

Abstract
Secretagogin (SCGN), a Ca2+-binding protein having six EF-hands, is selectively expressed in pancreatic β-cells and neuroendocrine cells. Previous studies suggested that SCGN enhances insulin secretion by functioning as a Ca2+-sensor protein, but the underlying mechanism has not been elucidated. The present study explored the mechanism by which SCGN enhances glucose-induced insulin secretion in NIT-1 insulinoma cells. To determine whether SCGN influences the first or second phase of insulin secretion, we examined how SCGN affects the kinetics of insulin secretion in NIT-1 cells. We found that silencing SCGN suppressed the second phase of insulin secretion induced by glucose and H2O2, but not the first phase induced by KCl stimulation. Recruitment of insulin granules in the second phase of insulin secretion was significantly impaired by knocking down SCGN in NIT-1 cells. In addition, we found that SCGN interacts with the actin cytoskeleton in the plasma membrane and regulates actin remodelling in a glucose-dependent manner. Since actin dynamics are known to regulate focal adhesion, a critical step in the second phase of insulin secretion, we examined the effect of silencing SCGN on focal adhesion molecules, including FAK (focal adhesion kinase) and paxillin, and the cell survival molecules ERK1/2 (extracellular-signal-regulated kinase 1/2) and Akt. We found that glucose- and H2O2-induced activation of FAK, paxillin, ERK1/2 and Akt was significantly blocked by silencing SCGN. We conclude that SCGN controls glucose-stimulated insulin secretion and thus may be useful in the therapy of Type 2 diabetes.
Keywords: actin, calcium-binding protein, focal adhesion, insulin secretion, secretagogin
INTRODUCTION
Impaired insulin secretion in pancreatic β-cells is a major feature of Type 2 diabetes. It is therefore important to understand the mechanism by which insulin secretion is regulated in β-cells. Glucose-stimulated insulin secretion (GSIS) from pancreatic β-cells occurs in two phases: (i) a rapidly and transiently initiated triggering pathway of pre-docked insulin granules near the plasma membrane induced by KCl; and (ii) a gradually developed and sustained amplifying pathway of newly recruited insulin granules to the plasma membrane, from intracellular reserve pool of granules, by glucose stimulation [1,2]. Because F-actin (filamentous actin) forms a dense web underneath the plasma membrane as a negative barrier, cortical F-actin remodelling is required for the recruitment of the reserve pool granules to the release site at the plasma membrane in the second phase of insulin exocytosis [3]. In this phase, glucose triggers the Ca2+ influx which controls the dynamic structure of the actin cytoskeleton and thereby regulates the release of insulin granules [4,5].
FAK (focal adhesion kinase) plays a critical role in the regulation of F-actin remodelling and insulin secretion [6,7]. In pancreatic β-cells, integrin β1-mediated intracellular signalling activates and phosphorylates FAK and paxillin upon glucose stimulation. Additionally, these molecules form focal adhesion complexes at the specific focal contact sites and activate ERK1/2 (extracellular-signal-regulated kinase) and Akt signalling pathways acting downstream of glucose-induced FAK activation [7]. This signalling contributes to the regulation of the insulin secretion by inducing actin cytoskeletal remodelling. An in vivo study using β-cell-specific FAK-knockout mice confirmed the essential role of the FAK-mediated pathway in GSIS [8]. Furthermore, remodelling of focal adhesion is also inhibited by agents such as jasplakinolide and latrunculin B that respectively block actin cytoskeleton polymerization and depolymerization [7].
In pancreatic β-cells, intracellular Ca2+ plays an essential role in insulin secretion as a second messenger [9,10], and proteins that bind to intracellular Ca2+ function as Ca2+ signal transducers [11]. Secretagogin (SCGN), a recently cloned Ca2+-binding protein having six EF-hands, is exclusively expressed in pancreatic β-cells and neuroendocrine cells [12]. SCGN is proposed as a Ca2+-sensor protein, because it has low Ca2+ affinity and undergoes conformational changes to control protein–protein interactions and cellular signalling processes [13]. The function of Ca2+-sensor proteins in regulating secretion is to transduce Ca2+ signals to exocytotic machinery during the release process in neuroendocrine and endocrine systems [14,15]. In pancreatic β-cells, intracellular Ca2+ concentration is rapidly increased in the first phase of insulin secretion, whereas the second phase requires oscillations of intracellular Ca2+ in addition to amplifying signals from glucose metabolism [16]. Recently, the expression level of SCGN in mouse insulinoma MIN6 cells was shown to control GSIS [17]. However, the exact biological function of SCGN as a Ca2+-sensor protein in pancreatic β-cells in exerting its positive effect on insulin secretion is not clear. In the present study, we tried to elucidate the molecular mechanisms underlying the regulation of insulin secretion by SCGN and the associated subcellular pathways, employing NIT-1 insulinoma cells as a model of insulin secretion [18–22].
MATERIALS AND METHODS
Antibodies and reagents
Anti-SCGN antibody was from AbFrontier. Anti-FAK, anti-paxillin, anti-phospho-paxillin (Tyr118), anti-ERK1/2, anti-phospho-ERK1/2 (Thr202/Tyr204), anti-Akt and anti-phospho-Akt (Ser473) antibodies were from Cell Signaling Technology. Anti-α-tubulin antibody, anti-β-actin antibody and normal rabbit IgG were from Santa Cruz Biotechnology. Anti-phospho-FAK (Tyr397) and anti-SCGN antibodies used in immunoprecipitation were from Abcam. Anti-paxillin antibody used in confocal microscopy was from Millipore Corporation. Anti-E-cadherin (epithelial cadherin) and anti-N-cadherin (neural cadherin) antibodies were from BD Biosciences. Horseradish peroxidase-conjugated goat anti-mouse IgG and goat anti-rabbit IgG were from Bio-Rad Laboratories. Rhodamine–phalloidin, Alexa Fluor® 488- or Alexa Fluor® 568-conjugated goat anti-rabbit IgG and Alexa Fluor® 488-conjugated goat anti-mouse IgG were from Invitrogen. Latrunculin B was from Calbiochem. Cytochalasin D, ionomycin and DMSO from Sigma–Aldrich. Penicillin G, streptomycin, FBS and trypsin were from Gibco Life Technologies. DMEM (Dulbecco's modified Eagle's medium) and 45% D-glucose were from WelGENE. SMARTpool siRNA and DharmaFECT1 transfection reagent were from Dharmacon. Insulin ELISA kit was from ALPCO. BCA protein assay was from Thermo Scientific. Protein G–Sepharose beads and silver staining kit were from GE Healthcare.
Cell culture
NIT-1 β-cells were grown and maintained in 5.6 mM glucose in DMEM supplemented with 10% (v/v) FBS, 100 μg/ml streptomycin and 100 units/ml penicillin G at 37°C under an atmosphere of 5% CO2 in air
Islet isolation and primary cell culture
Mouse islets were isolated from 8–10-week-old C57BL/6 mice by collagenase P perfusion and digestion as described previously [23]. Individual islets were hand-picked using micropipettes and cultured in RPMI 1640 medium supplemented with 10% (v/v) FBS and 100 μg/ml penicillin/streptomycin for 24 h before further experiments.
Knockdown of SCGN
ON-TARGETplus SMARTpool mouse SCGN siRNAs (25 nM) were used to knock down SCGN in NIT-1 insulinoma cells. ON-TARGETplus Non-targeting Pool siRNAs were used as control. Silencing was achieved using DharmaFECT 1 transfection reagent according to the manufacturer's recommendations. Changes in the expression of SCGN in NIT-1 cells were analysed 48 h after siRNA transfection. For mouse primary islet cells, Accell siRNAs (Dharmacon) were used. Dispersed mouse islet cells were treated with the non-targeting pool or Accell mouse SCGN siRNA SMART pool (1 μM) in RPMI 1640 medium containing 100 μg/ml penicillin/streptomycin and incubated for 96 h.
