

Abstract
We develop a novel approach to identify regions of the genome underlying population genetic differentiation in any genetic data where the underlying population structure is unknown, or where the interest is assessing divergence along a gradient. By combining the statistical framework for genome-wide association studies (GWASs) with eigenvector decomposition (EigenGWAS), which is commonly used in population genetics to characterize the structure of genetic data, loci under selection can be identified without a requirement for discrete populations. We show through theory and simulation that our approach can identify regions under selection along gradients of ancestry, and in real data we confirm this by demonstrating LCT to be under selection between HapMap CEU–TSI cohorts, and we then validate this selection signal across European countries in the POPRES samples. HERC2 was also found to be differentiated between both the CEU–TSI cohort and within the POPRES sample, reflecting the likely anthropological differences in skin and hair colour between northern and southern European populations. Controlling for population stratification is of great importance in any quantitative genetic study and our approach also provides a simple, fast and accurate way of predicting principal components in independent samples. With ever increasing sample sizes across many fields, this approach is likely to be greatly utilized to gain individual-level eigenvectors avoiding the computational challenges associated with conducting singular value decomposition in large data sets. We have developed freely available software, Genetic Analysis Repository (GEAR), to facilitate the application of the methods.
Introduction
In population genetics, eigenvectors have been routinely used to quantify genetic differentiation across populations and to infer demographic history (Cavalli-Sforza et al., 1996; Novembre et al., 2008; Reich et al., 2009). More recently, eigenvectors are commonly used as covariates in genome-wide association studies (GWASs) to adjust for population stratification (Price et al., 2006). Eigenvectors are usually estimated for each individual (individual-level eigenvectors, involving the inversion of a N × N matrix, where N is sample size). Theoretical studies have suggested that individual-level primary eigenvectors are measures of population differentiation reflecting Fst among subpopulations (Patterson et al., 2006; McVean, 2009; Bryc et al., 2013) and can be interpreted as the divergence of individuals from their most recent common ancestor. Eigenvectors can also be estimated for each single-nucleotide polymorphism (SNP; SNP-level eigenvectors that involve inversion of a M × M matrix; M is the number of SNPs) and these SNP-level eigenvectors can be interpreted as Fst metrics of each SNP (Weir, 1996). SNP-level eigenvectors from a reference population are useful for revealing the population structure of independent samples (Zhu et al., 2008) as they can be used to project, or predict, the eigenvector values of individuals. However, because of high-dimensional nature of GWAS data (commonly expressed as M>>N), direct estimation of SNP-level eigenvectors is nearly impossible when using millions of SNPs.
Singular value decomposition (SVD) enables SNP-level eigenvalues to be obtained in a computationally efficient manner for any set of genotype data (Chen et al., 2013); however, it is not possible to determine the SNPs that contribute most to the leading eigenvector, or to test whether specific SNPs are differentiated along the genetic gradient described by the eigenvector. Here, we propose an alternative, simple, fast approach for the estimation of SNP-level eigenvectors. By using individual-level eigenvectors as phenotypes in a linear regression, we demonstrate that the regression coefficients generated by single-SNP regression are equivalent to SVD SNP effects as proposed by Chen et al. (2013). As the single-SNP regression resembles the popular single-marker GWAS method, as implemented in PLINK (Purcell et al., 2007), we call this method EigenGWAS. We show that the EigenGWAS framework represents an alternative way for identifying regions under selection along gradients of ancestry.
Materials and methods
HapMap3 samples
HapMap3 samples were collected globally to represent genetic diversity of human population (Altshuler et al., 2010). HapMap3 contains representative samples from many continents: CEU and TSI represent population from north and south Europe, CHB and JPT from East Asia, and CHD Chinese from Denver, Colorado. Loci with palindrome alleles (A/T alleles or G/C alleles) were excluded, and 919 133 HapMap3 SNPs were used for the analysis.
1000 Genomes project
1000 Genomes project samples were used as a prediction set for projecting eigenvectors (The 1000 Genomes Project Consortium, 2012). We selected the Puerto Rico cohort (105 samples) and the Pakistan cohort (Punjabi from Lahore, Pakistan, 95 samples) for analysis.
POPRES samples
POPRES (Nelson et al., 2008) is a reference population for over 6000 samples from Asian, African and European nations. In this study, we selected 2466 European descendants. The POPRES genotype sample was imputed to a 1000 Genomes reference panel (The 1000 Genomes Project Consortium, 2012). Imputation for the POPRES was performed in two stages. First, the target data were haplotyped using HAPI-UR (Williams et al., 2012). Second, Impute2 was used to impute the haplotypes to the 1000 Genomes reference panel (Howie et al., 2011). We then selected SNPs that were present across all data sets at an imputation information score of >0.8. A full imputation procedure is described in https://github.com/CNSGenomics/impute-pipe. After quality control and removing loci with palindromic alleles (A/T alleles or G/C alleles), 643 995 SNPs for POPRES remained. In addition, we also conducted the analysis using nonimputed 234 127 common markers between POPRES and HapMap3. As the results between these two data sets were very similar, this report focussed on the results from 643 995 SNPs that were more informative.
Simulation scheme I: null model without population structure
A total of 2000 unrelated samples with 500 000 biallelic markers, which were in linkage equilibrium to each other, were simulated. The minor allele frequencies ranged from 0.01 to 0.5, and Hardy–Weinberg equilibrium was assumed for each locus. All individuals were simulated from a homogeneous population, with no population stratification. In order to calculate Fst at each locus, we divided the sample into subpopulations based upon eigenvectors that were estimated from a genetic relationship matrix calculated using all 500 000 markers (see below).
Simulation scheme II: null model with population structure
In general, this simulation scheme was followed Price et al., 2006. A total of 2000 unrelated samples with 10 000 biallelic markers, which were in linkage equilibrium to each other, were generated. For each marker, its ancestral allele frequency was sampled from a uniform distribution between 0.05 and 0.95, and its frequency in a subpopulation was sampled from β-distribution with parameters 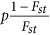 and
and 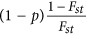 . The β-distribution had mean of P and sampling variance of P(1−P)Fst. Once the allele frequency for a subpopulation over a locus was determined as Ps, individuals were generated from a binomial distribution Binomil(2,Ps). It agreed with the quantity that measures the genetic distance between a pair of subpopulations (Cavalli-Sforza et al., 1996).
. The β-distribution had mean of P and sampling variance of P(1−P)Fst. Once the allele frequency for a subpopulation over a locus was determined as Ps, individuals were generated from a binomial distribution Binomil(2,Ps). It agreed with the quantity that measures the genetic distance between a pair of subpopulations (Cavalli-Sforza et al., 1996).
