M, Adnot S. Serotonin transporter overexpression is responsible for pulmonary artery smooth muscle hyperplasia in primary pulmonary hypertension. J Clin Invest. 2001;108:1141–1150. doi: 10.1172/JCI12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schermuly RT, Dony E, Ghofrani HA, Pullamsetti S, Savai R, Roth M, Sydykov A, Lai YJ, Weissmann N, Seeger W, et al. Reversal of experimental pulmonary hypertension by pdgf inhibition. J Clin Invest. 2005;115:2811–2821. doi: 10.1172/JCI24838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Izikki M, Guignabert C, Fadel E, Humbert M, Tu L, Zadigue P, Dartevelle P, Simonneau G, Adnot S, Maitre B, et al. Endothelial-derived fgf2 contributes to the progression of pulmonary hypertension in humans and rodents. J Clin Invest. 2009;119:512–523. doi: 10.1172/JCI35070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adnot S. Lessons learned from cancer may help in the treatment of pulmonary hypertension. J Clin Invest. 2005;115:1461–1463. doi: 10.1172/JCI25399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Houghton PJ, Huang S. Mtor as a target for cancer therapy. Curr Top Microbiol Immunol. 2004;279:339–359. doi: 10.1007/978-3-642-18930-2_20. [DOI] [PubMed] [Google Scholar]