Insulin secretion assay
NIT-1 cells were pre-incubated at 37°C for 2 h with glucose-free HBSS (Hanks balanced salt solution: 137 mM NaCl, 5.4 mM KCl, 1.26 mM CaCl2, 0.98 mM MgSO4, 0.44 mM KH2PO4, 0.36 mM Na2HPO4 and 4.2 mM NaHCO3, pH 7.4). Following glucose starvation, the cells were stimulated at 37°C for 5–45 min with HBSS containing various concentrations of D-glucose or H2O2. After stimulation for the indicated period, insulin secreted into the medium was measured using an insulin ELISA kit following the manufacturer's protocol. All medium extracts were normalized to cellular total protein concentration. The protein concentration of each sample was measured by a BCA protein assay performed according to the manufacturer's instructions.
Transmission electron microscopy
The cells were fixed with 2.5% (w/v) glutaraldehyde in HBSS overnight at 4°C and then stained with 2% (w/v) osmium tetroxide in 0.1 M cacodylate buffer for 1 h. The cells were dehydrated with ethanol series, infiltrated with Spurr's resin series, and polymerized at 60°C over 8 h. The cell embedded resin was cut with a diamond knife on ultramicrotome (Ultracuts). The sections were mounted directly on 150 mesh copper grids, stained with 2% (w/v) uranyl acetate in 50% methanol for 20 min and Reynold's lead citrate for 10 min. The grids were examined using a Tecnai F20 transmission electron microscope (FEI) at 200 kV.
SDS/PAGE and Western blot analysis
Proteins were separated by SDS/PAGE (12% gel) and immediately transferred on to a PVDF membrane. The membrane was blocked with 5% (w/v) BSA and 0.1% Tween 20-containing PBS for 1 h and sequentially incubated for 2 h at room temperature, with each primary antibody diluted in PBST (PBS buffer containing 3% BSA and 0.1% Tween 20) according to the manufacturer's instructions. In the case of some primary antibodies, the incubation was allowed overnight at 4°C. After washing three times with PBS containing 0.1% Tween 20 (10 min for each), the membrane was incubated with secondary goat anti-mouse or rabbit antibody, diluted 1:5000 in PBS containing 0.1% Tween 20 for 40 min at room temperature, and washed again three times. The immune complexes were detected with LAS-3000s (Fuji Photo Film) using a WEST-zol®plus Western Blot Detection System (iNtRON Biotechnology).
Immunoprecipitation from cell extracts
NIT-1 cells were harvested and lysed with immunoprecipitation buffer (150 mM NaCl, 50 mM Tris/HCl and 60 mM octyl β-D-glucopyranoside, pH 7.4) containing protease inhibitors (1 mM PMSF, 5 mM Na3VO4, 5 mM NaF, 5 μg/ml aprotinin, 10 μg/ml leupeptin and 10 μg/ml pepstatin A) and HDAC (histone deacetylase) inhibitors [10 μM TSA (trichostatin A) and 10 mM SB (sodium butyrate)] for 30 min on ice. The cell lysates were centrifuged at 16000 g for 15 min at 4°C. The supernatant was incubated with normal rabbit IgG or anti-SCGN antibody for 2 h at 4°C followed by 1 h of incubation with Protein G–Sepharose beads at 4°C. The beads were then washed three times with 1 ml of lysis buffer for removing non-specific binding and additionally twice with 1 ml of lysis buffer without any detergent. The immune complex was solubilized in non-reducing SDS gel sample buffer and then boiled at 95°C for 5 min. Samples separated on SDS/PAGE (12% gel) were visualized using a silver staining kit or by Western blot analysis.
Protein identification by nanoUPLC–ESI–q-TOF–tandem MS
In order to identify interacting proteins, the silver-stained protein bands of immune complex on SDS/PAGE were destained and digested with trypsin and the resulting peptides were extracted and analysed by nanoAcquity™ ESI-q-TOF tandem MS (SYNAPT™ HDMS™, Waters) as described previously [24].
Confocal microscopy
NIT-1 cells were grown on Secureslip™ (Sigma) cell culture glass coverslips to 70% confluence and gently washed twice with ice-cold HBSS, and then fixed with 4% (w/v) paraformaldehyde in HBSS for 10 min at room temperature. After washing with HBSS, cells were permeabilized with 0.1% Triton X-100 in HBSS for 10 min at room temperature. After washing with PBS, non-specific protein adsorption was blocked by incubation with 3% (w/v) BSA, 0.2% Tween 20 and 0.2% gelatin in PBS for 1 h at room temperature. For endogenous SCGN immunostaining, polyclonal anti-SCGN antibody was diluted at 1:500 in PBS containing 1% (w/v) BSA and 1% (w/v) sucrose. To visualize F-actin, actin was labelled with red fluorescent rhodamine–phalloidin. Cells were incubated with these primary antibodies for 2 h at 37°C. After washing three times with PBS for 10 min each, cells were stained for 1 h at 37°C with Alexa Fluor® 488- (green) or 568- (red) conjugated goat anti-rabbit IgG diluted 1:50. After incubation, cells were washed three times with PBS. ProLong Gold antifade reagent with DAPI (blue) was used for mounting and nuclear staining. After mounting, the cells were observed on a Zeiss LSM510 Meta laser-scanning microscope using an EC plan-Neofluar ×100 oil-immersion objective. Images were photographed and processed using LSM510 software (Carl Zeiss).
Sucrose gradient fractionation
For each assay, one 100-mm plate of NIT-1 cells (4×106 cells) was subjected to 5–42% (w/v) sucrose gradient in 10 mM HEPES buffer, pH 7.4. The cells were starved with glucose-free HBSS for 2 h and immediately treated with DMSO or 2 μM ionomycin for 30 min in 16.8 mM glucose in HBSS. The cells were then washed twice with ice-cold PBS and lysed with immunoprecipitation buffer containing protease inhibitors (detailed above). The cell lysates were passed through a 31 G needle five times using a 1 ml syringe and incubated on ice for 30 min. Subsequently, 500 μl of each lysate was layered on to 5–42% continuous sucrose gradients and centrifuged at 4°C in a swinging bucket SW41Ti rotor (Beckman Coulter) at 100000 g for 16 h. Fractions were collected from the top and subjected to Western blot analysis using anti-SCGN and anti-β-actin antibodies.
F-actin/G-actin ratio measurement
NIT-1 cells were washed once with PBS and then lysed in actin stabilization buffer (100 mM Pipes, pH 6.9, 5% glycerol, 5 mM MgCl2, 5 mM EGTA, 1% Triton X-100, 1 mM ATP and protease inhibitors) pre-warmed at 37°C and incubated on a microtube shaking incubator for 10 min at 37°C. The lysates were centrifuged at 1000 g for 5 min at room temperature using table top centrifuge. Immediately after removing non-lysed cells, lysates were centrifuged at 100000 g for 1 h at 37°C. The pellets containing F-actin were solubilized with actin depolymerization buffer (100 mM Pipes, pH 6.9, 1 mM MgCl2, 10 mM CaCl2 and 5 μM cytochalasin D) and incubated for 1 h on ice. The supernatant [G-actin (globular actin)] and pellet [F-actin] fractions were separated by SDS/PAGE and subsequently subjected to Western blot analysis with anti-β-actin antibody. The cellular F-actin/G-actin ratio was estimated from the Western blot analysis.
RESULTS
Secretagogin enhances glucose-stimulated insulin secretion in β-cells
SCGN is suggested to enhance pancreatic insulin secretion because tissues overexpressing SCGN increased insulin secretion in previous studies [12,17]. However, the underlying mechanism of SCGN's action in insulin secretion has not been explored. In the present study, we investigated whether SCGN is directly involved in insulin secretion by modulating SCGN expression level in NIT-1 β-cells, a mouse insulinoma cell line [25]. Since NIT-1 cells are too sensitive to overexpress proteins by transfection and it is not possible to generate a virus-infected stable cell line because glucose responsibility of insulin secretion is readily decayed depending on the cell passages [18], SCGN expression levels in NIT-1 cells were modulated by silencing with siRNA. We employed a mixture of four different siRNA sequences to target various regions of SCGN in this SCGN-depletion study. The mixture of siRNA oligonucleotides was transiently transfected via DharmaFECT1 reagent into NIT-1 cells to deplete endogenous SCGN.