Calculating individual-level eigenvectors
We assume that there is a reference sample consisting of N unrelated individuals and M markers. 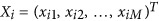 is a vector of the ith individual's genotypes along M loci, with x the number of the reference alleles. An N × N genetic relatedness (correlation) matrix A (matrix in bold font) for each pair of individuals is defined as
is a vector of the ith individual's genotypes along M loci, with x the number of the reference alleles. An N × N genetic relatedness (correlation) matrix A (matrix in bold font) for each pair of individuals is defined as 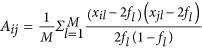 , in which fl is the frequency of the reference allele. The principal component analysis (PCA) is then implemented on the A matrix (Price et al., 2006), generating -, which is a N × K (K⩽N) matrix, in which Ek is the eigenvector corresponding to the kth largest eigenvector.
, in which fl is the frequency of the reference allele. The principal component analysis (PCA) is then implemented on the A matrix (Price et al., 2006), generating -, which is a N × K (K⩽N) matrix, in which Ek is the eigenvector corresponding to the kth largest eigenvector.
Unified framework for BLUP, SVD and EigenGWAS
Theoretically, PCA can also be implemented on a M × M matrix, but this is often infeasible because the M × M matrix is very large. However, for individual i, eigenvector k can also be written as:
in which βk is a M × 1 SNP-level vector of the SNP effects on Ek, and xi is the genotype of the ith individual across M loci. In the text below, we denote individual-level eigenvector as eigenvector (N × 1 vector), and SNP-level eigenvector (M × 1) as SNP effects.
We review three possible methods to estimate β given eigenvectors. The first method is best linear unbiased prediction (BLUP) that is commonly used in animal breeding and recently has been introduced to human genetics for prediction (Henderson, 1975; Goddard et al., 2009). The second method is to convert an individual-level eigenvector to SNP-level eigenvector using SVD, as proposed by Chen et al. (2013). The third method is the approach outlined here, EigenGWAS, that is a single-marker regression, as commonly used in GWAS analysis.
Methods 1 and 2: BLUP and SVD
For a quantitative trait, y=μ+βX+e, in which y is the phenotype, μ is the grand mean, β is the vector for additive effects, X is the genotype matrix and e is the residual. Without loss of generality, the BLUP equation can be expressed as:
in which 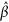 is the estimates of the SNP effects,
is the estimates of the SNP effects, 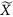 is the standardized genotype matrix, V is the variance covariance with
is the standardized genotype matrix, V is the variance covariance with 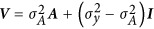 and y is the trait of interest (Henderson, 1975). Replacing y with individual-level eigenvector (Ek), Equation (2) can be written as
and y is the trait of interest (Henderson, 1975). Replacing y with individual-level eigenvector (Ek), Equation (2) can be written as
in which βk is the BLUP estimate of the SNP effects and Ek is the kth eigenvector estimated from the reference sample. The V matrix can be replaced with A because the eigenvector has no residual error (that is, h2=1). This method has also been proposed as an equivalent computing algorithm for genomic predictions (Maier et al., 2015).
In addition, the connection between PCA and SVD can be established through the transformation between the N × N matrix to the M × M matrix (McVean, 2009). Let A=PDP−1, in which D is a N × N diagonal matrix with λk, P is N × N matrix with the eigenvectors. B=XT(PDP−1)−1P=XTPD−1, in which B is M × N matrix. This is equivalent to the equation used in Chen et al. (2013) where BT=D−1(XTP)T. Thus, eigenvector transformation can be viewed as a special case of BLUP in which the heritability is 1 (Equation (3)). However, under SVD another analysis step is then required to evaluate the significance of the estimated SNP effect. In an EigenGWAS framework an empirical P-value is produced when estimating the regression coefficient.
Method 3: estimating SNP effects on eigenvectors with EigenGWAS
Given the realized genetic relationship matrix A, for unrelated homogeneous (i.i.d.) samples, E(Aij)=0 (i ≠ j), and consequently E(A)=I, an identity matrix. Because of sampling variance of the genetic relationship matrix A, the off diagonal is a number slightly different from zero even for unrelated samples (Chen, 2014). If we replace the matrix with its mathematical expectation—the identity matrix—Equation (3) can be further reduced to 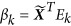 , equivalent to single-marker regression Ek=a + bx + e, as implemented in PLINK (Purcell et al., 2007). Furthermore, standardization for X is not required because it will not affect P-value. Thus, SNP effects can be estimated using the single-marker regression that is computationally much easier in practice and is implemented in many software packages. Each SNP effect,
, equivalent to single-marker regression Ek=a + bx + e, as implemented in PLINK (Purcell et al., 2007). Furthermore, standardization for X is not required because it will not affect P-value. Thus, SNP effects can be estimated using the single-marker regression that is computationally much easier in practice and is implemented in many software packages. Each SNP effect, 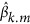 , is estimated independently, and the P-value of each marker can be estimated that requires additional steps in BLUP and SVD.
, is estimated independently, and the P-value of each marker can be estimated that requires additional steps in BLUP and SVD.
We summarize the properties and their transformation of SVD, BLUP and EigenGWAS as below:
E k is determined by the A matrix, or in another words, it is determined by the genotypes completely. If we consider each E k is the trait of interest—a quantitative trait—its heritability is 1.
h 2=1. SVD and BLUP are both computational tools in converting a vector from N × N matrix to a M × M matrix. SVD is a special case to BLUP when h 2=1 for BLUP.
h 2=1 and E( A )= I . When these two conditions are set, BLUP is further reduced to single-marker association studies that is EigenGWAS as suggested in this study.
Recently, in an independent work Galinsky et al. (2016) introduced an approximation to find the proper scaling for SNP effects (‘SNP weight' in Galinsky's terminology) estimated from SVD in order to produce accurate P-values. In our EigenGWAS framework, P-values for individual-level SNP eigenvector are automatically generated. In practice, it is conceptually easier to conduct EigenGWAS on eigenvectors than to conduct BLUP/SVD. In addition, if computational speed is of concern, EigenGWAS can be easily parallelized for each chromosome, each region or even each locus.
Interpretation for EigenGWAS signals
We can write a linear regression model Ek=a + βx + e, in which Ek is standardized. Assuming that a sample has two subdivisions that have sample size n1 and n2, 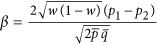 ,
, 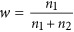 and the sampling variance for β is
and the sampling variance for β is 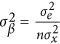 . The χ2 test for β is
. The χ2 test for β is
in which 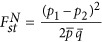 is Nei's estimator of genetic difference for a biallelic locus (Nei, 1973). Furthermore,
is Nei's estimator of genetic difference for a biallelic locus (Nei, 1973). Furthermore, 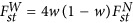 , in which
, in which 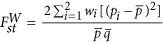 for a pair of subpopulations as defined in Weir (1996). Hence,
for a pair of subpopulations as defined in Weir (1996). Hence, 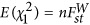 .
