

Abstract
This review presents concepts of scientific integrative medicine and relates them to the physiology of catecholamine systems and to the pathophysiology of catecholamine-related disorders. The applications to catecholamine systems exemplify how scientific integrative medicine links systems biology with integrative physiology. Concepts of scientific integrative medicine include (i) negative feedback regulation, maintaining stability of the body’s monitored variables; (ii) homeostats, which compare information about monitored variables with algorithms for responding; (iii) multiple effectors, enabling compensatory activation of alternative effectors and primitive specificity of stress response patterns; (iv) effector sharing, accounting for interactions among homeostats and phenomena such as hyperglycemia attending gastrointestinal bleeding and hyponatremia attending congestive heart failure; (v) stress, applying a definition as a state rather than as an environmental stimulus or stereotyped response; (vi) distress, using a noncircular definition that does not presume pathology; (vii) allostasis, corresponding to adaptive plasticity of feedback-regulated systems; and (viii) allostatic load, explaining chronic degenerative diseases in terms of effects of cumulative wear and tear. From computer models one can predict mathematically the effects of stress and allostatic load on the transition from wellness to symptomatic disease. The review describes acute and chronic clinical disorders involving catecholamine systems—especially Parkinson disease—and how these concepts relate to pathophysiology, early detection, and treatment and prevention strategies in the post-genome era.
Introduction
I describe here concepts of scientific integrative medicine and their applications to the physiology and pathophysiology of catecholamine systems.
These concepts have evolved over about four decades of patient-oriented clinical research on stress and a variety of catecholamine-related disorders—especially hypertension, chronic orthostatic intolerance, autonomic failure syndromes, and neurodegenerative diseases—and have been presented in several articles and books (123–125, 127, 129). They are based on classic teachings by Claude Bernard and Walter B. Cannon, but they also borrow heavily from modern perspectives such as by George Chrousos (50), Antonio Damasio (54), Bjorn Folkow (102), Philip W. Gold (122), Michael J. Joyner (165), Irwin J. Kopin (183), Richard Kvetnansky (191), Richard S. Lazarus (196), Bruce McEwen (220), and Denis Noble (238).
What is offered here is a framework and vocabulary to link systems biology with integrative clinical physiology and pathophysiology. The overall purpose is to make use of ever-expanding knowledge about catecholamines to increase understanding about how we humans meet the complex, dynamic, continual challenges to organismic integrity that we face throughout our lives and about what goes wrong in diseases and disorders. The main relevant ideas are negative feedback regulation, homeostats, multiple effectors, effector sharing, stress, distress, allostasis, and allostatic load.
Included are several kinetic models generated using the computer application, Stella (20), that predict levels of monitored variables as functions of effector activities over time. A section presents studies about responses of catecholamine systems in stress and distress, with the goal of illustrating how catecholaminergic responses can be understood in terms of the above concepts. In the third section of the review, scientific integrative medical concepts are used to help comprehend complex abnormalities in the functioning of catecholamine systems that contribute to acute disorders such as fainting and to chronic disorders such as Parkinson disease (PD). Abbreviations are in Table 1.
Table 1.
Abbreviations
| ACTH | Corticotropin |
| ADH | Antidiuretic hormone |
| ALDH | Aldehyde dehydrogenase |
| AR | Aldehyde aldose reductase |
| AVP | Arginine vasopressin |
| CHF | Congestive heart failure |
| CR | Conditioned response |
| CRH | Corticotropin-releasing hormone |
| CS | Conditioned stimulus |
| CSF | Cerebrospinal fluid |
| DA | Dopamine |
| DOPAC | Dihydroxyphenylacetic acid |
| DOPAL | Dihydroxyphenylacetaldehyde |
| DOPEGAL | Dihydroxyphenylglycolaldehyde |
| EPI | Epinephrine |
| 4-HNE | 4-hydroxynonenal |
| HPA | Hypothalamic-pituitary-adrenocortical |
| MAO | Monoamine oxidase |
| MSA | Multiple system atrophy |
| NE | Norepinephrine |
| OH | Orthostatic hypotension |
| PAF | Pure autonomic failure |
| PD | Parkinson disease |
| RAS | Renin-angiotensin-aldosterone system |
| PTSD | Posttraumatic stress disorder |
| SAS | Sympathetic adrenergic system |
| SCS | Sympathetic cholinergic system |
| SNS | Sympathetic noradrenergic system |
| UCR | Unconditioned response |
| UCS | Unconditioned stimulus |
| VMAT | Vesicular monoamine transporter |
Relationship between Scientific Integrative Medicine and Systems Biology
One might think at first that scientific integrative medicine is merely a specialized, applied form of systems biology. Actually, the term, “systems biology,” for which there are now about 5,000 PubMed listings yearly, was rarely used in medical scientific reports before the beginning of the 21st century, whereas the conceptual underpinnings of scientific integrative medicine originated with Claude Bernard in the mid-19th century and Walter B. Cannon in the early 20th century.
Systems biology has been defined variously. One definition is the study of dynamic interactions within biological networks. These interactions can give rise to “emergent” properties unpredicted by any of the components assessed in isolation, and in this sense systems biology can be viewed as “holistic” or “integrative.” Denis Noble has emphasized that in contrast with reductionist science, systems biology “is about putting together rather than taking apart, integration rather than reduction” (238). In the opinion of Michael J. Joyner, systems biology is a concept generated by reductionists who failed to build on theories that founded the field of integrative physiology (165, 167):
“We argue that a fundamentally narrow and reductionist perspective about the contribution of genes and genetic variants to disease is a key reason “omics” has failed to deliver the anticipated breakthroughs. We …. point out the critical utility of key concepts from physiology like homeostasis, regulated systems and redundancy as major intellectual tools to understand how whole animals adapt to the real world. We argue that a lack of fluency in these concepts is a major stumbling block for what has been narrowly defined as “systems biology” by some of its leading advocates.”
Scientific integrative medicine has the potential to link systems biology with integrative physiology and pathophysiology because of four distinguishing aspects.
First is the emphasis on regulation by negative feedback. This notion follows directly from Bernard’s milieu intérieur and Cannon’s homeostasis. Diseases and disorders can be understood in terms of loss of regulation of internal monitored variables because of disruption or declining efficiency at stations in negative feedback loops. Mathematical models incorporating afferent information, homeostats, effectors, etc., can be used to predict the roles of factors such as stress, adaptation, allostatic load, and resilience on the development and manifestations of acute and chronic disorders.
Second, scientific integrative medicine recognizes that in higher organisms the brain dominates in regulation of the body’s inner world. The brain controls levels of many internal monitored variables in parallel—analogous to a computer’s multitasking—each via a homeostatic system. Corollarily, pathophysiologic mechanisms of a variety of complex, mind-body, multisystem disorders involve—and may result from—altered central control.
Third, the brain’s plasticity enables modifications in the step-by-step instructions for organ and systemic processes. According to the concept of allostasis, set-points and other elements of response algorithms vary depending on instinct, imprinting, learning, perceptions, and even simulations of future events by the brain.
Fourth, scientific integrative medicine is medical. Its overall mission is to understand, rationally treat, retard the progression of, or even prevent disorders and diseases. The systems that maintain the stability of the inner world eventually degenerate, and as their efficiencies decline, the likelihood of deleterious, self-reinforcing positive feedback loops increases, threatening organismic stability and survival. Clinicians rarely cure patients. Rather, they manage patients, by exploiting negative feedback loops and attempting to forestall or counter positive feedback loops. Moreover, the medications and treatments clinicians prescribe interact with their patients’ internal systems. Multiple, simultaneous degenerations, combined with multiple effects of drugs and remedies and myriad interactions among the degenerations and the treatments constitute the bulk of modern medical practice. Scientific integrative medicine offers a schema and vocabulary for approaching the imposing complexity of managing patients.
“Integrative medicine” has also gained cachet recently. The word, “integrative,” has been used synonymously with holistic, complementary, or alternative. The scientific integrative medicine approach, however, actually fits quite well with conventional clinical science and integrative physiology. The emphasis is not on rationalizing or testing the efficacy of holistic or alternative treatment programs but on viewing the body as a coordinated system of systems.
An Example of Scientific Integrative Medical Thinking
To get a flavor for scientific integrative medical thinking, a specific example may help here. Suppose a person had a bicuspid aortic valve—the most common congenital valvular lesion in humans. The abnormal anatomy would cause turbulent blood flow across the valve. This might produce a “functional” heart murmur, but the individual could develop normally. Over the years of turbulent blood flow with each heartbeat, wear and tear on the valve would cause it to calcify and become stenotic, decreasing aortic filling. Via a negative feedback loop involving release of the sympathetic noradrenergic system (SNS) from baroreceptor restraint, the brain would direct a compensatory increase in cardiac sympathetic outflow. Increased delivery of norepinephrine (NE), the main sympathetic neurotransmitter in cardiovascular regulation, to myocardial cells would then help maintain cardiac function. Such adjustments in SNS outflows and NE delivery in the heart (95) and in the body as a whole (275) are typical of “normal” aging.
In the long run, however, these compensatory, adaptive responses could come at a cost. NE promotes myocardial hypertrophy (249), which increases the demand for oxygen and metabolic fuels delivered by coronary perfusion; it increases cardiac contractility (155, 158, 178), which in this case would maintain aortic filling at the expense of increased blood flow turbulence and wear and tear on the valve, accelerating the stenosis; and it reduces thresholds for arrhythmias (209, 223). We begin to see the potential for induction of deleterious positive feedback loops.
Especially in the setting of concurrent coronary artery disease, the increased demand for oxygen by the stimulated, hypertrophied heart could at times of stress exceed the supply—a kind of energy crisis, manifested clinically by easy fatigue and dyspnea on exertion among other symptoms. In sympathetic nerves, NE stored in vesicles leaks spontaneously continuously into the cytosol, and reuptake of NE back into the vesicles requires energy. One consequence of decreased energy availability would be decreased releasable stores of NE in sympathetic nerves. This would limit NE release during stress and escalate further the increases in SNS outflows. Inefficient sequestration of catecholamines that leak passively from the vesicles into the cytosol would result also in buildup of catecholamines in the cytosol, where they are “autotoxic” because of spontaneous oxidation to quinones (148) and chromes (287) and because of enzymatic oxidation to aldehydes (31). Destruction of sympathetic nerves due to autotoxicity would diminish further the stores of releasable NE. Reuptake of released NE back into the terminals would be attenuated concurrently, because neuronal reuptake is also an energy-requiring process. The patient would now have congestive heart failure, a state known to be characterized by myocardial NE depletion (48), increased NE release (82), and decreased neuronal reuptake of released NE (82).
Once cardiac pump function declined to below a certain level despite maximal SNS stimulation, blood would back up into the pulmonary veins, bringing on pulmonary edema. The patient would then become short of breath even at rest and, in a distress response, experience the classic “feeling of impending doom,” which has been associated from time immemorial with massive adrenomedullary release of adrenaline (epinephrine, EPI). Moreover, rather than augmenting left ventricular myocardial contractility, too much EPI is toxic to myocardial cells (188, 267). Myocardial contractility would decrease further, “stress cardiopathy” would develop, and the pulmonary edema would worsen. In several ways, physiologic negative feedback loops would have given way to pathophysiologic positive feedback loops. Within a sometimes surprisingly short period of time from the onset of symptoms, the patient could die—within minutes because of a catecholamine-evoked ventricular arrhythmia, hours because of intractable pulmonary edema, or days because of critically decreased perfusion of body organs such as the kidneys.
A goal of scientific integrative medicine is to detect early or even prevent such catastrophic outcomes mediated by positive feedback loops. Theoretically, one way to do so would be by tracking plasma NE levels, since it is well established that plasma NE levels are increased in heart failure (301), and high NE levels predict a poor outcome (159).
Scientific Integrative Medicine: Historical Context
A paradox of life
Within our bodies is an inner world characterized by apparent stability despite continuous change. We are born, we develop and mature, we reproduce, we live out our lives, we get old, we get sick, and we die, yet for most of our existence, we believe in our essential sameness day to day. Blood pressure, body temperature, blood glucose and oxygen levels, electrolyte concentrations, blood flows to vital organs, and many more variables normally do not vary by much or for very long. Even mood and personality remain about the same, typifying us to others and to ourselves. When levels of these variables do change and you feel sick, you do not feel “like yourself.”
In higher organisms, maintaining these steady states depends on complex coordination by the brain. Scientific integrative medicine is a way of thinking about how the brain regulates the body’s inner world to maintain organismic integrity so well for so long and about what goes wrong with that regulation in diseases.
In a single phrase, the brain controls the inner world via feedback-regulated systems. Just as the brain receives information from sense organs about and determines interactions with the outside world, the brain also receives information from internal sensors and acts on that information to maintain levels of monitored variables, via numerous effectors.
These effectors usually function unconsciously, involuntarily, and automatically. The body has three endogenous catecholamines—dopamine (DA), NE, and EPI. Each plays key roles in regulation of the inner world of the body. This review dwells on the effectors that use them.
Bernard’s milieu intérieur and Cannon’s homeostasis
The great French physiologist and experimentalist, Claude Bernard, propounded the founding concept of scientific integrative medicine when he theorized that body systems function as they do to maintain a constant internal environment—what he called the milieu intérieur. He taught that a fluid environment of nearly constant composition bathes and nourishes the cells. Near the end of his life, he postulated something even more profound—that the body maintains the constant internal environment by myriad, compensatory reactions. By restoring a state of equilibrium in response to outside changes, the compensatory reactions enable independence from the external environment.
Bernard therefore not only introduced the notion of an apparently constant inner world but also proposed a purpose for body processes. His Lectures on the Phenomena of Life Common to Animals and Vegetables (Vol. 1, translated by Hoff HE, Guillemin R, Guillemin L, Springfield, IL: Charles C Thomas Publisher, 1974) contains one of the most famous passages in the history of physiology: “The constancy of the internal environment is the condition for free and independent life” (p. 84). “All the vital mechanisms, however, varied they might be, always have one purpose, that of maintaining the integrity of the conditions of life within the internal environment” (p. 89). This view might seem straightforward or even simple-minded today, but it was revolutionary in the history of medical ideas.
Beginning about the turn of the 20th century, the highly influential American physiologist, Walter B. Cannon, expanded on Bernard’s notion of the milieu intérieur. Bernard’s theory addressed the “why” of bodily processes by postulating that they help maintain a constant internal environment. Based on a series of magnificent experiments over more than a quarter century (some of which this review highlights), Cannon’s work and ideas began to flesh out the “how.” Among other things, Cannon demonstrated for the first time many of the roles EPI plays in maintaining the constancy of the inner world.
Cannon introduced and popularized three ideas that by now are well known and widely accepted—homeostasis, fight-or-flight responses, and the functionally unitary sympathico-adrenal system. As well established as they are, each has required modification to take into account experimental realities. For the purposes of this introduction, Cannon invented the word, “homeostasis,” (36) by which he referred to the stability of the inner world. According to Cannon, the brain coordinates body systems with the aim of maintaining a set of goal values for internal variables. The core temperature is kept at 98.6°F (37°C), the serum sodium level at 140 mEq/L, the blood glucose level at 90 mg/dL, and so forth. Internal or external disturbances threatening homeostasis, by causing deviations from the goal values, arouse internal nervous and hormonal systems, induce emotional and motivational states, and generate externally observable behaviors, all of which help meet the goal of reestablishing homeostasis.
Concepts Of Scientific Integrative Medicine
One way that scientific integrative medicine expands on the ideas of Bernard and Cannon is by demonstrating via kinetic models that negative feedback regulation explains the maintenance of monitored variables of the body at steady-state levels. Each negative feedback loop regulating a monitored variable contains a comparator, a “homeostat,” which compares afferent information to the brain with settings for responding (Fig. 1). A discrepancy between what is sensed and what is set—the error signal—drives effectors in a manner that alters levels of the monitored variable and reduces the discrepancy. This section provides several examples of models using the commercially available computer application, Stella, to predict effects of stress, wear and tear, and aging on regulation of monitored variables.
Figure 1.
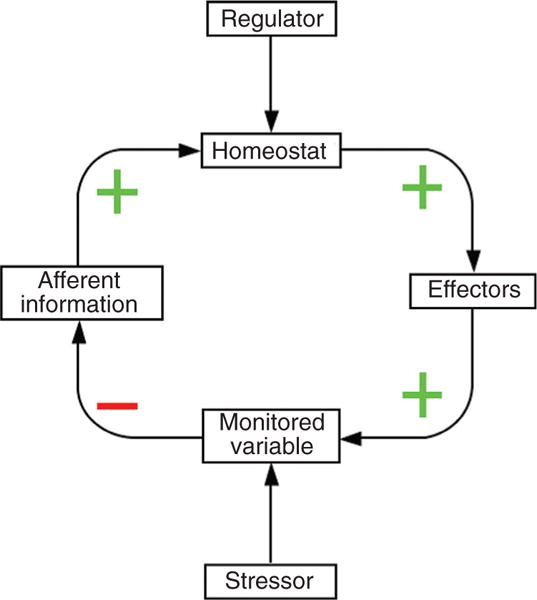
A homeostatic system. The monitored variable is regulated by negative feedback. Afferent information about the monitored variable reaches a comparator homeostat, which drives an effector that influences the monitored variable. (+) sign indicates a positive relationship and (−) a negative relationship.
Negative feedback—proportionate, integrated, derivative, and feed-forward control
Physiological homeostatic systems all entail negative feedback regulation of monitored variables such as blood pressure, core temperature, blood oxygen, and serum glucose and osmolality. Conceptually, each of these systems depends on a homeostat to compare afferent information about the monitored variable with settings for responding (Fig. 1).
By analogy to the system regulating the temperature inside your house, the thermostat has a temperature setting and receives information about ambient temperature. When there is a discrepancy between what is sensed and what is set (the error signal), the thermostat directs changes in activities of effectors (e.g., the furnace and heat pump), and the altered effector activities reduce the discrepancy by bringing the level of the monitored variable toward the thermostatic setting.
In the kinetic model in Figure 2, the rate of decline in the level of the monitored variable is a first-order process. The rate of decline depends on the level of the monitored variable at the time (i.e., Loss Rate = kLoss * Monitored Variable), and so the level of the monitored variable decreases exponentially.
Figure 2.
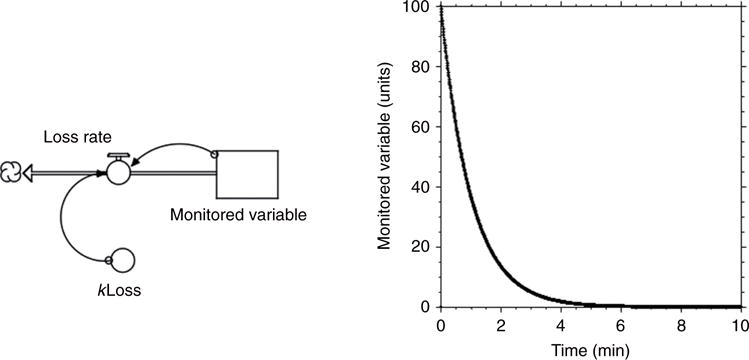
Monitored variable level in the absence of feedback regulation. In the computer model, the initial level of the “stock,” the monitored variable, is 100 units. The loss rate, indicated by the “pipe and valve,” depends on a rate constant, kLoss (in this case 1 per min), and on the level of the monitored variable (arrows). The level declines as a first order process, meaning the level falls exponentially.
In a negative feedback loop, there is an odd number of negative relationships (denoted by a “−” sign) in the loop (Fig. 1). Figure 3 shows a Stella model of a simple negative feedback loop. The level of the monitored variable is compared with a set point; when there is a sensed discrepancy between the two, the error signal drives an effector, and activation of the effector tends to restore the level of the monitored variable. In the analogy of the home heating system, when the thermostat detects a discrepancy between the sensed and set temperatures, the furnace turns on, and the temperature reaches a plateau level. The rate of attainment of the steady-state level depends on the power of the furnace (here denoted as kGain). The more powerful the furnace, the faster the temperature changes. The time to attain a steady-state level varies inversely with the rate constant (k) for the gain of heat. Notice in Figure 3 that when kGain is relatively high, the attained steady-state level is higher and the time to reach the steady-state level shorter than when kGain is relatively low.
Figure 3.
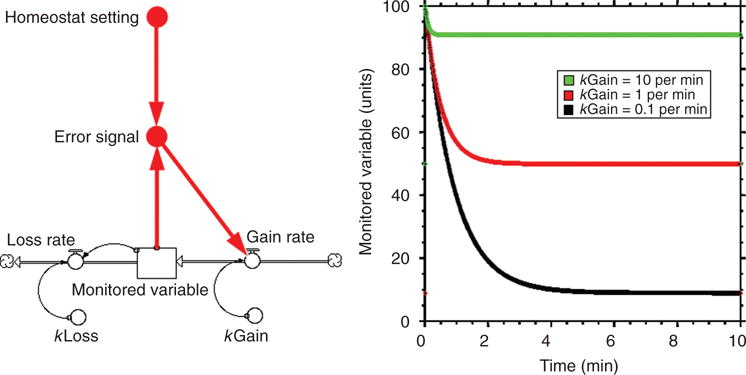
Negative feedback, with proportionate control. The difference between the level of the monitored variable and the homeostat setting, the error signal, determines the rate of increase (Gain Rate) of the monitored variable. Note that with negative feedback, the level of the monitored variable reaches a steady state. As the value for kGain increases, the plateau level of the monitored variable increases; however, with proportionate control the plateau level is below the homeostat setting.
This type of negative feedback loop uses “proportionate control.” That is, the response of the furnace is proportionate to the magnitude of the error signal. With proportionate control, the level of the monitored variable reaches a steady state, but the steady-state level never quite attains the homeostatic setting (unless the effector has infinite gain).
Negative feedback by proportionate control alone often does not simulate well what actually happens physiologically. More commonly, in response to a rapid but persistent perturbation, the level of the monitored variable decreases transiently but then returns to about the baseline level. For instance, when a person is tilted head-up from a supine position, the blood pressure can fall briefly, but then the blood pressure comes back up and thereafter stays at about the baseline level (Fig. 4).
Figure 4.
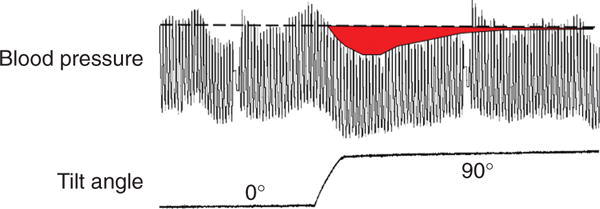
Effect of head-up tilting on beat-to-beat blood pressure in a healthy person. The blood pressure falls transiently but then returns to about the baseline level.
A way to simulate this rapid return to baseline is by adding “integrated control” (Fig. 5). With integrated control added to proportionate control, not only does the magnitude of the error signal itself drive the effector, so does the integral of the error signal. That is, the homeostat responds not only to the error signal but also to how the error signal has accumulated over time. If the furnace were very efficient, then the error signal would be reduced quickly, and the integral of the error signal over time would be relatively small; the furnace would not be on long. If the furnace were inefficient, then reducing the error signal to zero would take a longer time, and the integral of the error signal would be relatively large; the furnace would be on longer. Eventually, across a wide range of furnace powers, it can be shown mathematically that the level of the monitored variable will reach and stay at the set value.
Figure 5.

Computer model of a negative feedback loop with both proportionate and integrated control. The rate of increase in the monitored variable (Gain rate) is determined both by the error signal and the integrated error signal.
Modern control systems can include not only proportionate and integrated control but also control by a “derivative” factor, the triad constituting a “PID controller.” The derivative factor is the slope of the error signal. Inclusion of derivative control has the qualitative effect of damping oscillations introduced by integrated control, but at the expense of increased susceptibility to extraneous artifactual influences.
An even more sophisticated control system—characterizing actual human physiology—is proactive. The individual’s experiences and perceptions lead to predictions (simulations) about future conditions, and the system proactively adjusts activities of the effectors. This sort of proactive control is “feed forward.” In physiological terms, the brain, based on instinct, imprinting, classical conditioning, operant conditioning, and “fast and slow thinking” (170), directs changes in effector activities even in advance of the anticipated stress.
An example of this phenomenon is the well-known “central command” at the initiation of exercise (312). Another example of feed-forward control comes from the era of cardiovascular biofeedback research in the 1960s and 1970s. Using a shaping procedure, baboons were trained by operant conditioning to raise their diastolic blood pressure to a predetermined level and keep the pressure at that level in daily 12-h trials beginning at 12 noon (153). In trained animals, the gain of the cardiovagal component of the arterial baroreflex was decreased throughout the experimental trial but returned to normal between trials (133). Highly trained animals were found to have anticipatory decreases in baroreflex-cardiovagal gain at 11:45 AM before the trial began and anticipatory increases in gain at 11:45 PM before the trial ended—a remarkable example of an acquired circadian feed-forward mechanism influencing a physiological negative feedback loop.
Stress
Beginning in the 1930s, Hans Selye popularized the concept of stress as a medical idea (278). He defined stress as (or a state resulting in) “the nonspecific response of the body to any demand upon it” (279). His arguments were so persuasive that the notion of a unitary stress response persisted and remains widely used today. By “nonspecific” Selye meant a set of shared elements of responses, regardless of the nature of the causative agent, or stressor.
Selye proposed three stages of coping with a stressor—the “General Adaptation Syndrome”—consisting of an initial “alarm reaction” (corresponding to Cannon’s “fight or flight” response), a stage of adaptation associated with resistance to the stressor, and eventually a stage of exhaustion and organismic death. In Selye’s early experiments, after injection of any of a variety of tissue extracts or of formalin into rats, the animals developed a pathological triad of enlargement of the adrenal glands, atrophy of lymphoid tissue in the thymus, spleen, and lymph nodes, and bleeding gastrointestinal ulcers. It was later demonstrated that these changes are associated with, and to at least some extent result from, activation of the hypothalamic-pituitary-adrenocortical (HPA) axis. Steroids released into the circulation from the adrenal cortex contribute to resistance but may also be responsible for pathological changes. Selye’s concept that prolonged stress can produce physical disease and mental disorders is now widely accepted, and longitudinal studies have yielded results consistent with it (117, 207, 218). The area is notoriously difficult, however, and perennially contentious, at least partly because of the possibility of self-selection biases in a free society (124).
According to a homeostatic definition, stress is a condition in which expectations, whether genetically programmed, established by prior learning, or deduced from circumstances, do not match current or anticipated perceptions of the internal or external environment, and the discrepancy elicits patterned, compensatory responses (127). Stress reflects the difference between afferent information about conditions as sensed and the homeostatic set point for responding (140). One can readily conceptualize stress in terms of the error signal in a homeostatic negative feedback loop (Fig. 7 and 8), with the integrated error signal a measure of accumulated stress over time.
Figure 7.