We first confirmed that SCGN is involved in insulin secretion by modulating SCGN expression in NIT-1 cells. After transfection of the siRNA mixture, NIT-1 cells were pre-incubated with glucose-free HBSS for 2 h and then stimulated with various concentrations of glucose. Depletion of SCGN resulted in a significant inhibition of GSIS compared with the control in a glucose-dose-dependent manner (Figure 1A). SCGN depletion was confirmed by Western blot analysis with anti-SCGN using anti-α-tubulin antibody as a loading control. For further confirmation, we employed low concentrations of exogenous H2O2 (50 and 100 μM for 30 min) which increase insulin secretion by raising intracellular ROS (reactive oxygen species) levels as a glucose-mimicking stimulant [26–29]. As shown in Figure 1B, treatment with H2O2 after knocking down SCGN decreased insulin secretion. These results confirm that SCGN is involved in glucose- and H2O2-responsive insulin secretion. In order to validate the effects of SCGN under physiological conditions, we investigate glucose-stimulated insulin secretion using primary mouse islet cells. Isolated mouse islets were dissociated into single cells by trypsinization and then treated with either control or SCGN-specific Accell siRNA, a lipid-conjugated siRNA directed against mouse SCGN. GSIS was significantly impaired in the SCGN-depleted islet cells, in agreement with our observations in NIT-1 cells (Figure 1C). These results indicate that the GSIS response of NIT-1 cells parallels that of primary islet cells and that SCGN is required for GSIS in both the pancreatic β-cell line and primary islets.
Figure 1. Silencing secretagogin by siRNA leads to inhibition of GSIS in NIT-1 β-cells.

NIT-1 cells were transfected with commercial SCGN siRNA or non-targeting control oligonucleotides. After 48 h of transfection, cells were pre-incubated with glucose-free HBSS for 2 h followed by stimulation with the indicated concentration of glucose (A) or H2O2 (B) in HBSS for 30 min. Secreted insulin was measured and normalized to total cell protein concentration. Results are means±S.D. from triplicate experiments. *P<0.05, **P<0.01, ***P<0.005. (C) GSIS in islet cells isolated from mice. Control or SCGN siRNA-treated islet cells (105) were starved with basal glucose (3.3 mM) for 70 min and stimulated with glucose (16.7 mM) for 30 min keeping the temperature at 37°C. Secreted medium was analysed for insulin concentration. Whole-cell detergent lysates were subjected to Western blot analysis with anti-SCGN or anti-α-tubulin antibody as a loading control. All experiments were run in triplicates. Results are means±S.D. *P<0.005. CTL, control.
Secretagogin mediates the second phase of glucose-stimulated insulin secretion
To determine the point at which SCGN is involved in insulin secretion, we compared the effects of SCGN depletion on the first and second phase of insulin secretion. Cellular insulin secretion occurs in two phases: (i) a rapidly and transiently initiated triggering pathway of pre-docked insulin granules near the plasma membrane; and (ii) a gradually developed and sustained amplifying pathway of newly recruited insulin granules to the plasma membrane by glucose stimulation. Insulin secretion induced by KCl stimulation is both faster and greater in the initial phase than glucose stimulation, since KCl treatment causes only first-phase release through direct depolarization of the plasma membrane [30]. Second-phase insulin secretion is induced in response to glucose, not KCl. KCl has been used to induce the first phase of insulin secretion by membrane depolarization followed by increased intracellular Ca2+, whereas glucose is used to evoke both first- and second-phase insulin release through the KATP channel-independent amplification pathway [31–33]. To investigate the underlying defects of insulin secretion in SCGN-knockdown NIT-1 cells, we compared insulin secretion in response to KCl and glucose stimulation. SCGN-knockdown NIT-1 cells were pre-incubated with 5.4 mM KCl or glucose-free HBSS for 2 h, and then stimulated with 30 mM KCl or 16.8 mM glucose for 30 min respectively. The amounts of secreted insulin were measured. Figure 2A shows that SCGN depletion impaired GSIS (second phase), but not KCl-induced insulin secretion (first phase).
Figure 2. Secretagogin mediates the second phase of insulin secretion.

(A) SCGN depletion impairs insulin secretion induced by glucose, not by KCl. NIT-1 cells transfected with non-targeting control or SCGN siRNA were pre-incubated with 5.4 mM KCl in glucose-free HBSS at 37°C for 2 h and then stimulated with 30 mM KCl or 16.8 mM glucose in HBSS at 37°C for 30 min. Secreted insulin was measured using the mouse ultrasensitive insulin ELISA kit. Insulin content data were normalized to total cell protein concentration. Results are means±S.D. from triplicate samples. CTL, control. (B) Time course of insulin secretion and its inhibition in SCGN-depleted cells in the second phase of insulin secretion. NIT-1 cells transfected with control or SCGN siRNA were pre-incubated in glucose-free HBSS at 37°C for 2 h and then stimulated with 16.8 mM glucose in HBSS for the times indicated. Insulin secretion was determined using the insulin ELISA kit. Results are means±S.D. of triplicates. *P<0.05, ***P<0.005. Silencing of SCGN was confirmed by Western blot analysis with anti-SCGN antibody or anti-α-tubulin antibody as a loading control. CTL, control. (C) NIT-1 cells transiently transfected with control or SCGN siRNA were cultured on a glass-bottomed chamber slide dish. The cells were pre-incubated with glucose-free HBSS for 2 h and stimulated with 16.8 mM glucose for 35 min at 37°C, followed by fixation with 2.5% glutaraldehyde. Electron micrographs were taken in non-stimulated (upper) or glucose-stimulated (lower) control (left) and SCGN-knockdown (right) NIT-1 cells. White arrows indicate the pre-docked insulin granules and black arrows indicate the recruitment of insulin granules. Scale bar, 0.5 μm.
We then compared the kinetics of insulin secretion in glucose-stimulated control and SCGN depleted NIT-1 cells. Cells starved in glucose-free HBSS for 2 h were stimulated with high glucose (16.8 mM) for the times indicated and insulin secretion was measured. As shown in Figure 2B, there was no discernible difference in first-phase insulin secretion (<20 min) between control and SCGN-depleted cells, whereas extensive decreases in GSIS were detected in SCGN-knockdown cells in the second phase of secretion. These results indicate that, at least in vitro, SCGN is required for the second, glucose-stimulated, phase of insulin secretion, but not for the first, KCl/depolarization-induced, phase of insulin secretion in pancreatic β-cells.
We next examined insulin granule mobilization in the second phase of GSIS using TEM analysis (Figure 2C). There was no significant difference in the number of previously docked insulin granules between control and SCGN-knockdown cells under starved conditions. In contrast, the recruitment of insulin granules in response to glucose was significantly reduced in the cells lacking SCGN. These findings indicate that the glucose-driven refilling process of insulin granules in the second phase is impaired by silencing SCGN.
Secretagogin interacts with the actin cytoskeleton in response to glucose and Ca2+ signals
It is known that mitochondrial metabolism is critical for the second phase of insulin secretion, so we examined the mitochondrial function through monitoring the changes of mitochondrial membrane potential in response to glucose stimulation in control and SCGN-knockdown NIT-1 cells. We found that mitochondrial function in response to glucose was retained in SCGN-knockdown NIT-1 cells as well as control cells (Supplementary Figure S1). In order to determine how SCGN affects insulin secretion, we examined protein–protein interactions with endogenous SCGN in NIT-1 cells employing immunoprecipitation combined with peptide sequencing by nanoUPLC (nano-ultra-performance liquid chromatography)–ESI–q-TOF–tandem MS. NIT-1 cells were lysed with a lysis buffer containing octyl β-D-glucopyranoside, used in solubilization and isolation of membrane proteins in mild conditions [34]. The cell lysates were immunoprecipitated with anti-SCGN or control IgG antibody. The immune complexes were separated by SDS/PAGE and detected by silver staining (Figure 3A). Differentially appearing protein bands were identified after digestion with trypsin, by peptide sequencing with nanoUPLC–ESI–q-TOF–tandem MS. β-Actin (MS/MS spectrum in Figure 3B), was specifically identified in the SCGN lane after excluding the non-specific proteins in the control lane, and this was confirmed by Western blot analysis (Figure 3C). Also, the fact that actin was identified in the solubilized membrane proteins using octyl β-D-glucopyranoside as a detergent without protein denaturation suggests that actin–SCGN interaction occurs in the plasma membrane.
Figure 3. Secretagogin interacts with actin cytoskeleton in NIT-1 cells.

