

Summary
How the TCR repertoire, in concert with risk-associated MHC, imposes susceptibility for autoimmune diseases is incompletely resolved. Due largely to recombinatorial biases, a small fraction of TCR α or β chains are shared by most individuals, or public. If public TCR chains modulate a TCRαβ heterodimer’s likelihood of productively engaging autoantigen, because they are pervasive and often high frequency, they could also broadly influence disease risk and progression. Prior data, using low resolution techniques, have identified the heavy use of select public TCR in some autoimmune models. Here we assess public repertoire representation in mice with experimental autoimmune encephalomyelitis (EAE) at high resolution. Saturation sequencing was used to identify >18×106 TCRβ sequences from the central nervous systems, periphery, and thymi of mice at different stages of autoimmune encephalomyelitis and healthy controls. Analyses indicated the prominent representation of a highly diverse public TCRβ repertoire in the disease response. Preferential formation of public TCR implicated in autoimmunity was identified in pre-selection thymocytes and, consistently, public, disease-associated TCRβ were observed to be commonly oligoclonal. Increased TCR sharing and a focusing of the public TCR response was seen with disease progression. Critically, comparisons of peripheral and CNS repertoires and repertoires from pre-immune and diseased mice demonstrated that public TCR were preferentially deployed relative to non-shared, or private, sequences. Our findings implicate public TCR in skewing repertoire response during autoimmunity, and suggest that subsets of public TCR sequences may serve as disease-specific biomarkers or influence disease susceptibility or progression.
Keywords: T cell receptor, TCR repertoire, autoimmunity, encephalomyelitis, myelin, antigen, MHC
Introduction
The MHC is the principal genetic locus conveying susceptibility for tissue-specific autoimmunity. For example, homozygosity for HLA-DRB1*15:01 increases the probability of developing multiple sclerosis ~7-fold (1, 2). In contrast, few non-MHC polymorphisms impart more than a 1.2-fold increase in risk (3). Potential mechanisms for these allele specific effects include differential binding of self-antigen-derived epitopes to different MHC specificities and modulation of the selection and activation of pathologic effector and protective regulatory T cells (4, 5).
These risk-associated MHC engage a TCR repertoire that has the potential to generate 1015–1018 distinct receptors. A much smaller number of unique T cell clones is present in the circulation and represents only a small fraction of this potential diversity (6, 7). Nevertheless, due to the TCR’s capacity for diversification, each individual’s TCR repertoire is essentially unique. Considering the extensive variability of TCR repertoires and the large number of tissue specific antigens capable of binding MHC molecules that TCR may engage, it may be considered surprising that single MHC alleles so effectively impose population-wide disease risk.
Despite the theoretical diversity of the TCR repertoire, a small fraction of α and β monomers are shared by most individuals, or public (8–10). Public TCR α or β chains can pair with a vast array of independently rearranged and distinct β or α chains that contribute roughly equally to ligand recognition (11), and therefore would not be expected to bias TCR recognition. However, it has also been shown that certain TRAV and TRBV are preferentially employed in specific responses (12–15), and in one case structural data identified a binding “hotspot” between a single TRBV and antigen-MHC ligand that explained this preference (16). Considering this, it is possible that public α or β chains, which are fixed not only for the V region, but for J and CDR3 sequences, are capable of modulating a TCRαβ heterodimer’s likelihood of productively engaging autoantigen.
Public TCR largely result from recombinatorial biases in TCR α and β chain formation and, due to their preferential formation, are often present at high frequency (17). Public TCR have been found to be prevalent in some infectious, autoimmune, and other responses (18, 19). However, clarifying the role of public TCR in disease has been previously limited by several factors. These include prior assessments of responses with low levels of heterogeneity such as that in MBP-induced EAE in H-2u mice, low resolution sequencing or spectratyping analyses that though indicating heavy use of some public sequences are unable to robustly quantify this, absence of data defining differential publicity in the repertoires used by different T cell lineages, and, importantly, the general inability to establish whether public sequences are over-represented compared with expectations from pre-immune TCR frequencies.
Here we use saturation sequencing to assess in depth the use of public TCR in a model autoimmune disease, myelin oligodendroglial glycoprotein (MOG)35–55-induced experimental autoimmune encephalomyelitis (EAE). Analyses of greater than 18 million TCRβ from Foxp3+ regulatory and Foxp3− conventional T cells from different organs and time points and from diseased and pre-immune mice identified both a high level of TCR diversity as well as extensive use of public TCR within the autoimmune response. Comparisons of disease associated and unassociated repertoires from pre-immune and immune mice further demonstrated that public TCR were preferentially deployed from the pre-immune repertoire. Assessment of developing thymocytes identified a markedly increased formation of disease-associated public TCR in preselection thymocytes, and comparison of early and late disease repertoires indicated a focusing and enrichment of the public TCR response over time. These results indicate that public TCR are preferentially incorporated into the responding repertoire during MOG-EAE. They further support the hypothesis that recognition biases imposed by the public TCR repertoire directly bear on the autoimmune response.
Methods
Mice
C57BL/6J (B6) mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and bred with GFP-Foxp3 knock-in mice on a B6 background obtained from Dr. A. Rudensky (MSKCC) (20). Mice were bred under specific-pathogen-free conditions, and animal experiments were carried out in compliance with Institutional Animal Care and Use Committee guidelines.
Flow Cytometry
Red blood cells were lysed prior to staining. Fc receptors were blocked with FcR blocking reagent (Miltenyi Biotec, San Diego, CA). Cell surface staining was performed for 20 min at 4°C in PBS containing 0.1% sodium azide and 2% (vol/vol) fetal bovine serum (FBS). Monoclonal antibodies (Ab) specific for CD4 (clone RM4-5), CD8 (clone 53-6.7), TCRβ (clone H57-597), CD44 (clone 1M7), CD45RB (clone C363.16A), CD69 (clone H1-2F3), and TRBV13-2, 3 (KJ-16) were from BD Biosciences (San Jose, CA). Flow cytometric analysis was performed on an LSRFortessa (BD Biosciences) and analyzed by using FlowJo software (Tree Star, Ashland, OR).
EAE Induction
MOG35–55 peptide (MEVGWYRSPFSRVVHLYRNGK) was synthesized by the St. Jude Hartwell Center for Biotechnology and HPLC purified prior to use. EAE was induced and scored as described (21). B6 mice were immunized s.c. with 100 μg of MOG35–55 in complete Freund’s adjuvant containing 0.4 mg Mycobacterium tuberculosis H37RA (Difco, Lawrence, KS). Two hundred ng of pertussis toxin (List Biological Laboratories, Campbell, CA) was administered i.v. on days 0 and 2. Clinical scoring was: 1, limp tail; 2, hind limb paresis or partial paralysis; 3, total hind limb paralysis; 4, hind limb paralysis and body or front limb paresis or paralysis; 5, moribund.