Homeostatic definition of stress. Stress is defined as a condition or state in which there is a sensed discrepancy between afferent information and the homeostatic setting. The sensed discrepancy corresponds to the “error signal” in the computer model of a negative feedback loop.
Figure 8.

Introduction of a stressor into the computer model. The stressor augments the loss rate. The computer model predicts return to the set level of the monitored variable, with the time to return depending on the severity of the stressor. The integrated error signal is a measure of the accumulated stress.
Allostasis and allostatic load
Cannon’s idea of homeostasis implies that for each monitored variable there is an optimal setting or goal level; however, even in normal physiology values of acceptable levels are decidedly inconstant. Among other things, there are diurnal variations in body temperature, heart rate, blood pressure, etc., and appropriate responses to stressors such as exercise require temporary alterations in what is defined as acceptable.
Selye invented the term “heterostasis” (from the Greek heteros = other) to describe the establishment of a new steady state by changing the “set-point” to resist unusually high demands (278). This new steady state would be attained by treatment with remedies that have no direct curative action but enhance the body’s natural defenses (e.g., vitamins or antioxidant dietary supplements). The concept of changes in the homeostatic set point as a natural adaptive mechanism awaited the introduction of the notion of allostasis.
Sterling and Eyer introduced “allostasis” to describe the attainment of stability by alterations in acceptable ranges of variables attending adjustments during rest and activity (294). Steady-state levels of a monitored variable can be modified by changing the set point or other instructions for responding. Allostasis refers to this “other sameness.” Again from the analogy of the thermostat, the “sameness” is the attained steady-state temperature; the “other” is the change in the thermostatic setting. Chances are your thermostat is set lower in the winter and higher in the summer. The attained internal temperature is held at the setting, but the setting varies depending on the season.
The fever attending a viral infection probably exemplifies an allostatic state (Fig. 9). The thermostat is reset, and the level of the monitored variable reaches a new steady-state value. The rate of effector activity increases suddenly because of the large error signal but then declines to a new steady-state rate that is increased from the level before the infection; and so the core temperature rises to a new plateau level. Core temperature is now regulated around that level.
Figure 9.

Fever as an allostatic state. Changing the set-point of the homeostat (in this case at 12 h) increases the steady-state value for the monitored variable, the core temperature.
A price of allostasis is wear and tear—allostatic load (Fig. 10). With repeated, prolonged use of the furnace, wear, and tear builds up on components of the furnace. If there were design flaws or manufacturing defects in those components, or if there were a long series of unusually cold winters, or if the thermostat were set at a high temperature throughout the winter, the rate of accumulation of allostatic load would be increased.
Figure 10.

Stress, allostasis, and allostatic load in the computer model of negative feedback regulation of temperature by a thermostat. Allostasis refers to regulation of the level of the monitored variable at different steady-state values by adjusting the thermostat setting. Allostatic load refers to accumulated wear and tear on the furnace.
If allostatic load decreased efficiency of the effector, then eventually a positive feedback loop would cause breakdown of the effector and failure of the homeostatic system (Fig. 11). Suppose you went on sabbatical for a year and you forgot to close a large window in your house before you left. The air conditioner would be on more in the summer and the furnace more in the winter. With these effectors being on more of the time, there would be more wear and tear on them, and they would eventually become less efficient. For the thermostatic system to maintain the internal temperature via negative feedback regulation, the effectors would have to be on more of the time, but this would accelerate the wear and tear on them, which would decrease their efficiencies further, and so forth—positive feedback loops. When you returned, you might even find that all the HVAC components had ceased functioning, and there was no longer any control of the house temperature. If you set your thermostat relatively high in the winter and relatively low in the summer, you would also produce more wear and tear on the furnace and the air conditioner. According to this view of allostatic load, chronic stress may contribute to the development of degenerative diseases by way of prolonged activation of effectors to maintain allostasis and declining effector efficiency as allostatic load accumulates. (An analogous argument applies for efficiencies at all stations of negative feedback loops.) This can be modeled mathematically using Stella (Fig. 12).
Figure 11.
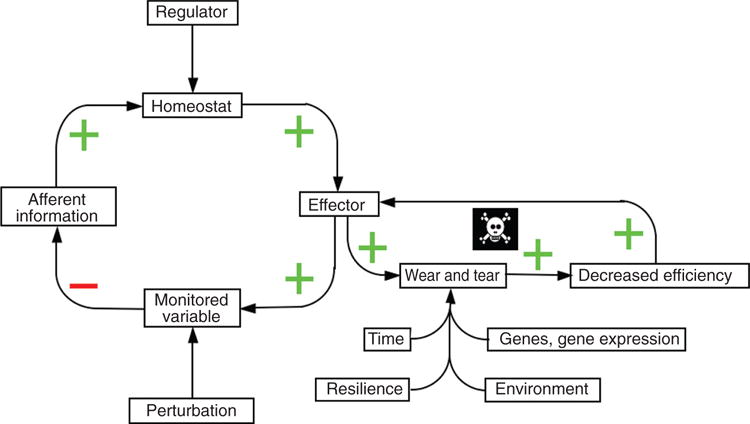
Inherited and acquired determinants of allostatic load. These determinants include genes and gene expression, environmental influences, resilience, and time. Note that decreased effector efficiency from allostatic load can induce a positive feedback loop, with all the relationships within the loop having a “+” sign.
Figure 12.

Predicted effects of allostatic load on wellness. Because of wear and tear on the effector, the effector becomes less efficient, and because it is less efficient it has to be “on” more in order to maintain the level of the monitored variable; however, the more it is “on,” the more wear and tear (allostatic load). This positive feedback loop results in accelerated decline in wellness, early onset of symptomatic system failure (arbitrarily placed at 40% of ideal), and premature death.
According to Selye, stress is not necessarily deleterious. He coined the term, “eustress,” to refer to stress that is not harmful and possibly is helpful to the body, whereas “distress” was defined in terms of damaging or unpleasant stress (279). Excessive, repeated, or inappropriate stress responses were viewed as maladaptive, and Selye coined the phrase, “diseases of adaptation,” to refer to situations in which the General Adaptation Syndrome is “derailed” (278). The contributions of stress to diseases of adaptation were suggested mainly from effects of large doses of glucocorticoids or mineralocorticoids. If abnormal (hyper-, hypo-, or dys-adaptive) responses did not directly cause these disorders, then they were thought to predispose the individual to develop them, based on tendencies he called “conditioning factors.”
Selye proposed an immense list of diseases of adaptation. Hyperfunctional and dysfunctional conditions included Cushing’s disease, adrenal tumors, chromaffinomas, renal artery stenosis, hypertension, periarteritis nodosa, nephrosclerosis, nephritis, rheumatic and inflammatory diseases, gouty arthritis, peptic ulceration, eclampsia, diabetes, allergic and hypersensitivity disorders, and psychosomatic disorders. Hypofunctional conditions included Addison’s disease, Waterhouse-Fredrichsen syndrome, cancer, and diseases of resistance in general (277). The most severely affected targets were thought to be the cardiovascular system, the joints, and metabolism.
A key deficiency in Selye’s stress theory is circularities (124), and one circularity is in the definition of distress. The theory defines distress as stress that is unpleasant or harmful to the body—but these are not the same things. If the latter criterion were used, then the only means to determine whether a particular stress were a distress or eustress would be the occurrence of observable tissue damage or shortened survival, and the explanation for observable tissue damage or shortened survival in the above disorders would be distress. Noncircular definitions are required to enable experimental testing about the health consequences of distress or eustress.
Distress
A noncircular definition of distress is that it is a form of stress with additional characteristics—consciousness, aversiveness, observable signs, and adrenal gland activation (139). Each of these aspects receives attention below.
Consciousness
The occurrence of stress does not require consciousness. Selye would have agreed with this assertion, because he claimed that stress reactions can occur in anesthetized animals, in lower animals without nervous systems or undergoing mechanical damage to denervated limbs, and even in cells cultured outside the body. In contrast, distress does require consciousness [or at least “core consciousness” as conceptualized by Antonio Damasio (55)], because distress involves not only a challenge to homeostasis but also a perception by the organism that homeostatic mechanisms may not suffice—that is, interpretation of afferent information and simulation of future events. This is a direct extension of the concept of psychological stress as a consequence of a perceived inability to cope. The sense of an inability to cope or of a lack of controllability is basic to psychological theories about feelings associated with distress (47, 324). An organism experiences distress when it perceives the inadequacy of compensatory adjustments to either a psychological or physiological stressor.
In keeping with this view, glucoprivation by 2-deoxyglucose administration produces smaller plasma EPI responses when people are sedated with alprazolam than when they are alert (24). The sympathetic adrenergic system (SAS) is a shared effector for the glucostat and “psychostat.” The total EPI response includes a homeostatic stress response to glucoprivation, which does not require consciousness, and distress, which does require consciousness. Sedation attenuates the portion of the EPI response resulting from distress.
Aversiveness
Distressed organisms avoid situations that are perceived as likely to reproduce the same aversive experience. Distress therefore is negatively reinforcing and motivates escape and avoidance learning. The experience of distress would be expected to enhance vigilance behavior and long-term memory of the distressing event. All these are adaptive adjustments that must have offered tremendous survival advantages in evolution. They also may involve catecholamines in the brain (120, 221, 292). In considering potential long-term health consequences of distress, one must bear in mind its important survival advantages. This relates to the notion of pleiotropy, discussed later.
Most animals can react instinctively not only to a stressor but also to symbolic substitutes that resemble the natural stimulus. Monkeys become agitated upon exposure to a snake, without ever having seen one before; rabbits freeze when a hawk-shaped shadow glides by; and male stickleback fish attack any red object in their territory (206).
The plasticity afforded by learning decreases the likelihood of inappropriate instinctive responses to symbolic cues. One definition of learning is modification of behavior based on experience. According to this definition, learning requires memory. Even primitive animals have the capacity to learn to withdraw or escape from noxious stimuli or to habituate after prolonged or repeated exposure to a stimulus (175). These forms of learning mirror each other, the former a sensitization and the latter a desensitization. The fact that primitive animals have these capabilities indicates the ancient and durable survival advantages of learning.
Classical (or Pavlovian) conditioning represents a refinement of these responses, in that habituation and sensitization are forms of nonassociative learning, where the organism learns about single stimuli, whereas classical conditioning (and operant conditioning, to be discussed shortly) involves learned associations between stimuli. In classical conditioning, repeated pairing of a neutral stimulus (e.g., a bell ringing) with an unconditioned stimulus (UCS) that elicits an instinctive unconditioned response (UCR) results eventually in the elicitation of the UCR (or components of it) by the previously neutral conditioned stimulus (CS). The CS elicits a conditioned response (CR). Although most classical conditioning experiments involve an external UCS, such as an electric shock to the skin, this does not imply that the UCS must be external. For instance, rats can acquire hyperglycemia as a CR after repeated pairing of a previously neutral cue with injections of insulin (285). Pavlov himself demonstrated classically conditioned nausea and vomiting after repeated pairing of a CS (approach of the experimenter) with an internal UCS (evoked by injected morphine).
Instrumental, or operant, conditioning represents a more advanced form of learning that requires a cerebral cortex. In instrumental conditioning, the likelihood of a behavior increases when the behavior leads to positive reinforcement (reward) and decreases when the behavior leads to negative reinforcement (punishment). Conversely—but circularly—reinforcement can be defined as an event that strengthens the response it follows. The conditioning is “operant” in that the individual’s behavior operates on the environment, determining the occurrence of reinforcement; and the conditioning is “instrumental” in that the learning is a means to an end, with the occurrence of reinforcement contingent on the behavior. Operant conditioning therefore differs from Pavlovian conditioning, in which the delivery of the reinforcement occurs independently of the individual’s behavior. Both forms of conditioning require remembering an association between reinforcement and behavior. In Pavlovian conditioning, behavior (the UCR and CR) depends on the reinforcement (the UCS), whereas in operant conditioning, reinforcement depends on the behavior.
In avoidance learning, a form of operant conditioning, the individual learns to avoid negative reinforcement by producing behaviors that decrease the likelihood of that reinforcement. If an organism experienced distress consistently in a given situation, subsequent perception of reexposure to the situation would elicit distress as a classically conditioned response. Situations evoking distress typically involve a complex interplay of classically conditioned and operantly conditioned behaviors, coupled with skeletal muscle and autonomic responses.
Instinctively communicated signs
A third characteristic of distress is evocation of signs that others can interpret as indicating the emotional state and intent of the organism. Darwin emphasized that the outward manifestations of emotion provide important means of communication that have had survival value (58). Darwin also proposed that physiological arousal intensifies emotions, amplifying the physiological stress responses that accompany those emotions—psychophysiological positive feedback loops. Perhaps this can explain flight degenerating to self-destructive panic, anger to frenzy, and fright to collapse.
Perceptions of signs of distress by other members of the species elicit involuntary, instinctive responses. Even in humans, the fiercest combat usually ends abruptly when one side shows a universally understood sign of surrender and submission. One such sign is waving a white flag—perhaps because of an instinctive association of pallor with defeat. In English, “wan,” “pallid,” and “pale” refer not only to skin turning white but also to weakness or feebleness. In contrast, waving a red flag is taken as an incitement and as an indicator of danger. We turn white with fright but red with rage. The communication value of external signs of distress helps to explain the continued elaboration of observable components of distress responses in modern society, despite the relative rarity of true fight-or-flight reactions in humans. During the course of human evolution, these signs originally may have been byproducts of genetically determined neurocirculatory adjustments supporting fleeing and fighting. In modern society, they continue to serve important signal functions.
Adrenal activation
A fourth characteristic of distress is adrenal gland activation. This involves enhanced release of catecholamines from the adrenal medulla and of glucocorticoids from the adrenal cortex.
Plasma levels of EPI constitute an extraordinarily rapid and sensitive chemical index of this activation and therefore of experienced distress. The EPI response is so rapid that when an animal is killed by decapitation, arterial EPI levels are increased by about 80-fold (193), while glucocorticoid levels are unchanged.
Cannon viewed the neural and hormonal components of the “sympathico-adrenal” system as functioning as a unit to preserve homeostasis in emergencies. According to the present conception, it is specifically the adrenomedullary hormonal component, the SAS, that characterizes distress. SNS outflows can increase, decrease, or stay the same, depending partly on whether there is a locomotor response (e.g., escape behavior), which entails increased skeletal muscle sympathetic outflows.
A fundamental aspect of scientific integrative medicine is the primacy of the brain in regulation of the body’s inner world. From this it seems reasonable to propose that just as the brain evokes relatively specific patterned responses to different stressors, in distress the brain directs evocation of relatively specific neuroendocrine, experiential, and behavioral allostatic changes. Just as there are relatively specific responses to orthostasis, altered environmental temperature, glucoprivation, salt deprivation, and so forth, there are also relatively specific distress responses, so that “fight” is not the same as “flight,” “fright,” “fume,” “fret,” or “defeat.”
“Eustress” revisited: Adaptation and resilience
The analogy to a home HVAC system is obviously limited in that organisms have capabilities to habituate, anticipate, heal, regenerate, and in general increase resilience. These processes may operate at multiple sites within homeostatic loops to increase the useful life of the effectors for the same amount of chronic exposure to a stressor. For instance, gating processes that decrease afferent nerve traffic and allostatic modifications of response algorithms reduce error signals; habituation attenuates effector activation; and learning and training exert proactive feed-forward effects.
Defining distress and eustress solely in terms of pathologic outcomes is circular and therefore unproductive scientifically. One can conceive of a noncircular definition of eustress that is a kind of mirror image of the noncircular definition of distress. Just as distress is negatively reinforcing, motivates escape and avoidance behavior, and enhances vigilance, eustress is positively reinforcing, motivates approach and appetitive behavior, and enhances self-centeredness. Both distress and eustress have offered survival advantages in evolution, but either can be pathogenic in the setting of modern humanity. That is, neither may be only good or only bad for health. Just as modern-day pathologic consequences of distress are thought to include panic/anxiety, melancholic depression, or posttraumatic stress disorder (PTSD), pathologic consequences of eustress might include drug and alcohol abuse, sex offenses, gambling and other risk-taking behaviors, and over-eating. At the risk of over-simplification, central NE may play a role in the experience of distress (322) and DA in the experience of eustress (273).
Adaptation, habituation, dishabituation, and responses to novel stressors
The ability of humans to adapt to altered environments is well known. People in Peru who live at high altitudes and consequently are exposed chronically to hypoxia and hypocapnia are polycythemic, as elevated hemoglobin increases the oxygen carrying capacity of the blood. [Ethiopians living at similar altitudes are not as polycythemic (51).] Polar explorers in Antarctica have increased body fat (12, 286). After exercise training, exertion at a level that previously would have been exhausting typically can be sustained longer and with less myocardial oxygen consumption (63), and the time required for return of postexercise heart rate to the baseline value is shortened. Such adaptations can be explained by improvements in effector efficiences that reduce integrated error signals and thereby the rate of accumulation of allostatic load.
With repeated exposure to a stressor, the magnitude of the response decreases. Habituation is a characteristic of even primitive animals such as Aplysia (7), Drosophila (91), and zebrafish larvae (15). The term, dishabituation, is used to refer to a return to the initial magnitude of response after habituation has taken place. A characteristic of the stimulus is modified (e.g., prolonged), and subsequent exposure to the initial stimulus yields the complete response.
A related phenomenon is exaggerated responsiveness of adapted organisms to a novel (“heterotypic”) stressor. For instance, mice with a model of chronic psychosocial stress have attenuated in vitro responses of adrenocortical secretion in response to corticotropin (ACTH) yet augmented corticosterone responses to the heterotypic stressor of exposure to being on an elevated platform (307). In rats exposed to different stressors (immobilization, glucoprivation evoked by 2-deoxyglucose, or cold), cold-adapted animals have enhanced adrenomedullary expression of PNMT, the gene encoding synthesis of EPI from NE, in response to immobilization or glucoprivation (191). On the other hand, immobilization-adapted rats do not have enhanced PNMT responses to heterotypic stressors. Thus, exposure of adapted animals to novel stressors can induce exaggerated responses, but this depends on the specific stressors. Since immobilization-adapted rats have exaggerated plasma catecholamine responses to glucoprivation or cold (73), observation of exaggerated responses to novel stressors in adapted animals also seems to depend on the type of dependent measure.
Resilience
Organisms can protect and repair themselves after stress and even learn to anticipate and proactively make feed-forward adjustments that mitigate damage from future stress exposures. The concept is emerging that certain aspects of lifestyle, such as exercise training and some psychological interventions, enhance resilience. Psychological interventions may increase resilience to subsequent emotional stressors (247). Moreover, it is well known that exercise training increases resilience to subsequent bouts of exercise, while a sedentary lifestyle is associated with increased risk factors for aging-related diseases (21).
There is also some evidence that repeated exposures may increase resilience to heterotypic stressors. Heart rate biofeedback training can modulate responses of heart rate and rate-pressure product (an index of myocardial oxygen consumption) to treadmill exercise (142). Exercise-trained humans have attenuated heart rate, diastolic blood pressure, and rate-pressure product responses to mental arithmetic (19); and people acclimated to cold have attenuated increases in heart rate and blood pressure during isometric handgrip exercise (212).
Homeostatic system disruption
Disruption of a negative feedback loop augments effects of a stressor on levels of the monitored variable. The hallmark of inactivation of a homeostatic system is fluctuating levels of the monitored variable. The mean level may or may not drift to a new value, but perturbations tending to increase the level of the monitored variable are no longer buffered and therefore are expressed more fully. The same holds for perturbations tending to decrease the level of the monitored variable.
For example, the issue of whether destruction of the baroreflex causes “neurogenic hypertension” was for several years a contentious issue in cardiovascular research; however, all cardiovascular researchers would agree that such disruption increases the lability of blood pressure. Thus, neck irradiation results in rigidification of carotid arteries and encasement of distortion-sensing baroreceptors in the carotid sinus walls. The resulting arterial baroreflex failure is associated with increased blood pressure variability (283) (Fig. 14).
Figure 14.

Labile blood pressure in patients with baroreflex failure as a late sequela of irradiation of the neck. Blood pressure lability in this setting exemplifies loss of control of the level of the monitored variable, by disruption of the barostatic negative feedback loop.
Disablement at any station in a negative feedback loop produces about the same effects on responses of the monitored variable to a perturbation. If there were no afferent information to the homeostat about the monitored variable, or the homeostat were destroyed by a disease process so that there was no error signal, or the effector were missing or dysfunctional, then the ability to mitigate by negative feedback the effects of a perturbation on the monitored variable would be impaired. On the other hand, the amount of cumulative wear and tear due to effector activation—allostatic load—would depend on the location of the broken connection in the feedback loop. For instance, if the homeostat became dysfunctional due to a disease process and no longer drove effector activity despite the error signal, then the extent of allostatic load on the effector would be dissociated from the integrated error signal (cumulative stress).
Positive feedback loops (all stations in the feedback loop having a “+” sign) are inherently unstable. Conversion from a negative to a positive feedback loop presages rapid decompensation of the system. As explained later, one can understand the transitions from orthostatic intolerance to fainting, emotional distress to takotsubo cardiopathy, compensated to decompensated heart failure, and presymptomatic to symptomatic PD in terms of positive feedback loops. In these situations, a positive feedback loop is added onto what had been a negative feedback-regulated system.
Multiple effectors and effector sharing
Multiple effectors regulate levels of monitored variables. The body has at its disposal a large array of effectors (Fig. 15), all of which have the characteristics of working automatically, unconsciously, and involuntarily. One may classify them arbitrarily in terms of the autonomic nervous system, hypothalamic-pituitary-endocrine system, and other. Having multiple effectors extends the range of control, allows at least some regulation of the monitored variable if a particular effector fails (compensatory activation, Figure 16), and enables elaboration of specific, adaptive effector patterns—all three offering clear and substantial survival advantages in evolution. One can model multiple effectors (Fig. 17) using Stella and from the model demonstrate compensatory activation of alternative effectors (Fig. 18).
Figure 15.

Some effectors regulating levels of monitored variables. The effectors are grouped arbitrarily into those of the autonomic nervous system (ANS), pituitary/endocrine (Pitu./Endo.) systems, and others. ANS effectors include the sympathetic noradrenergic system (SNS), sympathetic cholinergic system (SCS), sympathetic adrenergic system (SAS), parasympathetic nervous system (PNS), the DOPA-dopamine system (DDA), and the enteric nervous system (ENS). Pitu./Endo. systems include the hypothalamic-pituitary-adrenocortical (HPA) axis, renin-angiotensin-aldosterone system (RAS), thyroid hormone (THY), growth hormone (GH), gonadotrophic hormones (GON), prolactin/oxytocin (PRO), arginine vasopressin (AVP), insulin (INS), and glucagon (GLU). Other effectors include cytokines (CYT), endogenous opiate species (EOS), atrial natriuretic peptide (ANP), bradykinins (BRK), and nitric oxide (NO).
Figure 16.