NIT-1 cells were harvested and lysed with a lysis buffer containing protease inhibitors. Cell lysates were subjected to immunoprecipitation (IP) with anti-SCGN or control IgG antibody. (A) Immune complexes were separated by SDS/PAGE (12% gel) under reducing conditions and visualized with silver staining. Molecular masses are indicated in kDa. (B) Tandem MS spectrum of a peptide in β-actin. Differentially expressed protein bands between IgG and anti-SCGN antibody were extracted from gels, digested with trypsin and identified by peptide sequencing with nanoUPLC–ESI–q-TOF–tandem MS. (C) Immune complexes were confirmed by Western blotting with anti-β-actin or anti-SCGN antibody.
Since the actin cytoskeleton is known to play a principal regulatory role in the transport of insulin granules in the second phase of GSIS [35,36], we examined the SCGN–actin interaction further in response to glucose-stimulation. We found that the binding of SCGN to actin was increased in response to glucose stimulation (Figure 4A), which suggests that SCGN regulates insulin secretion by binding to the actin cytoskeleton. We also observed, employing confocal microscopy, that actin and SCGN co-localized in NIT-1 cells following H2O2 stimulation which mimics glucose signalling. Figure 4B shows that endogenous SCGN was distributed throughout the cells in the basal condition and was translocated from the cytosol to the plasma membrane after H2O2 stimulation, enhancing further the co-localization with the actin cytoskeleton in plasma membrane. Since glucose-induced Ca2+ signalling is a critical step for insulin secretion, we examined the effect of Ca2+ influx on the SCGN–actin interaction by stimulating the cells with ionomycin which induces actin polymerization with a rapid increase in the internal Ca2+ level [37]. We used sucrose gradient fractionation to directly analyse the distribution of the actin cytoskeleton and SCGN in NIT-1 cells. NIT-1 cells were pre-incubated with glucose-free HBSS for 2 h and subsequently treated with DMSO or 2 μM ionomycin in 16.8 mM glucose in HBSS for 30 min. We next fractionated the total cell lysates on 5–42% continuous sucrose gradients, and determined the protein level in each fraction by Western blotting (Figure 4C). We found that the distribution of actin became denser after ionomycin treatment compared with treatment with control DMSO, consistent with the fact that ionomycin induces polymerization of actin cytoskeleton and increases sedimentation of actin. The distribution of SCGN also became denser after treatment with ionomycin, parallel to the actin cytoskeleton (Figure 4C), indicating that Ca2+ influx induces co-sedimentation of SCGN and the actin cytoskeleton by potentiating actin polymerization.
Figure 4. Glucose and Ca2+ stimulation enhances the interaction of secretagogin with actin.
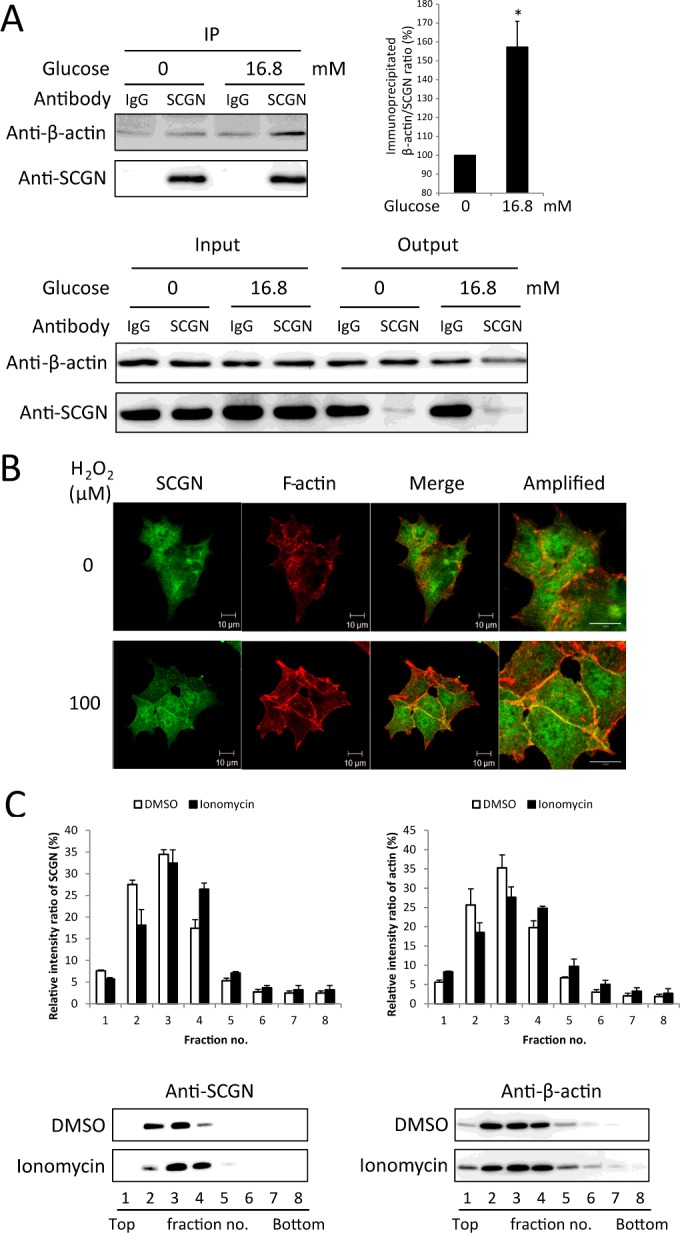
(A) NIT-1 cells were treated with 0 or 16.8 mM glucose HBSS for 30 min after pre-incubation with glucose-free HBSS for 2 h. The cell lysates were subjected to immunoprecipitation (IP) with anti-SCGN antibody or control IgG. Immune complexes were separated by SDS/PAGE (12% gel) under reducing conditions and detected by Western blotting with anti-β-actin and anti-SCGN antibodies. A representative blot from three independent experiments is shown. The relative intensities of the β-actin bands were normalized to SCGN bands and expressed as a ratio. Results are mean±S.D. intensity ratios from three independent experiments. *P<0.05. (B) NIT-1 cells were starved with glucose-free HBSS for 2 h and stimulated with 0 or 100 μM H2O2 in glucose-free HBSS for 30 min at 37°C. Cells were then fixed and stained for F-actin (red) and SCGN (green) using rhodamine–phalloidin and anti-SCGN antibody. Scale bar, 10 μm. (C) Sucrose gradient fractionation of total cell lysates from NIT-1 cells incubated with glucose-free HBSS for 2 h and stimulated with DMSO or 2 μM ionomycin for 30 min in 16.8 mM glucose in HBSS. Each fraction was separated by SDS/PAGE (12% gel) and visualized by Western blot analysis with anti-β-actin and anti-SCGN antibodies. For quantification of the distribution in the gradient, relative protein levels in each fraction were calculated as percentage of the total levels from all fractions using densitometry. Results are means±S.D. from three independent experiments.
Secretagogin modulates glucose-mediated actin dynamics
To determine whether SCGN plays a role in actin reorganization, we assessed both F-actin and G-actin in NIT-1 cells, because cytoskeletal remodelling is an essential step in insulin secretion, not only for eliminating the pre-existing F-actin barrier, but also for stabilizing actin polymerization in stress fibres serving as routes for insulin granules toward the plasma membrane [38,39]. To obtain the F-actin and G-actin fractions, NIT-1 cells lysed in warm actin stabilization buffer were centrifuged at 100000 g for 1 h, separating F-actin going into the pellet and G-actin remaining in the supernatant. The F-actin-containing pellet was then solubilized with actin depolymerization buffer. The cellular F-actin/G-actin ratio was estimated by Western blotting. The amount of F-actin in the SCGN-knockdown NIT-1 cells was discernibly decreased, indicating that SCGN affects the dynamics of actin cytoskeleton in the pancreatic β-cell (Figure 5A). Since F-actin reorganization plays a critical role in the regulation of GSIS [4], we examined H2O2-induced actin cytoskeletal remodelling in SCGN-knockdown NIT-1 cells using confocal microscopy. As shown in Figure 5B, H2O2 stimulation led to a partial depolymerization of the actin cytoskeleton at the cell periphery, resulting in assembly of F-actin stress fibres. However, NIT-1 cells depleted of SCGN showed collapsed actin stress fibres which accumulated at the cell periphery, and displayed a denser and more intense structure, which possibly blocks the secretion of insulin granules. These findings suggest that SCGN plays a regulatory role in actin remodelling, causing GSIS in NIT-1 cells.
