

Abstract
Metastatic urothelial carcinoma (mUC) has a very high mutational rate and is associated with an APOBEC mutation signature. We examined the correlation of APOBEC expression with overall survival (OS) and PD-L1 expression in a cohort of 73 mUC patients. mRNA expression of APOBEC3 family of genes (A3A, A3B, A3C, A3F_a, A3F_b, A3G, A3H) was measured using Nanostring. PD-L1 expression, evaluated by immunohistochemistry, on tumor infiltrating mononuclear cells (TIMCs) and tumor cells was scored from 0 to 4, with 2–4 being positive. Wilcoxon’s non-parametric tests assessed the association of APOBEC and PD-L1. The Cox regression model assessed the association of APOBEC with OS. All APOBEC genes were expressed in mUC. Increased A3A, A3D, and A3H expression associates with PD-L1 positive TIMCs (p = 0.0009, 0.009, 0.06). Decreased A3B expression was marginally associated with PD-L1 positive TIMCs expression (p = 0.05). Increased A3F_a and A3F_b expression was associated with increased expression of PD-L1 on tumor cells (p = 0.05). Increased expression of A3D and A3H was associated with longer OS (p = 0.0009). Specific APOBEC genes have different effects on mUC in terms of survival and PD-L1 expression. A3D and A3H may have the most important role in mUC as they are associated with OS and PD-L1 TIMC expression.
In the United States, there were more than 76,000 cases and more than 16,000 deaths from urothelial carcinoma (UC) in 20141. Cisplatin-based chemotherapy has improved clinical outcomes in metastatic UC (mUC), nonetheless the median overall survival is only 14 to 15 months, and mUC mostly remains an incurable disease2.
APOBEC deaminase enzyme family normally creates predicable mutations in viral DNA, limiting the replication ability of transposons and viruses3,4,5,6. Recently, APOBEC3 family has been shown to be a major source of somatic driver and passenger mutations in cancer7,8. The APOBEC3 family consists of seven members; APOBEC3A (A3A), APOBEC3B(A3B), APOBEC3C (A3C), APOBEC3D (A3D), APOBEC3F_a(A3F_a), APOBEC3F_b (A3F_b), APOBEC3G (A3G), and APOBEC3H (A3H).
Urothelial cancer has one of the highest mutational rates of all cancers, mean of 7.7 mutations per megabase7,9. The mechanism of this high mutation rate in UC is unknown. Although smoking has an epidemiologic association with UC, smoking carcinogens are not the cause of the majority of mutations based on mutational clustering analyses7,10,11. Mutational clustering observed in UC TCGA specimens were predominately TCW -> TTW or TGW changes, consistent with mutations caused by the APOBEC family of cytidine deaminases7,10,12,13,14. Also suggestive of APOBEC activity in UC was the high expression of APOBEC3B in almost all UC TCGA specimens7.
Multiple studies have demonstrated a correlation between A3B and A3A overexpression with mutational load, induction of DNA damage markers, and cell death15,16,17. It has been demonstrated that increased number of mutations correlate with better response rates to chemotherapy18. We hypothesized that increased APOBEC expression, which potentially plays a causative role in UC, would increase the mutation rate, thus increasing chemotherapy efficacy and OS as well as increasing PD-L1 positivity and increasing response to immunotherapy.
Assuming that increased APOBEC expression would cause more mutations, we inferred that increased expression of APOBEC correlates with PD-L1(CD274, B7-H1) expression in both tumor and immune cells. A higher mutational burden has been shown to lead to a higher neoantigen load. Since the anti-tumor immune response targets neoantigens, a higher neoantigen load means more reactive T cells and IFN-g production19. As a tumor evolves to evade the immune response, this IFN-g production may increase PD-L1 expression on tumor cells and TIMC, strengthening immune evasion. This could help to explain response to immunotherapy and will deserve further exploration in immunotherapy treated patients in future trials.