Compensatory activation. When a homeostatic system contains more than one effector, disabling of an effector leads to compensatory activation of the other effectors. Compensatory activation is one advantage of having multiple effectors.
Figure 17.
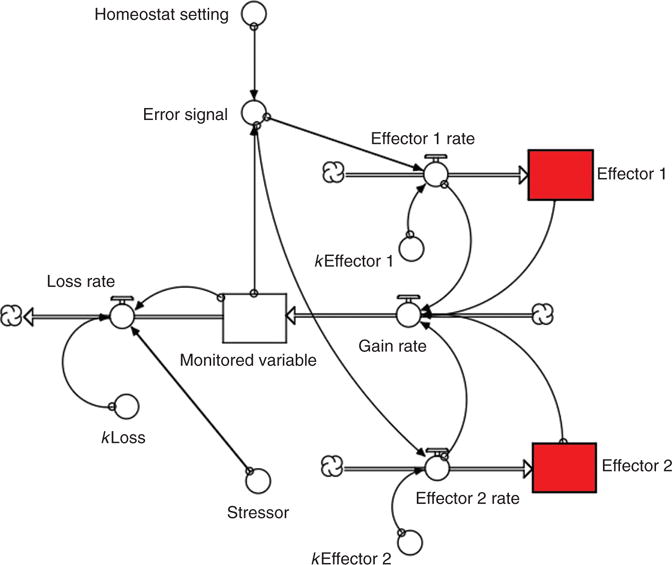
Computer model of multiple effectors.
Figure 18.
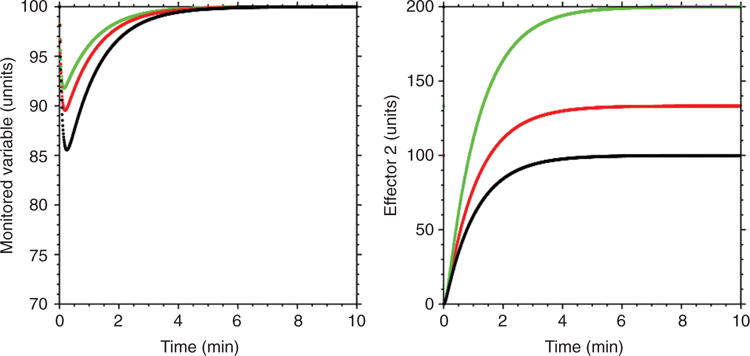
Computer-generated curves predicting effects of disabling one effector on activity of an alternative effector. As the rate constant for Effector 1 declines (green to red to black curves), the extent of activation of Effector 2 increases (compensatory activation).
Different homeostatic systems can share effectors (Figs. 19–21). In the setting of a shared effector, when one monitored variable is perturbed, the steady-state level is maintained at the set value by negative feedback, while the level of another monitored variable attains a new steady-state value. This phenomenon can also be demonstrated using Stella (Fig. 21).
Figure 19.

Effector sharing. Two homeostatic systems involving negative feedback loops share the same effector.
Figure 21.
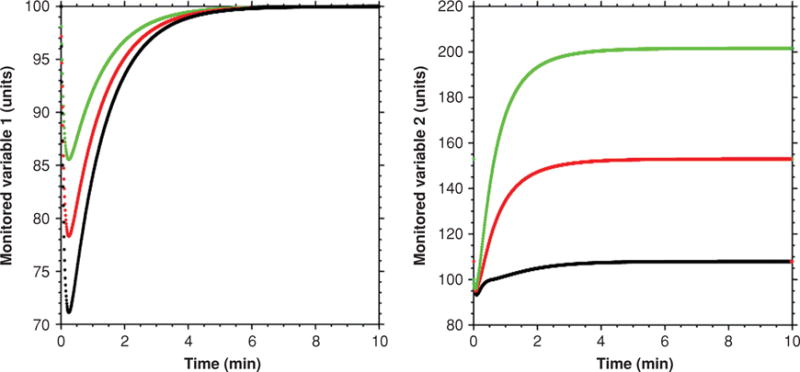
Predicted effects of effector sharing on levels of monitored variables. As the magnitude of stress increases in one homeostatic system (green to red to black curves), the level of the monitored variable for that homeostatic system returns to the baseline value, while the level of the monitored variable for the second homeostatic system reaches a different steady-state value. Increasing stress therefore results in maintenance of the first monitored variable at the set value, while levels of the second monitored variable increase to a new steady state. The extent of increase in the level of the second monitored variable depends on the extent of activation of the shared effector.
Based on the multiplicities of effectors and homeostats, responses to different stressors can be imposingly complex. Schematics for responses to orthostasis (Fig. 22) and to exercise (Fig. 23) provide examples of this complexity, yet experts in the field would probably note that even these schemas are overly simple. One should bear in mind also that the depicted networks are dynamic—magnitudes of responses change over time.
Figure 22.
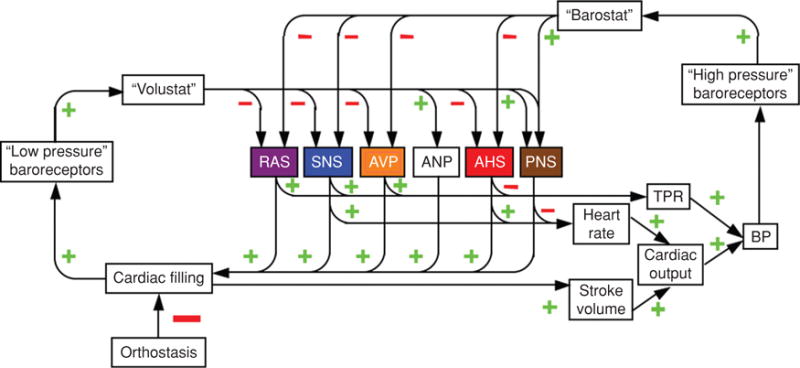
Complex involvement of multiple effectors and homeostats in the integrated response to orthostasis.
Figure 23.

Complex involvement of multiple effectors and homeostats in the integrated response to exercise.
Homeostats are theoretical entities. One may predict that with sophisticated mapping of brain pathways and advances in microscopic neurophysiologic and neurochemical measurements, these physiological comparators will be reified.
A minimum scientific integrative medical computer model includes at least two effectors regulating a monitored variable and sharing of an effector by at least two homeostats (Fig. 24). Although the diagram seems complex, it actually only glimpses at the situation in a living higher organism, because of the many systems regulating monitored variables by negative feedback and the many effectors shared by those systems.
Figure 24.

Minimum scientific integrative medicine model. The minimum model incorporates at least one monitored variable that is regulated by multiple effectors and at least one effector that is shared by multiple homeostats.
Cycling
Monitored variables of the body and activities of effectors determining their levels often change cyclically. The blood pressure is highest as the heart ejects blood (systolic pressure) and lowest just before the next heartbeat (diastolic pressure); correspondingly, activity of SNS outflow responsible for tightening blood vessels in skeletal muscle is pulse-synchronous. The blood glucose level goes up after meal ingestion, and you eat at particular times of the day every day (11); correspondingly, parasympathetic cholinergic system activity goes up during the “cephalic phase” of digestion, insulin levels rise, and the stomach secretes acid. The concentration of carbon dioxide at the nostrils increases with each exhalation; correspondingly, pacemaker neurons in the brainstem that drive breathing fire rhythmically. This periodicity complicates models about dynamic feedback regulation, because it can be difficult to determine which causes what, especially since these associations can be hard wired, instinctively acquired, or learned by classical (Pavlovian) conditioning (285).
Scientific Integrative Medical Concepts as Applied to the Physiology of Catecholamine Systems
Negative feedback regulation
Blood pressure is regulated by a negative feedback loop that incorporates a “barostat.” When the barostat senses a discrepancy between afferent information about blood pressure and the barostatic set-point, this drives multiple effectors, including the SNS, which rapidly increases the blood pressure to a plateau level about the same as the baseline blood pressure.
Although the barostat is a hypothetical entity, the functional neuroanatomy of baroreflexes has been worked out in some detail. Baroreceptor afferents traveling in the glossopharyngeal and vagus nerves synapse in the nucleus of the solitary tract in the medulla oblongata of the brainstem (276). Subsequent relay stations in the reflex arc include A1 noradrenergic neurons in the caudal ventrolateral medulla (4, 149); the nucleus ambiguus (a major source of descending cardiovagal outflow); and the rostral ventrolateral medulla (a major source of descending input to the sympathetic preganglionic neurons). Higher centers such as in the paraventricular nucleus of the hypothalamus modulate response characteristics of the medullary barostat (75, 112, 217, 280). Baroreflex pathways also ascend in the brain and might modulate consciousness, vigilance, nociception, or emotion (26, 256, 281, 311). Indeed, more than 30 years ago, it was proposed that high blood pressure reduces reactivity to noxious stimulation, via baroreceptor activation (78).
Stressors that decrease venous return to the heart increase SNS outflows reflexively. When a person blows against a resistance for several seconds (the Valsalva maneuver), venous return to the heart decreases and cardiac filling pressures fall. Since baroreceptors in the atria, pulmonary artery, and pulmonary veins are activated by mural stretch, the fall in cardiac filling decreases inhibitory baroreflex afferents, which travel in the vagus nerve via ganglia to the nucleus of the solitary tract in the dorsal medulla. Decreases in cardiac filling also lead complexly to decreased carotid sinus stretching, and the relative roles of “low pressure” cardiopulmonary and “high pressure” carotid sinus baroreceptors have been a long-standing topic of research (2, 27, 157). SNS outflows to skeletal muscle are disinhibited, resulting in increased pulse-synchronous bursts of nerve traffic, NE release, binding of NE to alpha-adrenoceptors on vascular smooth muscle cells, and skeletal muscle vasoconstriction.
During head-up tilt table testing, the extent of fall in cardiac filling depends on the tilt angle. As the severity of the orthostatic stressor increases, so does the rate of bursts of skeletal muscle SNS traffic (232). When a person is tilted from supine to 90° head up, plasma NE approximately doubles within 5 min. Skeletal muscle sympathetic nerve traffic also increases during i.v. infusion of nitroprusside (258), which relaxes blood vessels directly and decreases blood pressure. Because of disinhibition of baroreceptor afferents, both skeletal muscle sympathetic nerve traffic and plasma levels of NE increase in this setting.
The SNS is also a major effector in regulation of core temperature, via a central neural thermostat. The preoptic area of the anterior hypothalamus receives temperature information from two sources—temperature sensors in the skin, a key interface between the outside and the inner worlds, and sensors within the substance of the brain itself that monitor blood temperature. This duality corresponds to the two main determinants of heat dissipation and heat generation in the body—evaporative loss of heat from the skin’s surface and generation of heat by internal metabolic processes. One can dissociate these two determinants by infusing ice-cold saline into a central vein, with the room temperature unchanged. This induces marked activation of the SNS, and plasma NE increases (103, 104). Relaxation of cutaneous blood vessels upon exposure to increased environmental temperature is thought to result partly from SNS withdrawal but mainly from an active sympathetic vasodilator system (46).
Sympathetic neuronal activation does not accompany all stress responses equally (138). For instance, the body has three main effectors in glucose counterregulation—insulin, glucagon, and the SAS. Glucoprivation evokes heterogeneous increases in SNS outflows (228) and relatively small increments in plasma NE levels (25). In marked contrast, glucoprivation produced by insulin (253) or 2-deoxyglucose drastically increases plasma EPI levels (25, 315). Glucose sensors are found in the liver (168), with afferent information via the vagus nerve reaching the hypothalamus. Glucose sensors are also found at brainstem and hypothalamic sites; however, the exact pathways have not been mapped. It is thought that orexin/hypocretin-containing neurons enable sensing not only of absolute glucose concentrations but also changes in those concentrations (176)—corresponding to both proportionate and derivative control.
Dopamine (DA) is an effector in renal regulation of sodium homeostasis and thereby of extracellular fluid volume. Infused exogenous DA is well known to be a potent natriuretic drug. Endogenous DA in the kidneys is derived from uptake and decarboxylation of DOPA by proximal tubular cells, and all of urinary DA excretion is derived from circulating DOPA (321, 329). Dietary salt restriction decreases and dietary salt loading increases urinary excretion rates of DOPA and DA (145). These changes are relatively small, however, compared to changes in activity of the renin-angiotensin-aldosterone system (RAS). SNS outflow to the kidneys increases during sodium restriction (107), and this augments sodium reabsorption by renal proximal tubular cells (64). Thus, activities of the SNS and the renal DOPA-DA system change in opposite directions in response to alterations in dietary sodium intake. In line with multiple effectors participating in negative feedback regulation of sodium homeostasis, sodium restriction releases the sodium retention-promoting SNS and RAS from restraint by the “natristat,” while inhibiting the DOPA-DA system.
Disruption of a negative feedback loop, by blockade of afferent information or interference with the function of the homeostat, increases the variability of levels of the monitored variable. Thus, baroreceptor deafferentation increases the variability of blood pressure, as does bilateral destruction of the nucleus of the solitary tract, the likely anatomic correlate of the arterial barostat (235). Lesions of this region in humans can manifest clinically as baroreflex failure (17).
Compensatory activation
The availability of multiple effectors in negative feedback regulation of monitored variables enables compensatory activation of alternative effectors when one of the effectors is disabled. Examples of compensatory activation of catecholaminergic effectors abound in endocrinology and include augmentation of SNS activity by adrenalectomy, hypophysectomy, or thyroidectomy (108, 132, 306).
Hypothyroidism is associated with increased SNS outflows (213, 226, 252). Augmentation of SNS responses to cold in thyroidectomized animals fits with compensatory activation of the SNS (108). Analogously, hypopituitarism is associated with SNS activation (274, 295), and patients with isolated glucocorticoid deficiency have blunted EPI and augmented NE responses to cold pressor testing (331).
Under normal circumstances blockade of the SAS, RAS, or arginine vasopressin exerts little effect on blood pressure, but after chemical sympathectomy with 6-hydroxydopamine or guanethidine (sympatholytic drugs that spare the adrenal medulla), clamping of adrenal hilar vessels, administration of an angiotensin-converting enzyme inhibitor, or administration of a vasopressin receptor antagonist evokes severe decreases in blood pressure. As illustrated in Figure 25, via compensatory activation alternative effectors can maintain blood pressure when the main effector, the SNS, is disabled (13, 111).
Figure 25.

Compensatory activation of alternative effectors upon disabling of the SNS effector.
Primitive specificity
In addition to compensatory activation, another consequence of multiple effectors is patterning of stress responses— ”primitive specificity”—which one can comprehend in terms of the evolution of adaptively advantageous patterned adjustments (Figs. 26 and 27). During orthostasis or cold exposure, the SNS predominates; during manipulations of dietary salt intake, the RAS predominates; during manipulations of water availability, arginine vasopressin system (AVP) predominates; and during manipulations of glucose availability, responses of insulin, glucagon, and the SAS predominate. Small amounts of acute blood loss elicit mainly SNS responses, which maintains the output of blood by the heart and the flow of blood to the brain by redistributing blood volume; however, large amounts of acute blood loss sufficient to decrease blood pressure elicit a very complex and dynamic pattern of responses (14, 57), which can actually include sympathoinhibition (302).
Figure 26.

Catecholaminergic effectors associated with different homeostats. The different effector patterns result in “primitive specificity” of responses to different stressors. Effectors involving the catecholamines norepinephrine (sympathetic nervous system, SNS), epinephrine (sympathetic adrenergic system, SAS), or dopamine (DOPA-dopamine system, DDS) are in color. Other effectors depicted are the renin-angiotensin-aldosterone system (RAS), arginine vasopressin system (AVP), insulin (INS), glucagon (GLU), the parasympathetic nervous system (PNS), the hypothalamic-pituitary-thyroid system (HPT), and the sympathetic cholinergic system (SCS).
Figure 27.

Primitive specificity in different domains. For each stressor there is a particular pattern of autonomic, somatic changes, and experiential changes.
Stressors that pose global, metabolic challenges or are perceived as threats to well-being elicit SAS activation, even when the intensity of the stressor is mild. SAS activation is prominent in hypotensive hemorrhage, hypoglycemia, asphyxiation, circulatory collapse, and distress (14, 40, 105, 150, 224, 315). Stresses eliciting SAS activation typically also elicit HPA activation, as indicated by circulating levels of ACTH or cortisol (Fig. 13), and increases in release of endogenous opioids, as indicated by plasma levels of beta-endorphin, with small increases or even decreases in SNS outflows (139, 302).
Figure 13.

Relationship between extent of adrenaline and ACTH responses across multiple stressors, from a meta-analysis of literature (138).
In contrast, SNS activation is prominent in orthostasis, moderate exercise, regulation of core temperature, and the postprandial state (139, 214). Stresses associated with SNS activation often include a component of active movement (61). Patterned SNS activation during stress produces adaptive shifts in the distribution of blood volume or in glandular secretion. When these changes suffice to maintain homeostasis, they are not consciously experienced, but when the organism senses that these responses are not or will not mitigate effects of the stressor, the situation reaches consciousness, and SAS activation ensues.
The character and intensity of response patterns during distress depend on the perceptions by the organism about the stressor and about the available repertoire of coping responses (Fig. 27). HPA and SAS activation accompanies unanticipated, novel distress. At least three patterns of experiential, behavioral, hormonal, and physiological responses occur during distress—anger, which can develop into rage and fighting; fear, which can develop into terror and flight; and passivity, which can develop into “giving up,” decreased blood pressure, decreased blood flow to the brain, and even heart stoppage.
Physiological distinctions between fear and anger reflect differential changes in contraction of skeletal muscle, skin blood vessel and gastrointestinal smooth muscle, and smooth muscle in glands. The extent of skeletal muscle contraction, and the extent of recruitment of SNS activation to redistribute blood flows appropriately, generally varies with the intensity of the emotional experience.
It has long been thought that SAS activation is typically associated with fear and trembling (a form of ineffective skeletal muscle contraction) and SNS activation with coordinated skeletal muscle contraction and anger (109). Consistent with this view, in rats passive avoidance elicits large plasma EPI and corticosterone responses but small plasma NE responses, whereas active avoidance involves increases in all three measures (61).
For each stress there is an allostatic state in which neuroendocrine and physiological changes are coupled with behavioral changes (Fig. 27). For instance, regulation of total body water in humans depends on an interplay between behavior (the search for water and drinking), an internal experience or feeling (thirst), and the elicitation of a neurohormonal response pattern (in this case dominated by AVP, the antidiuretic hormone, and to a lesser extent angiotensin II, a potent stimulator of drinking).
Evoked changes in homeostat function often produce not only neuroendocrine and physiological effects but also behavioral responses; however, because of traditional boundaries among physiology, endocrinology, and psychology, interactions producing integrated patterns of response remain incompletely understood. Thus, studies about AVP and activity of the RAS during blood volume depletion rarely have included controls for or monitoring of thirst and salt hunger.
The notion of stressor-specific response patterns disagrees with the theories of both Cannon and Selye. Cannon, largely ignoring other systems, asserted that sympathico-adrenal system activation meets most or all important threats to the internal environment (38).
“The amazing feature of the role played by the sympathico-adrenal system is its applicability to the widespread range of possible disturbances that we have just noted. As stated earlier, the system commonly works as a unit. It is very remarkable indeed that such unified action can be useful in circumstances so diverse as low blood sugar, low blood pressure, and low temperature .… The appearance of inappropriate features in the total complex of sympathico-adrenal function is made reasonable, as I pointed out in 1928, if we consider, first, that it is, on the whole, a unitary system; second, that it is capable of producing effects in many different organs; and third, that among these effects are different combinations which are of the utmost utility in correspondingly different conditions of need (pp. 298–299).”
The current conception emphasizes separate regulation of the SNS and SAS, with if anything a closer association between adrenomedullary responses and responses of the HPA axis. Across a variety of stressors there is a closer link between SAS and HPA responses than between SAS and SNS responses, as illustrated in Figure 13 (138). Thus, in humans playing a video game, responses of ACTH levels correlate positively with responses of EPI levels but not with those of NE levels (131).
Differential regulation of the SNS and SAS during different forms of stress supports the concept of primitive specificity. Moreover, even within the domain of the SNS, studies based on microneurography have demonstrated differential activation of sympathetic outflows to the skin and skeletal muscle (312), and studies based on regional NE kinetics have demonstrated differential changes in rates of entry of NE into the venous drainage across different organs and disease states (93, 184).
This differential regulation also argues against Selye’s doctrine of nonspecificity. Students of Selye have emphasized responses of a single system—the HPA axis. Activation of this system may produce the pathological triad of the general adaptation syndrome—thymicolymphatic degeneration, adrenal hypertrophy, and gastrointestinal ulceration—but this only glimpses at the spectrum of systemic responses to stress.
The Swedish physiologist, Björn Folkow, proposed that patterns of nervous system activity associated with stress are always expressed in closely linked, situation-specific patterns, with behavioral, experiential (emotional), and automatic (autonomic and hormonal) facets (101). In terms of stress and distress, these facets correspond to externally observable and instinctively communicated movements and behaviors, mediated by the somatic nervous system; emotional feelings, resulting from cognitions about both the outside and inner worlds (269); and automatic, unconscious, involuntary changes in the inner world, mediated by several effectors including catecholamine systems.
One may speculate that stressor-specific triadic patterns of behavioral, experiential, and automatic activities during stress became intertwined so tightly in evolution that by now these patterns are expressed as units. If groups of genes can be selected as units (60), then perhaps groups of genes associated with these three facets of primitively specific stress responses evolved as units. Homeostatic regulation of blood pressure by the arterial baroreflex seems to contain a strong hereditary component (106, 243, 262). From this, one may hypothesize that people with relatively low baroreflex gain on a genetic basis have a tendency to disinhibition of SNS outflows and cardiovascular stimulation during distress.
Effector sharing
Different homeostatic systems can share the SNS or SAS effectors. For instance, the SNS is a shared effector for the thermostat and barostat. Thus, cooling of the skin augments plasma NE responses and improves measures of toleration of lower body negative pressure (53, 77). Vasoconstriction and thermogenesis in response to cold may involve different populations of sympathetic preganglionic neurons (230).
Sharing of the SAS by the barostat and glucostat explains hyperglycemia attending any of several emergencies such as hemorrhagic shock (42), stroke (187), sepsis (257), and myocardial infarction (79). Not surprisingly, treatment of the hyperglycemia by insulin infusion in these situations does not improve outcome.
Analogously, sharing of the AVP effector by the barostat and osmostat (255) can explain hyponatremia in heart failure (261, 296). From the principle of effector sharing one may predict that the most efficient means to reverse hyperglycemia in gastrointestinal hemorrhage and hyponatremia in heart failure is to treat the underlying cause—that is, blood transfusion for the hyperglycemia attending hemorrhage and perhaps an “unloading” vasodilator for the hyponatremia attending heart failure.
Stress and distress
In the early 20th century, Walter B. Cannon carried out ingenious experiments that for the first time demonstrated secretion of EPI by the adrenal glands during distress. A study published in 1911 reported experiments in which an instrumented cat was exposed to a barking dog. Blood taken from the vena cava of the stressed cat relaxed a rhythmically contracting intestinal strip in a bioassay preparation (40). This relaxation was not observed in adrenalectomized cats (Fig. 28). It was from these findings that Cannon first deduced that during stress a substance is secreted by the adrenal glands into the bloodstream.
Figure 28.

Cannon’s experiment in which he exposed an instrumented cat to a barking dog. Blood taken from the vena cava of the stressed cat relaxed a rhythmically contracting intestinal strip in a bioassay preparation (40). “Excited” blood was added at (b) and (f), and “quiet” blood from the same animal was added at (d).
Several years later, Cannon perfected a denervated heart preparation and used heart rate responses as a measure of the extent of adrenal secretion (39) (Fig. 29). Across several studies, a variety of severe stressors were found to increase heart rate in this preparation, with adrenalectomy blunting or preventing the effect.
Figure 29.