Figure 5. Intracellular actin remodelling is modulated by secretagogin.

(A) F-actin and G- actin in NIT-1 cells treated with control or SCGN siRNA were fractionated and subjected to Western blot analysis with anti-β-actin antibody. IP, immunoprecipitation. The ratio of F-actin to G-actin was measured by densitometry. Results are mean±S.D. intensity values from three independent experiments. *P<0.005. (B) NIT-1 cells transiently transfected with non-targeting control or SCGN siRNA were seeded on cell culture glass coverslips. After 48 h, cells were pre-incubated in glucose-free HBSS for 2 h and treated with 0 or 100 μM H2O2 in glucose-free HBSS for 30 min at 37°C. Cells were subsequently fixed and stained for F-actin (red) and SCGN (green) using rhodamine–phalloidin and anti-SCGN antibodies. Nucleus was visualized with DAPI (blue). Scale bar, 10 μm.
Secretagogin facilitates focal adhesion
On silencing endogenous SCGN, NIT-1 cells acquired a more round and less spread morphology, as shown in Figure 6A. We assessed the effects of this morphological change in NIT-1 cells on adherens junction components of the cadherin family. These changes in cell morphology up-regulated the expression of E-cadherin, a major component of adherens junctions, and down-regulated N-cadherin expression in SCGN-knockdown cells (Figure 6B). Such switches in E-cadherin and N-cadherin expression are in agreement with alterations in cell–cell adhesion molecules and junction organization predicted by morphological changes [40].
Figure 6. Secretagogin regulates cell morphology and focal adhesion.

(A) Morphology of control or SCGN siRNA-transfected NIT-1 cells in culture. (B) Western blot analysis of cell adhesion molecules. Control or SCGN-knockdown NIT-1 cells were lysed in gel sample buffer and subjected to Western blotting with the antibodies indicated. (C) Immunofluorescence analysis of the control or SCGN-knockdown NIT-1 cells starved with glucose-free HBSS for 2 h and stimulated with 0 or 100 μM H2O2 in glucose-free HBSS for 15 min at 37°C. Cells were subsequently fixed and stained for SCGN (red) and paxillin (green) using anti-SCGN and anti-paxillin antibodies. Nuclei (blue) were stained with DAPI. Scale bar, 10 μm.
Since changes in cellular morphology are known to modulate local focal adhesion [41] and focal adhesion regulated by actin cytoskeletal reorganization at the cell surface plays a crucial role in GSIS [7,8,42], we investigated whether SCGN, which stimulates insulin secretion, also affects focal adhesion formation. SCGN-depleted and control NIT-1 cells were starved with glucose-free HBSS and treated with H2O2 to induce insulin secretion, and focal adhesion assemblies in these cells were compared, employing immunofluorescence confocal microscopy. We used paxillin as a marker of focal adhesion assembly at the plasma membrane (Figure 6C). No focal adhesion assembly sites were seen in SCGN-knockdown NIT-1 cells stimulated by H2O2, whereas numerous dot-shaped focal adhesion sites were found near the plasma membrane in the H2O2-stimulated control cells (Figure 6C). These results suggest that SCGN plays a critical role in generating the focal adhesion assembly induced by H2O2 stimulation.
Secretagogin is essential for the activation of focal adhesion signalling
A previous study demonstrated that focal adhesion remodelling, mediated by activated FAK, is essential for GSIS [6]. Since depletion of SCGN inhibits the formation of focal adhesion assembly induced by H2O2, a glucose-mimicking stimulant, we examined the effects of silencing SCGN on the H2O2-induced focal adhesion activation pathway including phosphorylation of FAK (Tyr397) and paxillin (Tyr118) and its downstream signalling effectors, including ERK1/2 (Thr202/Tyr204) and Akt (Ser473). Since the focal adhesion signalling is a rapid and transient response, we first examined the kinetics of phosphorylation of focal adhesion signalling molecules. SCGN-knockdown NIT-1 cells were exposed to 100 μM H2O2 for various durations up to 30 min. The activation kinetics of FAK, paxillin, ERK1/2 and Akt are shown in Figure 7A. These activations are saturated upon short-term H2O2 stimulation (<15 min), and silencing SCGN obviously decreased these activations. Next we examined these activations at 15 min in a H2O2-dose-dependent manner. Silencing SCGN in NIT-1 cells inhibited H2O2-stimulated phosphorylation of focal adhesion molecules FAK and paxillin, as well as the phosphorylation of ERK1/2 and Akt, downstream targets of the focal adhesion proteins (Figure 7B). To confirm the effect of SCGN silencing on the activation of focal adhesion molecules, the experiments described above were performed after exposing the cells to various concentrations of glucose. The activation amplitudes of FAK, paxillin, ERK1/2 and Akt in SCGN-depleted NIT-1 cells were significantly lower than in control cells (Figure 7C). These results confirm that the activation of focal adhesion molecules in response to glucose-stimulation is regulated by SCGN in NIT-1 cells.
Figure 7. Focal adhesion signalling is significantly impaired by silencing secretagogin in NIT-1 cells.

(A) The activation kinetics of focal adhesion molecules are regulated by SCGN. NIT-1 cells were transfected with siRNA against SCGN or control siRNA and maintained for 2 days. Cells were incubated with glucose-free HBSS and stimulated with 100 μM H2O2 in glucose-free HBSS for the times indicated. Western blot analysis was carried out using the cell lysates with the antibodies indicated. (B and C) Silencing SCGN inhibits glucose-induced phosphorylation of FAK, paxillin and downstream effectors of focal adhesion molecules. NIT-1 cells were transiently transfected with control or SCGN siRNA. After 48 h, cells were incubated with glucose-free HBSS at 37°C for 2 h and stimulated with various concentration of H2O2 (B) or glucose (C) in HBSS at 37°C for 15 or 10 min respectively. Cells were then lysed with gel sample buffer. Proteins were resolved by SDS/PAGE (10% gel) under reducing conditions and analysed by Western blotting using the indicated antibodies. A representative blot from three independent experiments is shown. The relative intensities of the phosphorylated and total protein bands of starved and stimulated conditions were quantified by densitometry and expressed as a ratio. Ratios were normalized to the control siRNA-transfected cells without any stimulation. Results are mean±S.D. intensity values from three independent experiments. *P< 0.05, **P< 0.01.
Secretagogin plays a role in focal adhesion signalling by regulating the actin cytoskeleton
Actin cytoskeletal reorganization is known to be closely related to GSIS and focal adhesion signalling [7,36,43]. Previous studies found that the disruption of the dynamic equilibrium state of actin polymerization blunts the activation of focal adhesion signalling [5,7]. To determine whether SCGN affects focal adhesion signalling via the actin cytoskeleton, we compared glucose-induced activation of FAK and ERK in control and SCGN-knockdown cells, untreated or treated with latrunculin B, an actin-depolymerizing reagent. As illustrated in Figure 8, actin disruption induced by latrunculin B led to the blockage of the glucose-stimulated phosphorylation of focal adhesion molecules FAK and ERK, which was similar in both SCGN-knockdown and control cells. These results suggest that SCGN promotes focal adhesion signalling through the regulation of the actin cytoskeleton.
Figure 8. Effects of actin cytoskeleton disruption with latrunculin B on focal adhesion signalling are similar in SCGN-knockdown NIT-1 cells and control cells.

NIT-1 cells transfected with control or SCGN siRNA were starved with glucose-free HBSS containing DMSO or 1 μM latrunculin B at 37°C for 2 h and then stimulated with 0 or 16.8 mM glucose in HBSS with DMSO or 1 μM latrunculin B at 37°C for 10 min. The cell lysates were dissolved in gel sample buffer and separated by SDS/PAGE (10% gel) under reducing conditions. Western blot analysis using the indicated antibodies was performed to show protein expression.
DISCUSSION
In the present study, we have shown that SCGN, a Ca2+-binding protein having six EF-hands, regulates F-actin dynamics and focal adhesion remodelling in pancreatic NIT-1 β-cells, thereby promoting glucose- and H2O2-induced insulin secretion in these cells. We showed further that SCGN plays a predominant role in potentiating only the second phase of insulin secretion, without altering the first phase of insulin release. The present study is the first to demonstrate the involvement of SCGN in glucose-mediated insulin secretion by remodelling actin and activating focal adhesion molecules.