Cell Isolation and Sorting
T cells were isolated from spleen, CNS, and thymus as described (22). Splenic and CNS cells were stained with CD4 Ab and flow cytometrically sorted into CD4+GFP-Foxp3+ (Treg) and CD4+GFP-Foxp3− (Tconv) populations. Some splenic Tconv cells were further sorted into CD4+CD44hiCD45RBlo (memory/effector) and CD4+CD44loCD45RBhi (naïve) populations. Thymic T cells were stained with CD4, CD8, and TCR Abs and sorted into CD4+CD8+TCRlo double positive, CD4+CD8−Foxp3−, and CD4+CD8−Foxp3+ single positive T cell populations. Flow cytometric sorting used a Reflection (Sony Biotechnology, Champaign, IL) sorter.
High Throughput Sequencing and Raw Data Trim
DNA was prepared and high throughput sequencing performed as previously described (21). Sequencing was performed on an Illumina Genome Analyzer IIx sequencer (Illumina, San Diego, CA) and 125 bp reads covering the entire CDR3β region were obtained. Raw sequencing data was subsequently trimmed and the CDR3β sequences were filtered for quality using criteria we have previously described (23). Raw unprocessed data is accessible at European Nucleotide Archive (http://www.ebi.ac.uk/ena), study accession: PRJEB13451.
Hybridoma Production and Evaluation
T cell hybridomas were generated using isolated CNS-infiltrating T cells derived from 50 B6 mice with EAE. Cells were stimulated for three days with anti-CD3/anti-CD28 prior to fusion with partner cells as previously described and single cells cloned by limiting dilution (22). Resulting hybridomas were screened for KJ-16 (TRBV13-2, 13-3)-positivity prior to cDNA production and TCRβ amplification using TRBV13 and TCRα specific primers, and capillary sequencing. Responsiveness to MOG35–55 was measured by co-culture of the hybridomas with irradiated splenic APCs with or without 100 μg/ml MOG35–55 or with control anti-CD3. Sequences were considered “shared” if they were identified in the CNS repertoires of any of the 12 mice subjected to high throughput sequencing.
Cytokine Analysis
Culture supernatants from hybridomas were collected 24 hr after culture with irradiated splenic APCs in the absence of stimulation, in the presence of 100 μg/mL MOG35–55, or in the presence of anti-CD3, and analyzed for IL-2 using the Milliplex MAP mouse cytokine/chemokine immunoassay kit (Millipore, Billerica, MA) on a Luminex (Bio-Rad) instrument.
5′ RACE amplification of TRBV13-2+ TCR alpha chains
Foxp3-GFP reporter mice were immunized with MOG35–55 to induce EAE as described above. CNS tissue was harvested 17 days post injection, pooled from 9 mice, and CD4+Foxp3+ and CD4+Foxp3− TRBV13-2+ populations were isolated by flow cytometric sorting. 5′ RACE amplification of TCR alpha chains was performed as previously reported (24). Briefly mRNA was extracted using Oligotex Direct mRNA mini kit (QIAgen) and used as a template for cDNA amplification with the SMARTer PCR cDNA synthesis kit (Clontech). Amplified cDNA was cleaned using the PCR purification kit (QIAgen) and rearranged TCR products were amplified using a primer specific for the TCR alpha constant domain (5′ – TCAACTGGACCACAGCCTCAGCGTCA – 3′). Products were run on a 1% agarose gel and fragments between 500 and 700 bp were extracted using QIAgen gel purification kit. Extracted fragments were cloned into the PCR2.1-TOPO vector for miniprep and sequence analysis.
Statistics
Except as indicated below, means and SDs were calculated using Excel or PRISM. Plots demonstrate mean ± 1 SD. Two-tailed student t-tests were applied to compare any two groups and ANOVA for three or more groups. For multiple comparisons, significance is only shown for indicated groups. Bayesian analysis was conducted using the Fisher’s Exact Test using GraphPad. A p < 0.05 was considered statistically significant. For analysis of CDR3β length, the weighted linear regression method was applied to assess the correlation between CDR3β length and degree of publicity. The total number of available sequences identified as private, or public occurring in the specified number of mice was used as weight to determine the significance of this correlation. To assess preferential amino acid usage at specified positions of the CDR3β sequence, sequences were segregated into 3 groups: private, moderately public (occurring in 2–4 mice), and highly public (occurring in 5–9 mice). Chi-square tests were applied to compare the frequency of each amino acid at the indicated position across these groups. To offset the problem of multiple comparisons made across the same data set, the raw p values were adjusted using the Holm-Bonferroni method. Adjusted p values of <0.05 were considered significant. Significant differences in amino acid frequency were plotted as a heat map showing preferential usage in public or private TCRs, and the magnitude of the difference in the frequency. Significance levels are denoted in figures as: *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001.
Results
High prevalence of public TCRβ in the autoimmune repertoire
TRBV13-2 is the dominant TCRβ in MOG-specific T cells during EAE in C57BL/6 mice (25). To understand the composition and dynamics of autoimmune effector and regulatory repertoires, we flow cytometrically analyzed sorted TRBV13-2+ T cell populations from mice with MOG-induced EAE and healthy controls. Cells were isolated from the CNS and spleens of mice with early (day 15–18) and late (day 26) EAE, and the spleens and thymi of healthy controls. CNS infiltrating TRBV13-2+ T cell percentages were similar at the early and late time points. CD4+ Foxp3− (Tconv) and Foxp3+ (Treg) populations were acquired for most analyses. From some mice, the CD4+Foxp3− cells were further sorted into naïve (CD45RbhiCD44lo) and memory/effector (CD45RbloCD44hi) subsets. Saturation sequencing was performed using high throughput techniques, with >18×106 TCR studied (Supplemental Table SI) (8, 21, 23, 26, 27).
TRBJ utilization, an indicator of the heterogeneity of the sequenced populations, was overall similar in Tconv and Treg from the CNS and spleens of mice with EAE and spleens of controls (Fig. 1). This was true whether tallying unique amino acid sequences or total sequences acquired. Small perturbations, particularly an increased TRBJ2.1 representation in the CNS, were seen for both Foxp3+ and Foxp3− cells. However, the CNS TRBV13-2 response in MOG-EAE proved overall to be highly diverse, and TRBJ were incorporated roughly in proportion to their frequency in the pre-immune population. Not surprisingly, T cells infiltrating the autoimmune CNS were virtually exclusively memory/activated (not shown). Splenic TCR sequences that were also identified in the CNS were also heavily biased toward the memory/activated subset (Fig. 2a, b; 77.7±9.0% of unique, 99.0±0.7% of total TCRβ sequences acquired). Therefore a highly diverse, activated T cell population infiltrates the CNS in MOG-EAE, and this population shows concordant signs of activation in the spleen.
Figure 1. TCR TRBJ use during EAE.

TRBV13-2+ TCRβ repertoires were determined in the spleens of pre-immune mice (n=4) and spleens and CNS of mice with early (d 15–18, n=9) or late (d 26, n=3) EAE. TRBJ use was tabulated for acquired sequences as an indicator of repertoire diversity. Mean ± 1 S.D. percent of unique (A, B) or total (C, D) TRBJ use by CD4+GFP-Foxp3− (A, C) and CD4+GFP-Foxp3+ (B, D) derived TCRβ from individual mice is plotted.
Figure 2. Association of CNS TCR with the memory/effector T cell pool.