Illustration of Cannon’s use of the heart rate of a denervated heart as a measure of adrenal EPI secretion (35).
According to Cannon, a wide variety of threats to homeostasis, such as exposure to cold, hypotensive hemorrhage, traumatic pain, insulin-induced hypoglycemia, or emotional distress, elicit activation of the sympathico-adrenal system to restore homeostasis. The notion of a unitary sympathoadrenal system continues in medical thinking (52, 192, 282, 290).
Cannon also found that ordinary activities such as walking or changing position increased heart rate in his denervated heart model. He recognized that this challenged the notion of a purely emergency function of the sympathico-adrenal system, and he wrote,
“the emergency theory would have to be altered insofar as it might imply that the sympathico-adrenal mechanism is called into action only at times of violent emotion. According to the evidence now in hand, the greater the emergency, as measured by intensity of excitement and struggle, the more is that mechanism utilized (p. 463).”
That is, the extent of activation of the sympathico-adrenal system would vary with the intensity of the stress; however, the system would always function as a unit. Selye and his students had an analogous perspective about the relationship between HPA activation and the severity of stress. Neither Cannon nor Selye considered the possibility that for most day-to-day experiences of life, such as standing, eating, walking, and exposure to changes in environmental temperature, stress effectors are activated or inhibited in stressor-specific patterns. Because neither measured activities of multiple effectors simultaneously, neither had available the experimental data that would induce such a concept.
In 1939, Cannon formally proposed that EPI is not only the active principle of the adrenal gland but also the neurotransmitter of the SNS (41). He was wrong, and the mistake might have cost him a Nobel Prize. The identity of the substance released at sympathetic nerve terminals remained controversial until 1946, when US von Euler correctly identified the sympathetic neurotransmitter in mammals (313) as NE, and; for this discovery von Euler shared the Nobel Prize in Physiology or Medicine in 1970.
In the sheltered confines of a laboratory, with controlled temperature and ad libitum water, nutrients, and calories, mammals do not seem to require an intact SNS (37). It has become clear, however, that even under resting conditions, pulse-synchronous bursts of skeletal muscle sympathetic nerve activity and plasma levels of NE are detectable, and NE continuously enters the venous drainage of most organs. We also now recognize that activities of daily life, such as meal ingestion (248), public speaking (115), changing posture (194), and locomotion—that is, not only emergencies—are associated with rapid adjustments in SNS outflows. Each of these activities is associated with a somewhat different set of apparent steady states, directed by the brain and determined by coordinated actions of a variety of effectors—different allostatic states.
Selye acknowledged that responses to stressors have specific components that tend to reverse effects of the stressor; however, according to Selye, in addition to the specific responses, there is a nonspecific response, corresponding to stress. Chrousos and Gold (50) modified Selye’s doctrine of nonspecificity by proposing that above a threshold intensity any stressor elicits the nonspecific stress syndrome. More than a half century elapsed before Selye’s doctrine of non-specificity underwent experimental testing, which failed to confirm it (245). Nevertheless, modern lay and even scientific literature continues to accept the notion of a unitary stress response. For instance, a Google search yielded more than 10 million hits for “the stress response.”
Modern concepts view stress as a consciously or unconsciously sensed threat to homeostasis (140, 219), in which the response has a degree of specificity, depending among other things on the particular challenge to homeostasis, the organism’s perception of the stressor, the perceived ability to cope with it, and the repertoire of evocable neuroendocrine patterns by the brain.
Adaptation and resilience
Cannon alluded to what William James called “reservoirs of power” (35) in describing a direct antifatigue effect of EPI on repeatedly contracted skeletal muscle. Three chapters of Cannon’s book, Bodily Changes in Pain, Hunger, Fear, and Rage (35), are devoted to experiments demonstrating improved contraction of fatigued skeletal muscle after splanchnic nerve stimulation or addition of EPI to the bath. The experiments separately assessed effects of adrenal secretion and increased blood pressure on skeletal muscle fatigue.
Relatively little modern research has focused on antifatigue effects of catecholamines. Sympathetic stimulation itself produces relatively small increases in contraction of fatigued skeletal muscle, and isoproterenol a beta-adrenoceptor agonist, is without effect; however, concurrent administration of isoproterenol facilitates the antifatigue effect of sympathetic stimulation (162).
The augmentation of antifatigue by beta-adrenoceptor agonism in the setting of SNS activation leads directly to the view that inhalation of anti-asthma medications could offer an unfair advantage in competitive sports. On the other hand, athletes can have exercise-induced asthma. This poses an obvious dilemma. Article 4 of the World Antidoping Code prohibits beta-2 adrenoceptor agonists, but with the possibility of a Therapeutic Use Exemption. Inhaled formoterol, salbutamol, salmeterol, and terbutaline can be taken under an Abbreviated Therapeutic Use Exemption. Sympathomimetic amines are well known to exert antifatigue effects, and these drugs are of course banned in competitive sports.
Amphetamines are used routinely in modern military medicine. For instance, during the 2003 campaign Operation Iraqi Freedom, B-2 bomber pilots flew from one of two airfields with different distances from the targets. The pilots used dextroamphetamine for 97% of the relatively short and 58% of the relatively long sorties, and of those who took dextroamphetamine, 97% noted a benefit (180). There are many reports published in Chinese journals about antifatigue effects of various herbal remedies derived from plant extracts. Relative roles of central versus peripheral mechanisms of antifatigue action of sympathomimetic amines remain unknown.
Repeated exposures to stressors that alter SNS or SAS outflows may increase resilience to heterotypic stressors, although the literature on this is sparse. Exercise training augments plasma EPI responses to insulin-induced hypoglycemia (181), which could decrease the rate of accumulation of the error signal and allostatic load in the glucostatic system. In rats, long-term exercise or cold acclimation produce adaptive changes in SNS outflow, although cardiac sympathetic activation seems to contribute to cardiac hypertrophy attending these exposures (242). Repeated handling of rats results in habituation of ACTH and EPI responses but not of NE responses to subsequent handling (71). After repeated handling, handling by a different person dishabituates the ACTH and EPI responses and augments the NE response.
In humans, repetition of laboratory psychological challenges results in habituation of ACTH and cortisol responses but not of NE or EPI responses (115, 272). In a study of women exposed repeatedly to cold (winter swimming or cryotherapy), ACTH and cortisol responses habituated, whereas NE responses did not (197). On the other hand, men exposed repeatedly to cold had attenuated NE responses to subsequent exposure (212). Men and woman do not appear to differ in plasma catecholamine responses to acute cold (251), but whether there are gender differences in habituation of catecholamine systems to cold seems not to have been studied.
In mice, studies of strains inbred for occurrence of social defeat suggest a role of mesolimbic DA systems in psychological vulnerability versus resistance. The locus ceruleus-NE system is likely to play a role in phenomena such as vigilance, altered sleep, and facilitated memory of distressing events. According to the “stress inoculation” hypothesis, repeated episodes of psychological stress exposure in youth attenuates responsiveness in adulthood (152); however, whether central catecholamine systems are related to this form of resilience remains unknown.
Applications of Concepts of Scientific Integrative Medicine to Catecholamine-Related Disorders
In this section, concepts of scientific integrative medicine are applied to help understand pathophysiologic and pathogenetic mechanisms of catecholamine-related disorders—hypertension, heart failure, stress cardiopathy, PTSD, fainting, and PD.
Measures and effects of allostatic load
If repeated chronically, allostatic adjustments that are effective for short periods of time may increase long-term wear and tear on the effectors. In the computer model of a negative feedback-regulated system, the use-related, cumulative wear and tear on effectors corresponds to allostatic load. Allostatic load is related to—but not identical with—cumulative stress, which in the computer model is the integrated error signal.
With cumulative wear and tear on the effector, the efficiency of the effector declines. In the model, kGain decreases in a sigmoid manner. The model predicts an initially stable level of the monitored variable, followed by an accelerating decline (Fig. 12). When the level of the monitored variable decreases to below a certain value (the model uses 40% of ideal), the decrease becomes symptomatic. For the same amount of cumulative stress, factors augmenting allostatic load (e.g., vulnerability genes) shift to the left the relationship between the level of the monitored variable and time, with symptoms developing relatively early. Although not shown in the Figure, factors decreasing allostatic load (e.g., resilience from exercise training) shift the relationship to the right, so that symptoms develop relatively late.
Since the publication of several influential reports and essays by McEwen and colleagues in the early 2000s, the number of studies using the term, “allostatic load,” has risen exponentially (Fig. 30). Many recent articles have sought to validate a particular pattern of neuroendocrine responses as a measure of overall allostatic load. The notion of a unitary measure of allostatic load seems reminiscent of the notion of a unitary measure of stress. Given the multiple effectors used in negative feedback regulation of internal monitored variables, the overall risk of system failure is related to the amounts of allostatic load on the individual effectors. The body is a system of systems. In medical terms, it seems better practice to track the status of regulation of key monitored variables individually than to track the status of the “whole person” by a single index of risk.
Figure 30.
Articles culled from PubMed using the search term, “allostatic load,” as a function of 2-year periods since 1996. The number of articles on allostatic load increased exponentially.
From the discussions below, one gains the impression that several acute disorders (fainting and takotsubo cardiopathy are examples) are associated with particular neuroendocrine patterns because of effects of effector sharing overriding regulation by negative feedback. On the other hand, chronic disorders seem to reflect degenerative processes such as from autotoxicity outstripping compensatory activation. In both situations, decreased efficiency of regulation of monitored variables leads to conversion from negative to positive feedback loops.
Fainting and allostasis
Fainting (also called vasovagal syncope, reflex syncope, neurally mediated hypotension, and neurocardiogenic syncope) constitutes the most common cause of sudden loss of consciousness in the general population (1). Fainting reactions can occur in otherwise healthy people, especially in distressing circumstances where neither “fight” nor “flight” are options (90). Patients with frequent fainting can feel unwell between episodes, with an inability to tolerate prolonged standing, chronic fatigue, headache, heat intolerance, and a variety of nonspecific complaints (200).
Increased parasympathetic cardiovagal outflow explains the prominent bradycardia that often attends syncope; however, since cholinergic blockade by atropine fails to prevent the syncope, the hypotension does not depend on vagally induced bradycardia. In a famous study by Barcroft and Edholm, hemorrhage-evoked syncope was associated with a substantial increase in forearm blood flow (10). Because of the perceived importance of sympathetically mediated vasodilation in producing the hypotension, the authors introduced the term, “vaso-vagal syndrome.” In the same study, sympathectomized patients had a failure to evince skeletal muscle vasodilation but did have hemorrhage-evoked syncope, leading to the suggestion of sympathetically-mediated (as opposed to EPI-mediated) vasodilation.
At first glance, the occurrence of neurally mediated hypotension would seem to contradict the concept of negative feedback regulation of blood pressure by baroreflexes. According to the “collapse firing” hypothesis, the precipitant is a combination of decrease in cardiac filling and a sympathetically mediated increase in cardiac inotropy (Fig. 31). This would stimulate baroreceptors in the left ventricle (239) or veins (68) and evoke a reflex neuroendocrine pattern involving a drop in total peripheral resistance due to sympathoinhibition and bradycardia due to parasympathetic stimulation. Several lines of evidence have questioned the collapse firing hypothesis. There is no convincing evidence for an excessive fall in intracardiac volume (59) or an excessive increase in the inotropic state of the myocardium (201) prior to fainting. Heart transplant recipients, who have no or minimal nervous connections to the transplanted heart, nevertheless can faint (99, 118, 199, 227, 270). Moreover, rare patients can have syncope while supine (134).
Figure 31.

Diagrams of feedback loops that may be involved in fainting reactions (neurocardiogenic syncope, reflex syncope, vaso-vagal syncope). According to the collapse firing hypothesis, syncope results from a combination of SNS activation and decreased cardiac filling (such as from orthostasis or acute hemorrhage), which evokes a pattern of SNS withdrawal and PNS stimulation. According to a schema derived from concepts of scientific integrative medicine, syncope results from positive feedback loops and interference with negative feedback loops, at least partly due to sharing of the SNS and SAS effectors. The result is a specific neuroendocrine pattern that includes PNS activation and sympathoadrenal imbalance.
The right panel in Figure 31 provides a potential allostatic explanation for reflex syncope. According to this schema, distress and glucoprivation, via homeostats other than the barostat, increase activity of the SAS as a shared effector. Exposure to warm environmental temperature, also via a homeostat separate from the barostat, inhibits activity of the SNS as a shared effector. The net result is an altered neuroendocrine pattern from that anticipated from the barostatic negative feedback loop alone.
This centrally evoked pattern introduces the possibility of neurocirculatory positive feedback loops leading to hypotension and cerebral hypoperfusion. For instance, glucoprivation from skipping a meal could augment SAS-mediated skeletal vasodilation, while exposure to a warm environment could inhibit SNS-mediated reflexive vasoconstriction. If a person were standing in the heat, the gravitational fall in venous return to the heart might not be countered efficiently by baroreflexes, and if the brain perceived this failure to cope, the resulting distress response would augment SAS outflow further and aggravate the vasodilation. Meanwhile, EPI-related hyperventilation could decrease cerebral perfusion and further amplify the SAS response.
This schema might explain the neurally mediated hypotension (and forearm vasodilation, cutaneous vasoconstriction, sweating, and mydriasis) in fainting reactions but does not readily explain the parasympathetic stimulation and associated bradycardia and gastrointestinal symptoms. Possible explanations are an allostatic increase in barostatic gain, resulting in augmented parsympathetic outflow concurrent with SNS withdrawal, and distress-related activation of a “psychostat” that shares the PNS with the barostat, without an alteration in barostatic function per se.
Fainting reactions typically involve a particular, dynamic catecholamine pattern that is related to the neurocirculatory events. There is more prominent SAS than SNS activation (96, 134, 144, 160, 263, 264, 297), a phenomenon that has been called “sympathoadrenal imbalance.” The individual patient data in Figure 32 provide an example. Normally, when a person is tilted head-up, SNS outflows and forearm vascular resistance (FVR) increase reflexively, and mean arterial pressure is maintained. EPI levels in arterial plasma also increase, and the fractional increases in EPI and NE levels are about the same. In this patient, over time FVR began to fall, and EPI levels increased in a mirror image pattern. The greater fractional increase in EPI than NE indicated “sympathoadrenal imbalance” (134, 144, 160). When the FVR decreased to baseline, at about 30 min, a neurocirculatory positive feedback loop was initiated, which led to hypotension and syncope several minutes later.
Figure 32.

Mean arterial pressure (MAF), forearm vascular resistance (FVR), and arterial plasma levels of catecholamines in a patient with tilt-induced hypotension and syncope. The arrows emphasize the mirrored trends in FVR and plasma EPI. EPI becomes dissociated from NE (sympathoadrenal imbalance) and FVR falls below baseline several minutes before hypotension and syncope.
Studies have disagreed remarkably as to whether skeletal muscle SNS outflow falls abruptly (89, 229, 231, 232, 314) or is maintained (308, 309) at the time of fainting reactions. Moreover, it is possible that for a given amount of sympathetic nerve traffic, occupation of muscarinic receptors on sympathetic nerve terminals may attenuate NE release.
Neurocardiogenic syncope is attended by several other neuroendocrine changes, including elevated levels of beta-endorphin (160), atrial natriuretic peptide, corticotropin, and vasopressin (163, 299). The vasodilator effect of EPI may be mediated partly by increased production of nitric oxide (166). Since EPI is taken up avidly into non-neuronal cells via the Uptake-2 process (86), which involves a plasma membrane monoamine transporter and organic cation transporter 3 (74) but not the cell membrane NE transporter expressed by sympathetic nerves, it is also possible that the increase in nitric oxide production occurs independently of EPI binding to beta-2 adrenoceptors. Fainting reactions are associated with large increases in AVP levels (163, 299). AVP augments baroreflex restraint of SNS outflows in laboratory animals (154, 237) and might contribute to sympathoinhibition; however, whether AVP does so in humans in this setting is unclear (80). The adrenal medulla contains abundant angiotensin II receptors, and occupation of those receptors evokes EPI secretion from chromaffin cells (328). Children with a history of frequent fainting have elevated levels of plasma renin activity, and the extent of fall in blood pressure during fainting is positively correlated with the extent of elevation of plasma renin activity (305); however, angiotensin II levels have not been reported in this setting.
In terms of treatment, tilt training programs have been attempted to reduce vulnerability to fainting reactions (110). Repeated exposure to tilting attenuates plasma renin activity and aldosterone responses to subsequent tilt, exemplifying habituation; however, whether this actually decreases the frequency of spontaneous or evoked fainting reactions is unclear. Although EPI-induced relaxation of blood vessels in skeletal muscle could decrease vascular resistance in skeletal muscle and in the body as a whole, nonselective beta-adrenoceptor blockade has been disappointing in preventing tilt-induced neurocardiogenic syncope (88). Patients with substantial tachycardia prior to syncope might benefit, but in patients with bradycardia, such treatment might lead to prolonged asystole (56). Treatment with angiotensin-converting-enzyme inhibitor, enalapril, can prevent tilt-induced syncope, despite decreasing blood pressure during supine rest (325). Medications attenuating EPI release (e.g., benzodiazepines), blocking beta-2 adrenoceptors relatively selectively, augmenting SNS outflows (e.g., yohimbine), increasing occupation of alpha-adrenoceptors (e.g., sympathomimetic amines and midodrine), or increasing NE production independently of SNS outflows (e.g., droxidopa) merit consideration.
The allostatic model in Figure 31 leads straightforwardly to several suggestions about nondrug approaches to prevent neurally mediated hypotension. The likelihood of positive feedback loops would be decreased if the ambient temperature were low, if the person were not fasting, if distress were minimized, and if orthostatic blood pooling were decreased by an abdominal binder. In addition, ingestion of 16 ounces of water can increase vascular tone and blood pressure and improve orthostatic tolerance (208), via a form of osmopressor reflex not depicted in the schema.
It therefore appears that there are multiple determinants of the hypotension that characterizes fainting, those determinants change dynamically, and there are likely to be substantial individual differences in alterations of effector activities (69). These aspects may help explain perennial disagreements among investigators about mechanisms and treatment (70, 233). Experimental attempts to elucidate contributions of these determinants by blocking single effectors may yield false negative results because of compensatory activation of alternative effectors.
Hypertension treatment and negative feedback loops
The concept of multiple effectors in the negative feedback loops determining blood pressure can help resolve a long-standing controversy about the role of the SNS and other effectors in the development and maintenance of essential hypertension (92, 94, 100). Because multiple effectors determine blood pressure, blockade of single effectors such as the SNS to study its pathophysiologic role may yield false negative results because of compensatory activation of the alternative effectors. One would have to monitor or control activities of the alternative effectors.
Although stimulation of the carotid sinus nerve in humans evokes hypotension and bradycardia (43), researchers have debated for many years whether baroreceptor “debuffering” increases “resting” blood pressure—that is, whether debuffering produces a form of neurogenic hypertension (300). In humans, baroreceptor debuffering by local anesthesia of the glossopharyngeal and vagus nerves increases blood pressure acutely and concurrently increases values for indices of sympathetic nervous system activity (97). Irradiation-induced arterial baroreceptor denervation, while increasing blood pressure lability (Fig. 14) and predisposing to episodes of paroxysmal hypertension and high plasma NE levels, does not necessarily produce sustained hypertension (283). On the other hand, as noted below, carotid sinus electrical stimulation produces chronic decreases in blood pressure and SNS outflows in dogs (202–205).
Increased sympathetic outflow to the kidneys seems to be a major determinant of the renal function curve that relates natriuresis to renal perfusion pressure (65, 66). Release of renal sympathetic outflow from baroreceptor restraint could lead to a sustained increase in blood pressure by resetting the renal function curve and compensatorily activating the RAS.
Two relevant recent developments involve devices rather than drugs to treat chronic hypertension. One is based on afferent baroreflex activation and the other on renal sympathetic denervation. Chronic carotid sinus electrical stimulation produces decreases in blood pressure in hypertensive dogs (203–205), even during adrenergic blockade (202). Clinical trials of a carotid sinus stimulator for resistant hypertension are currently under way. A recent acute study of hypertensive patients reported a rapid depressor response to carotid stimulation that was associated with sympathoinhibition (156). Although muscle sympathetic nerve activity decreased sharply beginning soon after the start of the stimulation, sympathetic activity subsequently increased toward baseline.
A catheter-based system for radiofrequency ablation of renal sympathetic innervation has been introduced, also for resistant hypertension (186). This approach is derived from substantial preclinical literature that renal sympathetic nerves contribute to sodium retention and blood pressure, especially during stress. Renal adrenergic stimulation augments activity of the RAS, and in the central nervous system angiotensin modulates baroreflex regulation of renal sympathetic neural outflow (67). Although it was presumed that efferent renal sympathetic denervation would explain fully the antihypertensive effect of the procedure, skeletal muscle sympathetic outflow decreases, presumably from destruction of renal afferents (271). The procedure also increases insulin sensitivity (211), which offers the potential for a novel treatment of metabolic syndrome. Current and planned studies will assess whether renal artery radiofrequency ablation effectively treats milder or secondary forms of hypertension or benefits patients with congestive heart failure.
Given the multiplicity of effectors regulating blood pressure one may predict substantial inter-individual variability and complex determinants of efficacy of both these devices.
Heart failure and positive feedback loops
Heart failure entails markedly increased cardiac SNS traffic (82) and therefore increased delivery of NE to myocardial cells. High local catecholamine levels promote ventricular hypertrophy and predispose to arrhythmias. Both increased wall stiffness and arrhythmias worsen the heart’s pumping efficiency, in turn reflexively evoking further increases in sympathetic nerve traffic to the heart—a positive feedback loop.
Catecholamines increase the work of the heart. In a patient with coronary stenosis, the rate of oxygen utilization may exceed that of oxygen delivery via coronary perfusion. Lack of oxygen delivery to the sympathetic nerves themselves in the heart tends to render their storage vesicles “leaky,” augmenting NE release for the same rate of sympathetic nerve traffic (190). Moreover, as discussed in more detail below, buildup of catecholamines in the cytosol is potentially toxic, via conversion by MAO to catecholaldehydes. There is evidence that MAO-A contributes to heart failure (173,174, 222). No studies to date have actually measured levels of the catecholaldehyde produced by the action of MAO-A on NE, which is 3,4-dihydroxyphenylglycolaldehyde (DOPEGAL).
Increased SNS outflows augment cardiac filling because of decreased venous capacitance, direct and indirect sodium-retaining effects, and increased total peripheral resistance. Cardiac overfilling leads to accumulation of fluid in the lungs, producing hypoxemia, acidemia, and distress, all of which stimulate catecholamine systems, worsening cardiac overfilling. As noted for takotsubo cardiopathy, acutely severely increased catecholamine levels can decrease rather than increase cardiac pumping efficiency, precipitating a rapidly life-threatening positive feedback loop manifesting as fulminant pulmonary edema. One may reasonably propose that sustained high levels of catecholamines exert potentially reversible autotoxic effects in myocardial cells in other conditions, such as sepsis or sustained severe “ironman” events, which are associated with decreased cardiac ejection fraction (49, 72, 266).
Not surprisingly, in congestive heart failure, the plasma NE level constitutes an independent prognostic factor and correlates with functional status (159, 301). Compensatory activation of sympathetic nerves in the heart can for long periods be a major source of homeostasis in the face of intrinsic cardiovascular degeneration. Eventually, however, the same activation can induce neurocirculatory positive feedback loops, resulting in cardiovascular instability, rapid worsening of clinical status, and death. Associations of poor prognosis with a high rate of appearance of NE in the coronary sinus (cardiac NE spillover), a large arterial-cardiac venous increment in plasma NE, or decreased uptake and increased washout of cardiac 123I-metaiodobenzylguanidine-derived radioactivity occur in diverse disorders, including congestive heart failure (179, 303), ventricular arrhythmias (223), dilated cardiomyopathy (116), diabetes mellitus (119), metabolic syndrome (326), and chronic renal failure (189).
Hyponatremia occurs commonly in heart failure. One way to conceptualize the basis for this association is from sharing of the vasopressin/antidiuretic hormone (ADH) effector by two homeostats, the barostat and the osmostat. Decreased efficiency of cardiac ejection releases the vasopressin system from baroreflex restraint, and vasopressin levels in the blood-stream increase. Because of increased vasopressin levels, the kidneys retain “free water,” and serum osmolality and serum sodium concentrations fall. The most appropriate treatment for hyponatremia attending heart failure therefore is not hypertonic saline (which could precipitate pulmonary edema) or water restriction but alleviation of the heart failure.
The renal DOPA-DA system is upregulated in heart failure (98), possibly as part of a compensatory process. Renalase, a relatively recently described amine oxidase, metabolizes urinary catecholamines, and it has been proposed that renalase in luminal fluid might alter DA metabolism and proximal tubular sodium transport (62).
Stress cardiopathy and multiple effectors
Takotsubo cardiomyopathy refers to a relatively recently described form of acute, reversible, stress-related cardiomyopathy in which apical akinesia gives the heart the shape of a takotsubo, a Japanese fishing pot for trapping octopus (5). Takotsubo cardiomyopathy occurs with relatively high incidence in elderly women soon after exposure to severe emotional distress (320). Symptoms mimic acute myocardial infarction; however, coronary angiography fails to demonstrate coronary occlusion. The condition can trigger sudden cardiac failure or death, yet in survivors cardiac function typically normalizes within a few weeks. Survivors may be susceptible to subsequent episodes.
Takotsubo cardiomyopathy features remarkably high levels of catecholamines—especially EPI (320)—and depressed myocardial contractile function. Hypothesized pathogenetic mechanisms include coronary microvascular spasm (resulting at least partly from cardiac SNS activation) and cardiotoxicity from neuronal NE and hormonal EPI (5), which may interact to precipitate multiple positive feedback loops. The heart stands out among organs of the body in terms of dependence on neuronal uptake for inactivating catecholamines in the extracellular fluid (130). High circulating catecholamine levels such as in pheochromocytoma interfere with the neuronal uptake process (87) and augment occupation of adrenoceptors on myocardial cells. These findings help to explain how distress can exert selective cardiac toxicity as a result of high circulating catecholamine levels despite EPI being delivered to all organs equally by the arterial blood. It has also been proposed that high circulating EPI triggers a switch in cardiomyocyte intracellular signaling after occupation of beta-2 adrenoceptors, from stimulatory Gs to inhibitory Gi (210). Another possibility is that after non-neuronal uptake of a high circulating concentration of EPI by myocardial cells, the catecholamine is converted intracellularly to the toxic catecholaldehyde, DOPEGAL, via monoamine oxidase (MAO).
The reason takotsubo cardiomyopathy occurs especially in postmenopausal women remains unclear. Studies involving exposure of castrated female rats to immobilization, which can produce cardiomyopathy with apical ballooning (5), have supported the view that estrogen deficiency, via effects both in the central nervous system and the heart, increases susceptibility to takotsubo cardiomyopathy. Since estrogens normally inhibit extraneuronal uptake of catecholamines (45, 172), one may speculate that low estrogen levels augment myocardial cell uptake of circulating EPI, in turn amplifying toxicity exerted by DOPEGAL. This hypothesis predicts that, just as inhibition of MAO attenuates heart pump failure from pressure overload (174) and ischemia/reperfusion injury, MAO inhibition might ameliorate or prevent takotsubo cardiomyopathy.
Posttraumatic stress disorder: When distress really is bad
If one accepts the definition of distress given above as a conscious, aversive experience associated with instinctively communicated signs and adrenal activation, and does not define distress as pathologic, then one can assess by observation or experimentation the health consequences of distress. By motivating escape and avoidance behavior, increasing vigilance, and promoting long-term memory of associated stimuli, distress offers important survival advantages in evolution and protection of individuals. In PTSD, these adaptive, beneficial aspects might become pathologic.
The neuroendocrine pattern attending PTSD does not fit well with the notion of a unitary measure of allostatic load, nor—at least at first glance—with monolithic activation of the adrenal cortex and adrenal medulla as a functional unit in distress. In Vietnam veterans with PTSD, exposure to auditory combat-related stimuli was reported to increase plasma NE levels, a measure of SNS activation (18), and in PTSD urinary excretion rates of NE and EPI are chronically increased (185); however, cortisol levels are if anything decreased. This seems to pose a paradox (215), because if the adrenal cortex and medulla worked as a functional unit in distress, why would there be differential changes in HPA and SAS activities in PTSD?
Although there is an effective blood-brain barrier for catecholamines, an exception is in the hypothalamus (316), and infusion of the beta-adrenoceptor agonist, isoproterenol, decreases ACTH levels in humans (84). It seems reasonable to propose that by negative feedback exerted by EPI, the main endogenous beta-2 adrenoceptor agonist, chronically elevated circulating levels of catecholamines in PTSD may modify regulation of the HPA axis by a hypothalamic homeostat. Conversely, recent findings in a laboratory animal model of PTSD suggest that treatment with high doses of glucocorticoid might be beneficial (330). In healthy humans, prednisone treatment decreases skeletal muscle sympathetic outflow and plasma NE levels to about the same extent as the drug decreases ACTH levels (121). Perhaps glucocorticoid treatment of PTSD exerts effects at the level of the hypothalamic homeostat.
The findings of elevated CSF NE levels in PTSD patients (113) and augmented CSF NE responses to relevant emotional stimuli (114) indicate increased central noradrenergic activity and reactivity in PTSD. The main source of NE in the brain is the pontine locus ceruleus, and this region is well known to participate in vigilance (8), rapid eye movement sleep, and memory of distressing events. The hypothalamus, which receives innervation from noradrenergic centers lower in the brainstem, also is well known to play roles in vigilance (76) and distress responses, and medullary noradrenergic centers contribute to HPA responses to stressors (244). Central noradrenergic activation increases CRH release (182), which activates the HPA axis, stimulates adrenomedullary secretion (29), and contributes to adrenomedullary responses to distress (151, 164).
Parkinson disease and scientific integrative medicine
Disruption of negative feedback loops
The arterial baroreflex effectively buffers short-term changes in blood pressure, by reflexive changes in cardiovagal and sympathetic noradrenergic outflows. In PD, heart rate variability, a measure of baroreflex-cardiovagal outflow, is diminished (22, 171, 216, 265, 304, 310).
According to Braak’s concept for the pathogenetic sequence of PD (23), caudal medullary centers mediating baroreflex-cardiovagal outflow should be affected before rostral medullary centers mediating baroreflex-sympathoneural outflow. Consistent with this view, in PD without OH, baroreflex-cardiovagal function is decreased, while baroreflex-sympathoneural function often is normal (126, 161).
OH occurs fairly commonly in PD, and it is becoming increasingly recognized that OH can come on early in the disease process (128, 177). Since most patients with PD who do not have OH nevertheless have at least some loss of cardiac sympathetic nerves (136), PD appears to be not only a movement disorder but also a form of dysautonomia. Cardiac sympathetic denervation can precede the movement disorder by several years (143), providing a potential biomarker to detect the pathogenetic process in at-risk individuals and to track effects of putative neuroprotective agents.
Studies during the past decade have found consistently that all patients with PD+OH have neuroimaging evidence of cardiac sympathetic denervation. PD+OH patients also have evidence of extra-cardiac sympathetic noradrenergic denervation (141, 284), including decreased density of renal SNS innervation.
Recent findings suggest increased carotid stiffness in PD (unpublished observations), which would decrease baroreceptor afferent traffic. The combination of cardiac and extra-cardiac sympathetic denervation with afferent or central baroreflex failure may constitute a “triple whammy” that explains the OH attending PD (Fig. 33).
Figure 33.
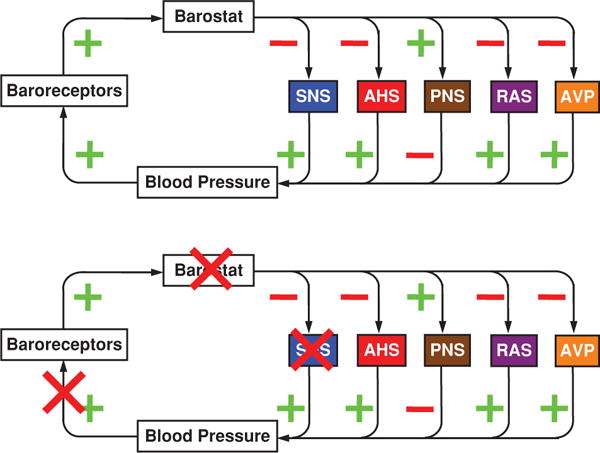
Multiple sites of interference with baroreflex regulation in Parkinson disease (PD) with orthostatic hypotension. Carotid wall thickening interferes with transduction of blood pressure information into baroreceptor afferent traffic. Alpha-synucleinopathy or neuronal loss in brainstem nuclei interferes with central barostatic function. Neuroimaging and neurochemical evidence indicates substantial noradrenergic denervation or dysfunction in the left ventricular myocardium, renal cortex, and other extra-cranial sites.
Autotoxicity and positive feedback loops
A reasonable societal goal of medical research is to enable each individual to live as long and productive a life as his or her genes endow, and then all body systems at the same time. In a way, Oliver Wendell Holmes expressed this wish in his famous poem about the one-horse shay:
Have you heard of the wonderful one-hoss shay,
That was built in such a logical way
It ran a hundred years to a day, ….
… it went to pieces all at once,—
All at once, and nothing first,—
Just as bubbles do when they burst.
If you had a heart that would last 90 years, and a 90 year liver, 90 year kidneys, and 90 year lungs—but a brain that would last only 70 years due to a neurodegenerative disease—then your goal would be to retard progression of the neurodegeneration sufficiently so that at 90 years old, all the organ systems would fail together. This sort of approach probably is better for the individual and surely is better for society in terms of the cost and other resources spent on health care.
The movement disorder in PD results from profound depletion of DA in the striatum (putamen and caudate), and all forms of Parkinsonism are associated with this depletion. Given that DA terminal loss is already far advanced by the time a patient manifests motor symptoms of PD, the future of experimental therapeutics in this area is in detection of the disease process in the presymptomatic phase, with the goal of retarding the neurodegeneration. It might even be possible to identify predispositions to catecholamine neuronal death that if dealt with effectively and early enough might prevent the disease process from developing at all during the person’s lifetime.
Positive feedback loops from autotoxicity
Randolph M. Nesse and George C. Williams, in their thought-provoking book, Why We Get Sick (236) pose the following question: “If senescence so devastates our fitness, why has not natural selection eliminated it?” Williams provided an answer in 1957 in his pleiotropic theory (318), according to which genes causing senescence have early benefits. In lay terms, “senescence is the price we pay for the vigor of youth.” The pleiotropic theory is the basis for the “getaway car analogy” (Figs. 34 and 35) as a potential explanation for the death of catecholamine neurons that causes motor and nonmotor manifestations of PD.
Figure 34.