.
As the proportion of the variance explained by the largest eigenvalue is equal to FstW in PCA (McVean, 2009), 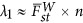 , in which
, in which 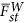 characterizes the average divergence for a pair of subpopulations. When the test statistic, Equation (4), is adjusted by the largest eigenvalue λ1, an equivalent technique in GWAS for the correction of population stratification,
characterizes the average divergence for a pair of subpopulations. When the test statistic, Equation (4), is adjusted by the largest eigenvalue λ1, an equivalent technique in GWAS for the correction of population stratification, 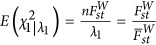 . Hence,
. Hence,
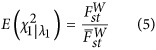 |
after the adjustment of the largest eigenvalue, the EigenGWAS test statistic on E1 immunes of population stratification, at least for a divergent sample.
For a locus under selection, it should have a greater Fst than 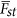 the background divergence. Hence, the statistical power for detecting whether a locus is under selection is determined by the strength of selection, and this can be defined as the ratio between Fst of a particular locus and
the background divergence. Hence, the statistical power for detecting whether a locus is under selection is determined by the strength of selection, and this can be defined as the ratio between Fst of a particular locus and 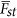 the average divergent in the sample. It is analogous to consider the χ2 test with noncentrality parameter (NCP),
the average divergent in the sample. It is analogous to consider the χ2 test with noncentrality parameter (NCP), 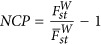 . Otherwise specified, in this study Fst is referred to the one defined in Weir (1996).
. Otherwise specified, in this study Fst is referred to the one defined in Weir (1996).
Validation and prediction for population structure
Once βk is estimated, it is straightforward to get genealogical profile for an independent target sample. In general, it is equivalent to genomic prediction, and the theory for prediction can be applied (Daetwyler et al., 2008; Dudbridge, 2013). The predicted genealogical score can be generated as
in which 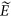 k is the predicted kth eigenvector,
k is the predicted kth eigenvector, 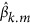 is the estimated SNP effects and X is the genotype for the target sample. We focus on the correlation between the predicted eigenvectors and the direct eigenvectors, and thus it does not matter whether X or
is the estimated SNP effects and X is the genotype for the target sample. We focus on the correlation between the predicted eigenvectors and the direct eigenvectors, and thus it does not matter whether X or 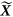 is used.
is used.
In contrast to conventional prediction studies that focus on a metric phenotype of interest, prediction of population structure is focussed on a ‘latent' variable. This latent variable is the genetic structure of population that is shaped by allele frequency and linkage disequilibrium of markers. Thus, expectations of prediction accuracy differ from what has been established for conventional prediction (Daetwyler et al., 2008; Dudbridge, 2013) 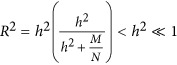 . We therefore assess prediction of accuracy for E1 across markers when using different prediction thresholding (Purcell et al., 2009).
. We therefore assess prediction of accuracy for E1 across markers when using different prediction thresholding (Purcell et al., 2009).
Here we proposed an equation for prediction accuracy, especially for E1
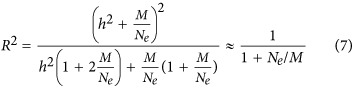 |
when there is no heritability, the predictor can be simplified to 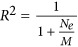 , meaning that as the number of markers increases prediction accuracy should rapidly reach 1. Here the h2 is interpreted as the genetic difference in the source population, or real ancestry informative markers. For a homogeneous population, the genetic difference is large because of genetic drift, and h2 ≈ 0.
, meaning that as the number of markers increases prediction accuracy should rapidly reach 1. Here the h2 is interpreted as the genetic difference in the source population, or real ancestry informative markers. For a homogeneous population, the genetic difference is large because of genetic drift, and h2 ≈ 0.
For this study, the genetic relationship matrix, PCA and BLUP estimation were conducted using GCTA software (Yang et al., 2011a). Single-marker GWAS was conducted using PLINK (Purcell et al., 2007) or GEAR (https://github.com/gc5k/GEAR/wiki/EigenGWAS; https://github.com/gc5k/GEAR/wiki/ProPC).
Results
Properties of the estimating SNP effects for eigenvectors
We applied EigenGWAS to the HapMap cohort, a known structured population. Eigenvectors were estimated via PCA based on the A matrix using all 919 133 SNPs. We conducted EigenGWAS for HapMap, using Ek, the kth eigenvector, as the phenotype and investigated the performance of EigenGWAS from E1 to E10. From E1 to E10, we found 546 716 significant signals (231 677 quasi-independent signals after clumping) on E1 and gradually reduced to 236 (163 after clumping) selection signals on E10 (Figure 1). The large numbers of genome-wide significant loci are likely because HapMap3 comprised samples from different ethnicities, and these loci can be interpreted as ancestry informative marker (AIM). For each Ek, its associated eigenvalue was highly correlated with the λGC, the genomic inflation factor that is commonly used in adjusting population stratification for GWAS (Devlin and Roeder, 1999), resulted from its EigenGWAS. The top five eigenvalues associated to HapMap samples were 100.14, 47.66, 7.168, 5.92 and 4.40, and the corresponding λGC of EigenGWAS were 103.72, 44.69, 6.47, 5.17, and 3.96, respectively (Table 1). The large eigenvalues observed were consistent with previous theory that the magnitude of eigenvalues indicate structured population (Patterson et al., 2006). The connection between λGC and eigenvalues provides a straightforward interpretation: a large λGC indicates underlying population structure (Equation (5)). Therefore, correction for λGC will filter out signals due to population stratification, allowing loci under selection to be identified. For example, after correction for λGC, the number of GWAS hits was reduced for each EigenGWAS. For HapMap, EigenGWAS hits on E1 dropped from 548 716 to 6 loci only, and for POPRES from 10 885 to 152, indicating that AIMs were largely driven by the genetic drift. These observations agreed well with our theory (see Materials and methods).
Figure 1.

Manhattan plots for EigenGWAS for top 10 eigenvectors for HapMap. Using Ei as the phenotype, the single-marker association was conducted for nearly 919 133 markers. The left panel illustrates from E1 to E5 and the right panel from E6 to E10. The horizontal lines indicate genome-wide significance after Bonferroni correction.