In a clinically annotated cohort of metastatic bladder cancer patients treated with platinum based therapy, we analyzed the association of APOBEC mRNA expression with PD-L1 expression, and overall survival (OS).
Methods
Patients and samples
73 mUC patients were identified from Brigham and Women’s Hospital and Hospital del Mar in Barcelona (Spain). Formalin fixed paraffin-embedded (FFPE) specimens from radical cystectomy or transurethral resection of bladder tumors were retrieved from the departments of pathology. All patients subsequently developed metastatic disease and received platinum based first line therapy. Prognostic factors including ECOG PS at initiation of chemotherapy, and whether patients had visceral site of metastasis, and clinical follow up data were collected. All subjects provided written informed consent. Institutional Review Board approval was obtained at Hospital del Mar and Dana-Farber/Harvard Cancer Centerbefore data acquisition and tumor staining, and all research was performed in accordance with the approved guidelines.
mRNA expression and mutational analysis
mRNA expression profile of 300 genes, chosen based on their known or potential role in UC, was measured using Nanostring technology. Oligonucleotide probes for all genes analyzed were synthesized by Nanostring, and transcripts were counted using the automated Nanostring nCounter system. Counts were normalized with the nSolver Analysis Software (v1.0) in which mRNA expression was compared to internal Nanostring controls, several housekeeping genes, and invariant genes in bladder cancer identified by analyzing gene expression variances in several published datasets20,21,22. For this analysis, we only looked at APOBEC mRNA expression. Throughput mutation profiling was performed by using both mass spectroscopy-based genotyping (Oncomap 3 platform) and confirmed with hME sequencing (Supplementary Table 1).
Immunohistochemistry and scoring of PD-L1 Expression
A tissue micro array (TMA) was constructed from treatment-naïve primary UC tissue. PD-L1 expression was evaluated by immunohistochemistry using a mouse monoclonal anti-PD-L1 antibody (405.9A11) developed in Dr. Gordon Freeman’s laboratory (Dana-Farber Cancer Institute, Boston, MA)23,24,25 Tumor-infiltrating mononuclear cells (TIMCs), PD-L1 expression on tumor cells and TIMCs with membranous expression was determined by two independent pathologists (MC, SS). PD-L1 tumor positivity was defined as ≥5% of tumor cell membrane staining. The extent of TIMCs was assessed in hematoxylin and eosin-stained slides and recorded as absent (0), focal (1), mild (2), moderate (3) and high (4) with score 0 or 1 considered negative. The extent of PD-L1-positive TIMCs was also assessed using the same scoring scale (0–4) and samples with a score of 2–4 were considered PD-L1 positive. For additional information, please see ref. 25.
Statistical Analysis
Overall survival (OS) was defined from the start of first line chemotherapy to the date of death or censored on the last known date alive. Cox regression model was used to assess the association of APOBEC expression with OS in multivariable analysis adjusting for ECOG status and whether patients had visceral disease. Hazard ratio and 95% CI are also listed. Wilcoxon’s non-parametric tests were used to summarize the associations of expression of APOBEC genes, and PD-L1 expression on TIMCs and tumor cells. Hotspot mutations correlation with APOBEC gene expression was assessed using Wilcoxon’s non-parametric test treating gene expression as continuous variables. All statistical analyses were performed using SAS 9.4 (SAS Institute, NC). All tests were two-sided and a p-value of <0.05 was considered statistically significant.
Results
Patient characteristics (n = 73) are presented in Table 1. All patients were included in the phase I clinical trial of cisplatin, gemcitabine, and paclitaxel (TCG) or phase II clinical trial comparing TCG vs. GC26.Median OS is 13 months and 41 patients died at time of data collection. Median follow up is 21 months.