The getaway car analogy. A car’s engine uses energy for locomotion. The bank robber’s getaway car is kept idling, so that the driver can rapidly shift from “park” to “drive.” As the engine idles, toxic combustion products are produced, which are detoxified by a catalytic converter. The oil lubricates the pistons. Eventually, the engine fails, and deposits are found in the engine and oil.
Figure 35.

Catecholamine neurons are like the idling getaway car engine. Catecholamines such as dopamine leak from storage vesicles into the cytoplasm, where they undergo enzymatic oxidative deamination catalyzed by MAO-A to form toxic catecholaldehydes such as DOPAL. DOPAL is detoxified by aldehyde dehydrogenase (ALDH). Eventually the catecholaminergic neurons die, and deposits of alpha-synuclein are found in Lewy bodies.
Suppose you were a bank robber, with your getaway car idling at the curb outside the bank. The car would be idling because when the time came you would have to get away, and fast. There are obvious advantages of having a getaway car idling, but there is a cost. Eventually, the engine could break down—especially if there were design flaws or manufacturing defects, or the engine were “revved” continually, or the fuel were contaminated or the oil formulated wrong. In the vocabulary of scientific integrative medicine, allostatic load on the engine would be the price paid for keeping the getaway car idling.
Let us consider two specific first causes of allostatic load in the getaway car—a faulty catalytic converter and a faulty fuel recovery system. For the first, suppose that combustion of the fuel resulted in formation of toxic byproducts. If the catalytic converter were dysfunctional, the toxic byproducts could build up and damage the engine. For the second, suppose that the fuel injector worked by injecting fuel into the combustion chamber at a high rate, but with an almost equally high rate of fuel recovery back into the injector, so that in the idling situation the actual rate of combustion were low. If the fuel recovery system were dysfunctional, so that in the idling situation there were too rapid a net rate of injection of fuel, the combustion rate would be high. The rate of production of toxic byproducts might exceed the capacity of even a normally functioning catalytic converter, and this would also damage the engine.
As a final component of the getaway car analogy, consider the oil lubricating the pistons. Toxic combustion products would increase the rate of accumulation of deposits in the piston chamber and the oil. At first, these deposits might not cause harm themselves, but eventually there would so much gunk buildup that the pistons would freeze up, even with a normal rate of fuel combustion, normal fuel recycling, and normal detoxification of the combustion byproduct by the catalytic converter.
Catecholamine neurons are like the idling engine of the getaway car (Fig. 35)
They are “on” all the time, in that the vesicular stores leak passively continuously into the cytoplasm. Most of the cytosolic catecholamine is recycled actively by reuptake into the vesicles via the vesicular monoamine transporter (VMAT); however, a small proportion undergoes oxidation—”combustion.” In the cytoplasm of dopaminergic neurons, oxidative deamination of DA is catalyzed by MAO-A in the outer mitochondrial membrane, to form DOPAL. In noradrenergic neurons, the same leakage process goes on, with MAO-A catalyzing conversion of NE to DOPEGAL. Indeed, all of the enzymatic metabolism of cytosolic catecholamines is channeled through catecholaldehydes, which are formed continuously in neuronal life.
One may ask, what good does it do to have leaky vesicular stores of catecholamines? My colleague Graeme Eisenhofer provided an insightful answer—”gearing down” (85). He referred to NE in sympathetic nerves, but an identical argument can be made for DA in nigrostriatal terminals.
Turnover of catecholamines, representing the constant loss and replenishment of neurotransmitter by synthesis, is usually considered to be driven exclusively by catecholamine release. This is incorrect … at least 75% of norepinephrine turnover is due to intraneuronal metabolism without prior release at sympathetic nerve endings …. The large contribution of leakage to catecholamine turnover may seem inconsistent with cellular economy. In fact, this contribution provides an important mechanism for “gearing down” the requirement for increases in catecholamine synthesis to match increases in catecholamine release, and thereby provides sympathetic nerves with a capacity for a more extended range of sustainable release rates in response to stress than would otherwise be possible.
Catecholamine neurons are rare in the brain and periphery. What makes these neurons susceptible to loss in neurodegenerative diseases like PD? An answer comes from the hypothesis that the immediate products of oxidative deamination of intra-neuronal catecholamines, catecholaldehydes, are toxic. This is the centerpiece of the “catecholaldehyde hypothesis” (Fig. 36). Ongoing production of DOPAL in striatal dopaminergic neurons is a necessary consequence of the “gearing down” that has provided survival advantages in evolution. The cost is eventual loss of those neurons, due to accumulation of allostatic load and precipitation of positive feedback loops.
Figure 36.

The “catecholaldehyde hypothesis.” According to this hypothesis, decreased vesicular sequestration of cytosolic catecholamines and impaired catecholaldehyde detoxification cause the death of catecholamine neurons that characterizes Parkinson disease. Under resting conditions, most of the irreversible loss of dopamine (DA) from the neurons is due to passive leakage from vesicles (DAv) into the cytosol (DAc) and efficient but imperfect vesicular uptake mediated by the type 2 vesicular monoamine transporter (VMAT2). This loss is balanced by ongoing catecholamine biosynthesis from the action of L-aromatic-amino-acid decarboxylase (LAAAD) on 3,4-dihydroxyphenylalanine (DOPA) produced from tyrosine (TYR) by tyrosine hydroxylase (TH). Release by exocytosis is followed by reuptake mediated by the cell membrane DA transporter (DAT). Intra-neuronal metabolism of DA is channeled through enzymatic deamination catalyzed by monoamine oxidase (MAO), producing the catecholaldehyde 3,4-dihydroxyphenylacetaldehyde (DOPAL). DOPAL is detoxified mainly by aldehyde dehydrogenase (ALDH), to form the acid, 3,4-dihydroxyphenylacetic acid (DOPAC), with 3,4-dihydroxyphenylethanol (DOPET) a minor metabolite formed via aldose/aldehyde reductase (AR). Both DAc and DOPAL spontaneously auto-oxidize to quinones, which augment generation of reactive oxygen species (ROS), resulting in lipid peroxidation. 4-Hydroxynonenal (4HNE), a major lipid peroxidation product, inhibits ALDH. DOPAL cross-links with proteins, augmenting oligomerization of alpha-synuclein and inhibiting TH.
Organisms have faced the challenge of dealing with aldehydes for millions of years. The main enzyme detoxifying DOPAL, the deaminated metabolite of DA, is aldehyde dehydrogenase (ALDH), with aldehyde/aldose reductase (AR) playing a minor alternative role; and the main enzyme detoxifying DOPEGAL, the deaminated metabolite of NE, is AR, with ALDH playing a minor alternative role. This difference in metabolic fates explains why the main end-product of intra-neuronal DA metabolism is the acid, dihydroxyphenylacetic acid (DOPAC), whereas the main end-product of intraneuronal NE metabolism is the glycol, dihydroxyphenylglycol (DHPG).
According to the catecholaldehyde hypothesis, the aldehydes of DA and NE cause or contribute to the death of catecholamine neurons in the brain and heart in PD. That catecholaldehydes are indeed toxic is well established (32, 246). The toxicity occurs by several routes, including protein cross-linking (260), auto-oxidation to form quinones (6), generation of free radicals (198), and oligomerization of alpha-synuclein (30) (more on this below).
Is there evidence that DOPAL actually does build up in PD? In postmortem tissue of the putamen, the site of the most severe loss of dopaminergic terminals, DOPAL concentrations are indeed higher with respect to DA in PD patients than in controls (147).
What causes the buildup of DOPAL? One potential cause is decreased ALDH activity. This would correspond to a faulty catalytic converter (enzymes literally are catalytic converters). Mice with double knockout of the genes encoding ALDH1A1 and ALDH2 have abnormalities mimicking those in PD (317). We developed and validated a method to measure tissue ALDH activity from the ratio of DOPAC:DOPAL, and by this measure ALDH activity is decreased in the putamen in PD (147). Blockade of ALDH (and of the alternative enzyme AR) augments intracellular DOPAL levels (146).
A second potential cause of DOPAL buildup is decreased activity of VMAT2, which is responsible for sequestering cytosolic DA into the vesicles in dopaminergic neurons. This would correspond to faulty fuel recovery in the getaway car. Mice with inherited very low VMAT2 activity have neuropathologic and neurobehavioral abnormalities reminiscent of motor and nonmotor features of PD (44, 298). We have found that vesicular uptake is decreased in Lewy body diseases (135). Moreover, we now know that blockade of vesicular uptake augments intracellular DOPAL levels (146).
What of the “gunk” buildup in the failing getaway car engine? The protein, alpha-synuclein, normally is dissolved in the neuronal cytoplasm. The functions of alpha-synuclein remain incompletely understood; however, alpha-synucleinopathy predisposes to PD. The first identified genotypic abnormality found to cause familial PD (PARK1) was mutation of the gene encoding this protein (254); genome-wide association studies have consistently identified other genotypic abnormalities of the alpha-synuclein gene in PD patients (81, 268); and deposits of alpha-synuclein are found characteristically in Lewy bodies, the pathologic hallmark of PD (293). Inherited replication of the normal gene encoding alpha-synuclein (289) increases alpha-synuclein protein content (33). Synuclein gene replication produces not only a familial form of PD but also cardiac sympathetic denervation (288). DOPAL potently oligomerizes alpha-synuclein (30), and it is thought that oligomerized alpha-synuclein is the pathogenetically significant form of the protein (28, 319). In other words, the toxic byproducts of combustion in the getaway car engine not only harm the engine directly but also accelerate the buildup of the gunk.
In summary, due to the pleiotropic effect of improved resilience and antifatigue in youth at the cost of aging-related autotoxicity, accumulation of allostatic load in catecholaminergic neurons may lead eventually to multiple positive feedback loops and death of those neurons in PD.
Compensatory activation and timing of neuroprotective treatment
As striatal dopaminergic terminals are lost, a variety of compensatory adjustments take place, such as increased DA release from remaining terminals (34, 327), decreased reuptake of released DA (291), increased DA synthesis via tyrosine hydroxylase (16), up-regulation of postsynaptic DA receptors (169, 250), and increased excitability of the target striatal medium spiny neurons (9). These adaptive changes, which exemplify compensatory activation, maintain dopaminergic functions until the loss of the terminals is advanced.
Because of compensatory activation in PD, DA delivery to the extracellular fluid can be maintained despite decreased intra-neuronal stores. It can be shown mathematically that compensatory activation prolongs the time before the disease process manifests clinically (Fig. 38).
Figure 38.
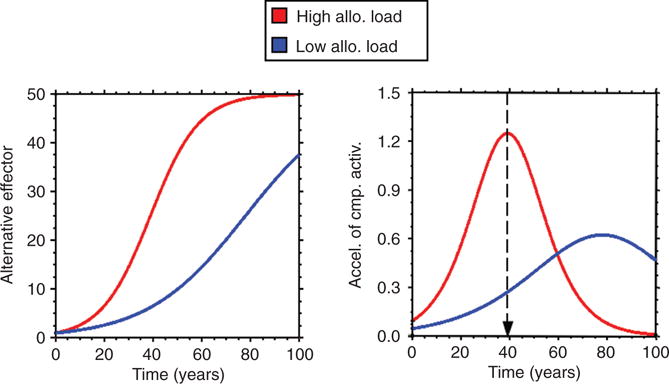
Computer model-generated curves illustrating that compensatory activation prolongs the time before a disease process manifests clinically. Tracking the rate of compensatory activation may inform decision-making about appropriate timing for initiation of neuroprotective treatment.
The catecholaldehyde hypothesis predicts straightforwardly that MAO inhibition should attenuate or prevent death of catecholamine neurons. This would be analogous to decreasing the idle rate of the getaway car. Indeed, MAO-B inhibitors are used currently to treat symptomatic PD, and there is evidence—albeit controversial—that MAO-B inhibition may retard the neurodegenerative process (240,241). The MAO isoform in catecholaminergic neurons is MAO-A, not MAO-B, and so why MAO-B inhibitors should be neuroprotective, if they are, remains mysterious (234). In humans, treatment with L-deprenyl (selegiline), a selective MAO-B inhibitor, decreases plasma levels of both DOPAC and DHPG—that is, MAO-B inhibition may indirectly decrease oxidative deamination of DA and NE (83). Since DA is a better substrate than NE is for MAO-A (unpublished observations), a potential treatment of people at risk for PD who have biomarkers of loss of catecholamine neurons is treatment with an L-deprenyl patch at the lowest dose that decreases plasma DOPAC levels. MAO-B in nondopaminergic cells is important for converting 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) to MPP, which is well established to produce Parkinsonism via DA neuronal death (195). Accordingly, MAO-B inhibition might interfere with production of other toxins of exogenous origin. Whether MAO-B inhibition interferes with autotoxic DOPAL formation in nondopaminergic cells is unknown.
A potential neurochemical correlate of compensatory activation in PD is the concentration ratio of DA:DOPAC in cerebrospinal fluid (CSF). This is because of likely differential dependence of CSF DA and CSF DOPAC on release and reuptake of DA vs. leakage of vesicular stores into the cytoplasm. Consistent with this view, across individual PD patients, CSF DOPAC is less than expected for CSF DA (137); however, since decreased ALDH activity would produce the same abnormal pattern, whether CSF DA:DOPAC provides a valid index of compensatory activation in PD remains unproven.
The lipid peroxidation product, 4-hydroxynonenal (4-HNE), is an aldehyde, and 4-HNE inhibits ALDH, resulting in increased DOPAL production (259). Another therapeutic strategy is to increase the efficiency of ALDH, analogous to increasing the efficiency of the catalytic converter in the getaway car, or to develop an aldehyde scavenger that could substitute for ALDH in detoxifying 4-HNE and DOPAL.
When would one initiate treatment for a person with presymptomatic disease who has demonstrable loss of catecholamine neurons? Developing means to detect compensatory activation could provide a biomarker by which to decide on appropriate timing for initiation of a neuroprotective strategy (Fig. 38). In the home heating system, if there were a furnace and a heat pump, and overhauling the furnace were very expensive, when would you decide on the timing of the overhaul? The earliest measure of decreased furnace efficiency from wear and tear would not be a fall in internal temperature during the winter but an acceleration in the activity of the alternative effector, the heat pump. In a patient at risk for developing PD, the earliest measure of loss of DA terminals would not be symptoms of motor impairment but compensatory activation, perhaps as measured by a neurochemical index of the balance of nigrostriatal pathway traffic versus stored DA. From the notion of compensatory activation discussed above, treatment would begin when the index of compensatory activation reached its peak velocity of change.
What about preventing the disease process entirely? In the new era of individualized medicine, it may be possible to identify genetic predispositions, where “life counseling” might prevent the death of catecholamine neurons and thereby prevent PD from developing during the person’s lifetime. For instance, ALDH1A1 gene expression is decreased in the blood of patients with PD (225), mice with combined ALDH1A1,2 knockout have congenitally increased striatal DOPAL levels and aging-related neurobehavioral and neuropathological findings mimicking PD (317), PD patients have decreased putamen ALDH activity (147), and ALDH inhibition augments DOPAL responses vesicular uptake blockade (146). From these findings one may predict that in the future people with congenitally low ALDH activity detected by neurochemical testing after early postnatal genetic profiling may be counseled to avoid careers involving high rates of exposure to agents such as pesticides that inhibit ALDH or vesicular uptake. Meanwhile, indirect epidemiological evidence suggests that exercise training might increase resilience and retard development of PD (3, 323), although one cannot exclude the possibility that less participation in physical activity is an early manifestation of the disease process.
In conclusion, about treatment or prevention, by tracking compensatory activation in presymptomatic individuals one might be able to optimize timing of initiation of neuroprotective treatment with drugs that inhibit MAO, detoxify catecholaldehydes, attenuate generation of reactive oxygen species related to DOPAL, or interfere with DOPAL-induced oligomerization of alpha-synuclein.
Summary and Conclusions
Concepts of scientific integrative medicine help link current systems biology with classic integrative physiology. Four distinguishing aspects of scientific integrative medicine are the emphasis on via negative feedback regulation of monitored variables mediated by homeostats; dominance of the brain in coordinated, parallel processing of afferent information and determination of effector activities; plasticity afforded by allostatic adjustments of homeostat settings; and allostatic load as applied to mechanisms and treatments of chronic diseases and disorders.
Stress can be defined as a condition in which a sensed discrepancy between afferent information about a monitored variable and a homeostatic set-point for responding leads to changes in effector activities in a manner that reduces the discrepancy—the error signal in the negative feedback loop. A noncircular definition of distress is that it is a form of stress that is consciously experienced, aversive, entails instinctively communicated signs, and features adrenal activation.
Catecholamines are key chemical agents for effectors in many of the negative feedback loops that maintain levels of internal monitored variables. Studies of catecholaminergic responses to stressors illustrate principles such as negative feedback regulation, compensatory activation, and effector sharing.
Allostasis refers to adjustments in homeostatic settings. Allostatic load—cumulative wear and tear on an effector—relates stress with degenerative disorders. The timing and rapidity of system failure from positive feedback loops depend on dynamic interactions between usage experience of the system, built-in manufacturing and design characteristics, compensatory activation of alternative effectors, and processes such as habituation and proactive feed-forward adjustments that increase resilience.
Scientific integrative medicine focuses on prevention and treatment in the presymptomatic phase of degenerative disorders. The goal is to prevent positive feedback loops that cause premature system failure. In adults, common, complex, modern-day diseases are mainly disorders of regulation, only indirectly related to genetic changes.
Pleiotropy can explain “leaky” vesicular stores of catecholamines, and the getaway car analogy can explain aging-related autotoxic death of catecholamine neurons in PD. Applying the principle of compensatory activation may help identify the best time to initiate neuroprotective treatment in premotor PD.
The genes are life’s blueprint, but continuous information processing by the brain and myriad dynamic, compensatory, and proactive adjustments in activities of multiple effectors enable life to go on. Scientific integrative medicine provides a framework and lexicon for undertstanding how that processing and those adjustments can go awry and on what might be done to retard or prevent degenerative loss of control of internal monitored variables, so that we live as long and productive lives as our genes endow, until, like in Holmes’s poem about the hundred year old one-hoss shay, the bubble bursts.
Figure 6.

Predicted values for levels of the monitored variable as a function of time, with negative feedback by both proportionate and integrated control. The level of the monitored variable returns to the baseline level. The rate of attainment of the baseline level depends on the rate constant for the effector (kEffector). The predicted curve fits well the blood pressure response to tilting in Figure 4.
Figure 20.

Computer model of effector sharing. In this model, two homeostats determine the state of activity of the same effector, which in turn affects levels of two monitored variables. Via effector sharing, a stressor affecting levels of one monitored variable results in altered levels of a different monitored variable.
Figure 37.