CNS TCRβ sequences were compared with sequences from sorted splenic CD45RbhiCD44lo (naïve) or CD45RbloCD44hi (memory) T cells from the same mice (n=5) to determine the association of CNS-infiltrating sequences with the naïve and memory populations in the spleen. (A) Percent of unique CNS sequences that were identified exclusively in the splenic memory or naïve populations, or in both memory and naïve populations are plotted. Also plotted (far left bar), is the percent of unique CNS sequences identified anywhere within the splenic memory pool (union of percent of sequences only identified in the memory cells and identified in both memory and naïve cells). (B) Percent of total CNS sequences acquired were plotted as in (A).
Due to the limited size of the CNS infiltrates, substantially fewer unique sequences were identified in the CNS compared with splenic repertoires (Supplemental Table SI). Of these, 9.2% of the unique Foxp3− CNS sequences present in mice with early EAE (d 15–18) were shared by ≥2 of 9 mice (Fig. 3a). However, these shared sequences comprised 50.8% of total CNS sequences acquired, indicating a high prevalence of the shared TCRβ. Similarly, in 3 mice analyzed with late disease (d 26), 8.1% of unique sequences were shared, but these comprised 49.6% of total CNS Foxp3− sequences (Fig. 3d). Progressively smaller numbers of unique Foxp3− TCRβ were shared in the CNS by increasing numbers of mice (Fig. 3b, e), but these shared TCRβ were heavily utilized (Fig. 3c, f). For example, 0.05% of unique CD4+ Foxp3−-derived TCRβ sequences were identified in ≥7 of the 9 early EAE CNS, but comprised >1.5% of total sequences acquired, a 30-fold enrichment. A similar pattern of highly shared TCR representation was evident with CNS-infiltrating Treg cells (Fig. 3g–l).
Figure 3. Representation of shared TRBV13-2+ TCRβ among CNS-infiltrating T cells.

CNS TRBV13-2+ Foxp3− and Foxp3+ TCRβ repertoires from mice with early (d 15–18, n=9; A–C, G–I) or late (d 26, n=3, D–F, J–L) EAE were determined by high throughput sequencing. (A, D, G, J) Individual Foxp3+ and Foxp3− TCRβ amino acid sequences were tabulated. The proportion of unique TCRβ amino acid sequences identified and of total TCRβ sequences acquired that were cumulatively identified in the indicated number of mice is plotted. (B, E, H, K) The number of unique amino acid sequences identified in the CNS’ of the indicated number of mice with early or late EAE is plotted. (C, F, I, L) To visualize the relative prevalence of sequences with different publicities, the frequency of identification of each unique amino acid sequence relative to that of an average sequence in the same CNS, or representation ratio, was calculated. The representation ratio is therefore a measure of enrichment of a sequence compared with an average sequence. The mean representation ratio for sequences shared by the indicated number of mice with early or late EAE is plotted.
To further characterize private and shared TCR CDR3β sequences, we compared their average lengths. A slight but highly significant decrease in the average length of the CDR3β loops of public TCRs was observed, and this was proportional to the degree of publicity (Figure 4a). In addition, differences were observed in amino acid use at positions 1–12 of private and public TCR CDR3βs (Figure 4b). Public TCRs had a preference for a glycine at position 3, aspartic acid at position 4, alanine at position 5, and glycine at position 6. The magnitude of the increased frequency of these amino acids at these positions also increased with the degree of publicity (not shown). The selection of the glycine at P3 and the aspartic acid at P4 indicated that public TCRs preferentially used the germline encoded C-terminal TRBV13-2 sequence (P1–P4 = ASGD), whereas private TCRs were more likely to have non-germline encoded amino acids at these positions. Increased use of a tyrosine residue toward the end of public CDR3 sequence was also seen. This likely resulted from the slightly shorter average length of public TCRs, and thus an increased probability of the CDR3 terminal tyrosine residue locating to positions 10–12. The trends in public TCR CDR3β sequence length and amino acid use were observed in both Foxp3− and Foxp3+ sequences, as well as in both spleen and CNS samples, suggesting that they are not related to regulatory properties or the presence of EAE, but are biases more generally associated with public TCR.
Figure 4. Changes in CDR3β length and amino acid composition with TCR publicity.

(A) Weighted linear regression analysis was applied to compare the CDR3β length with regard to TCR publicity in Foxp3− and Foxp3+ spleen and CNS samples. The p value and estimated regression coefficient for each sample is shown. (B) Sequences were segregated into private, moderately public (occurring in 2–4 mice), and highly public (occurring in 5–9 mice). The frequency of each amino acid at CDR3β positions 1–12 was calculated as a percentage of the total number of sequences in each set. Chi-squared analysis modified for multiple comparisons was used to determine if there were significant differences between private and each set of public sequences at each position for each amino acid. The heat map indicates positions at which statistical analyses indicated that specific amino acids were more common in highly public or private TCR. Similar trends were observed when comparing moderately public and private TCR (not shown). Coloration in the heat map indicates the magnitude difference observed between private and highly public sequences. Boxes without color did not have significant differences in amino acid frequency among the three populations.
Sabatino and colleagues, using 2D-based affinity analyses previously estimated that ~60% of CNS infiltrating T cells in MOG-EAE recognize the MOG35–55 autoantigen (28). To assess fractional response here and determine if there is a differential responsiveness to autoantigen among T cells expressing CNS shared or non-shared TCR, we produced T cell hybridomas from the pooled CNS’ of 50 mice with EAE. TCRβ cDNA from KJ-16 antibody (TRBV13-2, 13-3 specific)-positive hybridomas were sequenced, and results were correlated with the reactivity of the hybridomas to MOG35–55 antigen. Concordant with Fig. 3a and d demonstrating that approximately one half of CNS sequences are shared, 42.5% of 73 TRBV13-2+ hybridoma TCRβ sequences identified were also found in the CNS-infiltrating repertoire data sets described above (Fig. 5a, b). An identical value, 42.5%, of the 73 hybridomas responded to MOG35–55. A modest and non-significant increase was seen in the proportion of hybridomas incorporating shared versus non-shared TCRβ sequences that demonstrated MOG35–55-responsiveness (45.2% shared vs. 40.5% non-shared, p>0.05 Fisher’s exact test). This indicates that a large proportion of CNS-infiltrating T cells are specific for cognate autoantigen and that publicity does not demonstrably alter this. As it is possible that not all hybridomas derived from T cells responding to autoantigen showed detectable reactivity in the in vitro assay here, these results set only a minimum threshold for the extent of MOG responsiveness among CNS-infiltrating T cells.
Figure 5. MOG-specificity of public and private CNS-infiltrating T cells.

T cell hybridomas were generated and cloned from CNS-infiltrating T cells from 50 mice with EAE. cDNA was generated from hybridomas staining positively with the KJ-16 (TRBV13-2, 3-specific) antibody. Full TCRβ sequence was obtained on 86 hybridomas (73 TRBV13-2+, 13 TRBV13-3+). The hybridomas were further assessed for MOG35–55 reactivity by culture in the absence of stimulation, with 100 μg/ml MOG35–55 and APCs, or with anti-CD3, and assaying IL-2 production. Sample IL-2 production from a single representative analysis showing identification of four MOG-reactive clones (360, 376, 598, 623) and three non-reactive TRBV13-2+ clones (425, 492, 523) is shown (A). (B) The table shows the numbers of isolated TRBV13-2+ hybridomas that were MOG35–55 reactive or non-reactive and whose TCRβ sequences were identified in the CNS of the 12 mice with early or late EAE analyzed by high throughput sequencing (Supplemental Table SI). Differences in the proportion of shared and non-shared TCR functionally responding to MOG35–55 were not identified (p>0.05, Fisher’s Exact Test).