Ca2+-binding proteins having EF-hand domains are effective as buffers against increases in intracellular Ca2+, and as Ca2+ sensors that promote conformational changes triggering protein–protein interactions and induce cell-state-specific signalling events by Ca2+ [44]. This superfamily of 45 kDa Ca2+-binding proteins, which includes calretinin, calumenin, reticulocalbin and SCGN, has been shown using bioinformatics analysis to contain six EF-hands (results not shown). Of these, SCGN was distinguished for acting in Ca2+-dependent release mechanisms such as insulin secretion from pancreatic β-cells [12,13,17,45] and neurotransmitter secretion from nerve endings [12]. SCGN is expressed exclusively in pancreatic β-cells and in neuroendocrine cells. However, before the present study, little was known of the molecular mechanisms underlying SCGN's action in pancreatic β-cells or its role in insulin secretion. The present study demonstrates (i) that SCGN controls the glucose-stimulated second phase of insulin secretion, the pathway that mediates ROS generation, but has no influence on the KCl-stimulated pathway which provokes only the first phase of insulin secretion [1,46]; (ii) that SCGN interacts with actin, which is potentiated by glucose and intracellular Ca2+ signalling; and (iii) that SCGN and actin co-localize near the plasma membrane in response to glucose stimulation. However, the binding mechanism of SCGN and actin or the significance of their co-localization remains to be elucidated.
GSIS in pancreatic β-cells involves remodelling of cortical actin which transiently disrupts the interaction between polymerized actin with the plasma membrane t-SNARE (target soluble N-ethylmaleimide-sensitive fusion protein-attachment protein receptor) complex. F-actin can have both positive and negative roles in the process of insulin secretion depending on the nature of F-actin remodelling [36]. After the rapid and ready release of insulin granules beneath the plasma membrane in the first phase of insulin secretion [30,47], the second, glucose-stimulated, phase of insulin secretion requires reorganization of the filamentous actin network for recruitment of an intracellular storage pool of insulin granules towards the plasma membrane [30,39,48]. Glucose stimulation induces localized depolymerization of cortical F-actin, which acts as a physical barrier impeding the access of insulin granules to the cell periphery [35,38,48,49], as well as polymerization of F-actin, which provides a cytoskeletal track for insulin granule transport [50,51]. Silencing SCGN impairs glucose-induced depolymerization of actin at the membrane, and significantly reduces polymerization of actin in the central part of β-cells. In accordance with this, F-actin was decreased in SCGN-knockdown cells, indicating that the second phase of insulin secretion caused by F-actin polymerization was blunted [52,53]. Our findings suggest further that SCGN, which interacts with actin to promote actin dynamics and stress fibre formation in response to glucose, is necessary for the second phase of insulin secretion.
Actin polymerization is essential for cell adhesion [54,55] and is closely involved in focal adhesion activation. Actin dynamics and focal adhesion remodelling influence the second phase of release of insulin granules [5–8]. Upon glucose stimulation, FAK is activated by integrin β1-mediated intracellular signalling and phosphorylates paxillin. The activated focal adhesion molecules translocate to the plasma membrane, providing a molecular scaffold for the actin cytoskeleton, and act in co-ordination with F-actin remodelling to amplify the second phase of insulin secretion [6,7,56]. In the present study, silencing SCGN in NIT-1 cells blocked glucose-induced focal adhesion signalling in a dose- and time-dependent manner. Knocking down SCGN caused disruption of actin dynamics which in turn attenuated focal adhesion and impaired insulin granule replenishment during glucose stimulation. Considered together, these findings suggest that SCGN modulates the activation of the upstream signal of FAK-mediated focal adhesion by regulating actin cytoskeletal reorganization and influences insulin secretion upon glucose stimulation. Actin cytoskeletal reorganization via interaction between SCGN and actin is required for the regulation of the FAK-mediated signalling pathway, which in turn enhances the second phase of insulin secretion in pancreatic β-cells. The present study suggests that SCGN regulates F-actin dynamics and focal adhesion remodelling in pancreatic β-cells, and thereby the second phase of insulin secretion. Actin remodelling has been demonstrated to play critical roles in vesicle trafficking and the exocytotic process in many other secretory cells as well [57–59]. Impaired secretion of insulin in pancreatic β-cells is a major feature of Type 2 diabetes. The present study advances our understanding of the mechanisms regulating insulin secretion by β-cells, and our finding that SCGN plays a role in insulin secretion suggests possible new approaches useful in the therapy of Type 2 diabetes.
In summary, the present study is the first to show that SCGN performs a central regulatory function in the second phase of glucose-stimulated insulin secretion and advances our understanding of the basis for impaired glucose-stimulated insulin secretion in islet β-cells. On the basis of the significant regulatory role played by SCGN in insulin secretion, SCGN may be potentially useful in the treatment of Type 2 diabetes.
Acknowledgments
We thank Ludwig Wagner of the Medical University of Vienna (Vienna, Austria) for providing the secretagogin clone.
Abbreviations
- DMEM
Dulbecco's modified Eagle's medium
- E-cadherin
epithelial cadherin
- ERK
extracellular-signal-regulated kinase
- F-actin
filamentous actin
- FAK
focal adhesion kinase
- G-actin
globular actin
- GSIS
glucose-stimulated insulin secretion
- HBSS
Hanks balanced salt solution
- nanoUPLC
nano-ultra-performance liquid chromatography
- N-cadherin
neural cadherin
- ESI
elextrospray ionization
- q-TOF tandem MS
quadrupole-time of flight tandem mass spectrometry
- ROS
reactive oxygen species
- SCGN
secretagogin
AUTHOR CONTRIBUTION
Kong-Joo Lee and Seo-Yun Yang designed the study and wrote the paper. Jin-Hee Lee prepared the reagents. Seo-Yun Yang performed biological experiments and Jae-Jin Lee analysed MS results. Kyungeun Lee provided technical help for EM analysis, and Seung Hoon Oh, Yu-Mi Lim and Myung-Shik Lee provided the primary islet cells isolated from mice and the idea of the perifusion assay of insulin secretion.
FUNDING
This work was supported by the Global Research Lab Program [grant number 2012K1A1A2045441] of the National Research Foundation (NRF). S.-Y.Y. was supported by the Brain Korea 21 Plus (BK21 Plus) Project. K.L. was supported by the Korea Institute of Science and Technology (KIST) Institutional Project [grant number 2V04081].