Table 1. Patient Characteristics.
| N | % | |
|---|---|---|
| ECOG PS | ||
| 0 | 24 | 33% |
| 1 | 47 | 64% |
| 2 | 2 | 3% |
| Visceral disease | ||
| Without | 42 | 58% |
| With | 31 | 42% |
| Stage | ||
| 0 | 5 | 7% |
| 1 | 5 | 7% |
| 2 | 35 | 48% |
| 3 | 23 | 32% |
| 4 | 4 | 5% |
| Unknown | 1 | 1% |
PD-L1 tumor cell and PD-L1 MNC expression across the entire cohort is described in Table 2. mRNA expression level of APOBEC3A (A3A), APOBEC3B (A3B), APOBEC3C (A3C), APOBEC3D (A3D), APOBEC3F_a (A3F_a), APOBEC3F_b (A3F_b), APOBEC3G (A3G), and APOBEC3H (A3H) were measured using Nanostring and expression levels were dichotomized at the median. Median and quartile values are presented in Table 3.
Table 2. PD-L1 Expression.
| N | % | |
|---|---|---|
| Mononuclear Cell presence (score = 2, 3, 4) | ||
| 0 | 1 | 1% |
| 1 | 24 | 33% |
| 2 | 21 | 29% |
| 3 | 17 | 23% |
| 4 | 3 | 4% |
| Unknown | 7 | 10% |
| PD-L1 mononuclear presence (score = 2, 3, 4) | ||
| 0 | 19 | 26% |
| 1 | 21 | 29% |
| 2 | 18 | 25% |
| 3 | 6 | 8% |
| 4 | 2 | 3% |
| Unknown | 7 | 10% |
| PD-L1 tumor (≥5%) | ||
| Negative | 63 | 86% |
| Positive | 10 | 14% |
Table 3. APOBEC expression.
| Gene | Median Expression | Quartile 1 Expression | Quartile 3 Expression |
|---|---|---|---|
| APOBEC3A | 14.09 | 6.79 | 26.27 |
| APOBEC3B | 43.13 | 23.37 | 83.28 |
| APOBEC3C | 162.04 | 100.51 | 264.17 |
| APOBEC3D | 59.48 | 44.61 | 97.2 |
| APOBEC3F_b | 67.91 | 50.43 | 98.51 |
| APOBEC3F_a | 30.51 | 19.31 | 51.87 |
| APOBEC3G | 97.03 | 71.81 | 163.87 |
| APOBEC3H | 13.26 | 7.37 | 21.79 |
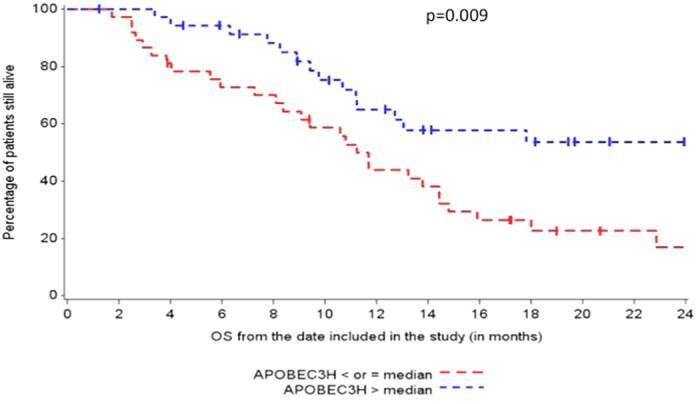
We initially explored the correlation between APOBEC expression and OS, as increased mutations correlate with response to platinum in UC18 (Table 4). High expression of A3A, A3D, and A3H were correlated with longer OS in multivariate analysis (p = 0.01 [HR:0.45 (0.23–0.85)], p = 0.02 [HR = 0.46 (0.24, 0.88), p = 0.004 [HR = 0.38(0.19, 0.73)], respectively).