Preferential deployment of public versus private TCRβ to the autoimmune repertoire
The high prevalence of shared TCRβ in the CNS repertoire (Fig. 3) did not necessarily indicate that shared sequences were preferentially deployed from the peripheral immune repertoire. Public sequences are often present at high frequency. The frequency of public sequences incorporated into the CNS repertoire may therefore have been proportionate to their representation in the larger repertoire and to that of all public sequences, whether involved or uninvolved in the autoimmune response (29). To assess this and establish the extent to which CNS-infiltrating T cells are disproportionately public, we analyzed the more abundant TCRβ sequences present in the spleen, where TCR from cells involved and uninvolved in the CNS response could be directly compared. If TCR engaged in the CNS response were randomly and equivalently deployed from the public and private peripheral repertoires, the proportions of splenic sequences that are identified and not identified in the CNS would be expected to be similarly public (Supplemental Fig. S1). In contrast, if public TCRβ were preferentially incorporated into the CNS response, the splenic sequences also seen in the CNS should be enriched in public sequences.
Consistent with prior reports in other systems, few Foxp3+ and Foxp3− TCRβ from the spleens of mice with EAE were shared (Fig. 6). However, TCRβ sequences also seen in the CNS were markedly more likely to be shared than sequences not observed in the CNS (Fig. 7a). For example, 28.5% of the unique CNS Foxp3− sequences from mice with early disease were identified in all spleens analyzed compared with 2.7% of non-CNS sequences, indicating that many CNS sequences were derived from the public repertoire. Pairwise analysis of the sharing of CNS and non-CNS sequences between individual mice verified this finding (Fig. 7c). Results were similar in mice with late disease (Fig. 7b, c) and for Foxp3+ T cells (Fig. 7d–f). This indicates that the high prevalence of shared sequences within the CNS reflects a selectively elevated use of public TCR among CNS-infiltrating T cells compared with that anticipated if public and private sequences were randomly used.
Figure 6. TCRβ sharing within the spleens of mice with EAE.

CD4+ GFP-Foxp3− and CD4+ GFP-Foxp3+ TRBV13-2+ TCRβ repertoires were determined in the spleens of mice with early (d 15–18, n=4) and late (d 26, n=3) EAE. Percent of unique amino acid sequences that were identified in the indicated number of mice is plotted.
Figure 7. Over-representation of unique public sequences in the CNS-infiltrating repertoire.

Splenic CD4+GFP-Foxp3− and CD4+GFP-Foxp3+ TRBV13-2+ TCRβ repertoires from mice with early (d 15–18; n=4) or late (d 26; n=3) EAE were segregated into sequences identified or not within the CNS. Percent of unique TCRβ sequences that were shared by the indicated number of mice with early (A, D) and late (B, E) disease is shown. (C, F) The overlap of the repertoire from each mouse was compared pairwise with that of every other mouse in a cohort.
The increased incorporation of unique public TCRβ sequences in the CNS may have resulted from a greater TCRα heterogeneity among T cell clones bearing public versus private TCRβ. This would lead to greater clonal diversity of cells expressing public compared with private TCRs. Individual public TCRβ sequences would then be associated with a larger number of specificities and therefore an increased probability of being incorporated into the autoimmune response. To exclude this potential confounding factor and determine whether public TCRβ were more generally over-represented among CNS-infiltrating T cell clones, the analyses above enumerating unique TCRβ sequences was replicated tabulating total splenic TCRβ sequences acquired. This analysis, however, also showed a dramatically enhanced identification of CNS compared with non-CNS sequences among the shared sequences (Fig. 8). Therefore, measurements of both unique and total TCR sequences indicate that public TCRβ are present at a markedly elevated frequency within the CNS repertoire compared with that anticipated if public and private sequences were randomly selected from the repertoire.
Figure 8. Over-representation of total public sequences in the CNS-infiltrating repertoire.

Analyses were performed on the same data sets as in Fig. 7, however overlap among total, rather than unique, TCRβ sequences acquired was assessed. For this, frequencies were normalized so total sequence number was effectively equal in each mouse. This ensured that the weighting of each mouse in calculations was equivalent. Percent of total TCRβ sequences that were shared by the indicated number of mice with early (A, D) and late (B, E) disease is shown. (C, F) The overlap of the repertoire from each mouse was compared pairwise with that of every other in a cohort.
Over-representation of CNS T cell-associated TCRβ in the pre-immune public repertoire
If public T cells were preferentially recruited and expanded in the CNS, CNS infiltrating TCRβ should be disproportionately public in pre-immune mice too. To test this, we mapped CNS infiltrating and uninvolved splenic TCRβ sequences from mice with EAE onto pre-immune repertoires from four unimmunized mice. For all samples analyzed, either Foxp3− or Foxp3+ and early or late disease, substantially more CNS-infiltrating than non-infiltrating TCRβ sequences were identified as public in the pre-immune repertoire (Fig. 9). This again was true both for unique and total TCRβ sequences analyzed. Together, these data support a heavily biased public TCRβ usage in the EAE repertoire, with this preference traceable to the pre-immune repertoire.
Figure 9. Over-representation of unique and total CNS-associated Foxp3+ and Foxp3− public TCRβ in the pre-immune repertoire.

Splenic TRBV13-2+ TCRβ amino acid repertoires from CD4+GFP-Foxp3− and CD4+GFP-Foxp3+ T cells of mice with early (d 15–18; n=4) or late (d 26, n=3) EAE were mapped onto the splenic CD4+GFP-Foxp3− or CD4+GFP-Foxp3+ repertoires from pre-immune mice (CTRL; n=4). The sequences were categorized as to whether or not they were also present in the CNS to determine the relative extent to which CNS and non-CNS sequences present in mice with EAE were also shared with pre-immune mice. Percent of unique CD4+GFP-Foxp3− sequences that were or were not identified in the CNS from mice with early (A) or late (B) EAE and also present in the CD4+GFP-Foxp3− repertoires of the indicated number of pre-immune spleens is plotted. (C) As in (A, B), but the overlap of repertoires from individual mice with EAE and individual pre-immune mice is plotted. (D–F) Analyses are equivalent to (A–C), but assessing unique sequence overlap between CD4+GFP-Foxp3+ repertoires. (G–L) Parallel analyses of total CD4+GFP-Foxp3− and CD4+GFP-Foxp3+ sequences acquired from mice with early or late EAE and pre-immune mice. As in figure 8, sequence events were normalized for comparisons of total sequence overlap between mice, effectively equalizing the total number of sequences in each mouse.