Table 1. EigenGWAS signals for HapMap and POPRES.
|
HapMap |
POPRES |
|||||||
|---|---|---|---|---|---|---|---|---|
| Eigenvector (Ei) | Eigenvalue | GWAS λGCa | No. of GWAS hitsb | No. after clumpingc | Eigenvalue | GWAS λGCa | No. of GWAS hitsb | No. after clumpingc |
| 1 | 100.135 | 103.715 | 546 716 (6) | 231 677 | 5.104 | 5.005 | 10 885 (152) | 3004 |
| 2 | 47.658 | 44.686 | 382 867 (7) | 161 022 | 2.207 | 1.929 | 1254 (802) | 289 |
| 3 | 7.168 | 6.471 | 33 317 (36) | 15 344 | 2.157 | 1.910 | 1201 (713) | 340 |
| 4 | 5.923 | 5.173 | 21 935 (51) | 12 401 | 2.077 | 1.464 | 1353 (1,074) | 331 |
| 5 | 4.402 | 3.964 | 9554 (66) | 4727 | 1.971 | 1.866 | 781 (273) | 76 |
| 6 | 2.449 | 1.982 | 1113 (36) | 567 | 1.871 | 1.295 | 1162 (878) | 111 |
| 7 | 2.285 | 1.986 | 593 (22) | 389 | 1.843 | 1.337 | 1239 (1027) | 130 |
| 8 | 2.107 | 1.742 | 236 (6) | 171 | 1.818 | 1.486 | 1259 (940) | 152 |
| 9 | 2.056 | 1.729 | 268 (3) | 174 | 1.807 | 1.503 | 1701 (1086) | 113 |
| 10 | 2.0217 | 1.661 | 236 (14) | 163 | 1.798 | 1.492 | 1639 (1287) | 90 |
Abbreviation: GWAS, genome-wide association study.
HapMap has 988 samples and 919 133 single-nucleotide polymorphisms (SNPs); its GWAS hits had P-values <5.44e−08 given α=0.05. POPRES has 2466 European samples and 643 995 SNPs; its GWAS hits had P-value <7.76e−08 given α=0.05.
λGC was calculated as the ratio between the median of observed χ2 from EigenGWAS to the median of χ2 value that is 0.455.
The GWAS hits were counted without λGC correction, and with λGC correction in parentheses.
After clumping, the reported numbers were quasi-independent GWAS hits. Within 250K bp and linkage disequilibrium of r2 >0.5 only the most significant GWAS hit was counted as a GWAS hit (see PLINK —clump default option). See Supplementary Tables 2 and 3 for pruned POPRES results.
We demonstrate theoretically that for EigenGWAS, the estimated SNP effects using single-marker GWAS are approximately equivalent to the estimates from BLUP, and the correlation between the estimates from these two methods was very high (greater than R2 >0.98 on average; Figure 2), even in HapMap samples that consist of a mix of ethnicities where the A matrix is non-zero for off-diagonal elements (Supplementary Figure 1). This confirms that our EigenGWAS approach provides an accurate representation of the SNP effects on eigenvalues.
Figure 2.

Linear correlation for the SNP effects estimated using EigenGWAS and BLUP for HapMap3. The x axis represents EigenGWAS estimation for SNP effects, and the y axis represents BLUP estimation for SNP effects. The left panel illustrates from E1 to E5 and the right panel from E6 to E10. As illustrated at top left in each plot, the correlation, measured in R2, is nearly 1.
We also conducted EigenGWAS on the POPRES samples, from which we selected 2466 European samples. On E1, there were 10 885 (3004 quasi-independent signals after clumping) genome-wide significant signals, and reduced to 1639 (90 after clumping) on E10 (Table 1). As in the HapMap sample, we observed a concordance between eigenvalues and λGC in POPRES. The top five eigenvalues were 5.104, 2.207, 2.157, 2.077 and 1.971, and their associated EigenGWAS λGC were 5.005, 1.929, 1.910, 1.464 and 1.866, respectively (Table 1), indicating population structure. The genetic relationship matrix estimated from the POPRES data resembled a diagonal matrix that had off-diagonal elements close to zero, suggesting that POPRES is a more homogenous samples as compared with HapMap (Supplementary Figure 1). Correlations between the estimates from EigenGWAS and BLUP were high, with an average of >0.999 from E1 to E10 (Supplementary Figure 2), close to one as expected.
The χ2 statistics of the estimated SNP effects on eigenvectors from EigenGWAS were correlated with Fst for each SNP, such as on HapMap samples E1 (Supplementary Figure 3), consistent with previous established relationship between eigenvectors and Fst (Patterson et al., 2006; McVean, 2009). Using naive threshold of Ek > 0, 2466 POPRES samples were divided into nearly two even groups that would be served as two subgroups in calculating Fst. E1 > 0 split the POPRES samples into North and South Europe; samples from UK, Ireland, Germany, Austria and Australia were in one group, and samples from Italy, Spain and Portugal were in the other group; samples from Switzerland and France were nearly evenly split into two groups. Fst for each SNP was consequently calculated based on these two groups. For every eigenvector until E10, we observed strong correlations between Fst and the χ2 test statistics for EigenGWAS signals (Figure 3), and the averaged correlation was 0.925 (s.d., 0.067). For example, the correlation was 0.89 (P-value <1e−16) between χ2 test statistics and Fst for E1 in POPRES (Supplementary Table 1). This correlation is consistent with our theory where Fst has a strong linear relationship with its EigenGWAS χ2 test statistic.
Figure 3.

The correlation between Fst and 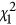 for EigenGWAS SNP effects for POPRES. For each eigenvector, upon Ei >0 or Ei ⩽0, POPRES samples were split into two groups, upon which Fst was calculated for each locus. The correlation, at top left in each plot, was measured in R2.
for EigenGWAS SNP effects for POPRES. For each eigenvector, upon Ei >0 or Ei ⩽0, POPRES samples were split into two groups, upon which Fst was calculated for each locus. The correlation, at top left in each plot, was measured in R2.
We also validated our results in the simulation scheme I, in which there was neither selection nor population stratification. Given 2000 simulated samples, each of which had 500 000 unlinked SNPs, the EigenGWAS showed few GWAS signals, 2 genome-wide significant signals on E2 (Supplementary Figure 4). As expected, λGC ranged from ∼1.124 to 1.130, with a mean of 1.127 for EigenGWAS on the top 10 eigenvectors, indicating little population stratification for the simulated data. After correction for λGC, no loci were significant, indicating that the type-I error rate was controlled. After splitting the samples into two groups depending on Ei > 0, the correlation between χ2 test statistics and Fst is ∼0.67 from E1 to E10 (Supplementary Figure 5). Furthermore, as expected, after correction for λGC, the P-values followed a uniformed distribution for EigenGWAS from E1 to E10 (Supplementary Figure 6).