Table 4. APOBEC overexpression correlation with PD-L1/PD-1 and OS.
| Gene | MNC presence (score = 2, 3, 4) | PDL1 MNC presence (score = 2, 3, 4) |
Longer OS |
PDL1 tumor expression ( ≥ 5%) |
|||||
|---|---|---|---|---|---|---|---|---|---|
| P-vlaue | P-vlaue | HR, CI | P-vlaue | P-vlaue | |||||
| APOBEC3A | 0.007 | High Expression | 0.0009 | High expression | 0.45 (CI:0.23–0.86) | 0.01 | High expression | 0.87 | |
| APOBEC3B | 0.13 | 0.05 | Low expression | 0.89 | 0.18 | ||||
| APOBEC3C | 0.80 | 0.2 | 0.6 | 0.06 | |||||
| APOBEC3D | 0.05 | High Expression | 0.009 | High expression | 0.49 (0.25–0.95) | 0.02 | High expression | 0.23 | |
| APOCEC3F_a | 0.40 | 0.49 | 0.43 | 0.05 | Low expression | ||||
| APOBEC3F_b | 0.46 | 0.31 | 0.15 | 0.04 | Low expression | ||||
| APOBEC3G | 0.59 | 0.65 | 0.55 | 0.47 | |||||
| APOBEC3H | 0.19 | 0.06 | High expression | 0.36 (0.19–0.71) | 0.004 | High expression | 0.8 | ||
Correlation of APOBEC expression with OS was performed using a Cox regression model in multivariate analysis adjusting for ECOG PS and visceral disease. Wilcoxon’s non-parametric tests were used to summarize the associations of expression of APOBEC genes, and PD-L1 expression on TIMCs and tumor cells. *HR: Hazard Ratio; *CI: Confidence Interval.
Subsequently, we investigated the association between APOBEC3 family expression and PD-L1 positivity in tumor cells and TIMCs, as previous evidence indicated specific members of the APOBEC3 family increases the number of mutations, and an increased mutational burden was associated with increased positive PD-L1 staining27.
Increased expression of A3A and A3D were significantly correlated with presences of TIMCs (p = 0.007, p = 0.05, respectfully) (Table 3).Increased expression of A3A, A3D, and A3H was associated with increased expression of PD-L1 on TIMCs (p = 0.0009, 0.0009, 0.06, respectively)_(Figs 1 and 2). Decreased expression of A3B was marginally associated with increased expression of PD-L1 on TIMCs (p = 0.05) (Table 4).
Figure 1. Association of APOBEC3D and OS.
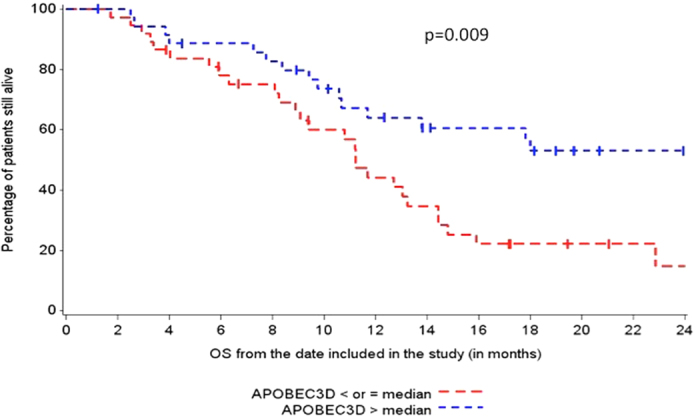
Figure 2. Association of APOBEC3H and OS.
While PD-L1 expression on TIMCs has been associated with OS and an improved response to the checkpoint inhibitors in mUC28,29,30,31, we also analyzed the association between PD-L1 expression on tumor cells and APOBEC expression, as it may provide insight about how the expression of these two proteins within the same tumor affect each other. While increased expression of A3A, A3D, and A3H correlated with increased PD-L1 expression in TIMCs, low expression of A3F_a and A3F_b was associated with increased expression of PD-L1 on tumor cells (p = 0.04). No other APOBEC gene expression was associated with PD-L1 expression in tumor cells.