Temporal focusing of the public TCR repertoire
To determine whether the extent of sharing of autoimmune repertoires differed in early and late EAE, we next compared CNS repertoires from individual mice. Overlap of CNS sequences was lowest when pairs of d 15–18 mice were compared with each other. Repertoire overlap increased when d 15–18 mice were compared with d 26 mice, and was greatest when d 26 mice were compared (Fig. 10a, b). Splenic TCR overlap, unlike that of CNS TCR, did not change with disease stage (Fig. 10c, d). Therefore the extent of sharing increases with time in the disease-associated repertoire. That mice with early disease shared more of their CNS repertoire with late disease mice than with each other further indicates a temporal focusing of the shared CNS repertoire.
Figure 10. Public autoimmune repertoire focusing and formation.

CNS TCRβ repertoires of CD4+GFP-Foxp3− and CD4+GFP-Foxp3+ T cells were compared between individual mice with early and late disease to determine changes in the overlap between repertoires with disease progression. Percent CNS CD4+GFP-Foxp3− (A) and Foxp3+ (B) CNS TCRβ repertoire overlap between individual mice with early disease, mice with early versus late disease, and mice with late disease is plotted. Comparable analyses were performed on splenic CD4+GFP-Foxp3− (C) and CD4+GFP-Foxp3+ (D) TCRβ repertoires.
Recombinatorial bias and oligoclonality in autoimmune repertoire formation
Considering the relatedness of the d 15–18 and d 26 repertoires, we combined the 12 CNS data sets we acquired to identify common TCRβ sequences. Forty eight TCRβ were identified in ≥10 of the 12 CNS repertoires (Supplemental Table SII). These receptors were oligoclonal with multiple nucleotide sequences used for each CNS amino acid sequence cumulatively (range 2–59, median=13) and within single mice (mean 2.8±1.3). Consistent with their oligoclonal presence, in individual CNS 46.7±19.9% of the 48 public TCRβ were identified in both the Foxp3+ and Foxp3− lineages compared with 2.9±1.9% of private sequences (p<10−5).
It is hypothesized that public TCRs have a high probability of forming in the thymus. Biases in V(D)J recombination selects for TCRs with fewer nucleotide additions at the junctions, and public TCRs often have CDR3 sequences that more closely resemble germline V, D, and J sequences relative to private TCRs. Consistently, we observed a trend toward slightly shorter CDR3 lengths in autoimmune public TCRs, as well as preferential use of the germline TRBV sequence relative to private TCRs (Figure 4). Additionally, public TCRs with a single amino acid sequence are often found to be encoded by multiple nucleotide sequences, indicating that they are formed repeatedly in the thymus through multiple independent rearrangements, also providing an explanation for their commonality (9, 17, 30).
To test for a preferential thymic formation of public TRBV13-2+ TCR sequences in MOG-EAE, we assessed their nucleotide use in pre-selection CD4+CD8+TCRlo (DP) thymocytes from pre-immune mice (5). These cells were further identified as CD5 and CD69 negative, verifying their pre-selection status. Nucleotide sequence diversity in pre-selection thymocytes for each of the 48 public TCRβ was compared with that of paired control private TCRβ from the same mouse. These paired private sequences were selected based on having identical Vβ, Jβ and CDR3 length, and similar identification frequencies (>0.25 – <4 fold). Average nucleotide sequence numbers across 4 mice for each of the 48 sequences and their controls was calculated. If individual public sequences had a high probability of forming due to independent pre-selection recombination events, multiple nucleotide sequences encoding a single public amino acid sequence would be expected within an individual thymus. The public sequences indeed displayed markedly greater mean pre-selection nucleotide variability than control private sequences (Fig. 11; 6.2±2.9 vs 1.1±0.1 nt sequences), indicating that public sequences have an increased probability of forming and that pre-selection recombinatorial biases foster disease-associated public repertoire formation.
Figure 11. Increased pre-selection formation of disease associated public TCRβ.
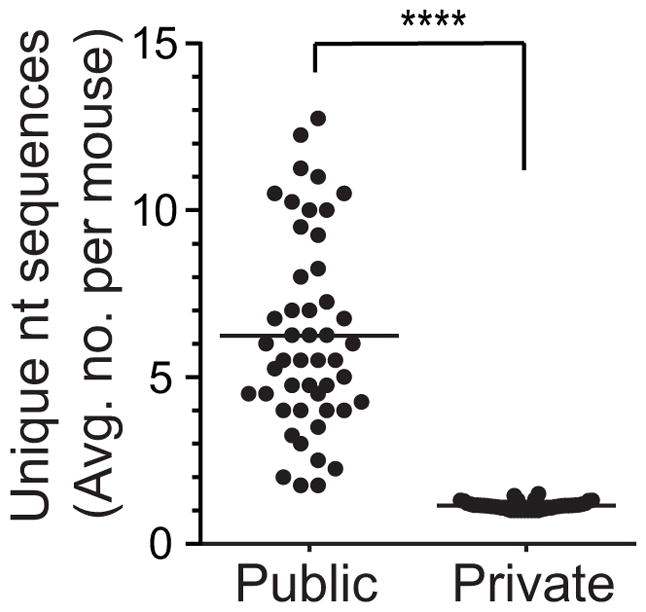
To determine the diversity of the public autoimmune repertoire in pre-selection thymocytes, nucleotide (nt) sequences identified for 48 highly public CNS TCRβ amino acid sequences were assessed in sorted pre-selection CD4+CD8+TCRlo thymocytes from healthy mice (n=4). The mean number of unique nt sequences per thymus for each of the 48 sequences is plotted. For each CNS sequence, paired control private sequences were identified in each mouse with the same TRBV, TRBJ, and CDR3β length, and similar frequency (>0.25–<4 fold) as the public sequence for comparison. Mean unique nt sequences is similarly plotted.
TRAV and TRAJ use by TRBV13-2+ CNS-infiltrating TCRs
In preliminary studies, we assessed TCRα chain use in TRBV13-2+ CNS infiltrating TCR. We utilized 5′ RACE amplification of rearranged TCRs in sorted Foxp3+ and Foxp3− TRBV13-2+ CD4+ T cells followed by template-switch anchored RT-PCR to isolate and then sequence recombined TCR alpha products. TRAV7 was preferentially used among Foxp3− clones, both in total sequences and unique sequences (Supplemental Fig S2). A more moderate use of TRAV7 and an increase in TRAV12 sequences was observed among Foxp3+ clones. The most common TRAJ gene detected, TRAJ23, exhibited relatively consistent use between the Foxp3− and Foxp3+ clones examined here. This difference in TRAV use between Foxp3− and Foxp3+ clones suggests that TRAV selection may be skewed depending on the regulatory status of the cells. However, these results are based on a small data set, and more extensive analyses are needed to define the role of TCRα in this model system.
Discussion
Using saturation sequencing of disease-associated and unassociated TRBV13-2 TCR repertoires, we demonstrate a selective and prominent role of public TCR in the CNS autoimmune response. TRBV13-2+ TCR were analyzed because of this Vβ chain’s documented prominence in the MOG-specific T cell response. We find that the frequencies at which unique and total CNS-infiltrating TCRβ sequences were shared among mice was markedly elevated when compared with TCRβ unengaged in autoimmunity both from mice with EAE and from pre-immune mice. The extent of use of public TCR did not substantially differ in Foxp3+ and Foxp3− T cells, indicating that publicity is intrinsic to the response regardless of lineage assignment.