Furthermore, we also validated the theory in the simulation scheme II, in which there was population stratification. We wanted to know whether the adjustment of the test statistic with the greatest eigenvalue could render the distribution of the test statistics immunes of population stratification. Given various sample sizes for two subdivisions, after the adjustment for the test statistic with the largest eigenvalue, the test statistic followed the null distribution that was the χ2 distribution of 1 degree of freedom (Supplementary Figure 7), indicating a well control of population stratification after correction.
The statistical power of EigenGWAS was also evaluated. As demonstrated, the power of EigenGWAS in detecting a locus under selection was determined by the ratio between the specific Fst of a locus and the averaged population stratification in the sample (Supplementary Figure 8).
Using EigenGWAS to identify loci under selection in structured populations
We propose EigenGWAS as a method of finding loci differentiated among populations, or across a gradiant of ancestry. Intuitively, every EigenGWAS hit is an AIM that differs in allele frequency along an eigenvector because of genetic drift or selection. A locus under selection should be more differed across populations than genetic drift can bring out. Thus, correction for λGC controls for background population structure, providing a test of whether an AIM shows greater allelic differentiation than expected under the process of genetic drift.
We pooled together CEU (112 individuals) and TSI (88 individuals) that represent Northwestern and Southern European populations in HapMap. EigenGWAS was conducted on E1 that partitioned CEU and TSI into two groups accurately using E1 > 0 as threshold (Supplementary Figure 9). We corrected for λGC, which was 1.723, for CEU and TSI. Adjustment for λGC significantly reduced population stratification (Supplementary Figure 10), and was consequently possible to filter out the baseline difference between these two cohorts. After correction, we found evidence of selection at the lactose persistence locus, LCT (P-value=1.21e−20, Figure 4). Because of hitchhiking effect, the region near LCT also showed divergent allele frequencies. For example, the DARS gene, 0.15 M away from LCT, was also significantly associated with E1 (P-values=1.51e−23). HERC2 was slightly below genome-wide significance level (P-value=8.22e−08), indicating that anthropological difference reflected geographic locations of two cohorts but not under selection as strong as LCT. For the TSI and CEU example, given 200 individuals and 919 133 markers, it used ∼6 min and 1.9G RAM to complete the following three steps involved in EigenGWAS, developed in Java: (1) generate correlation matrix for 200 individuals using 919 133 markers; (2) estimate the first eigenvector, 919 133 elements, from the correlation matrix; and (3) run the linear regression model for every marker.
Figure 4.

EigenGWAS for CEU (112 samples) and TSI (88 samples) from HapMap. (a) Manhattan plot for EigenGWAS on E1 without correction for λGC. When there was no correction, we found LCT on chromosome 2, MICA on chromosome 6 (HMC region), HIF1A on chromosome 14 and HERC2 on chromosome 15. The line in the middle was genome-wide significant level at α=0.05 given multiple correction. (b) Manhattan plot for EigenGWAS on E1 with λGC correction; LCT was still significant, and HERC2 was slightly below whole genome-wide significance level. The genome-wide significance threshold was P-value=5.44e−08 for α=0.05.
We then conducted EigenGWAS in the POPRES sample by treating E1 as a quantitative trait, and calculated the approximate Fst for each SNP given two groups split by the threshold of E1 > 0 (Supplementary Figure 11). Given 643 995 SNPs, the genome-wide threshold was P-value <7.76e−08 for the significance level of α=0.05 (Figure 5). As expected for POPRES, λGC=5.00 indicated substantial population stratification. Correcting for λGC systematically reduced the EigenGWAS χ2 test statistics (Supplementary Figure 12), and we replicated the significance of LCT (P-value=1.23e−22) and DARS (P-value=8.99e−22) (Table 2), suggesting selection at these regions. HERC2 was also replicated with P-value 8.15e−09, and with Fst of 0.041. The results were consistent after SNP pruning for POPRES (Supplementary Tables 2 and 3).
Figure 5.

EigenGWAS for POPRES samples on eigenvector 1. (a) Manhattan plot for EigenGWAS without correction for λGC. (b) After correction for λGC, we found LCT on chromosome 2, SLC44A4 on chromosome 6 and HERC2 on chromosome 15. The genome-wide significance level was P-value=7.76e−08 given α=0.05.
Table 2. Gene discovery using EigenGWAS.
| Gene | Lead SNP | Position | Allele | P-valuea | MAF (TSI:CEU) | Fstb | Annotation |
|---|---|---|---|---|---|---|---|
| CEU and TSI samples | |||||||
| LCT | rs6719488 | 2:135817629 | G/T | 6.68e−34 (1.21e−20) | 0.733:0.206 | 0.558 | Lactose persistent locus |
| DARS | rs13404551 | 2:135964425 | C/T | 8.18e−39 (1.51e−23) | 0.756:0.206 | 0.604 | Genetic hitchhiking because of LCT |
| MICA | rs2256175 | 6:31412672 | T/C | 8.94e−10 (2.60e−6) | 0.665:0.360 | 0.183 | MHC class I polypeptide-related sequence A |
| HIF1A | rs2256205 | 14:61670944 | A/G | 1.51e−10 (8.86e−7) | 0.464:0.179 | 0.192 | HIF-1A thus plays an essential role in embryonic vascularization, tumour angiogenesis and pathophysiology of ischaemic disease. |
| HERC2 | rs8039195 | 15:26189679 | C/T | 2.75e−12 (8.22e−08) | 0.403:0.122 | 0.212 | Genetic variations in this gene are associated with skin/hair/eye pigmentation variability |
| Gene | Lead SNP | Position | Allele | P-valuea | MAF (Southern Europeans: Northern Europeans) | Fstb | Annotation |
| POPRES European samples | |||||||
| LCT | rs3754686 | 2:135817629 | T/C | 3.30e−106 (1.23e−22) | 0.514:0.279 | 0.110 | |
| DARS | rs13404551 | 2:135964425 | C/T | 6.32e−102 (8.99e−22) | 0.518:0.293 | 0.106 | |
| SLC4A4 | rs605203 | 6:31819235 | C/A | 8.94e−44 (5.77e−10) | 0.214:0.343 | 0.040 | Defects in this gene can cause sialidosis, a lysosomal storage disease |
| HERC2 | rs1667394 | 15:26189679 | C/T | 3.90e−38 (8.15e−09) | 0.276:0.173 | 0.041 | |
Abbreviations: GWAS, genome-wide association study; MAF, minor allele frequency; SNP, single-nucleotide polymorphism.
The P-value cutoff for CEU and TSI was 5.44e−08 (919 133 SNPs) and for POPRES was 7.76e−08 (643 995 SNPs) at genome-wide significance level of α=0.05. λGC=1.725 for CEU and TSI and λGC=5.00 for POPRES.