We summarized number of hotspot mutations, using Oncomapv3 (Table 4) in a subset of patients. We grouped number of mutations as 0 vs. 1(Supplementary Table 2). There was no significant association found between having hotspot mutations in these select genes and APOBEC gene expression (p-values-A3A: 0.09, A3B: 0.80, A3C: 0.79, A3D: 0.21, A3F: 0.64, A3G: 0.39, A3H: 0.20).
Discussion
Many cancers are triggered by genomic instability. Instability can be induced by external factors, including UV, carcinogens, or smoking, or it can be induced by internal factors including mutations in MSH1 and BRCA132,33 genes. Recently, evidence has emerged that the APOBEC family promotes genomic instability in cancer by causing specific mutations in tumors10,13,16,34,35. UC has one of the highest mutational rates of all cancers in the TCGA analysis7,9. It is predicted, based on mutational clustering, that the high mutation rate in UC is largely caused by the APOBEC enzyme family10.
In this study, we examined the association between APOBEC expression with OS and PD-L1 expression on TIMCs and tumor cells. Increased expression of A3A and A3D were correlated with increased TIMC presence. All of these values became more significant when correlating APOBEC expression with increased PD-L1 expression in TIMCs, indicating APOBEC expression may increase TIMC presence along PD-L1 expression. We also saw decreased expression of A3B with increased PD-L1 expression in TIMCs.
A significant association between A3A, A3D, and A3H with OS was observed, which might be driven by these APOBECs causing a high mutational burden. A3D and A3H both affect cell cycle regulation6. A3H is small, nuclear bound and interacts with DNA in interphase and telophase, whereas A3D is known to cause cell cycle profile changes in HIV36. These APOBEC enzymes may cause a high mutational burden and might be responsible for chemotherapy18 response leading to survival benefit in a similar way to what is seen with immunotherapy27.
We observed the strongest association between A3A and PD-L1 expression. Higher expression of A3A was also correlated with TIMC presence and longer OS. Recently Chan et al described that, based on the mutational pattern, A3A is more likely than A3B to be responsible for the majority of mutations in UC and other tumor types7,34, potentially explaining the strong association we observed. Overexpression of A3A is also known to cause more mutations than other APOBEC enzymes34, whereas A3B was the highest expressed APOBEC gene in the TCGA analysis and is also correlated with increased mutations in breast and UC7,37. In our study, A3B expression was higher than A3A, indicating that the expression level in which APOBEC enzymes produce mutation may be different for the different enzymes.
Decreased expression of A3F_a and A3F_b were associated with increased PD-L1 expression in tumor cells. It is known that A3F lacks efficacy in causing mutations, as demonstrated by work done in HIV38,39, however it is unknown why we observed this opposite correlation. It is also possible that A3F and other APOBEC genes cause additional changes in the cell immune response, such as up regulating other immune checkpoint inhibitors, which down regulate PD-L1 expression.
Increased expression of A3G and A3C did not associate with increased PD-L1 expression in TIMCs, tumor cells, or OS. A3G has a different binding motif and is thought to not act in the same fashion as other APOBECs10. While A3C had the highest expression in our analysis, it has been shown to not be as potent at creating mutations compared to the other APOBEC genes40. Based on our observations, these genes might not play a strong role in UC mutagenesis.
Recently, immune checkpoint inhibitors, anti-PD1 and anti-PD-L1, have emerged as promising treatment strategies in UC41. Increased PD-L1 expression on tumor infiltrating immune cells is correlated with improved response to checkpoint inhibitors and with potentially better OS in mUC24,41. However, presently, there are no well-established predictive response biomarkers42 to either chemotherapy or checkpoint inhibitors. Several hypotheses have been presented to describe why some diseases are more responsive to these agents and why only select patients respond. Response to anti-PD-L1 therapy has been correlated with PD-L1 expression in tumor and immune cells, alterations in PIK3/AKT pathway, STAT3/JAK3 pathway, specific neoantigens expression, and mutational load27,43,44,45,46,47. In UC, it is hypothesized that responses to checkpoint inhibitors is due to the high mutational rate or frequent alterations in the PIK3/AKT pathway48,49,50. We can hypothesize based on our findings, that APOBEC expression may be a predictor of response to immunotherapy, due to the likely increase in mutation rate27,45,51, however this needs to be confirmed in further studies.