Recombinatorial biases in preselection thymocytes serve as the primary source for the generation of the broad public repertoire (10, 31, 32). Predispositions in VDJ use and activity levels of TdT and nucleases modifying junctional sequences increase the probability these sequences will form. We implicate similar pre-selection biases in also generating the public autoimmune repertoire, and identify substantial ongoing oligoclonal production of public autoimmune-associated TCR within the thymus. Indeed, many public TCRβ were identified in both regulatory and effector T cells in the CNS of individual mice, consistent with their increased formation in pre-selection thymocytes. As previously documented by Dyson and colleagues with the larger public repertoire (33), analyses here of the frequency of individual receptors in pre- and post-selection thymocytes and splenic T cells in pre-immune mice further failed to indicate that, once formed, disease-associated public TCR are preferentially selected (data not shown).
Most prior studies exploring public TCR representation have used low resolution techniques that identify small numbers of response-enriched public TCR. Our results with high resolution sequencing indicate that public TCR-produced repertoire distortions need not result from idiosyncratic effects due to the presence of rare high frequency dominant clonotypes. Rather, the TCR used by public CNS-infiltrating T cells proved to be highly heterogeneous. Indeed, the most common public TCR identified, was found in a median of only 0.2% of TRBV13-2+ TCR sequenced.
Our results also do not imply that public TCR possess unique structural properties that distinguish them from private TCR. Indeed, despite comprising a small fraction of the total repertoire, public sequences remain diverse. It would therefore seem unlikely that their preferential recruitment into the autoimmune response when compared with private sequences is due to a distinct biochemistry. Rather, one possibility is that because public TCR are pervasive across a population, specificity distortions they introduce within the repertoire will be introduced into all individuals bearing relevant MHC alleles. Thus, we can speculate that MOG35–55 serves as the dominant autoantigen in C57BL/6 EAE precisely because the public repertoire in conjunction with the restricting MHC, IAb, augments repertoire reactivity toward this autoantigen. Other neuroantigens transparent to the public repertoire and preferentially engaged by private TCR would only be capable of mediating autoimmunity in the small number of individuals that stochastically possess adequate numbers and subsets of antigen-specific private T cells.
An alternative explanation for the preferential incorporation of public sequences into the autoimmune repertoire is that these sequences more generally predispose TCRαβ toward self reactivity. Other repertoire studies, though more limited in scope and definition compared with these, have also identified public sequences among autoreactive T cells (18, 34, 35). However, it would seem more likely that over-representation of public TCR is not specific to self-reactive populations, but rather reflects a generically increased responsiveness of public TCR to antigen. Public TCR use has also been found in the responses to several pathogens and other antigens (10, 17). Although the extent to which public TCR are actually over-represented rather than just prevalent in immune responses requires further study, a number of analyses demonstrate a strong presence of public TCRs, implying a central role in specific epitope responses (36, 37). It is reasonable to expect that the recombinase machinery is tuned to preferentially generate sequences well adapted to support MHC engagement. If so, public sequences, which have a high likelihood of forming, may be particularly well suited to support TCR associations with MHC-antigen. The majority of the TCR interface with antigen-MHC binds the MHC rather than antigenic peptide, and MHC-specific associations are critical to orienting the TCR and effective recognition (11, 38). An overall enhanced compatibility with MHC may thereby lead to the increased utilization of public TCR in diverse responses. An element of enhanced self-reactivity would be expected to accompany any generically increased binding fitness of TCR for MHC, and indeed this has been observed in mutational analyses increasing TCR affinity (39, 40).
Although this latter model remains speculative, in potential support we find that though the representation of public TCR among the autoimmune response in EAE is elevated, the fraction of CNS infiltrating cells that functionally respond to MOG35–55 is similar among shared and non-shared sequences. This suggests that publicity in and of itself did not increase the proportion of activated CNS infiltrating T cells with threshold reactivity to cognate MOG35–55 autoantigen. However, the increased presence of public TCR in the CNS implicates these cells as having an increased likelihood of more generally responding to the immunizing stimulus. Ultimately, further functional assessments of the public and private immune response here as well as to additional immunizing events will be necessary to establish patterns of response distinguishing the public and private repertoires and the generality of these findings.
The capacity to interrogate the repertoire continues to increase with improving sequencing technologies (26). Data sets are being collected in several disease models and these may be linked to functional analyses of individual TCR, similar to those we describe here, to identify the relevance of specific sequences. Indeed, the identification of preferential TRBV and TRAV usage in several diseases may imply that public TCRα or TCRβ sequences bearing pre-defined V, J, and CDR3 sequences will strongly skew response characteristics as here. If so, specific public sequences may prove useful for the longitudinal monitoring of immune responses during autoimmune diseases. Likewise, if public TCR ultimately prove not only to be over-represented, but drivers of the autoimmune response as originally hypothesized by Sercarz and colleagues (41), it may be possible to modulate the autoimmune response by targeting the selection or activity of T cells bearing public TCR sequences.
Supplementary Material
Acknowledgments
Supported by the National Institutes of Health Grant AI056153 and AI06600 (to TLG) and ALSAC/SJCRH.
We thank Richard Cross, Greig Lennon, and Parker Ingle for assistance with flow cytometric sorting, and Jackie Wright and Bofeng Li for assistance with hybridoma production and DNA sequencing.
Footnotes
Author Contributions
Y.Z. helped with study design, performed experiments, analyzed data, prepared figures, and assisted with manuscript composition. P.N., L.L.J., and T.W. assisted with study design, and performed experiments and data analyses. J.M., D.P., and C.C. provided bioinformatics support and assisted with data analyses. T.L.G. provided project supervision, designed studies, analyzed data, and prepared the manuscript and figures with Y.Z. and L.L.J.