References
- 1.Rorsman P., Eliasson L., Renstrom E., Gromada J., Barg S., Gopel S. The cell physiology of biphasic insulin secretion. News Physiol. Sci. 2000;15:72–77. doi: 10.1152/physiologyonline.2000.15.2.72. [DOI] [PubMed] [Google Scholar]
- 2.Rorsman P., Renstrom E. Insulin granule dynamics in pancreatic β cells. Diabetologia. 2003;46:1029–1045. doi: 10.1007/s00125-003-1153-1. [DOI] [PubMed] [Google Scholar]
- 3.Nevins A.K., Thurmond D.C. Glucose regulates the cortical actin network through modulation of Cdc42 cycling to stimulate insulin secretion. Am. J. Physiol. Cell Physiol. 2003;285:C698–C710. doi: 10.1152/ajpcell.00093.2003. [DOI] [PubMed] [Google Scholar]
- 4.Henquin J.C., Mourad N.I., Nenquin M. Disruption and stabilization of β-cell actin microfilaments differently influence insulin secretion triggered by intracellular Ca2+ mobilization or store-operated Ca2+ entry. FEBS Lett. 2012;586:89–95. doi: 10.1016/j.febslet.2011.11.030. [DOI] [PubMed] [Google Scholar]
- 5.Tomas A., Yermen B., Min L., Pessin J.E., Halban P.A. Regulation of pancreatic β-cell insulin secretion by actin cytoskeleton remodelling: role of gelsolin and cooperation with the MAPK signalling pathway. J. Cell Sci. 2006;119:2156–2167. doi: 10.1242/jcs.02942. [DOI] [PubMed] [Google Scholar]
- 6.Rondas D., Tomas A., Halban P.A. Focal adhesion remodelling is crucial for glucose-stimulated insulin secretion and involves activation of focal adhesion kinase and paxillin. Diabetes. 2011;60:1146–1157. doi: 10.2337/db10-0946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rondas D., Tomas A., Soto-Ribeiro M., Wehrle-Haller B., Halban P.A. Novel mechanistic link between focal adhesion remodeling and glucose-stimulated insulin secretion. J. Biol. Chem. 2012;287:2423–2436. doi: 10.1074/jbc.M111.279885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cai E.P., Casimir M., Schroer S.A., Luk C.T., Shi S.Y., Choi D., Dai X.Q., Hajmrle C., Spigelman A.F., Zhu D., et al. In vivo role of focal adhesion kinase in regulating pancreatic β-cell mass and function through insulin signaling, actin dynamics, and granule trafficking. Diabetes. 2012;61:1708–1718. doi: 10.2337/db11-1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ashcroft F.M., Proks P., Smith P.A., Ammala C., Bokvist K., Rorsman P. Stimulus-secretion coupling in pancreatic β cells. J. Cell. Biochem. 1994;55(Suppl.):54–65. doi: 10.1002/jcb.240550007. [DOI] [PubMed] [Google Scholar]
- 10.Henquin J.C., Ravier M.A., Nenquin M., Jonas J.C., Gilon P. Hierarchy of the β-cell signals controlling insulin secretion. Eur. J. Clin. Invest. 2003;33:742–750. doi: 10.1046/j.1365-2362.2003.01207.x. [DOI] [PubMed] [Google Scholar]
- 11.Niki I., Hidaka H. Roles of intracellular Ca2+ receptors in the pancreatic β-cell in insulin secretion. Mol. Cell. Biochem. 1999;190:119–124. doi: 10.1023/A:1006997822987. [DOI] [PubMed] [Google Scholar]
- 12.Wagner L., Oliyarnyk O., Gartner W., Nowotny P., Groeger M., Kaserer K., Waldhausl W., Pasternack M.S. Cloning and expression of secretagogin, a novel neuroendocrine- and pancreatic islet of Langerhans-specific Ca2+-binding protein. J. Biol. Chem. 2000;275:24740–24751. doi: 10.1074/jbc.M001974200. [DOI] [PubMed] [Google Scholar]
- 13.Rogstam A., Linse S., Lindqvist A., James P., Wagner L., Berggard T. Binding of calcium ions and SNAP-25 to the hexa EF-hand protein secretagogin. Biochem. J. 2007;401:353–363. doi: 10.1042/BJ20060918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoshihara M., Littleton J.T. Synaptotagmin I functions as a calcium sensor to synchronize neurotransmitter release. Neuron. 2002;36:897–908. doi: 10.1016/S0896-6273(02)01065-6. [DOI] [PubMed] [Google Scholar]
- 15.Gustavsson N., Han W. Calcium-sensing beyond neurotransmitters: functions of synaptotagmins in neuroendocrine and endocrine secretion. Biosci. Rep. 2009;29:245–259. doi: 10.1042/BSR20090031. [DOI] [PubMed] [Google Scholar]
- 16.Rorsman P., Braun M. Regulation of insulin secretion in human pancreatic islets. Ann. Rev. Physiol. 2013;75:155–179. doi: 10.1146/annurev-physiol-030212-183754. [DOI] [PubMed] [Google Scholar]
- 17.Hasegawa K., Wakino S., Kimoto M., Minakuchi H., Fujimura K., Hosoya K., Komatsu M., Kaneko Y., Kanda T., Tokuyama H., et al. The hydrolase DDAH2 enhances pancreatic insulin secretion by transcriptional regulation of secretagogin through a Sirt1-dependent mechanism in mice. FASEB J. 2013;27:2301–2315. doi: 10.1096/fj.12-226092. [DOI] [PubMed] [Google Scholar]
- 18.Hamaguchi K., Gaskins H.R., Leiter E.H. NIT-1, a pancreatic β-cell line established from a transgenic NOD/Lt mouse. Diabetes. 1991;40:842–849. doi: 10.2337/diab.40.7.842. [DOI] [PubMed] [Google Scholar]
- 19.Kao G., Nordenson C., Still M., Ronnlund A., Tuck S., Naredi P. ASNA-1 positively regulates insulin secretion in C. elegans and mammalian cells. Cell. 2007;128:577–587. doi: 10.1016/j.cell.2006.12.031. [DOI] [PubMed] [Google Scholar]
- 20.Xia H.Q., Pan Y., Peng J., Lu G.X. Over-expression of miR375 reduces glucose-induced insulin secretion in Nit-1 cells. Mol. Biol. Rep. 2011;38:3061–3065. doi: 10.1007/s11033-010-9973-9. [DOI] [PubMed] [Google Scholar]
- 21.Weinhaus A.J., Poronnik P., Tuch B.E., Cook D.I. Mechanisms of arginine-induced increase in cytosolic calcium concentration in the β-cell line NIT-1. Diabetologia. 1997;40:374–382. doi: 10.1007/s001250050690. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y., Xu M., Zhang S., Yan L., Yang C., Lu W., Li Y., Cheng H. The role of G protein-coupled receptor 40 in lipoapoptosis in mouse β-cell line NIT-1. J. Mol. Endocrinol. 2007;38:651–661. doi: 10.1677/JME-06-0048. [DOI] [PubMed] [Google Scholar]
- 23.Chang I., Cho N., Kim S., Kim J.Y., Kim E., Woo J.E., Nam J.H., Kim S.J., Lee M.S. Role of calcium in pancreatic islet cell death by IFN-γ/TNF-α. J. Immunol. 2004;172:7008–7014. doi: 10.4049/jimmunol.172.11.7008. [DOI] [PubMed] [Google Scholar]
- 24.Jeong J., Jung Y., Na S., Lee E., Kim M.S., Choi S., Shin D.H., Paek E., Lee H.Y., Lee K.J. Novel oxidative modifications in redox-active cysteine residues. Mol. Cell. Proteomics. 2011;10:M110.000513. doi: 10.1074/mcp.M110.000513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saini D.K., Karunarathne W.K., Angaswamy N., Saini D., Cho J.H., Kalyanaraman V., Gautam N. Regulation of Golgi structure and secretion by receptor-induced G protein βγ complex translocation. Proc. Natl. Acad. Sci. U.S.A. 2010;107:11417–11422. doi: 10.1073/pnas.1003042107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pi J., Bai Y., Zhang Q., Wong V., Floering L.M., Daniel K., Reece J.M., Deeney J.T., Andersen M.E., Corkey B.E., Collins S. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes. 2007;56:1783–1791. doi: 10.2337/db06-1601. [DOI] [PubMed] [Google Scholar]
- 27.Maechler P., Jornot L., Wollheim C.B. Hydrogen peroxide alters mitochondrial activation and insulin secretion in pancreatic β cells. J. Biol. Chem. 1999;274:27905–27913. doi: 10.1074/jbc.274.39.27905. [DOI] [PubMed] [Google Scholar]
- 28.Llanos P., Contreras-Ferrat A., Barrientos G., Valencia M., Mears D., Hidalgo C. Glucose-dependent insulin secretion in pancreatic β-cell islets from male rats requires Ca2+ release via ROS-stimulated ryanodine receptors. PLoS One. 2015;10:e0129238. doi: 10.1371/journal.pone.0129238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leloup C., Tourrel-Cuzin C., Magnan C., Karaca M., Castel J., Carneiro L., Colombani A.L., Ktorza A., Casteilla L., Penicaud L. Mitochondrial reactive oxygen species are obligatory signals for glucose-induced insulin secretion. Diabetes. 2009;58:673–681. doi: 10.2337/db07-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Z., Thurmond D.C. Mechanisms of biphasic insulin-granule exocytosis: roles of the cytoskeleton, small GTPases and SNARE proteins. J. Cell Sci. 2009;122:893–903. doi: 10.1242/jcs.034355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henquin J.C. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes. 2000;49:1751–1760. doi: 10.2337/diabetes.49.11.1751. [DOI] [PubMed] [Google Scholar]