Pathogenetic mechanisms resulting in loss of catecholaminergic neurons may reflect induction of a variety of positive feedback loops.
Footnotes
This article is a U.S. government work and is in the public domain in the U.S.A.
References
- 1.Abboud FM. Neurocardiogenic syncope. N Engl J Med. 1993;328:1117–1120. doi: 10.1056/NEJM199304153281510. [DOI] [PubMed] [Google Scholar]
- 2.Abboud FM, Eckberg DL, Johannsen UJ, Mark AL. Carotid and cardiopulmonary baroreceptor control of splanchnic and forearm vascular resistance during venous pooling in man. J Physiol. 1979;286:173–184. doi: 10.1113/jphysiol.1979.sp012612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahlskog JE. Does vigorous exercise have a neuroprotective effect in Parkinson disease? Neurology. 2011;77:288–294. doi: 10.1212/WNL.0b013e318225ab66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aicher SA, Kurucz OS, Reis DJ, Milner TA. Nucleus tractus solitarius efferent terminals synapse on neurons in the caudal ventrolateral medulla that project to the rostral ventrolateral medulla. Brain Res. 1995;693:51–63. doi: 10.1016/0006-8993(95)00660-i. [DOI] [PubMed] [Google Scholar]
- 5.Akashi YJ, Goldstein DS, Barbaro G, Ueyama T. Takotsubo cardiomyopathy: A new form of acute, reversible heart failure. Circulation. 2008;118:2754–2762. doi: 10.1161/CIRCULATIONAHA.108.767012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson DG, Mariappan SV, Buettner GR, Doorn JA. Oxidation of 3,4-dihydroxyphenylacetaldehyde, a toxic dopaminergic metabolite, to a semiquinone radical and an ortho-quinone. J Biol Chem. 2011;286:26978–26986. doi: 10.1074/jbc.M111.249532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Antonov I, Kandel ER, Hawkins RD. Presynaptic and postsynaptic mechanisms of synaptic plasticity and metaplasticity during intermediate-term memory formation in Aplysia. J Neurosci. 2010;30:5781–5791. doi: 10.1523/JNEUROSCI.4947-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aston-Jones G, Rajkowski J, Cohen J. Role of locus coeruleus in attention and behavioral flexibility. Biol Psychiatry. 1999;46:1309–1320. doi: 10.1016/s0006-3223(99)00140-7. [DOI] [PubMed] [Google Scholar]
- 9.Azdad K, Chavez M, Don Bischop P, Wetzelaer P, Marescau B, De Deyn PP, Gall D, Schiffmann SN. Homeostatic plasticity of striatal neurons intrinsic excitability following dopamine depletion. PloS one. 2009;4:e6908. doi: 10.1371/journal.pone.0006908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barcroft H, Edholm OG. On the vasodilatation in human skeletal muscle during post-haemorrhagic fainting. J Physiol. 1945;104:161–175. doi: 10.1113/jphysiol.1945.sp004113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bechtold DA, Loudon AS. Hypothalamic clocks and rhythms in feeding behaviour. Trends Neurosci. 2013;36:74–82. doi: 10.1016/j.tins.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 12.Belkin V, Karasik D. Anthropometric characteristics of men in Antarctica. Int J Circumpolar Health. 1999;58:152–169. [PubMed] [Google Scholar]
- 13.Berecek KH, Work J, Mitchum TN, Ram S. Effects of chronic peripheral sympathectomy on plasma levels of, and the pressor response to, vasopressin. J, Hypertens. 1985;3:225–230. doi: 10.1097/00004872-198506000-00005. [DOI] [PubMed] [Google Scholar]
- 14.Bereiter DA, Zaid AM, Gann DS. Effect of rate of hemorrhage on sympathoadrenal catecholamine release in cats. Am J Physiol. 1986;250:E69–E75. doi: 10.1152/ajpendo.1986.250.1.E69. [DOI] [PubMed] [Google Scholar]
- 15.Best JD, Berghmans S, Hunt JJ, Clarke SC, Fleming A, Goldsmith P, Roach AG. Non-associative learning in larval zebrafish. Neuropsychopharmacology. 2008;33:1206–1215. doi: 10.1038/sj.npp.1301489. [DOI] [PubMed] [Google Scholar]
- 16.Bezard E, Jaber M, Gonon F, Boireau A, Bloch B, Gross CE. Adaptive changes in the nigrostriatal pathway in response to increased 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurodegeneration in the mouse. Eur J Neurosci. 2000;12:2892–2900. doi: 10.1046/j.1460-9568.2000.00180.x. [DOI] [PubMed] [Google Scholar]
- 17.Biaggioni I, Whetsell WO, Jobe J, Nadeau JH. Baroreflex failure in a patient with central nervous system lesions involving the nucleus tractus solitarii. Hypertension. 1994;23:491–495. doi: 10.1161/01.hyp.23.4.491. [DOI] [PubMed] [Google Scholar]
- 18.Blanchard EB, Kolb LC, Prins A, Gates S, McCoy GC. Changes in plasma norepinephrine to combat-related stimuli among Vietnam veterans with posttraumatic stress disorder. J Nerv Ment Dis. 1991;179:371–373. doi: 10.1097/00005053-199106000-00012. [DOI] [PubMed] [Google Scholar]
- 19.Blumenthal JA, Fredrikson M, Kuhn CM, Ulmer RL, Walsh-Riddle M, Appelbaum M. Aerobic exercise reduces levels of cardiovascular and sympathoadrenal responses to mental stress in subjects without prior evidence of myocardial ischemia. Am J Cardiol. 1990;65:93–98. doi: 10.1016/0002-9149(90)90032-v. [DOI] [PubMed] [Google Scholar]
- 20.Bogen DK. Simulation software for the Macintosh. Science. 1989;246:138–142. doi: 10.1126/science.246.4926.138. [DOI] [PubMed] [Google Scholar]
- 21.Booth FW, Laye MJ, Roberts MD. Lifetime sedentary living accelerates some aspects of secondary aging. J Appl Physiol. 2011;111:1497–1504. doi: 10.1152/japplphysiol.00420.2011. [DOI] [PubMed] [Google Scholar]
- 22.Bouhaddi M, Vuillier F, Fortrat JO, Cappelle S, Henriet MT, Rumbach L, Regnard J. Impaired cardiovascular autonomic control in newly and long-term-treated patients with Parkinson’s disease: Involvement of L-dopa therapy. Auton Neurosci. 2004;116:30–38. doi: 10.1016/j.autneu.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 23.Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004;318:121–134. doi: 10.1007/s00441-004-0956-9. [DOI] [PubMed] [Google Scholar]
- 24.Breier A, Davis O, Buchanan R, Listwak S, Holmes C, Pickard D, Goldstein D. Effects of alprazolam on pituitary-adrenal and catecholaminergic responses to metabolic stress in humans. Biol Psychiatry. 1992;32:880–890. doi: 10.1016/0006-3223(92)90177-2. [DOI] [PubMed] [Google Scholar]
- 25.Breier A, Davis O, Buchanan R, Listwak SJ, Holmes C, Pickar D, Goldstein DS. Effects of alprazolam on pituitary-adrenal and catecholaminergic responses to metabolic stress in humans. Biol Psychiatry. 1992;32:880–890. doi: 10.1016/0006-3223(92)90177-2. [DOI] [PubMed] [Google Scholar]
- 26.Bridgers SL, Spencer SS, Spencer DD, Sasaki CT. A cerebral effect of carotid sinus stimulation. Observation during intraoperative electroencephalographic monitoring. Arch Neurol. 1985;42:574–577. doi: 10.1001/archneur.1985.04060060076012. [DOI] [PubMed] [Google Scholar]
- 27.Brown CM, Hecht MJ, Neundorfer B, Hilz MJ. Effects of lower body negative pressure on cardiac and vascular responses to carotid baroreflex stimulation. Physiol Res. 2003;52:637–645. [PubMed] [Google Scholar]
- 28.Brown DR. Oligomeric alpha-synuclein and its role in neuronal death. IUBMB Life. 2010;62:334–339. doi: 10.1002/iub.316. [DOI] [PubMed] [Google Scholar]
- 29.Brown MR, Fisher LA, Webb V, Vale WW, Rivier JE. Corticotropin-releasing factor: A physiologic regulator of adrenal epinephrine secretion. Brain Res. 1985;328:355–357. doi: 10.1016/0006-8993(85)91048-0. [DOI] [PubMed] [Google Scholar]
- 30.Burke WJ, Kumar VB, Pandey N, Panneton WM, Gan Q, Franko MW, O’Dell M, Li SW, Pan Y, Chung HD, Galvin JE. Aggregation of alpha-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol. 2008;115:193–203. doi: 10.1007/s00401-007-0303-9. [DOI] [PubMed] [Google Scholar]
- 31.Burke WJ, Li SW, Chung HD, Ruggiero DA, Kristal BS, Johnson EM, Lampe P, Kumar VB, Franko M, Williams EA, Zahm DS. Neurotoxicity of MAO metabolites of catecholamine neurotransmitters: Role in neurodegenerative diseases. Neurotoxicology. 2004;25:101–115. doi: 10.1016/S0161-813X(03)00090-1. [DOI] [PubMed] [Google Scholar]
- 32.Burke WJ, Li SW, Williams EA, Nonneman R, Zahm DS. 3,4-Dihydroxyphenylacetaldehyde is the toxic dopamine metabolite in vivo: Implications for Parkinson’s disease pathogenesis. Brain Res. 2003;989:205–213. doi: 10.1016/s0006-8993(03)03354-7. [DOI] [PubMed] [Google Scholar]
- 33.Byers B, Cord B, Nguyen HN, Schule B, Fenno L, Lee PC, Deisseroth K, Langston JW, Pera RR, Palmer TD. SNCA triplication Parkinson’s patient’s iPSC-derived DA neurons accumulate alpha-synuclein and are susceptible to oxidative stress. PloS One. 2011;6:e26159. doi: 10.1371/journal.pone.0026159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Calne DB, Zigmond MJ. Compensatory mechanisms in degenerative neurologic diseases. Insights from parkinsonism. Arch Neurol. 1991;48:361–363. doi: 10.1001/archneur.1991.00530160025009. [DOI] [PubMed] [Google Scholar]
- 35.Cannon WB. Bodily Changes in Pain, Hunger, Fear and Rage. New York: D. Appleton & Co; 1929a. [Google Scholar]
- 36.Cannon WB. Organization for physiological homeostasis. Physiol Rev. 1929b;9:399–431. [Google Scholar]
- 37.Cannon WB. The effects of progressive sympathectomy on blood pressure. Am J Physiol. 1931;97:592–595. [Google Scholar]
- 38.Cannon WB. The Wisdom of the Body. New York: W.W. Norton; 1939. [Google Scholar]
- 39.Cannon WB, Britton SW. The influence of motion and emotion on medulliadrenal secretion. Am J Physiol. 1927;79:433–465. [Google Scholar]
- 40.Cannon WB, de la Paz D. Emotional stimulation of adrenal gland secretion. Am J Physiol. 1911;28:64–70. [Google Scholar]
- 41.Cannon WB, Lissak K. Evidence for adrenaline in adrenergic neurones. Am J Physiol. 1939;125:765–777. [Google Scholar]
- 42.Carey LC, Curtin R, Sapira JD. Influence of hemorrhage on adrenal secretion, blood glucose and serum insulin in the awake pig. Ann Surg. 1976;183:185–192. doi: 10.1097/00000658-197602000-00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carlsten A, Folkow B, Grimby G, Hamberger CA, Thulesius O. Cardiovascular effects of direct stimulation of the carotid sinus nerve in man. Acta Physiol Scand. 1958;44:138–145. doi: 10.1111/j.1748-1716.1958.tb01615.x. [DOI] [PubMed] [Google Scholar]
- 44.Caudle WM, Richardson JR, Wang MZ, Taylor TN, Guillot TS, McCormack AL, Colebrooke RE, Di Monte DA, Emson PC, Miller GW. Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J Neurosci. 2007;27:8138–8148. doi: 10.1523/JNEUROSCI.0319-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chan CC, Kalsner S. Termination of responses to sympathetic nerve stimulation and to noradrenaline in a perfused arterial preparation: The role of neuronal and extraneuronal uptake. J Pharmacol Exp Ther. 1982;222:731–740. [PubMed] [Google Scholar]
- 46.Charkoudian N. Skin blood flow in adult human thermoregulation: How it works, when it does not, and why. Mayo Clin Proc. 2003;78:603–612. doi: 10.4065/78.5.603. [DOI] [PubMed] [Google Scholar]
- 47.Charles ST. Strength and vulnerability integration: A model of emotional well-being across adulthood. Psychol Bull. 2010;136:1068–1091. doi: 10.1037/a0021232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chidsey CA, Braunwald E. Sympathetic activity and neurotransmitter depletion in congestive heart failure. Pharmacol Rev. 1966;18:685–700. [PubMed] [Google Scholar]
- 49.Chopra M, Das P, Golden H, Dostal DE, Watson LE, Sharma AC. Norepinephrine induces systolic failure and inhibits antiapoptotic genes in a polymicrobial septic rat model. Life sciences. 2010;87:672–678. doi: 10.1016/j.lfs.2010.09.029. [DOI] [PubMed] [Google Scholar]
- 50.Chrousos GP, Gold PW. The concepts of stress and stress system disorders. Overview of physical and behavioral homeostasis. J Am Med Assoc. 1992;267:1244–1252. [PubMed] [Google Scholar]
- 51.Claydon VE, Gulli G, Slessarev M, Appenzeller O, Zenebe G, Gebremedhin A, Hainsworth R. Cerebrovascular responses to hypoxia and hypocapnia in Ethiopian high altitude dwellers. Stroke. 2008;39:336–342. doi: 10.1161/STROKEAHA.107.491498. [DOI] [PubMed] [Google Scholar]
- 52.Cryer PE. Physiology and pathophysiology of the human sympathoadrenal neuroendocrine system. N Engl J Med. 1980;303:436–444. doi: 10.1056/NEJM198008213030806. [DOI] [PubMed] [Google Scholar]
- 53.Cui J, Durand S, Levine BD, Crandall CG. Effect of skin surface cooling on central venous pressure during orthostatic challenge. Am J Physiol Heart Circ Physiol. 2005;289:H2429–H2433. doi: 10.1152/ajpheart.00383.2005. [DOI] [PubMed] [Google Scholar]
- 54.Damasio A. Descartes’ Error. Emotion, Reason, and the Human Brain. New York: Avon Books, Inc; 1994. [Google Scholar]
- 55.Damasio A. The Feeling of What Happens. New York: Harcourt Brace & Company; 1999. [Google Scholar]
- 56.Dangovian MI, Jarandilla R, Frumin H. Prolonged asystole during head-up tilt table testing after beta blockade. Pacing Clin Electrophysiol. 1992;15:14–16. doi: 10.1111/j.1540-8159.1992.tb02895.x. [DOI] [PubMed] [Google Scholar]
- 57.Darlington DN, Shinsako J, Dallman MF. Responses of ACTH, epinephrine, norepinephrine, and cardiovascular system to hemorrhage. Am J Physiol. 1986;251:H612–H618. doi: 10.1152/ajpheart.1986.251.3.H612. [DOI] [PubMed] [Google Scholar]
- 58.Darwin C. The Expression of the Emotions in Man and Animals. Chicago: Univ. of Chicago Press; 1965. [Google Scholar]
- 59.Davrath LR, Gotshall RW, Tucker A, Sadeh WZ, Luckasen GJ, Downes TR, Coonts CC. The heart is not necessarily empty at syncope. Aviat Space Environ Med. 1999;70:213–219. [PubMed] [Google Scholar]
- 60.Dawkins R. The Selfish Gene. New York: Oxford University Press; 1989. [Google Scholar]
- 61.De Boer SF, Slangen JL, Van der Gugten J. Plasma catecholamine and corticosterone levels during active and passive shock-prod avoidance behavior in rats: Effects of chlordiazepoxide. Physiol Behav. 1990;47:1089–1098. doi: 10.1016/0031-9384(90)90357-a. [DOI] [PubMed] [Google Scholar]
- 62.Desir GV. Role of renalase in the regulation of blood pressure and the renal dopamine system. Curr Opin Nephrol Hypertens. 2011;20:31–36. doi: 10.1097/MNH.0b013e3283412721. [DOI] [PubMed] [Google Scholar]
- 63.Deuster PA, Chrousos GP, Luger A, DeBolt JE, Bernier LL, Trostmann UH, Kyle SB, Montgomery LC, Loriaux DL. Hormonal and metabolic responses of untrained, moderately trained, and highly trained men to three exercise intensities. Metabolism. 1989;38:141–148. doi: 10.1016/0026-0495(89)90253-9. [DOI] [PubMed] [Google Scholar]
- 64.DiBona GF. Neural regulation of renal tubular sodium reabsorption and renin secretion. Fed Proc. 1985;44:2816–2822. [PubMed] [Google Scholar]
- 65.DiBona GF. Neural mechanisms in body fluid homeostasis. Fed Proc. 1986;45:2871–2877. [PubMed] [Google Scholar]
- 66.DiBona GF. Sympathetic nervous system and the kidney in hypertension. Curr Opin Nephrol Hypertens. 2002;11:197–200. doi: 10.1097/00041552-200203000-00011. [DOI] [PubMed] [Google Scholar]
- 67.DiBona GF. Central angiotensin modulation of baroreflex control of renal sympathetic nerve activity in the rat: Influence of dietary sodium. Acta Physiol Scand. 2003;177:285–289. doi: 10.1046/j.1365-201X.2003.01074.x. [DOI] [PubMed] [Google Scholar]
- 68.Dickinson CJ. Fainting precipitated by collapse-firing of venous baroreceptors. Lancet. 1993;342:970–972. doi: 10.1016/0140-6736(93)92008-h. [DOI] [PubMed] [Google Scholar]
- 69.Dietz NM, Halliwill JR, Spielmann JM, Lawler LA, Papouchado BG, Eickhoff TJ, Joyner MJ. Sympathetic withdrawal and forearm vasodilation during vasovagal syncope in humans. J Appl Physiol. 1997;82:1785–1793. doi: 10.1152/jappl.1997.82.6.1785. [DOI] [PubMed] [Google Scholar]
- 70.Dietz NM, Joyner MJ, Shepherd JT. Vasovagal syncope and skeletal muscle vasodilatation: The continuing conundrum. Pacing Clin Electrophysiol. 1997;20:775–780. doi: 10.1111/j.1540-8159.1997.tb03903.x. [DOI] [PubMed] [Google Scholar]
- 71.Dobrakovova M, Kvetnansky R, Oprsalova Z, Jezova D. Specificity of the effect of repeated handling on sympathetic-adrenomedullary and pituitary-adrenocortical activity in rats. Psychoneuroendocrinology. 1993;18:163–174. doi: 10.1016/0306-4530(93)90001-2. [DOI] [PubMed] [Google Scholar]
- 72.Douglas PS, O’Toole ML, Katz SE. Prolonged exercise alters cardiac chronotropic responsiveness in endurance athletes. J Sports Med Phys Fitness. 1998;38:158–163. [PubMed] [Google Scholar]
- 73.Dronjak S, Jezova D, Kvetnansky R. Different effects of novel stressors on sympathoadrenal system activation in rats exposed to long-term immobilization. Ann N Y Acad Sci. 2004;1018:113–123. doi: 10.1196/annals.1296.013. [DOI] [PubMed] [Google Scholar]
- 74.Duan H, Wang J. Selective transport of monoamine neurotransmitters by human plasma membrane monoamine transporter and organic cation transporter 3. J Pharmacol Exp Ther. 2010;335:743–753. doi: 10.1124/jpet.110.170142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Duan YF, Kopin IJ, Goldstein DS. Stimulation of the paraventricular nucleus modulates firing of neurons in the nucleus of the solitary tract. Am J Physiol. 1999;277:R403–R411. doi: 10.1152/ajpregu.1999.277.2.R403. [DOI] [PubMed] [Google Scholar]
- 76.Duan YF, Winters R, McCabe PM, Green EJ, Huang Y, Schneiderman N. Behavioral characteristics of defense and vigilance reactions elicited by electrical stimulation of the hypothalamus in rabbits. Behav Brain Res. 1996;81:33–41. doi: 10.1016/s0166-4328(96)00042-3. [DOI] [PubMed] [Google Scholar]
- 77.Durand S, Cui J, Williams KD, Crandall CG. Skin surface cooling improves orthostatic tolerance in normothermic individuals. Am J Physiol Regul Integr Comp Physiol. 2004;286:R199–R205. doi: 10.1152/ajpregu.00394.2003. [DOI] [PubMed] [Google Scholar]
- 78.Dworkin BR, Filewich RJ, Miller NE, Craigmyle N, Pickering TG. Baroreceptor activation reduces reactivity to noxious stimulation: Implications for hypertension. Science. 1979;205:1299–1301. doi: 10.1126/science.472749. [DOI] [PubMed] [Google Scholar]
- 79.Dziewierz A, Giszterowicz D, Siudak Z, Rakowski T, Dubiel JS, Dudek D. Admission glucose level and in-hospital outcomes in diabetic and non-diabetic patients with acute myocardial infarction. Clin Res Cardiol. 2010;99:715–721. doi: 10.1007/s00392-010-0175-1. [DOI] [PubMed] [Google Scholar]
- 80.Ebert TJ, Cowley AW., Jr Baroreflex modulation of sympathetic outflow during physiological increases of vasopressin in humans. Am J Physiol. 1992;262:H1372–H1378. doi: 10.1152/ajpheart.1992.262.5.H1372. [DOI] [PubMed] [Google Scholar]
- 81.Edwards TL, Scott WK, Almonte C, Burt A, Powell EH, Beecham GW, Wang L, Zuchner S, Konidari I, Wang G, Singer C, Nahab F, Scott B, Stajich JM, Pericak-Vance M, Haines J, Vance JM, Martin ER. Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet. 2010;74:97–109. doi: 10.1111/j.1469-1809.2009.00560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Eisenhofer G, Friberg P, Rundqvist B, Quyyumi AA, Lambert G, Kaye DM, Kopin IJ, Goldstein DS, Esler MD. Cardiac sympathetic nerve function in congestive heart failure. Circulation. 1996;93:1667–1676. doi: 10.1161/01.cir.93.9.1667. [DOI] [PubMed] [Google Scholar]
- 83.Eisenhofer G, Goldstein DS, Stull R, Keiser HR, Sunderland T, Murphy DL, Kopin IJ. Simultaneous liquid-chromatographic determination of 3,4-dihydroxyphenylglycol, catecholamines, and 3,4-dihydroxyphenylalanine in plasma, and their responses to inhibition of monoamine oxidase. Clin Chem. 1986;32:2030–2033. [PubMed] [Google Scholar]
- 84.Eisenhofer G, Goldstein DS, Stull RW, Gold PW, Keiser HR, Kopin IJ. Dissociation between corticotrophin and catecholamine responses to isoprenaline in humans. Clin Exp Pharmacol Physiol. 1987;14:337–341. doi: 10.1111/j.1440-1681.1987.tb00980.x. [DOI] [PubMed] [Google Scholar]
- 85.Eisenhofer G, Kopin IJ, Goldstein DS. Leaky catecholamine stores: Undue waste or a stress response coping mechanism? Ann N Y Acad Sci. 2004;1018:224–230. doi: 10.1196/annals.1296.027. [DOI] [PubMed] [Google Scholar]
- 86.Eisenhofer G, Rundqvist B, Aneman A, Friberg P, Dakak N, Kopin IJ, Jacobs MC, Lenders JW. Regional release and removal of catecholamines and extraneuronal metabolism to metanephrines. J Clin Endocrinol Metab. 1995;80:3009–3017. doi: 10.1210/jcem.80.10.7559889. [DOI] [PubMed] [Google Scholar]
- 87.Eldadah BA, Pacak K, Eisenhofer G, Holmes C, Kopin IJ, Goldstein DS. Cardiac uptake-1 inhibition by high circulating norepinephrine levels in patients with pheochromocytoma. Hypertension. 2004;43:1227–1232. doi: 10.1161/01.HYP.0000127305.87552.d6. [DOI] [PubMed] [Google Scholar]
- 88.Eldadah BA, Pechnik SL, Holmes CS, Moak JP, Saleem AM, Goldstein DS. Failure of propranolol to prevent tilt-evoked systemic vasodilatation, adrenaline release and neurocardiogenic syncope. Clin Sci. 2006;111:209–216. doi: 10.1042/CS20060017. [DOI] [PubMed] [Google Scholar]
- 89.Ellenbogen KA, Morillo CA, Wood MA, Gilligan DM, Eckberg DL, Smith ML. Neural monitoring of vasovagal syncope. Pacing Clin Electrophysiol. 1997;20:788–794. doi: 10.1111/j.1540-8159.1997.tb03905.x. [DOI] [PubMed] [Google Scholar]
- 90.Engel GL. Psychologic distress, vasodepressor (vasovagal) syncope, and sudden death. Ann Int Med. 1978;89:403–412. doi: 10.7326/0003-4819-89-3-403. [DOI] [PubMed] [Google Scholar]
- 91.Engel JE, Wu CF. Neurogenetic approaches to habituation and dishabituation in Drosophila. Neurobiol Learn Mem. 2009;92:166–175. doi: 10.1016/j.nlm.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Esler M. The sympathetic system and hypertension. Am J Hypertens. 2000;13:99S–105S. doi: 10.1016/s0895-7061(00)00225-9. [DOI] [PubMed] [Google Scholar]