P-values without λGC correction, and with λGC correction in parentheses.
Fst is calculated by partitioning the sample into two groups upon E1 >0. For TSI and CEU set, partitioning on E1 perfectly separated TSI (88 samples) and CEU (112 samples). For POPRES, partitioning on E1 separated southern European population (1092 samples) and northern European population (1374 samples).
Prediction accuracy for projected eigenvector
We investigated three aspects of EigenGWAS prediction: (1) the number of loci needed to achieve high accuracy for the projected eigenvectors; (2) the required sample size of the training set; (3) the importance of matching the population structure between the training and the test sets.
Using the POPRES samples, we split 5% (125 individuals), 10% (250 individuals), 20% (500 individuals), 30% (750 individuals), 40% (1000 individuals) and 50% (1250 individuals) of the sample as the training set, and used the remainder of the samples as the test set. Eigenvectors were estimated using all markers in each training set. As predicted by our theory (Equation (7)), the prediction accuracy of the projected eigenvector was consistent with 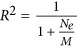 in which Ne=1000 for E1 empirically. If only 100 and 1000 random SNPs were sampled as predictors, the expected maximal is R2=0.091 and 0.5, respectively, and accuracy reached almost 1 if >100 000 SNPs were sampled. In agreement with our theory (Figure 6), if the number of predictors were too small, the prediction accuracy was poor, with prediction accuracy increasing with the addition of more markers for E1. When the sample size of the discovery was ⩾1000, maximal prediction accuracy was achieved, as predicted in our theory. Therefore, a discovery with a sample size of >1000 should be sufficient to predict the first eigenvector of an independent set, provided that population structure is the same across the discovery and prediction samples (Figure 6). In contrast, the prediction accuracy for prediction eigenvectors decreased (Figure 6) quickly for eigenvectors other than E1. For example, the prediction accuracy for E2 was below R2 <0.2 and R2 <0.15 for E3. For E4–E10, the prediction accuracy dropped down to nearly zero. This is consistent with the top 2–3 eigenvectors explaining the majority of variation (McVean, 2009), if the training and the test sets had their population structure matched.
in which Ne=1000 for E1 empirically. If only 100 and 1000 random SNPs were sampled as predictors, the expected maximal is R2=0.091 and 0.5, respectively, and accuracy reached almost 1 if >100 000 SNPs were sampled. In agreement with our theory (Figure 6), if the number of predictors were too small, the prediction accuracy was poor, with prediction accuracy increasing with the addition of more markers for E1. When the sample size of the discovery was ⩾1000, maximal prediction accuracy was achieved, as predicted in our theory. Therefore, a discovery with a sample size of >1000 should be sufficient to predict the first eigenvector of an independent set, provided that population structure is the same across the discovery and prediction samples (Figure 6). In contrast, the prediction accuracy for prediction eigenvectors decreased (Figure 6) quickly for eigenvectors other than E1. For example, the prediction accuracy for E2 was below R2 <0.2 and R2 <0.15 for E3. For E4–E10, the prediction accuracy dropped down to nearly zero. This is consistent with the top 2–3 eigenvectors explaining the majority of variation (McVean, 2009), if the training and the test sets had their population structure matched.
Figure 6.

Prediction accuracy of the projected eigenvectors for POPRES samples. Given 2466 POPRES samples, the data were split to 5:95%, 10:90%, 20:80%, 30:70%, 40:60% and 50:50%, as training and test sets. The left columns represent prediction accuracy (R2) using randomly selected numbers (100, 1000, 10 000, 100 000, all) of markers, and the 95% confidence interval were calculated from 30 replication for resampling given number of markers. In contrast, the right columns represent the predicted accuracy for 8 P-value thresholds (1e−6, 1e−5, 1e−4, 1e−3, 1e−2, 1e−1, 0.5 and 1) for EigenGWAS SNPs.
If EigenGWAS SNPs of low P-value were likely to be AIMs, we would hypothesize that AIM markers would be more efficient in giving high accuracy for the predicted eigenvectors (Figure 6). For E1, the prediction accuracy reached 1 more quickly by using markers selected by P-value thresholds. The prediction accuracy for projected E2 was dependent upon the threshold. For projected E2 given a 50:50 split of POPRES sample, applying the threshold of P-value <1e−6 (927 SNPs), R2=0.136, as high as using all markers. For other projected eigenvectors, the pattern of accuracy did not change much after applying P-value thresholds because, in general, the prediction accuracy was low. This indicated that eigenvectors other than the first two eigenvectors capture little replicable population structure in POPRES.
In practice, the training and the test set may not match perfectly on population structure, and this will likely lead to a reduction in prediction accuracy. To demonstrate this, we split the POPRES samples into two sets: pooling Swiss (991 samples) and French (96 samples) samples into one group (SF), and the rest of the samples into the other group (NSF). We used SF as the training and the NSF as the testing. As SF was almost an average of North European and South European gene flow, making a less stratified population, its EigenGWAS effects would be consequently small and less ‘heritable'. When using all SNPs effects estimated from SF set, the observed prediction accuracy for NSF set was R2=0.33 and 0.005 for E1 and E2, respectively. These results indicate that a matched training and test set is important for prediction accuracy of the projected eigenvectors.
Ancestry information may still be elucidated well even if the training set and the test set do not match well in their population structure. Using HapMap3 as the training set, we also tried to infer the ancestry of the Puerto Rican cohort (105 individuals) and Pakistani cohort (95 individuals) from 1000 Genomes project (The 1000 Genomes Project Consortium, 2012). A total of 907 614 common SNPs were found between HapMap3 and 1000 Genomes project. As illustrated, using these SNPs between HapMap3 and 1000 Genome projects, the projected eigenvectors accurately revealed the demographic history of Puerto Rican cohort, an admixture of African and European gene flows, and Pakistan cohort, an admixture of Asian and European gene flows (Figure 7).
Figure 7.

Projected eigenvectors for Puerdo Rican cohort (PUR) and Pakistan cohort (PJL) in 1000 Genomes project. The training set was HapMap3 samples build on 919 133 SNPs. The eigenvectors 1 and 2 were generated on the 907 614 common SNPs. PUR showed an admixture of African and European gene flows, and PJL Asian and European gene flows.
As a negative control, we replicated the prediction study for simulated data used in the previous section. The simulated data were split to two equal sample size. As there was no population structure in the simulated data, the prediction accuracy was poor, R2 <0.01 from E1 to E10. This demonstrates that prediction can be used to validate whether population structure exists within a genotype sample.