There were multiple limitations to our study. First, we did not have next generation sequencing data on these samples, thus we had to assume the APOBEC expression is associated with the mutational signatures and burdens as has been previously reported8,10,34,52. The expression of APOBEC is likely driven by tumor cells, however single-cell sequencing will provide additional evidence to this hypothesis.
In a brief exploration of mass spectrometry based hot spot sequencing of these samples, hot spot mutation burden was not correlated with any APOBEC expression. Tissue sample analyzed were obtained from local tumors at the time of diagnosis and not from distant metastatic locations. It is known that PD-L1 expression and mutational burden can differ between primary and metastatic sites in other tumor types25,53. It is also unknown about how other clinical variables or treatment variables affect PDL1 expression. This may be different in different tumor types, like the differences seen between smoking and PD-L1 expression in bladder versus NSCLC24,54. There is considerably more research that needs to be completed. To confirm these results, a large prospective cohort of uniformly treated mUC patients, ideally, comparing primary and metastatic tissue. Our findings should be confirmed in chemotherapy treated patients and expanded into immunotherapy treated patients. If confirmed, APOBEC expression may be used to identify patients who respond well to metastatic chemotherapy. Of great interest would be to comparing APOBEC expression, APOBEC signatures, mutational load, and response to chemotherapy and immunotherapy in UC.
Overall, we observed increased expression of APOBEC genes that also associated with increased PD-L1 expression and OS. This may indicate that patients with increased expression of these proteins derive a survival benefit when receiving platinum based chemotherapy and we can hypothesize that they might be more likely to respond to checkpoint inhibitors. These observations require prospective validation and warrant future study in the ongoing checkpoint inhibitors trials in UC that are looking for better predictive factors of response.
Additional Information
How to cite this article: Mullane, S. A. et al. Correlation of Apobec Mrna Expression with overall Survival and pd-l1 Expression in Urothelial Carcinoma. Sci. Rep. 6, 27702; doi: 10.1038/srep27702 (2016).
Supplementary Material
Acknowledgments
This work has been supported by FIS PI10/02112 (microRNAS) (Spanish Health Ministry Grant “Fondo de Investigacion Sanitaria”) and RTICC 06/0020/19 grants. Fundació Cellex (Barcelona) provided a generous donation to the Group of Molecular Therapeutics and Biomarkers, Hospital del Mar. The Tumor Bank of the Department of Pathology of Hospital del Mar (RD09/0076/0036) and the Xarxa de Bancs de Tumors sponsored by Pla Director d’Oncologia de Catalunya (XBTC) provided tissue samples. This study was also possible thanks to a grant from Friends of Dana-Farber, the Retired Professional Fire Fighters Cancer Fund, and the generous support of Whole Foods Golf Classic for bladder cancer research in memoriam of Christopher Snell. Also a private donation in the memory of Rich Beaudoin. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Joaquim Bellmunt: Consultant for Pierre Fabre, Astellas, Pfizer; Merck; Genentech. Gordon Freedman: Research funding from Takeda, Sanofi Gordon Freedman: Pending patent royalties on PD-1 pathway from BMS, Roche, Merck, EMD-Serono, Boehringer-Ingelheim, AstraZeneca, Novartis. Advisory Borad: CoStim, Novartis, Roche, BMS. Toni K. Choueiri: Consulting or Advisory for: Pfizer, GSK, Novartis, Merck, Bayer. Research funding: BMS, Merck, Exelixis, Roche, AstraZeneca, Tracon. Other coauthors: none.
Author Contributions J.B. and S.M. conceived of the study and participated in its design and coordination. S.M. drafted the manuscript. L.W., S.S., M.C. and J.R. performed the study. L.W., S.S., M.C., G.F., J.R. and T.K.C. participated in its design, and coordination and assisted in editing of manuscript. All authors read and approved the final manuscript.