Reference List
- 1.Hoppenbrouwers IA, Hintzen RQ. Genetics of multiple sclerosis. Biochimica et biophysica acta. 2011;1812:194–201. doi: 10.1016/j.bbadis.2010.09.017. [DOI] [PubMed] [Google Scholar]
- 2.Tsai S, Santamaria P. MHC Class II Polymorphisms, Autoreactive T-Cells, and Autoimmunity. Frontiers in immunology. 2013;4:321. doi: 10.3389/fimmu.2013.00321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sawcer S, Hellenthal G, Pirinen M, Spencer CC, Patsopoulos NA, Moutsianas L, Dilthey A, Su Z, Freeman C, Hunt SE, Edkins S, Gray E, Booth DR, Potter SC, Goris A, Band G, Oturai AB, Strange A, Saarela J, Bellenguez C, Fontaine B, Gillman M, Hemmer B, Gwilliam R, Zipp F, Jayakumar A, Martin R, Leslie S, Hawkins S, Giannoulatou E, D’Alfonso S, Blackburn H, Martinelli Boneschi F, Liddle J, Harbo HF, Perez ML, Spurkland A, Waller MJ, Mycko MP, Ricketts M, Comabella M, Hammond N, Kockum I, McCann OT, Ban M, Whittaker P, Kemppinen A, Weston P, Hawkins C, Widaa S, Zajicek J, Dronov S, Robertson N, Bumpstead SJ, Barcellos LF, Ravindrarajah R, Abraham R, Alfredsson L, Ardlie K, Aubin C, Baker A, Baker K, Baranzini SE, Bergamaschi L, Bergamaschi R, Bernstein A, Berthele A, Boggild M, Bradfield JP, Brassat D, Broadley SA, Buck D, Butzkueven H, Capra R, Carroll WM, Cavalla P, Celius EG, Cepok S, Chiavacci R, Clerget-Darpoux F, Clysters K, Comi G, Cossburn M, Cournu-Rebeix I, Cox MB, Cozen W, Cree BA, Cross AH, Cusi D, Daly MJ, Davis E, de Bakker PI, Debouverie M, D’Hooghe BM, Dixon K, Dobosi R, Dubois B, Ellinghaus D, Elovaara I, Esposito F, Fontenille C, Foote S, Franke A, Galimberti D, Ghezzi A, Glessner J, Gomez R, Gout O, Graham C, Grant SF, Guerini FR, Hakonarson H, Hall P, Hamsten A, Hartung HP, Heard RN, Heath S, Hobart J, Hoshi M, Infante-Duarte C, Ingram G, Ingram W, Islam T, Jagodic M, Kabesch M, Kermode AG, Kilpatrick TJ, Kim C, Klopp N, Koivisto K, Larsson M, Lathrop M, Lechner-Scott JS, Leone MA, Leppa V, Liljedahl U, Bomfim IL, Lincoln RR, Link J, Liu J, Lorentzen AR, Lupoli S, Macciardi F, Mack T, Marriott M, Martinelli V, Mason D, McCauley JL, Mentch F, Mero IL, Mihalova T, Montalban X, Mottershead J, Myhr KM, Naldi P, Ollier W, Page A, Palotie A, Pelletier J, Piccio L, Pickersgill T, Piehl F, Pobywajlo S, Quach HL, Ramsay PP, Reunanen M, Reynolds R, Rioux JD, Rodegher M, Roesner S, Rubio JP, Ruckert IM, Salvetti M, Salvi E, Santaniello A, Schaefer CA, Schreiber S, Schulze C, Scott RJ, Sellebjerg F, Selmaj KW, Sexton D, Shen L, Simms-Acuna B, Skidmore S, Sleiman PM, Smestad C, Sorensen PS, Sondergaard HB, Stankovich J, Strange RC, Sulonen AM, Sundqvist E, Syvanen AC, Taddeo F, Taylor B, Blackwell JM, Tienari P, Bramon E, Tourbah A, Brown MA, Tronczynska E, Casas JP, Tubridy N, Corvin A, Vickery J, Jankowski J, Villoslada P, Markus HS, Wang K, Mathew CG, Wason J, Palmer CN, Wichmann HE, Plomin R, Willoughby E, Rautanen A, Winkelmann J, Wittig M, Trembath RC, Yaouanq J, Viswanathan AC, Zhang H, Wood NW, Zuvich R, Deloukas P, Langford C, Duncanson A, Oksenberg JR, Pericak-Vance MA, Haines JL, Olsson T, Hillert J, Ivinson AJ, De Jager PL, Peltonen L, Stewart GJ, Hafler DA, Hauser SL, McVean G, Donnelly P, Compston A. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–219. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hsieh CS, Lee HM, Lio CW. Selection of regulatory T cells in the thymus. Nature reviews Immunology. 2012;12:157–167. doi: 10.1038/nri3155. [DOI] [PubMed] [Google Scholar]
- 5.Stritesky GL, Jameson SC, Hogquist KA. Selection of self-reactive T cells in the thymus. Annual review of immunology. 2012;30:95–114. doi: 10.1146/annurev-immunol-020711-075035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Casrouge A, Beaudoing E, Dalle S, Pannetier C, Kanellopoulos J, Kourilsky P. Size estimate of the alpha beta TCR repertoire of naive mouse splenocytes. Journal of immunology. 2000;164:5782–5787. doi: 10.4049/jimmunol.164.11.5782. [DOI] [PubMed] [Google Scholar]
- 7.Arstila TP, Casrouge A, Baron V, Even J, Kanellopoulos J, Kourilsky P. A direct estimate of the human alphabeta T cell receptor diversity. Science. 1999;286:958–961. doi: 10.1126/science.286.5441.958. [DOI] [PubMed] [Google Scholar]
- 8.Robins HS, Srivastava SK, Campregher PV, Turtle CJ, Andriesen J, Riddell SR, Carlson CS, Warren EH. Overlap and effective size of the human CD8+ T cell receptor repertoire. Science translational medicine. 2010;2:47ra64. doi: 10.1126/scitranslmed.3001442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Venturi V, Price DA, Douek DC, Davenport MP. The molecular basis for public T-cell responses? Nature reviews Immunology. 2008;8:231–238. doi: 10.1038/nri2260. [DOI] [PubMed] [Google Scholar]
- 10.Li H, Ye C, Ji G, Han J. Determinants of public T cell responses. Cell research. 2012;22:33–42. doi: 10.1038/cr.2012.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annual review of immunology. 2006;24:419–466. doi: 10.1146/annurev.immunol.23.021704.115658. [DOI] [PubMed] [Google Scholar]
- 12.Cibotti R, Cabaniols JP, Pannetier C, Delarbre C, Vergnon I, Kanellopoulos JM, Kourilsky P. Public and private V beta T cell receptor repertoires against hen egg white lysozyme (HEL) in nontransgenic versus HEL transgenic mice. The Journal of experimental medicine. 1994;180:861–872. doi: 10.1084/jem.180.3.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mendel Kerlero de Rosbo N, Ben-Nun A. Delineation of the minimal encephalitogenic epitope within the immunodominant region of myelin oligodendrocyte glycoprotein: diverse V beta gene usage by T cells recognizing the core epitope encephalitogenic for T cell receptor V beta b and T cell receptor V beta a H-2b mice. European journal of immunology. 1996;26:2470–2479. doi: 10.1002/eji.1830261030. [DOI] [PubMed] [Google Scholar]
- 14.Osman GE, Toda M, Kanagawa O, Hood LE. Characterization of the T cell receptor repertoire causing collagen arthritis in mice. The Journal of experimental medicine. 1993;177:387–395. doi: 10.1084/jem.177.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simone E, Daniel D, Schloot N, Gottlieb P, Babu S, Kawasaki E, Wegmann D, Eisenbarth GS. T cell receptor restriction of diabetogenic autoimmune NOD T cells. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:2518–2521. doi: 10.1073/pnas.94.6.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ishizuka J, Stewart-Jones GB, van der Merwe A, Bell JI, McMichael AJ, Jones EY. The structural dynamics and energetics of an immunodominant T cell receptor are programmed by its Vbeta domain. Immunity. 2008;28:171–182. doi: 10.1016/j.immuni.2007.12.018. [DOI] [PubMed] [Google Scholar]