- 32.Henquin J.C. Regulation of insulin secretion: a matter of phase control and amplitude modulation. Diabetologia. 2009;52:739–751. doi: 10.1007/s00125-009-1314-y. [DOI] [PubMed] [Google Scholar]
- 33.Straub S.G., Sharp G.W. Hypothesis: one rate-limiting step controls the magnitude of both phases of glucose-stimulated insulin secretion. Am. J. Physiol. Cell Physiol. 2004;287:C565–C571. doi: 10.1152/ajpcell.00079.2004. [DOI] [PubMed] [Google Scholar]
- 34.Arachea B.T., Sun Z., Potente N., Malik R., Isailovic D., Viola R.E. Detergent selection for enhanced extraction of membrane proteins. Protein Expr. Purif. 2012;86:12–20. doi: 10.1016/j.pep.2012.08.016. [DOI] [PubMed] [Google Scholar]
- 35.Jewell J.L., Luo W., Oh E., Wang Z., Thurmond D.C. Filamentous actin regulates insulin exocytosis through direct interaction with Syntaxin 4. J. Biol. Chem. 2008;283:10716–10726. doi: 10.1074/jbc.M709876200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kalwat M.A., Thurmond D.C. Signaling mechanisms of glucose-induced F-actin remodeling in pancreatic islet β cells. Exp. Mol. Med. 2013;45:e37. doi: 10.1038/emm.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilder J.A., Ashman R.F. Actin polymerization in murine B lymphocytes is stimulated by cytochalasin D but not by anti-immunoglobulin. Cell. Immunol. 1991;137:514–528. doi: 10.1016/0008-8749(91)90098-V. [DOI] [PubMed] [Google Scholar]
- 38.Orci L., Gabbay K.H., Malaisse W.J. Pancreatic β-cell web: its possible role in insulin secretion. Science. 1972;175:1128–1130. doi: 10.1126/science.175.4026.1128. [DOI] [PubMed] [Google Scholar]
- 39.Thurmond D.C., Gonelle-Gispert C., Furukawa M., Halban P.A., Pessin J.E. Glucose-stimulated insulin secretion is coupled to the interaction of actin with the t-SNARE (target membrane soluble N-ethylmaleimide-sensitive factor attachment protein receptor protein) complex. Mol. Endocrinol. 2003;17:732–742. doi: 10.1210/me.2002-0333. [DOI] [PubMed] [Google Scholar]
- 40.Christiansen J.J., Rajasekaran A.K. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res. 2006;66:8319–8326. doi: 10.1158/0008-5472.CAN-06-0410. [DOI] [PubMed] [Google Scholar]
- 41.Chen C.S., Alonso J.L., Ostuni E., Whitesides G.M., Ingber D.E. Cell shape provides global control of focal adhesion assembly. Biochem. Biophys. Res. Commun. 2003;307:355–361. doi: 10.1016/S0006-291X(03)01165-3. [DOI] [PubMed] [Google Scholar]
- 42.Vicente-Manzanares M., Choi C.K., Horwitz A.R. Integrins in cell migration: the actin connection. J. Cell Sci. 2009;122:199–206. doi: 10.1242/jcs.018564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Turner C.E. Paxillin and focal adhesion signalling. Nat. Cell Biol. 2000;2:E231–E236. doi: 10.1038/35046659. [DOI] [PubMed] [Google Scholar]
- 44.Alpar A., Attems J., Mulder J., Hokfelt T., Harkany T. The renaissance of Ca2+-binding proteins in the nervous system: secretagogin takes center stage. Cell. Signal. 2012;24:378–387. doi: 10.1016/j.cellsig.2011.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maj M., Gartner W., Ilhan A., Neziri D., Attems J., Wagner L. Expression of TAU in insulin-secreting cells and its interaction with the calcium-binding protein secretagogin. J. Endocrinol. 2010;205:25–36. doi: 10.1677/JOE-09-0341. [DOI] [PubMed] [Google Scholar]
- 46.Olofsson C.S., Gopel S.O., Barg S., Galvanovskis J., Ma X., Salehi A., Rorsman P., Eliasson L. Fast insulin secretion reflects exocytosis of docked granules in mouse pancreatic β-cells. Pflugers Arch. 2002;444:43–51. doi: 10.1007/s00424-002-0781-5. [DOI] [PubMed] [Google Scholar]
- 47.Daniel S., Noda M., Straub S.G., Sharp G.W. Identification of the docked granule pool responsible for the first phase of glucose-stimulated insulin secretion. Diabetes. 1999;48:1686–1690. doi: 10.2337/diabetes.48.9.1686. [DOI] [PubMed] [Google Scholar]
- 48.Mourad N.I., Nenquin M., Henquin J.C. Metabolic amplifying pathway increases both phases of insulin secretion independently of β-cell actin microfilaments. Am. J. Physiol. Cell Physiol. 2010;299:C389–C398. doi: 10.1152/ajpcell.00138.2010. [DOI] [PubMed] [Google Scholar]
- 49.Aunis D., Bader M.F. The cytoskeleton as a barrier to exocytosis in secretory cells. J. Exp. Biol. 1988;139:253–266. doi: 10.1242/jeb.139.1.253. [DOI] [PubMed] [Google Scholar]
- 50.Varadi A., Ainscow E.K., Allan V.J., Rutter G.A. Involvement of conventional kinesin in glucose-stimulated secretory granule movements and exocytosis in clonal pancreatic β-cells. J. Cell Sci. 2002;115:4177–4189. doi: 10.1242/jcs.00083. [DOI] [PubMed] [Google Scholar]
- 51.Swanston-Flatt S.K., Carlsson L., Gylfe E. Actin filament formation in pancreatic β-cells during glucose stimulation of insulin secretion. FEBS Lett. 1980;117:299–302. doi: 10.1016/0014-5793(80)80966-5. [DOI] [PubMed] [Google Scholar]
- 52.Heaslip A.T., Nelson S.R., Lombardo A.T., Beck Previs S., Armstrong J., Warshaw D.M. Cytoskeletal dependence of insulin granule movement dynamics in INS-1 β-cells in response to glucose. PLoS One. 2014;9:e109082. doi: 10.1371/journal.pone.0109082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Uenishi E., Shibasaki T., Takahashi H., Seki C., Hamaguchi H., Yasuda T., Tatebe M., Oiso Y., Takenawa T., Seino S. Actin dynamics regulated by the balance of neuronal Wiskott–Aldrich syndrome protein (N-WASP) and cofilin activities determines the biphasic response of glucose-induced insulin secretion. J. Biol. Chem. 2013;288:25851–25864. doi: 10.1074/jbc.M113.464420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bailly M. Connecting cell adhesion to the actin polymerization machinery: vinculin as the missing link? Trends Cell Biol. 2003;13:163–165. doi: 10.1016/S0962-8924(03)00030-8. [DOI] [PubMed] [Google Scholar]
- 55.Lee G.H., Ahn T., Kim D.S., Park S.J., Lee Y.C., Yoo W.H., Jung S.J., Yang J.S., Kim S., Muhlrad A., et al. Bax inhibitor 1 increases cell adhesion through actin polymerization: involvement of calcium and actin binding. Mol. Cell. Biol. 2010;30:1800–1813. doi: 10.1128/MCB.01357-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nakamura K., Yano H., Uchida H., Hashimoto S., Schaefer E., Sabe H. Tyrosine phosphorylation of paxillin α is involved in temporospatial regulation of paxillin-containing focal adhesion formation and F-actin organization in motile cells. J. Biol. Chem. 2000;275:27155–27164. doi: 10.1074/jbc.M000679200. [DOI] [PubMed] [Google Scholar]
- 57.Smythe E., Ayscough K.R. Actin regulation in endocytosis. J. Cell Sci. 2006;119:4589–4598. doi: 10.1242/jcs.03247. [DOI] [PubMed] [Google Scholar]
- 58.Malacombe M., Bader M.F., Gasman S. Exocytosis in neuroendocrine cells: new tasks for actin. Biochim. Biophys. Acta. 2006;1763:1175–1183. doi: 10.1016/j.bbamcr.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 59.Lopez J.A., Burchfield J.G., Blair D.H., Mele K., Ng Y., Vallotton P., James D.E., Hughes W.E. Identification of a distal GLUT4 trafficking event controlled by actin polymerization. Mol. Biol. Cell. 2009;20:3918–3929. doi: 10.1091/mbc.E09-03-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]