References
- Siegel R. L., Miller K. D. & Jemal A.. Cancer statistics, 2015. CA Cancer J Clin 65, 5–29 (2015). [DOI] [PubMed] [Google Scholar]
- Kaufman D. S., Shipley W. U. & Feldman A. S.. Bladder cancer. Lancet 374, 239–249 (2009). [DOI] [PubMed] [Google Scholar]
- Liu P., Carvalho C. M., Hastings P. J. & Lupski J. R.. Mechanisms for recurrent and complex human genomic rearrangements. Curr Opin Genet Dev 22, 211–220 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suspène R. et al. Somatic hypermutation of human mitochondrial and nuclear DNA by APOBEC3 cytidine deaminases, a pathway for DNA catabolism. Proc Natl Acad Sci USA 108, 4858–4863 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beale R. C. et al. Comparison of the differential context-dependence of DNA deamination by APOBEC enzymes: correlation with mutation spectra in vivo. J Mol Biol 337, 585–596 (2004). [DOI] [PubMed] [Google Scholar]
- Refsland E. W. et al. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res 38, 4274–4284 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- C. G. A. R. Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 507, 315–322 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M. S. et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts S. A. et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet 45, 970–976 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shea J. P. et al. pLogo: a probabilistic approach to visualizing sequence motifs. Nat Methods 10, 1211–1212 (2013). [DOI] [PubMed] [Google Scholar]
- Nik-Zainal S. et al. Association of a germline copy number polymorphism of APOBEC3A and APOBEC3B with burden of putative APOBEC-dependent mutations in breast cancer. Nat Genet 46, 487–491 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts S. A. et al. Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol Cell 46, 424–435 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S. et al. Mutational processes molding the genomes of 21 breast cancers. Cell 149, 979–993 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns M. B. et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 494, 366–370 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara M. et al. APOBEC3B can impair genomic stability by inducing base substitutions in genomic DNA in human cells. Sci Rep 2, 806 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor B. J. et al. DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. Elife 2, e00534 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen E. M. et al. Somatic ERCC2 mutations correlate with cisplatin sensitivity in muscle-invasive urothelial carcinoma. Cancer Discov 4, 1140–1153 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascierto M. L. et al. An immunologic portrait of cancer. J Transl Med 9, 146 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. et al. Development of a multiplex quantitative PCR signature to predict progression in non-muscle-invasive bladder cancer. Cancer Res 69, 3810–3818 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riester M. et al. Combination of a novel gene expression signature with a clinical nomogram improves the prediction of survival in high-risk bladder cancer. Clin Cancer Res 18, 1323–1333 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Carbayo M., Socci N. D., Lozano J., Saint F. & Cordon-Cardo C.. Defining molecular profiles of poor outcome in patients with invasive bladder cancer using oligonucleotide microarrays. J Clin Oncol 24, 778–789 (2006). [DOI] [PubMed] [Google Scholar]
- Choueiri T. K. et al. PD-L1 expression in nonclear-cell renal cell carcinoma. Ann Oncol 25, 2178–2184 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellmunt J. et al. Association of PD-L1 expression on tumor-infiltrating mononuclear cells and overall survival in patients with urothelial carcinoma. Ann Oncol (2015). [DOI] [PubMed] [Google Scholar]
- Callea M. et al. Differential Expression of PD-L1 between Primary and Metastatic Sites in Clear-Cell Renal Cell Carcinoma. Cancer Immunol Res 3, 1158–1164 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellmunt J. et al. Phase I–II study of paclitaxel, cisplatin, and gemcitabine in advanced transitional-cell carcinoma of the urothelium. Spanish Oncology Genitourinary Group. J Clin Oncol 18, 3247–3255 (2000). [DOI] [PubMed] [Google Scholar]
- Rizvi N. A. et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plimack E.. (Journal of Clinical Oncology, ASCO, 2015), vol. 33.