- 17.Venturi V, Kedzierska K, Price DA, Doherty PC, Douek DC, Turner SJ, Davenport MP. Sharing of T cell receptors in antigen-specific responses is driven by convergent recombination. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:18691–18696. doi: 10.1073/pnas.0608907103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Menezes JS, van den Elzen P, Thornes J, Huffman D, Droin NM, Maverakis E, Sercarz EE. A public T cell clonotype within a heterogeneous autoreactive repertoire is dominant in driving EAE. The Journal of clinical investigation. 2007;117:2176–2185. doi: 10.1172/JCI28277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fazilleau N, Delarasse C, Sweenie CH, Anderton SM, Fillatreau S, Lemonnier FA, Pham-Dinh D, Kanellopoulos JM. Persistence of autoreactive myelin oligodendrocyte glycoprotein (MOG)-specific T cell repertoires in MOG-expressing mice. European journal of immunology. 2006;36:533–543. doi: 10.1002/eji.200535021. [DOI] [PubMed] [Google Scholar]
- 20.Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 21.Nguyen P, Liu W, Ma J, Manirarora JN, Liu X, Cheng C, Geiger TL. Discrete TCR repertoires and CDR3 features distinguish effector and Foxp3+ regulatory T lymphocytes in myelin oligodendrocyte glycoprotein-induced experimental allergic encephalomyelitis. Journal of immunology. 2010;185:3895–3904. doi: 10.4049/jimmunol.1001550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alli R, Nguyen P, Geiger TL. Retrogenic modeling of experimental allergic encephalomyelitis associates T cell frequency but not TCR functional affinity with pathogenicity. Journal of immunology. 2008;181:136–145. doi: 10.4049/jimmunol.181.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nguyen P, Ma J, Pei D, Obert C, Cheng C, Geiger TL. Identification of errors introduced during high throughput sequencing of the T cell receptor repertoire. BMC genomics. 2011;12:106. doi: 10.1186/1471-2164-12-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quigley MF, Almeida JR, Price DA, Douek DC. Unbiased molecular analysis of T cell receptor expression using template-switch anchored RT-PCR. In: Coligan John E, et al., editors. Current protocols in immunology. Unit10. Chapter 10. 2011. p. 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mendel I, Kerlero de Rosbo N, Ben-Nun A. A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2b mice: fine specificity and T cell receptor V beta expression of encephalitogenic T cells. European journal of immunology. 1995;25:1951–1959. doi: 10.1002/eji.1830250723. [DOI] [PubMed] [Google Scholar]
- 26.Robins H. Immunosequencing: applications of immune repertoire deep sequencing. Current opinion in immunology. 2013;25:646–652. doi: 10.1016/j.coi.2013.09.017. [DOI] [PubMed] [Google Scholar]
- 27.Freeman JD, Warren RL, Webb JR, Nelson BH, Holt RA. Profiling the T-cell receptor beta-chain repertoire by massively parallel sequencing. Genome research. 2009;19:1817–1824. doi: 10.1101/gr.092924.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sabatino JJ, Jr, Huang J, Zhu C, Evavold BD. High prevalence of low affinity peptide-MHC II tetramer-negative effectors during polyclonal CD4+ T cell responses. J Exp Med. 2011;208:81–90. doi: 10.1084/jem.20101574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jenkins MK, Moon JJ. The role of naive T cell precursor frequency and recruitment in dictating immune response magnitude. Journal of immunology. 2012;188:4135–4140. doi: 10.4049/jimmunol.1102661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huseby ES, White J, Crawford F, Vass T, Becker D, Pinilla C, Marrack P, Kappler JW. How the T cell repertoire becomes peptide and MHC specific. Cell. 2005;122:247–260. doi: 10.1016/j.cell.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 31.Ndifon W, Gal H, Shifrut E, Aharoni R, Yissachar N, Waysbort N, Reich-Zeliger S, Arnon R, Friedman N. Chromatin conformation governs T-cell receptor Jbeta gene segment usage. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:15865–15870. doi: 10.1073/pnas.1203916109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nishana M, Raghavan SC. Role of recombination activating genes in the generation of antigen receptor diversity and beyond. Immunology. 2012;137:271–281. doi: 10.1111/imm.12009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Furmanski AL, Ferreira C, Bartok I, Dimakou S, Rice J, Stevenson FK, Millrain MM, Simpson E, Dyson J. Public T cell receptor beta-chains are not advantaged during positive selection. Journal of immunology. 2008;180:1029–1039. doi: 10.4049/jimmunol.180.2.1029. [DOI] [PubMed] [Google Scholar]
- 34.Madi A, Shifrut E, Reich-Zeliger S, Gal H, Best K, Ndifon W, Chain B, Cohen IR, Friedman N. T-cell receptor repertoires share a restricted set of public and abundant CDR3 sequences that are associated with self-related immunity. Genome research. 2014;24:1603–1612. doi: 10.1101/gr.170753.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Madakamutil LT, Maricic I, Sercarz EE, Kumar V. Immunodominance in the TCR repertoire of a [corrected] TCR peptide-specific CD4+ Treg population that controls experimental autoimmune encephalomyelitis. Journal of immunology. 2008;180:4577–4585. doi: 10.4049/jimmunol.180.7.4577. [DOI] [PubMed] [Google Scholar]
- 36.Iglesias MC, Almeida JR, Fastenackels S, van Bockel DJ, Hashimoto M, Venturi V, Gostick E, Urrutia A, Wooldridge L, Clement M, Gras S, Wilmann PG, Autran B, Moris A, Rossjohn J, Davenport MP, Takiguchi M, Brander C, Douek DC, Kelleher AD, Price DA, Appay V. Escape from highly effective public CD8+ T-cell clonotypes by HIV. Blood. 2011;118:2138–2149. doi: 10.1182/blood-2011-01-328781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Price DA, Asher TE, Wilson NA, Nason MC, Brenchley JM, Metzler IS, Venturi V, Gostick E, Chattopadhyay PK, Roederer M, Davenport MP, Watkins DI, Douek DC. Public clonotype usage identifies protective Gag-specific CD8+ T cell responses in SIV infection. The Journal of experimental medicine. 2009;206:923–936. doi: 10.1084/jem.20081127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marrack P, Scott-Browne JP, Dai S, Gapin L, Kappler JW. Evolutionarily conserved amino acids that control TCR-MHC interaction. Annual review of immunology. 2008;26:171–203. doi: 10.1146/annurev.immunol.26.021607.090421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Udyavar A, Alli R, Nguyen P, Baker L, Geiger TL. Subtle affinity-enhancing mutations in a myelin oligodendrocyte glycoprotein-specific TCR alter specificity and generate new self-reactivity. Journal of immunology. 2009;182:4439–4447. doi: 10.4049/jimmunol.0804377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holler PD, Kranz DM. Quantitative analysis of the contribution of TCR/pepMHC affinity and CD8 to T cell activation. Immunity. 2003;18:255–264. doi: 10.1016/s1074-7613(03)00019-0. [DOI] [PubMed] [Google Scholar]
- 41.Wilson SS, van den Elzen P, Maverakis E, Beech JT, Braciak TA, Kumar V, Sercarz EE. Residual public repertoires to self. Journal of neuroimmunology. 2000;107:233–239. doi: 10.1016/s0165-5728(00)00218-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.