- 93.Esler M, Jennings G, Korner P, Blombery P, Sacharias N, Leonard P. Measurement of total and organ-specific norepinephrine kinetics in humans. Am J Physiol. 1984;247:E21–E28. doi: 10.1152/ajpendo.1984.247.1.E21. [DOI] [PubMed] [Google Scholar]
- 94.Esler M, Julius S, Randall O, DeQuattro V, Zweifler A. High-renin essential hypertension: Adrenergic cardiovascular correlates. Clin Sci Mol Med Suppl. 1976;3:181s–184s. doi: 10.1042/cs051181s. [DOI] [PubMed] [Google Scholar]
- 95.Esler MD, Turner AG, Kaye DM, Thompson JM, Kingwell BA, Morris M, Lambert GW, Jennings GL, Cox HS, Seals DR. Aging effects on human sympathetic neuronal function. Am J Physiol. 1995;268:R278–R285. doi: 10.1152/ajpregu.1995.268.1.R278. [DOI] [PubMed] [Google Scholar]
- 96.Evans JM, Leonelli FM, Ziegler MG, McIntosh CM, Patwardhan AR, Ertl AC, Kim CS, Knapp CF. Epinephrine, vasodilation and hemoconcentration in syncopal, healthy men and women. Auton Neurosci. 2001;93:79–90. doi: 10.1016/S1566-0702(01)00323-X. [DOI] [PubMed] [Google Scholar]
- 97.Fagius J, Wallin BG, Sundlof G, Nerhed C, Englesson S. Sympathetic outflow in man after anaesthesia of the glossopharyngeal and vagus nerves. Brain. 1985;108(Pt 2):423–438. doi: 10.1093/brain/108.2.423. [DOI] [PubMed] [Google Scholar]
- 98.Ferreira A, Bettencourt P, Pimenta J, Frioes F, Pestana M, Soares-da-Silva P, Cerqueira-Gomes M. The renal dopaminergic system, neurohumoral activation, and sodium handling in heart failure. Am Heart J. 2002;143:391–397. doi: 10.1067/mhj.2002.120292. [DOI] [PubMed] [Google Scholar]
- 99.Fitzpatrick AP, Banner N, Cheng A, Yacoub M, Sutton R. Vasovagal reactions may occur after orthotopic heart transplantation. J Am Coll Cardiol. 1993;21:1132–1137. doi: 10.1016/0735-1097(93)90235-s. [DOI] [PubMed] [Google Scholar]
- 100.Flaa A, Eide IK, Kjeldsen SE, Rostrup M. Sympathoadrenal stress reactivity is a predictor of future blood pressure: An 18-year follow-up study. Hypertension. 2008;52:336–341. doi: 10.1161/HYPERTENSIONAHA.108.111625. [DOI] [PubMed] [Google Scholar]
- 101.Folkow B. Physiological aspects of primary hypertension. Physiol Rev. 1982;62:347–504. doi: 10.1152/physrev.1982.62.2.347. [DOI] [PubMed] [Google Scholar]
- 102.Folkow B. Stress, hypothalamic function and neuroendocrine consequences. Acta Med Scand. 1988;(Suppl 723):61–69. doi: 10.1111/j.0954-6820.1987.tb05929.x. [DOI] [PubMed] [Google Scholar]
- 103.Frank SM, Higgins MS, Fleisher LA, Sitzmann JV, Raff H, Breslow MJ. Adrenergic, respiratory, and cardiovascular effects of core cooling in humans. Am J Physiol. 1997;272:R557–R562. doi: 10.1152/ajpregu.1997.272.2.R557. [DOI] [PubMed] [Google Scholar]
- 104.Frank SM, Raja SN, Bulcao CF, Goldstein DS. Relative contribution of core and cutaneous temperatures to thermal comfort and autonomic responses in humans. J Appl Physiol. 1999;86:1588–1593. doi: 10.1152/jappl.1999.86.5.1588. [DOI] [PubMed] [Google Scholar]
- 105.Fredholm BB, Farnebo LO, Hamberger B. Plasma catecholamines, cyclic AMP and metabolic substrates in hemorrhagic shock of the rat. The effect of adrenal demedullation and 6-OH-dopamine treatment. Acta Physiol Scand. 1979;105:481–495. doi: 10.1111/j.1748-1716.1979.tb00113.x. [DOI] [PubMed] [Google Scholar]
- 106.Frenzel H, Bohlender J, Pinsker K, Wohlleben B, Tank J, Lechner SG, Schiska D, Jaijo T, Ruschendorf F, Saar K, Jordan J, Millan JM, Gross M, Lewin GR. A genetic basis for mechanosensory traits in humans. PLoS Biol. 2012;10:e1001318. doi: 10.1371/journal.pbio.1001318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Friberg P, Meredith I, Jennings G, Lambert G, Fazio V, Esler M. Evidence for increased renal norepinephrine overflow during sodium restriction in humans. Hypertension. 1990;16:121–130. doi: 10.1161/01.hyp.16.2.121. [DOI] [PubMed] [Google Scholar]
- 108.Fukuhara K, Kvetnansky R, Cizza G, Pacak K, Ohara H, Goldstein DS, Kopin IJ. Interrelations between sympathoadrenal system and hypothalamo-pituitary-adrenocortical/thyroid systems in rats exposed to cold stress. J Neuroendocrinol. 1996;8:533–541. doi: 10.1046/j.1365-2826.1996.04877.x. [DOI] [PubMed] [Google Scholar]
- 109.Funkenstein DH. Nor-epinephrine-like and epinephrine-like substances in relation to human behavior. J Mental Dis. 1956;124:58–68. doi: 10.1097/00005053-195607000-00009. [DOI] [PubMed] [Google Scholar]
- 110.Gajek J, Zysko D, Krzeminska S, Mazurek W. The influence of a tilt training programme on the renin-angiotensin-aldosterone system activity in patients with vasovagal syncope. Acta Cardiol. 2009;64:505–509. doi: 10.2143/AC.64.4.2041616. [DOI] [PubMed] [Google Scholar]
- 111.Gauthier P, Nadeau R, De Champlain J. Acute and chronic cardiovascular effects of 6-hydroxydopamine in dogs. Circ Res. 1972;31:207–217. doi: 10.1161/01.res.31.2.207. [DOI] [PubMed] [Google Scholar]
- 112.Gebber GL, Snyder DW. Hypothalamic control of baroreceptor reflexes. Am J Physiol. 1969;218:124–131. doi: 10.1152/ajplegacy.1970.218.1.124. [DOI] [PubMed] [Google Scholar]
- 113.Geracioti TD, Jr, Baker DG, Ekhator NN, West SA, Hill KK, Bruce AB, Schmidt D, Rounds-Kugler B, Yehuda R, Keck PE, Jr, Kasckow JW. CSF norepinephrine concentrations in posttraumatic stress disorder. Am J Psychiatry. 2001;158:1227–1230. doi: 10.1176/appi.ajp.158.8.1227. [DOI] [PubMed] [Google Scholar]
- 114.Geracioti TD, Jr, Baker DG, Kasckow JW, Strawn JR, Jeffrey Mulchahey J, Dashevsky BA, Horn PS, Ekhator NN. Effects of trauma-related audiovisual stimulation on cerebrospinal fluid norepinephrine and corticotropin-releasing hormone concentrations in post-traumatic stress disorder. Psychoneuroendocrinology. 2008;33:416–424. doi: 10.1016/j.psyneuen.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 115.Gerra G, Zaimovic A, Mascetti GG, Gardini S, Zambelli U, Timpano M, Raggi MA, Brambilla F. Neuroendocrine responses to experimentally-induced psychological stress in healthy humans. Psychoneuroendocrinology. 2001;26:91–107. doi: 10.1016/s0306-4530(00)00046-9. [DOI] [PubMed] [Google Scholar]
- 116.Gerson MC, McGuire N, Wagoner LE. Sympathetic nervous system function as measured by I-123 metaiodobenzylguanidine predicts transplant-free survival in heart failure patients with idiopathic dilated cardiomyopathy. J Card Fail. 2003;9:384–391. doi: 10.1054/s1071-9164(03)00134-9. [DOI] [PubMed] [Google Scholar]
- 117.Gianaros PJ, Jennings JR, Sheu LK, Greer PJ, Kuller LH, Matthews KA. Prospective reports of chronic life stress predict decreased grey matter volume in the hippocampus. Neuroimage. 2007;35:795–803. doi: 10.1016/j.neuroimage.2006.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Giannattasio C, Grassi G, Mancia G. Vasovagal syncope with bradycardia during lower body negative pressure in a heart transplant recipient. Blood Press. 1993;2:309–311. doi: 10.3109/08037059309077173. [DOI] [PubMed] [Google Scholar]
- 119.Giordano A, Calcagni ML, Verrillo A, Pellegrinotti M, Frontoni S, Spallone V, Gambardella S. Assessment of sympathetic innervation of the heart in diabetes mellitus using 123I-MIBG. Diabetes Nutr Metab. 2000;13:350–355. [PubMed] [Google Scholar]
- 120.Goddard AW, Ball SG, Martinez J, Robinson MJ, Yang CR, Russell JM, Shekhar A. Current perspectives of the roles of the central norepinephrine system in anxiety and depression. Depress Anxiety. 2010;27:339–350. doi: 10.1002/da.20642. [DOI] [PubMed] [Google Scholar]
- 121.Golczynska A, Lenders JW, Goldstein DS. Glucocorticoid-induced sympathoinhibition in humans. Clin Pharmacol Ther. 1995;58:90–98. doi: 10.1016/0009-9236(95)90076-4. [DOI] [PubMed] [Google Scholar]
- 122.Gold PW, Goodwin FK, Chrousos GP. Clinical and biochemical manifestations of depression: Relation to the neurobiology of stress (Part 1 of 2 parts) N Engl J Med. 1988;319:348–353. doi: 10.1056/NEJM198808113190606. [DOI] [PubMed] [Google Scholar]
- 123.Goldstein D, Fink D, Mettee DR. Cognition of arousal and actual arousal as determinants of emotion. J Pers Soc Psychol. 1972;21:41–51. doi: 10.1037/h0031873. [DOI] [PubMed] [Google Scholar]
- 124.Goldstein DS. Stress, Catecholamines, and Cardiovascular Disease. New York: Oxford University Press; 1995. [Google Scholar]
- 125.Goldstein DS. The Autonomic Nervous System in Health and Disease. New York, NY: Marcel Dekker, Inc; 2001. [Google Scholar]
- 126.Goldstein DS. Dysautonomia in Parkinson’s disease: Neurocardiological abnormalities. Lancet Neurol. 2003;2:669–676. doi: 10.1016/s1474-4422(03)00555-6. [DOI] [PubMed] [Google Scholar]
- 127.Goldstein DS. Adrenaline and the Inner World: An Introduction to Scientific Integrative Medicine. Baltimore, MD: The Johns Hopkins University Press; 2006. [Google Scholar]
- 128.Goldstein DS. Orthostatic hypotension as an early finding in Parkinson disease. Clin Auton Res. 2006;16:46–64. doi: 10.1007/s10286-006-0317-8. [DOI] [PubMed] [Google Scholar]
- 129.Goldstein DS. Stress, allostatic load, catecholamines, and other neurotransmitters in neurodegenerative diseases. Cell Mol Neurobiol. 2012;32:661–666. doi: 10.1007/s10571-011-9780-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Goldstein DS, Eisenhofer G, Kopin IJ. Sources and significance of plasma levels of catechols and their metabolites in humans. J Pharmacol Exp Ther. 2003;305:800–811. doi: 10.1124/jpet.103.049270. [DOI] [PubMed] [Google Scholar]
- 131.Goldstein DS, Eisenhofer G, Sax FL, Keiser HR, Kopin IJ. Plasma norepinephrine pharmacokinetics during mental challenge. Psychosom Med. 1987;49:591–605. doi: 10.1097/00006842-198711000-00004. [DOI] [PubMed] [Google Scholar]
- 132.Goldstein DS, Garty M, Bagdy G, Szemeredi K, Sternberg EM, Listwak S, Deka-Starosta A, Hoffman A, Chang PC, Stull R, Gold PW, Kopin IJ. Role of CRH in glucopenia-induced adrenomedullary activation in rats. J Neuroendocrinol. 1993;5:475–486. doi: 10.1111/j.1365-2826.1993.tb00511.x. [DOI] [PubMed] [Google Scholar]
- 133.Goldstein DS, Harris AH, Brady JV. Baroreflex sensitivity during operant blood pressure conditioning. Biofeedback Self-Regul. 1977;2:127–138. doi: 10.1007/BF00998663. [DOI] [PubMed] [Google Scholar]
- 134.Goldstein DS, Holmes C, Frank SM, Naqibuddin M, Dendi R, Snader S, Calkins H. Sympathoadrenal imbalance before neurocardiogenic syncope. Am J Cardiol. 2003;91:53–58. doi: 10.1016/s0002-9149(02)02997-1. [DOI] [PubMed] [Google Scholar]
- 135.Goldstein DS, Holmes C, Kopin IJ, Sharabi Y. Intra-neuronal vesicular uptake of catecholamines is decreased in patients with Lewy body diseases. J Clin Inv. 2011;121:3320–3330. doi: 10.1172/JCI45803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Goldstein DS, Holmes C, Li ST, Bruce S, Metman LV, Cannon RO., III Cardiac sympathetic denervation in Parkinson disease. Ann Intern Med. 2000;133:338–347. doi: 10.7326/0003-4819-133-5-200009050-00009. [DOI] [PubMed] [Google Scholar]
- 137.Goldstein DS, Holmes C, Sharabi Y. Cerebrospinal fluid biomarkers of central catecholamine deficiency in Parkinson’s disease and other synucleinopathies. Brain. 2012;135:1900–1913. doi: 10.1093/brain/aws055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Goldstein DS, Kopin IJ. Adrenomedullary, adrenocortical, and sympathoneural responses to stressors: A meta-analysis. Endo Regul. 2008;42:111–119. [PMC free article] [PubMed] [Google Scholar]
- 139.Goldstein DS, Kopin IJ. Evolution of concepts of stress. Stress. 2007;10:109–120. doi: 10.1080/10253890701288935. [DOI] [PubMed] [Google Scholar]
- 140.Goldstein DS, McEwen B. Allostasis, homeostats, and the nature of stress. Stress. 2002;5:55–58. doi: 10.1080/102538902900012345. [DOI] [PubMed] [Google Scholar]
- 141.Goldstein DS, Orimo S. Cardiac sympathetic neuroimaging: Summary of the First International Symposium. Clin Auton Res. 2009;19:133–136. doi: 10.1007/s10286-009-0002-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Goldstein DS, Ross RS, Brady JV. Biofeedback heart rate training during exercise. Biofeedback Self-Regul. 1977;2:107–125. doi: 10.1007/BF00998662. [DOI] [PubMed] [Google Scholar]
- 143.Goldstein DS, Sharabi Y, Karp BI, Bentho O, Saleem A, Pacak K, Eisenhofer G. Cardiac sympathetic denervation preceding motor signs in Parkinson disease. Clin Auton Res. 2007;17:118–121. doi: 10.1007/s10286-007-0396-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Goldstein DS, Spanarkel M, Pitterman A, Toltzis R, Gratz E, Epstein S, Keiser HR. Circulatory control mechanisms in vasodepressor syncope. Am Heart J. 1982;104:1071–1075. doi: 10.1016/0002-8703(82)90442-2. [DOI] [PubMed] [Google Scholar]
- 145.Goldstein DS, Stull R, Eisenhofer G, Gill JR., Jr Urinary excretion of dihydroxyphenylalanine and dopamine during alterations of dietary salt intake in humans. Clin Sci. 1989;76:517–522. doi: 10.1042/cs0760517. [DOI] [PubMed] [Google Scholar]
- 146.Goldstein DS, Sullivan P, Cooney A, Jinsmaa Y, Sullivan R, Gross DJ, Holmes C, Kopin IJ, Sharabi Y. Vesicular uptake blockade generates the toxic dopamine metabolite 3,4-dihydroxyphenylacetaldehyde in PC12 Cells: Relevance to the pathogenesis of Parkinson disease. J Neurochem. 2012;123:932–943. doi: 10.1111/j.1471-4159.2012.07924.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Goldstein DS, Sullivan P, Holmes C, Kopin IJ, Basile MJ, Mash DC. Catechols in post-mortem brain of patients with Parkinson disease. Eur J Neurol. 2011;18:703–710. doi: 10.1111/j.1468-1331.2010.03246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Graham DG. Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol Pharmacol. 1978;14:633–643. [PubMed] [Google Scholar]
- 149.Granata AR, Numao Y, Kumada M, Reis DJ. A1 noradrenergic neurons tonically inhibit sympathoexcitatory neurons of C1 area in rat brainstem. Brain Res. 1986;377:127–146. doi: 10.1016/0006-8993(86)91198-4. [DOI] [PubMed] [Google Scholar]
- 150.Greenough A, Lagercrantz H, Pool J, Dahlin I. Plasma catecholamine levels in preterm infants. Effect of birth asphyxia and Apgar score. Acta Paediatr Scand. 1987;76:54–59. doi: 10.1111/j.1651-2227.1987.tb10414.x. [DOI] [PubMed] [Google Scholar]
- 151.Habib KE, Weld KP, Rice KC, Pushkas J, Champoux M, Listwak S, Webster EL, Atkinson AJ, Schulkin J, Contoreggi C, Chrousos GP, McCann SM, Suomi SJ, Higley JD, Gold PW. Oral administration of a corticotropin-releasing hormone receptor antagonist significantly attenuates behavioral, neuroendocrine, and autonomic responses to stress in primates. Proc Natl Acad Sci U S A. 2000;97:6079–6084. doi: 10.1073/pnas.97.11.6079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Haglund ME, Nestadt PS, Cooper NS, Southwick SM, Charney DS. Psychobiological mechanisms of resilience: Relevance to prevention and treatment of stress-related psychopathology. Dev Psychopathol. 2007;19:889–920. doi: 10.1017/S0954579407000430. [DOI] [PubMed] [Google Scholar]
- 153.Harris AH, Gilliam WJ, Findley JS, Brady JV. Instrumental conditioning of large-magnitude, daily, 12-hour blood pressure elevations in the baboon. Science. 1973;182:175–177. doi: 10.1126/science.182.4108.175. [DOI] [PubMed] [Google Scholar]
- 154.Hasser EM, Bishop VS, Hay M. Interactions between vasopressin and baroreflex control of the sympathetic nervous system. Clin Exp Pharmacol Physiol. 1997;24:102–108. doi: 10.1111/j.1440-1681.1997.tb01791.x. [DOI] [PubMed] [Google Scholar]
- 155.Hein L, Altman JD, Kobilka BK. Two functionally distinct alpha2-adrenergic receptors regulate sympathetic neurotransmission. Nature. 1999;402:181–184. doi: 10.1038/46040. [DOI] [PubMed] [Google Scholar]
- 156.Heusser K, Tank J, Engeli S, Diedrich A, Menne J, Eckert S, Peters T, Sweep FC, Haller H, Pichlmaier AM, Luft FC, Jordan J. Carotid baroreceptor stimulation, sympathetic activity, baroreflex function, and blood pressure in hypertensive patients. Hypertension. 2010;55:619–626. doi: 10.1161/HYPERTENSIONAHA.109.140665. [DOI] [PubMed] [Google Scholar]
- 157.Ichinose M, Nishiyasu T. Arterial baroreflex control of muscle sympathetic nerve activity under orthostatic stress in humans. Front Physiol. 2012;3:314. doi: 10.3389/fphys.2012.00314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Imrich R, Eldadah BA, Bentho O, Pechnik S, Sharabi Y, Holmes C, Goldstein DS. Attenuated pre-ejection period response to tyramine in patients with cardiac sympathetic denervation. Ann N Y Acad Sci. 2008;1148:486–489. doi: 10.1196/annals.1410.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Isnard R, Pousset F, Trochu J, Chafirovskaia O, Carayon A, Golmard J, Lechat P, Thomas D, Bouhour J, Komajda M. Prognostic value of neurohormonal activation and cardiopulmonary exercise testing in patients with chronic heart failure. Am J Cardiol. 2000;86:417–421. doi: 10.1016/s0002-9149(00)00957-7. [DOI] [PubMed] [Google Scholar]
- 160.Jacobs MC, Goldstein DS, Willemsen JJ, Smits P, Thien T, Dionne RA, Lenders JW. Neurohumoral antecedents of vasodepressor reactions. Eur J Clin Invest. 1995;25:754–761. doi: 10.1111/j.1365-2362.1995.tb01954.x. [DOI] [PubMed] [Google Scholar]
- 161.Jain S, Goldstein DS. Cardiovascular dysautonomia in Parkinson disease: From pathophysiology to pathogenesis. Neurobiol Dis. 2012;46:572–580. doi: 10.1016/j.nbd.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Jami L, Laporte Y, Scott JJ. Some effects of sympathetic stimulation and isoprenaline on fatigued tetanic contractions of skeletal muscle in the cat. Brain Res. 1984;321:386–389. doi: 10.1016/0006-8993(84)90199-9. [DOI] [PubMed] [Google Scholar]
- 163.Jardine DL, Melton IC, Crozier IG, Bennett SI, Donald RA, Ikram H. Neurohormonal response to head-up tilt and its role in vasovagal syncope. Am J Cardiol. 1997;79:1302–1306. doi: 10.1016/s0002-9149(9x)00084-9. [DOI] [PubMed] [Google Scholar]
- 164.Jeong KH, Jacobson L, Pacak K, Widmaier EP, Goldstein DS, Majzoub JA. Impaired basal and restraint-induced epinephrine secretion in corticotropin-releasing hormone-deficient mice. Endocrinology. 2000;141:1142–1150. doi: 10.1210/endo.141.3.7370. [DOI] [PubMed] [Google Scholar]
- 165.Joyner MJ. Giant sucking sound: Can physiology fill the intellectual void left by the reductionists? J Appl Physiol. 2011;111:335–342. doi: 10.1152/japplphysiol.00565.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Joyner MJ, Dietz NM. Sympathetic vasodilation in human muscle. Acta Physiol Scand. 2003;177:329–336. doi: 10.1046/j.1365-201X.2003.01090.x. [DOI] [PubMed] [Google Scholar]
- 167.Joyner MJ, Pedersen BK. Ten questions about systems biology. J Physiol. 2011;589:1017–1030. doi: 10.1113/jphysiol.2010.201509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Jungermann K, Stumpel F. Role of hepatic, intrahepatic and hepatoenteral nerves in the regulation of carbohydrate metabolism and hemodynamics of the liver and intestine. Hepatogastroenterology. 1999;46(Suppl 2):1414–1417. [PubMed] [Google Scholar]
- 169.Kaasinen V, Ruottinen HM, Nagren K, Lehikoinen P, Oikonen V, Rinne JO. Upregulation of putaminal dopamine D2 receptors in early Parkinson’s disease: A comparative PET study with [11C] raclopride and [11C]N-methylspiperone. J Nucl Med. 2000;41:65–70. [PubMed] [Google Scholar]
- 170.Kahneman D. Thinking, Fast and Slow. New York: Farrar, Strauss and Giroux; 2011. [Google Scholar]
- 171.Kallio M, Haapaniemi T, Turkka J, Suominen K, Tolonen U, Sotaniemi K, Heikkila VP, Myllyla V. Heart rate variability in patients with untreated Parkinson’s disease. Eur J Neurol. 2000;7:667–672. doi: 10.1046/j.1468-1331.2000.00127.x. [DOI] [PubMed] [Google Scholar]
- 172.Kalsner S. Role of extraneuronal mechanisms in the termination of contractile responses to amines in vascular tissue. Br J Pharmacol. 1975;53:267–277. doi: 10.1111/j.1476-5381.1975.tb07358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kaludercic N, Carpi A, Menabo R, Di Lisa F, Paolocci N. Monoamine oxidases (MAO) in the pathogenesis of heart failure and ischemia/reperfusion injury. Biochim Biophys Acta. 2011;1813:1323–1332. doi: 10.1016/j.bbamcr.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kaludercic N, Takimoto E, Nagayama T, Feng N, Lai EW, Bedja D, Chen K, Gabrielson KL, Blakely RD, Shih JC, Pacak K, Kass DA, Di Lisa F, Paolocci N. Monoamine oxidase A-mediated enhanced catabolism of norepinephrine contributes to adverse remodeling and pump failure in hearts with pressure overload. Circ Res. 2010;106:193–202. doi: 10.1161/CIRCRESAHA.109.198366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kandel ER, Tauc L. Mechanism of heterosynaptic facilitation in the giant cell of the abdominal ganglion of Aplysia depilans. J Physiol. 1965;181:28–47. doi: 10.1113/jphysiol.1965.sp007743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Karnani M, Burdakov D. Multiple hypothalamic circuits sense and regulate glucose levels. Am J Physiol Regul Integr Comp Physiol. 2011;300:R47–R55. doi: 10.1152/ajpregu.00527.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kaufmann H, Goldstein DS. Dysautonomia in Parkinson disease. Handbook Clin Neurol. 2007;83:343–363. doi: 10.1016/S0072-9752(07)83014-4. [DOI] [PubMed] [Google Scholar]
- 178.Kawada T, Yamazaki T, Akiyama T, Shishido T, Miyano H, Sato T, Sugimachi M, Alexander J, Jr, Sunagawa K. Interstitial norepinephrine level by cardiac microdialysis correlates with ventricular contractility. Am J Physiol. 1997;273:H1107–H1112. doi: 10.1152/ajpheart.1997.273.3.H1107. [DOI] [PubMed] [Google Scholar]