We concluded that to achieve high prediction accuracy of projected eigenvectors for independent samples, there are several conditions to be met: (1) the training set should harbour sufficient population stratification; (2) the sample size of the training should be sufficiently large; (3) the test sets should be as concordant as possible in its population structure; (4) when there is no real population structure, the prediction accuracy is very low close to zero; and (5) depending on the population, high prediction was largely achievable for the projected E1.
Discussion
Eigenvectors have been routinely employed in population genetics, and various approaches have been proposed to offer interpretation and efficient algorithms (Patterson et al., 2006; Rokhlin et al., 2009; McVean, 2009; Chen et al., 2013; Galinsky et al., 2016). In this study, we created a GWAS framework for studying and validating population structure, and offer an interpretation of the GWAS signals for eigenvectors within this framework. Without prior information about the structure/grouping of the sample, the EigenGWAS framework (least square) identifies ancestry informative markers and loci under selection across gradients of ancestry. Although EigenGWAS resembles the conventional GWAS, it should be noted that EigenGWAS has its phenotype generated from the genotypes via PCA. Hence, EigenGWAS is a GWAS but does not use phenotypes in conventional sense. The effects estimated in EigenGWAS can be alternatively estimated via BLUP or SVD (Equation (3)), but two major differences should be noticed: (1) BLUP or SVD will be computationally infeasible when there are many markers involved, whereas EigenGWAS reduces the computation into simple regression and largely avoids this problem; (2) it is not obviously how to obtain the P-values for BLUP or SVD estimates, but EigenGWAS makes it possible to conduct a standard z-score test for an estimated SNP effect.
We integrated SVD, BLUP and single-marker regression into a unified framework for the estimation of SNP-level eigenvectors. SVD is a special case of BLUP when heritability is of 1 for the trait and the target phenotype is an eigenvector. Furthermore, the BLUP is equivalent to the commonly used GWAS method for estimating SNP effects. As demonstrated, the correlation between BLUP and GWAS is almost 1 for the estimated SNP effects. EigenGWAS offers an alternative way in estimating Fst that can replace conventional Fst when population labels are unknown, populations are admixed or differentiation occurs across a gradient. As demonstrated for CEU–TSI samples, EigenGWAS brings out nearly identical estimation of Fst compared with conventional estimation.
Different from conventional GWAS, which requires conventional phenotypes, the proposed EigenGWAS provides a novel method for finding loci under selection based on eigenvectors that are generated from the genotype data itself. An EigenGWAS hit may reflect the consequence of process and thus additional evidence is needed to differentiate selection from drift. LCT is a known locus under selection that differs in its allele frequency as indicated by Fst statistic between Northern and Southern Europeans (Bersaglieri et al., 2004). We replicated the significance of LCT in CEU and TSI samples and POPRES European samples. DARS has been found in association with hypomyelination with brainstem and spinal cord involvement and leg spasticity (Taft et al., 2013). In addition, we also found HERC2 locus independently that may indicate the existence of anthropological difference in certain characters, such as hair, skin or eye colour across European nations (Voight et al., 2006; Visser et al., 2012).
Although by definition selection and genetic drift are different biological processes, both lead to allele frequency differentiation across populations and it is often difficult to tear them apart. In this study, with and without adjustment for λGC, EigenGWAS offers a straightforward way to filter out population stratification. For example, with adjustment for λGC, LCT and DARS were still significant in both EigenGWAS, whereas HERC2 was only significant in POPRES. If adjustment for λGC removed the average genetic drift since the most recent common ancestor for the whole sample, it might indicate that HERC2 reflected the anthropological difference between subsamples but not under selection as strong as that for LCT. Nevertheless, LCT was differentiated because of selection that was on top of genetic drift, and for DARS, it might be significant because of hitchhiking effect. Hence, LCT, DARS and HERC2 were significant in EigenGWAS for different mechanisms.
In EigenGWAS application, it provides a clear scenario that λGC is necessary if genetic drift/population stratification should be filtered out. It has been debated whether correction for λGC is necessary for GWAS (Yang et al., 2011b). If the inflation is because of population stratification, as initially λGC introduced, it seems necessary to control for it. In contrast, if it is because of polygenic genetic architecture, then correction for λGC will be a overkilling for GWAS signals. Interestingly, Patterson et al. (2006) found that the top eigenvalues reflect population stratification, and in our study we found λGC from EigenGWAS was numerically so similar to its corresponding eigenvalues. It in another aspect indicates that λGC captures population stratification. Hence, in concept and implementation, the correction for λGC is technically reasonable. Of note, Galinsky et al. (2016). also proposed a similar procedure to filter out population stratification in a study similar to ours, but we believe our framework is much easier to understand and implement in practice.
Once we have EigenGWAS SNP effects estimated, it is straightforward to project those effects onto an independent sample. The prediction of population structure was that of recent studies (Chen et al., 2013). We found that the prediction accuracy for the top eigenvector could be as high as almost 1. Given a training set of ∼1000 samples, the prediction accuracy could be very high if there were a reasonable number of common markers in the order of 100 000. This number, which needs to be available in both reference set and the target set, is achievable. Further investigation may be needed to check whether this number of markers is related to effective number or markers after correction for linkage disequilibrium for GWAS data. When the population structure of the test sample resembles the training sample, high accuracy will be achieved for the leading projected eigenvectors. Therefore, this approach is likely to be extremely beneficial for extremely large samples, such as UK Biobank samples and 23andMe, both of which have more than half million samples where direct eigenvector analysis may be infeasible. Our results suggest that sampling ∼1000 individuals from the whole sample as the training set and subsequently project EigenGWAS SNP effects to the reminding samples will be sufficient to reach a reasonable high resolution of the population structure.
Many improvements to the inference of ancestry using projected eigenvectors have been suggested (Chen et al., 2013). As the concordance of population structure between the training and test sets is often unknown (population structure, upon from genetic or social–cultural perspectives, its definition can be difficult or controversial), improvement of the inference of ancestry may or may not be achieved dependent upon the scale of the precision required for a sample. However, for classification of samples at ethnicity level, projected eigenvectors are likely to have high accuracy, as demonstrated in the Puerto Rican cohort and the Pakistani cohort. Therefore, when identifying ethnic outliers, using projected eigenvectors from HapMap is likely to be sufficient in practice.
Eigenvector analysis of GWAS data is an important well-utilized data technique, and here we show that its interpretation depends on many factors, such as proportion of different subpopulations and Fst between subpopulations. Our EigenGWAS approach provides intuitive interpretation of population structure, enabling AIMs to be identified, and potentially loci under selection to be identified. To facilitate the use of projected eigenvectors, we provide estimated SNP effects from HapMap samples and POPRES and software that can largely reduce the logistics involved in conventional way in generating eigenvectors, such as reference allele match, and strand flips.