- Petrylak D.. (Journal of Clinical Oncology, ASCO, 2015), chap. (suppl 7; abstr 297).
- Plimack E. R., Gupta S., Bellmunt J., Berger R., Montgomery B., Gonzalez E. J., Pulini J. et al. (Annals of Oncology 25, 2014), vol. no. suppl 4.
- Hoffman-Censits J., Grivas P. . Ed. (J Clin Oncol 34, 2016 (suppl 2S; abstr 355), IMvigor 210, a phase II trial of atezolizumab (MPDL3280A) in platinum-treated locally advanced or metastatic urothelial carcinoma (mUC)).
- Negrini S., Gorgoulis V. G. & Halazonetis T. D.. Genomic instability–an evolving hallmark of cancer. Nat Rev Mol Cell Biol 11, 220–228 (2010). [DOI] [PubMed] [Google Scholar]
- Sieber O. M., Heinimann K. & Tomlinson I. P.. Genomic instability–the engine of tumorigenesis? Nat Rev Cancer 3, 701–708 (2003). [DOI] [PubMed] [Google Scholar]
- Chan K. et al. An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nat Genet (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts S. A. & Gordenin D. A.. Hypermutation in human cancer genomes: footprints and mechanisms. Nat Rev Cancer 14, 786–800 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultquist J. F. et al. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J Virol 85, 11220–11234 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- T. C. G. A. Network. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaipan C., Smith J. L., Hu W. S. & Pathak V. K.. APOBEC3G restricts HIV-1 to a greater extent than APOBEC3F and APOBEC3DE in human primary CD4+ T cells and macrophages. J Virol 87, 444–453 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagi E. et al. Stably expressed APOBEC3F has negligible antiviral activity. J Virol 84, 11067–11075 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlois M. A., Beale R. C., Conticello S. G. & Neuberger M. S.. Mutational comparison of the single-domained APOBEC3C and double-domained APOBEC3F/G anti-retroviral cytidine deaminases provides insight into their DNA target site specificities. Nucleic Acids Res 33, 1913–1923 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powles T. et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 515, 558–562 (2014). [DOI] [PubMed] [Google Scholar]
- Rosenberg J.. (Annals of Oncology, 2015).
- Parsa A. T. et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med 13, 84–88 (2007). [DOI] [PubMed] [Google Scholar]
- Xue G. et al. Integrated Akt/PKB Signaling in Immunomodulation and Its Potential Role in Cancer Immunotherapy. J Natl Cancer Inst 107, (2015). [DOI] [PubMed] [Google Scholar]
- Snyder A., Wolchok J. D. & Chan T. A.. Genetic basis for clinical response to CTLA-4 blockade. N Engl J Med 372, 783 (2015). [DOI] [PubMed] [Google Scholar]
- Van Allen E. M. et al. Long-term benefit of PD-L1 blockade in lung cancer associated with JAK3 activation. Cancer Immunol Res (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le D. T. et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med 372, 2509–2520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- C. G. A. R. Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513, 202–209 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. et al. Genomic analyses reveal mutational signatures and frequently altered genes in esophageal squamous cell carcinoma. Am J Hum Genet 96, 597–611 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellmunt J. et al. Somatic Copy Number Abnormalities and Mutations in PI3K/AKT/mTOR Pathway Have Prognostic Significance for Overall Survival in Platinum Treated Locally Advanced or Metastatic Urothelial Tumors. PLoS One 10, e0124711 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen E. M. et al. Genomic correlates of response to CTLA4 blockade in metastatic melanoma. Science (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker B. A. et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat Commun 6, 6997 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran H. et al. Whole-Exome Sequencing of Metastatic Cancer and Biomarkers of Treatment Response. JAMA Oncol 1, 466–474 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Incecco A. et al. PD-1 and PD-L1 expression in molecularly selected non-small-cell lung cancer patients. Br J Cancer 112, 95–102 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.