Acknowledgments
This research was funded by ARC (DE130100614 and DP160102126 to SHL) and NHMRC (APP1080157 to SHL, APP1084417 and APP1079583 to BB and APP1050218 to MRR) and G-BC was supported by IAP P7/43-BeMGI from the Belgian Science Policy Office Interuniversity Attraction Poles (BELSPO-IAP) program. We thank Peter M Visscher for discussion, helpful comments and for proposing the name EigenGWAS. Robert Maier assisted with ggplot and Alex Holloway helped with Github. We also thank the Information Technology Group, Queensland Brain Institute. The POPRES data set was obtained from dbGaP at http://www.ncbi.nlm.nih.gov/gap through accession number phs000145.v4.p2.
Author contributions
G-BC, SHL and BB conceived study; G-BC, SHL, BB and MRR designed the experiment; G-BC and SHL developed the theory and methods; BB conducted the quality control for HapMap data, and MRR conducted quality control for POPRES data; G-BC performed the analyses of the study; GBC and Z-XZ developed GEAR software; GBC, MRR, SHL and BB wrote the paper.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on Heredity website (http://www.nature.com/hdy)
Web resource and data availability
GEAR is available at http://cnsgenomics.com/; GCTA at http://cnsgenomics.com/; PLINK at http://pngu.mgh.harvard.edu/~purcell/plink/index.shtml; and 1000 Genomes Project at http://www.1000genomes.org/.
Supplementary Material
References
- Altshuler DM, Gibbs RA, Peltonen L, Dermitzakis E, Schaffner SF, Yu F et al. (2010). Integrating common and rare genetic variation in diverse human populations. Nature 467: 52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersaglieri T, Sabeti PC, Patterson N, Vanderploeg T, Schaffner SF, Drake JA et al. (2004). Genetic signatures of strong recent positive selection at the lactase gene. Am J Hum Genet 74: 1111–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryc K, Bryc W, Silverstein JW. (2013). Separation of the largest eigenvalues in eigenanalysis of genotype data from discrete subpopulations. Theor Popul Biol 89: 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli-Sforza LL, Menozzi P, Piazza A. (1996) The History and Geography of Human Genes. Princeton University Press. [Google Scholar]
- Chen C-Y, Pollack S, Hunter DJ, Hirschhorn JN, Kraft P, Price AL. (2013). Improved ancestry inference using weights from external reference panels. Bioinformatics 29: 1399–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G-B. (2014). Estimating heritability of complex traits from genome-wide association studies using IBS-based Haseman-Elston regression. Front Genet 5: 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daetwyler HD, Villanueva B, Woolliams JA. (2008). Accuracy of predicting the genetic risk of disease using a genome-wide approach. PLoS One 3: e3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin B, Roeder K. (1999). Genomic control for association studies. Biometrics 55: 997–1004. [DOI] [PubMed] [Google Scholar]
- Dudbridge F. (2013). Power and predictive accuracy of polygenic risk scores. PLoS Genet 9: e1003348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galinsky KJ, Bhatia G, Loh P-R, Georgiev S, Mukherjee S, Patterson NJ et al. (2016). Fast principal components analysis reveals independent evolution of ADH1B gene in Europe and East Asia. Am J Hum Genet 98: 456–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard ME, Wray NR, Verbyla K, Visscher PM. (2009). Estimating effects and making predictions from genome-wide marker data. Stat Sci 24: 517–529. [Google Scholar]
- Henderson CR. (1975). Best linear unbiased estimation and prediction under a selection model. Biometrics 31: 423–447. [PubMed] [Google Scholar]
- Howie B, Marchini J, Stephens M, Chakravarti A. (2011). Genotype imputation with thousands of genomes. G3 1: 457–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier R, Moser G, Chen G-B, Ripke S Cross-Disorder Working Group of the Psychiatric Genomics Consortium Coryell W et al. (2015). Joint analysis of psychiatric disorders increases accuracy of risk prediction for schizophrenia, bipolar disorder, and major depressive disorder. Am J Hum Genet 96: 283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean G. (2009). A genealogical interpretation of principal components analysis. PLoS Genet 5: e1000686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M. (1973). Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci USA 70: 3321–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MR, Bryc K, King KS, Indap A, Boyko AR, Novembre J et al. (2008). The population reference sample, POPRES: a resource for population, disease, and pharmacological genetics research. Am J Hum Genet 83: 347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novembre J, Johnson T, Bryc K, Kutalik Z, Boyko AR, Auton A et al. (2008). Genes mirror geography within Europe. Nature 456: 98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, Price AL, Reich D. (2006). Population structure and eigenanalysis. PLoS Genet 2: e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick N A, Reich D. (2006). Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38: 904–909. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81: 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, Wray NR, Stone JL, Visscher PM, O'Donovan MC, Sullivan PF et al. (2009). Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460: 748–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich D, Thangaraj K, Patterson N, Price AL, Singh L. (2009). Reconstructing Indian population history. Nature 461: 489–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokhlin V, Szlam A, Tygert M. (2009). A randomized algorithm for principal component analysis. SIAM J Matrix Anal Appl 31: 1100–1124. [Google Scholar]
- Taft RJ, Vanderver A, Leventer RJ, Damiani S A, Simons C, Grimmond SM et al. (2013). Mutations in DARS cause hypomyelination with brain stem and spinal cord involvement and leg spasticity. Am J Hum Genet 92: 774–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium. (2012). An integrated map of genetic variation from 1,092 human genomes. Nature 491: 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser M, Kayser M, Palstra R-J. (2012). HERC2 rs12913832 modulates human pigmentation by attenuating chromatin-loop formation between a long-range enhancer and the OCA2 promoter. Genome Res 22: 446–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voight BF, Kudaravalli S, Wen X, Pritchard JK. (2006). A map of recent positive selection in the human genome. PLoS Biol 4: e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir BS. (1996) Genetic Data Analysis 2nd edn Sinauer Associates, Inc.: Sunderland, MA, USA. [Google Scholar]
- Williams AL, Patterson N, Glessner J, Hakonarson H, Reich D. (2012). Phasing of many thousands of genotyped samples. Am J Hum Genet 91: 238–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Lee SH, Goddard ME, Visscher PM. (2011. a). GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 88: 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Weedon MN, Purcell S, Lettre G, Estrada K, Willer CJ et al. (2011. b). Genomic inflation factors under polygenic inheritance. Eur J Hum Genet 19: 807–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Li S, Cooper RS, Elston RC. (2008). A unified association analysis approach for family and unrelated samples correcting for stratification. Am J Hum Genet 82: 352–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.