- 179.Kaye DM, Lefkovits J, Jennings GL, Bergin P, Broughton A, Esler MD. Adverse consequences of high sympathetic nervous activity in the failing human heart. J Am Coll Cardiol. 1995;26:1257–1263. doi: 10.1016/0735-1097(95)00332-0. [DOI] [PubMed] [Google Scholar]
- 180.Kenagy DN, Bird CT, Webber CM, Fischer JR. Dextroamphetamine use during B-2 combat missions. Aviat Space Environ Med. 2004;75:381–386. [PubMed] [Google Scholar]
- 181.Kjaer M, Mikines KJ, Christensen NJ, Tronier B, Vinten J, Sonne B, Richter EA, Galbo H. Glucose turnover and hormonal changes during insulin-induced hypoglycemia in trained humans. J Appl Physiol. 1984;57:21–27. doi: 10.1152/jappl.1984.57.1.21. [DOI] [PubMed] [Google Scholar]
- 182.Koob GF. Corticotropin-releasing factor, norepinephrine, and stress. Biol Psychiatry. 1999;46:1167–1180. doi: 10.1016/s0006-3223(99)00164-x. [DOI] [PubMed] [Google Scholar]
- 183.Kopin IJ. Definitions of stress and sympathetic neuronal responses. Ann N Y Acad Sci. 1995;771:19–30. doi: 10.1111/j.1749-6632.1995.tb44667.x. [DOI] [PubMed] [Google Scholar]
- 184.Kopin IJ, Rundqvist B, Friberg P, Lenders J, Goldstein DS, Eisenhofer G. Different relationships of spillover to release of norepinephrine in human heart, kidneys, and forearm. Am J Physiol. 1998;275:R165–R173. doi: 10.1152/ajpregu.1998.275.1.R165. [DOI] [PubMed] [Google Scholar]
- 185.Kosten TR, Mason JW, Giller EL, Ostroff RB, Harkness L. Sustained urinary norepinephrine and epinephrine elevation in post-traumatic stress disorder. Psychoneuroendocrinology. 1987;12:13–20. doi: 10.1016/0306-4530(87)90017-5. [DOI] [PubMed] [Google Scholar]
- 186.Krum H, Schlaich M, Whitbourn R, Sobotka PA, Sadowski J, Bartus K, Kapelak B, Walton A, Sievert H, Thambar S, Abraham WT, Esler M. Catheter-based renal sympathetic denervation for resistant hypertension: A multicentre safety and proof-of-principle cohort study. Lancet. 2009;373:1275–1281. doi: 10.1016/S0140-6736(09)60566-3. [DOI] [PubMed] [Google Scholar]
- 187.Kruyt ND, Biessels GJ, Devries JH, Roos YB. Hyperglycemia in acute ischemic stroke: Pathophysiology and clinical management. Nat Rev Neurol. 2010;6:145–155. doi: 10.1038/nrneurol.2009.231. [DOI] [PubMed] [Google Scholar]
- 188.Kubota T, Yamazaki N, Sudo J, Monma Y, Kaku T, Okuyama T, Tanabe T. Protective effects of adrenoceptor-blocking agents on myocardial injury induced by epinephrine in mice. J Toxicol Sci. 1990;15:1–13. doi: 10.2131/jts.15.1. [DOI] [PubMed] [Google Scholar]
- 189.Kurata C, Uehara A, Ishikawa A. Improvement of cardiac sympathetic innervation by renal transplantation. J Nucl Med. 2004;45:1114–1120. [PubMed] [Google Scholar]
- 190.Kurz T, Richardt G, Seyfarth M, Schomig A. Nonexocytotic noradrenaline release induced by pharmacological agents or anoxia in human cardiac tissue. Naunyn Schmiedeberg’s Arch Pharmacol. 1996;354:7–16. doi: 10.1007/BF00168700. [DOI] [PubMed] [Google Scholar]
- 191.Kvetnansky R. Stressor specificity and effect of prior experience on catecholamine biosynthetic enzyme phenylethanolamine N-methyltransferase. Ann N Y Acad Sci. 2004;1032:117–129. doi: 10.1196/annals.1314.009. [DOI] [PubMed] [Google Scholar]
- 192.Kvetnansky R, Pacak K, Fukuhara K, Viskupic E, Hiremagalur B, Nankova B, Goldstein DS, Sabban EL, Kopin IJ. Sympathoadrenal system in stress. Interaction with the hypothalamic-pituitary-adrenocortical system. Ann N Y Acad Sci. 1995;771:131–158. doi: 10.1111/j.1749-6632.1995.tb44676.x. [DOI] [PubMed] [Google Scholar]
- 193.Kvetnansky R, Sun CL, Lake CR, Thoa N, Torda T, Kopin IJ. Effect of handling and forced immobilization on rat plasma levels of epinephrine, norepinephrine, and dopamine-beta-hydroxylase. Endocrinology. 1978;103:1868–1874. doi: 10.1210/endo-103-5-1868. [DOI] [PubMed] [Google Scholar]
- 194.Lake CR, Ziegler MG, Kopin IJ. Use of plasma norepinephrine for evaluation of sympathetic neuronal function in man. Life sciences. 1976;18:1315–1325. doi: 10.1016/0024-3205(76)90210-1. [DOI] [PubMed] [Google Scholar]
- 195.Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–980. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- 196.Lazarus RS. Emotion and Adaptation. New York: Oxford University Press; 1991. [Google Scholar]
- 197.Leppaluoto J, Westerlund T, Huttunen P, Oksa J, Smolander J, Dugue B, Mikkelsson M. Effects of long-term whole-body cold exposures on plasma concentrations of ACTH, beta-endorphin, cortisol, catecholamines and cytokines in healthy females. Scand J Clin Lab Invest. 2008;68:145–153. doi: 10.1080/00365510701516350. [DOI] [PubMed] [Google Scholar]
- 198.Li SW, Lin TS, Minteer S, Burke WJ. 3,4-Dihydroxyphenylacetaldehyde and hydrogen peroxide generate a hydroxyl radical: Possible role in Parkinson’s disease pathogenesis. Brain Res Mol Brain Res. 2001;93:1–7. doi: 10.1016/s0169-328x(01)00120-6. [DOI] [PubMed] [Google Scholar]
- 199.Lightfoot JT, Rowe SA, Fortney SM. Occurrence of presyncope in subjects without ventricular innervation. Clin Sci. 1993;85:695–700. doi: 10.1042/cs0850695. [DOI] [PubMed] [Google Scholar]
- 200.Linzer M, Pontinen M, Gold DT, Divine GW, Felder A, Brooks WB. Impairment of physical and psychosocial function in recurrent syncope. J Clin Epidemiol. 1991;44:1037–1043. doi: 10.1016/0895-4356(91)90005-t. [DOI] [PubMed] [Google Scholar]
- 201.Liu JE, Hahn RT, Stein KM, Markowitz SM, Okin PM, Devereux RB, Lerman BB. Left ventricular geometry and function preceding neurally mediated syncope. Circulation. 2000;101:777–783. doi: 10.1161/01.cir.101.7.777. [DOI] [PubMed] [Google Scholar]
- 202.Lohmeier TE, Hildebrandt DA, Dwyer TM, Iliescu R, Irwin ED, Cates AW, Rossing MA. Prolonged activation of the baroreflex decreases arterial pressure even during chronic adrenergic blockade. Hypertension. 2009;53:833–838. doi: 10.1161/HYPERTENSIONAHA.109.128884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Lohmeier TE, Iliescu R. Chronic lowering of blood pressure by carotid baroreflex activation: Mechanisms and potential for hypertension therapy. Hypertension. 2011;57:880–886. doi: 10.1161/HYPERTENSIONAHA.108.119859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Lohmeier TE, Iliescu R. Lowering of blood pressure by chronic suppression of central sympathetic outflow: Insight from prolonged baroreflex activation. J Appl Physiol. 2012;113:1652–1658. doi: 10.1152/japplphysiol.00552.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Lohmeier TE, Iliescu R, Dwyer TM, Irwin ED, Cates AW, Rossing MA. Sustained suppression of sympathetic activity and arterial pressure during chronic activation of the carotid baroreflex. Am J Physiol. 2010;299:H402–H409. doi: 10.1152/ajpheart.00372.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Lorenz K. On Aggression. New York: Bantam; 1963. [Google Scholar]
- 207.Low CA, Matthews KA, Kuller LH, Edmundowicz D. Psychosocial predictors of coronary artery calcification progression in post-menopausal women. Psychosom Med. 2011;73:789–794. doi: 10.1097/PSY.0b013e318236b68a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Lu CC, Diedrich A, Tung CS, Paranjape SY, Harris PA, Byrne DW, Jordan J, Robertson D. Water ingestion as prophylaxis against syncope. Circulation. 2003;108:2660–2665. doi: 10.1161/01.CIR.0000101966.24899.CB. [DOI] [PubMed] [Google Scholar]
- 209.Lukas A, Ferrier GR. Arrhythmic effects of norepinephrine in a model of cardiac ischemia and reperfusion. Can J Physiol Pharmacol. 1989;67:765–771. doi: 10.1139/y89-122. [DOI] [PubMed] [Google Scholar]
- 210.Lyon AR, Rees PS, Prasad S, Poole-Wilson PA, Harding SE. Stress (Takotsubo) cardiomyopathy–a novel pathophysiological hypothesis to explain catecholamine-induced acute myocardial stunning. Nat Clin Pract Cardiovasc Med. 2008;5:22–29. doi: 10.1038/ncpcardio1066. [DOI] [PubMed] [Google Scholar]
- 211.Mahfoud F, Schlaich M, Kindermann I, Ukena C, Cremers B, Brandt MC, Hoppe UC, Vonend O, Rump LC, Sobotka PA, Krum H, Esler M, Bohm M. Effect of renal sympathetic denervation on glucose metabolism in patients with resistant hypertension: A pilot study. Circulation. 2011;123:1940–1946. doi: 10.1161/CIRCULATIONAHA.110.991869. [DOI] [PubMed] [Google Scholar]
- 212.Makinen TM, Mantysaari M, Paakkonen T, Jokelainen J, Palinkas LA, Hassi J, Leppaluoto J, Tahvanainen K, Rintamaki H. Autonomic nervous function during whole-body cold exposure before and after cold acclimation. Aviat Space Environ Med. 2008;79:875–882. doi: 10.3357/asem.2235.2008. [DOI] [PubMed] [Google Scholar]
- 213.Manhem P, Bramnert M, Hallengren B, Lecerof H, Werner R. Increased arterial and venous plasma noradrenaline levels in patients with primary hypothyroidism during hypothyroid as compared to euthyroid state. J Endocrinol Invest. 1992;15:763–765. doi: 10.1007/BF03347648. [DOI] [PubMed] [Google Scholar]
- 214.Marino F, Sockler JM, Fry JM. Thermoregulatory, metabolic and sympathoadrenal responses to repeated brief exposure to cold. Scand J Clin Lab Invest. 1998;58:537–545. doi: 10.1080/00365519850186157. [DOI] [PubMed] [Google Scholar]
- 215.Marshall RD, Garakani A. Psychobiology of the acute stress response and its relationship to the psychobiology of post-traumatic stress disorder. Psychiatr Clin North Am. 2002;25:385–395. doi: 10.1016/s0193-953x(01)00005-3. [DOI] [PubMed] [Google Scholar]
- 216.Mastrocola C, Vanacore N, Giovani A, Locuratolo N, Vella C, Alessandri A, Baratta L, Tubani L, Meco G. Twenty-four-hour heart rate variability to assess autonomic function in Parkinson’s disease. Acta Neurol Scand. 1999;99:245–247. doi: 10.1111/j.1600-0404.1999.tb07355.x. [DOI] [PubMed] [Google Scholar]
- 217.Matsukawa K, Komine H, Nakamoto T, Murata J. Central command blunts sensitivity of arterial baroreceptor-heart rate reflex at onset of voluntary static exercise. Am J Physiol Heart Circ Physiol. 2006;290:H200–H208. doi: 10.1152/ajpheart.00013.2005. [DOI] [PubMed] [Google Scholar]
- 218.Matthews KA, Zhu S, Tucker DC, Whooley MA. Blood pressure reactivity to psychological stress and coronary calcification in the Coronary Artery Risk Development in Young Adults Study. Hypertension. 2006;47:391–395. doi: 10.1161/01.HYP.0000200713.44895.38. [DOI] [PubMed] [Google Scholar]
- 219.McEwen B, Stellar E. Stress and the individual. Mechanisms leading to disease. Arch Int Med. 1993;153:2093–2101. [PubMed] [Google Scholar]
- 220.McEwen BS. Interacting mediators of allostasis and allostatic load: Towards an understanding of resilience in aging. Metabolism. 2003;52:10–16. doi: 10.1016/s0026-0495(03)00295-6. [DOI] [PubMed] [Google Scholar]
- 221.McFall ME, Murburg MM, Ko GN, Veith RC. Autonomic responses to stress in Vietnam combat veterans with posttraumatic stress disorder. Biol Psychiatry. 1990;27:1165–1175. doi: 10.1016/0006-3223(90)90053-5. [DOI] [PubMed] [Google Scholar]
- 222.Menazza S, Blaauw B, Tiepolo T, Toniolo L, Braghetta P, Spolaore B, Reggiani C, Di Lisa F, Bonaldo P, Canton M. Oxidative stress by monoamine oxidases is causally involved in myofiber damage in muscular dystrophy. Hum Mol Genet. 2010;19:4207–4215. doi: 10.1093/hmg/ddq339. [DOI] [PubMed] [Google Scholar]
- 223.Meredith IT, Broughton A, Jennings GL, Esler MD. Evidence of a selective increase in cardiac sympathetic activity in patients with sustained ventricular arrhythmias. N Engl J Med. 1991;325:618–624. doi: 10.1056/NEJM199108293250905. [DOI] [PubMed] [Google Scholar]
- 224.Millar RA, Keener EB, Benfey BG. Plasma adrenaline and noradrenaline after phenoxybenzamine administration, and during haemorrhagic hypotension, in normal and adrenalectomized dogs. Br J Pharmacol Chemother. 1959;14:9–13. doi: 10.1111/j.1476-5381.1959.tb00921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Molochnikov L, Rabey JM, Dobronevsky E, Bonucelli U, Ceravolo R, Frosini D, Grunblatt E, Riederer P, Jacob C, Aharon-Peretz J, Bashenko Y, Youdim MB, Mandel SA. A molecular signature in blood identifies early Parkinson’s disease. Molec Neurodegen. 2012;7:26. doi: 10.1186/1750-1326-7-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Momose M, Inaba S, Emori T, Imamura K, Kawano K, Ueda T, Kobayashi H, Hosoda S. Increased cardiac sympathetic activity in patients with hypothyroidism as determined by iodine-123 metaiodobenzylguanidine scintigraphy. Eur J Nucl Med. 1997;24:1132–1137. doi: 10.1007/BF01254245. [DOI] [PubMed] [Google Scholar]
- 227.Montebugnoli L, Montanari G. Vasovagal syncope in heart transplant patients during dental surgery. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1999;87:666–669. doi: 10.1016/s1079-2104(99)70157-5. [DOI] [PubMed] [Google Scholar]
- 228.Morgan DA, Balon TW, Ginsberg BH, Mark AL. Nonuniform regional sympathetic nerve responses to hyperinsulinemia in rats. Am J Physiol. 1993;264:R423–R427. doi: 10.1152/ajpregu.1993.264.2.R423. [DOI] [PubMed] [Google Scholar]
- 229.Morillo CA, Eckberg DL, Ellenbogen KA, Beightol LA, Hoag JB, Tahvanainen KU, Kuusela TA, Diedrich AM. Vagal and sympathetic mechanisms in patients with orthostatic vasovagal syncope. Circulation. 1997;96:2509–2513. doi: 10.1161/01.cir.96.8.2509. [DOI] [PubMed] [Google Scholar]
- 230.Morrison SF. Differential regulation of sympathetic outflows to vasoconstrictor and thermoregulatory effectors. Ann N Y Acad Sci. 2001;940:286–298. doi: 10.1111/j.1749-6632.2001.tb03684.x. [DOI] [PubMed] [Google Scholar]
- 231.Mosqueda-Garcia R, Fernandez-Violante R, Tank J, Snell M, Cunningham G, Furlan R. Yohimbine in neurally mediated syncope. Pathophysiological implications. J Clin Invest. 1998;102:1824–1830. doi: 10.1172/JCI3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Mosqueda-Garcia R, Furlan R, Fernandez-Violante R, Desai T, Snell M, Jarai Z, Ananthram V, Robertson RM, Robertson D. Sympathetic and baroreceptor reflex function in neurally mediated syncope evoked by tilt. J Clin Invest. 1997;99:2736–2744. doi: 10.1172/JCI119463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Mosqueda-Garcia R, Furlan R, Tank J, Fernandez-Violante R. The elusive pathophysiology of neurally mediated syncope. Circulation. 2000;102:2898–2906. doi: 10.1161/01.cir.102.23.2898. [DOI] [PubMed] [Google Scholar]
- 234.Nagatsu T, Sawada M. Molecular mechanism of the relation of monoamine oxidase B and its inhibitors to Parkinson’s disease: Possible implications of glial cells. J Neural Transm (Suppl) 2006:53–65. doi: 10.1007/978-3-211-33328-0_7. [DOI] [PubMed] [Google Scholar]
- 235.Nathan MA, Reis DJ. Chronic labile hypertension produced by lesions of the nucleus tractus solitarii in the cat. Circ Res. 1977;40:72–81. doi: 10.1161/01.res.40.1.72. [DOI] [PubMed] [Google Scholar]
- 236.Nesse RM, Williams GC. Why We Get Sick. The New Science of Darwinian Medicine. New York: Times Books; 1994. [Google Scholar]
- 237.Nishida Y, Bishop VS. Vasopressin-induced suppression of renal sympathetic outflow depends on the number of baroafferent inputs in rabbits. Am J Physiol. 1992;263:R1187–R1194. doi: 10.1152/ajpregu.1992.263.6.R1187. [DOI] [PubMed] [Google Scholar]
- 238.Noble D. The Music of Life: Biology beyond the Genome. Oxford, UK: Oxford University Press; 2006. [Google Scholar]
- 239.Oberg B, Thorten P. Increased activity in left ventricular receptors during hemorrhage or occlusion of caval veins in the cat. A possible cause of vasovagal reaction. Acta Physiol Scand. 1972;85:164–173. doi: 10.1111/j.1748-1716.1972.tb05247.x. [DOI] [PubMed] [Google Scholar]
- 240.Olanow CW, Hauser RA, Gauger L, Malapira T, Koller W, Hubble J, Bushenbark K, Lilienfeld D, Esterlitz J. The effect of deprenyl and levodopa on the progression of Parkinson’s disease. Ann Neurol. 1995;38:771–777. doi: 10.1002/ana.410380512. [DOI] [PubMed] [Google Scholar]
- 241.Olanow CW, Rascol O, Hauser R, Feigin PD, Jankovic J, Lang A, Langston W, Melamed E, Poewe W, Stocchi F, Tolosa E. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med. 2009;361:1268–1278. doi: 10.1056/NEJMoa0809335. [DOI] [PubMed] [Google Scholar]
- 242.Ostman-Smith I. Adaptive changes in the sympathetic nervous system and some effector organs of the rat following long term exercise or cold acclimation and the role of cardiac sympathetic nerves in the genesis of compensatory cardiac hypertrophy. Acta Physiol Scand. 1980;108:1–118. [PubMed] [Google Scholar]
- 243.Osztovits J, Horvath T, Littvay L, Steinbach R, Jermendy A, Tarnoki A, Tarnoki D, Metneki J, Kollai M, Jermendy G. Effects of genetic vs. environmental factors on cardiovascular autonomic function: A twin study. Diabet Med. 2011;28:1241–1248. doi: 10.1111/j.1464-5491.2011.03363.x. [DOI] [PubMed] [Google Scholar]
- 244.Pacak K. Stressor-specific activation of the hypothalamic-pituitary-adrenocortical axis. Physiol Res. 2000;49:S11–S17. [PubMed] [Google Scholar]
- 245.Pacak K, Palkovits M, Yadid G, Kvetnansky R, Kopin IJ, Goldstein DS. Heterogeneous neurochemical responses to different stressors: A test of Selye’s doctrine of nonspecificity. Am J Physiol. 1998;275:R1247–R1255. doi: 10.1152/ajpregu.1998.275.4.R1247. [DOI] [PubMed] [Google Scholar]
- 246.Panneton WM, Kumar VB, Gan Q, Burke WJ, Galvin JE. The neurotoxicity of DOPAL: Behavioral and stereological evidence for its role in Parkinson disease pathogenesis. PLoS One. 2010;5:e15251. doi: 10.1371/journal.pone.0015251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Park ER, Traeger L, Vranceanu AM, Scult M, Lerner JA, Benson H, Denninger J, Fricchione GL. The development of a patient-centered program based on the relaxation response: The Relaxation Response Resiliency Program (3RP) Psychosomatics. 2013;54:165–174. doi: 10.1016/j.psym.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 248.Patel JN, Coppack SW, Goldstein DS, Miles JM, Eisenhofer G. Norepinephrine spillover from human adipose tissue before and after a 72-hour fast. J Clin Endocrinol Metab. 2002;87:3373–3377. doi: 10.1210/jcem.87.7.8695. [DOI] [PubMed] [Google Scholar]
- 249.Patel MB, Loud AV, King BD, Anversa P, Sack D, Hintze TH. Global myocardial hypertrophy in conscious dogs with chronic elevation of plasma norepinephrine levels. J Mol Cell Cardiol. 1989;21(Suppl 5):49–61. doi: 10.1016/0022-2828(89)90771-2. [DOI] [PubMed] [Google Scholar]
- 250.Perlmutter JS, Kilbourn MR, Raichle ME, Welch MJ. MPTP-induced upregulation of in vivo dopaminergic radioligand-receptor binding in humans. Neurology. 1987;37:1575–1579. doi: 10.1212/wnl.37.10.1575. [DOI] [PubMed] [Google Scholar]
- 251.Pettit SE, Marchand I, Graham T. Gender differences in cardiovascular and catecholamine responses to cold-air exposure at rest. Can J Appl Physiol. 1999;24:131–147. doi: 10.1139/h99-011. [DOI] [PubMed] [Google Scholar]
- 252.Polikar R, Kennedy B, Ziegler M, O’Connor DT, Smith J, Nicod P. Plasma norepinephrine kinetics, dopamine-beta-hydroxylase, and chromogranin-A, in hypothyroid patients before and following replacement therapy. J Clin Endocrinol Metab. 1990;70:277–281. doi: 10.1210/jcem-70-1-277. [DOI] [PubMed] [Google Scholar]
- 253.Polinsky RJ, Kopin IJ, Ebert MH, Weise V. The adrenal medullary response to hypoglycemia in patients with orthostatic hypotension. J Clin Endocrinol Metab. 1980;51:1401–1406. doi: 10.1210/jcem-51-6-1401. [DOI] [PubMed] [Google Scholar]
- 254.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 255.Quillen EW, Jr, Cowley AW., Jr Influence of volume changes on osmolality-vasopressin relationships in conscious dogs. Am J Physiol. 1983;244:H73–H79. doi: 10.1152/ajpheart.1983.244.1.H73. [DOI] [PubMed] [Google Scholar]
- 256.Randich A, Aicher SA. Medullary substrates mediating antinociception produced by electrical stimulation of the vagus. Brain Res. 1988;445:68–76. doi: 10.1016/0006-8993(88)91075-x. [DOI] [PubMed] [Google Scholar]
- 257.Rattanataweeboon P, Vilaichone W, Vannasaeng S. Stress hyperglycemia in patients with sepsis. J Med Assoc Thai. 2009;92(Suppl 2):S88–S94. [PubMed] [Google Scholar]
- 258.Rea RF, Eckberg DL, Fritsch JM, Goldstein DS. Relation of plasma norepinephrine and sympathetic traffic during hypotension in man. Am J Physiol. 1990;258:R982–R986. doi: 10.1152/ajpregu.1990.258.4.R982. [DOI] [PubMed] [Google Scholar]
- 259.Rees JN, Florang VR, Anderson DG, Doorn JA. Lipid peroxidation products inhibit dopamine catabolism yielding aberrant levels of a reactive intermediate. Chem Res Toxicol. 2007;20:1536–1542. doi: 10.1021/tx700248y. [DOI] [PubMed] [Google Scholar]
- 260.Rees JN, Florang VR, Eckert LL, Doorn JA. Protein reactivity of 3,4-dihydroxyphenylacetaldehyde, a toxic dopamine metabolite, is dependent on both the aldehyde and the catechol. Chem Res Toxicol. 2009;22:1256–1263. doi: 10.1021/tx9000557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Riegger GA, Liebau G, Kochsiek K. Antidiuretic hormone in congestive heart failure. Am J Med. 1982;72:49–52. doi: 10.1016/0002-9343(82)90576-9. [DOI] [PubMed] [Google Scholar]
- 262.Riese H, Rijsdijk FV, Ormel J, van Roon AM, Neeleman J, Rosmalen JG. Genetic influences on baroreflex sensitivity during rest and mental stress. J Hypertens. 2006;24:1779–1786. doi: 10.1097/01.hjh.0000242402.83709.27. [DOI] [PubMed] [Google Scholar]
- 263.Robertson DA, Johnson GA, Robertson RM, Nies AS, Shand DG, Oates JA. Comparative assessment of stimuli that release neuronal and adrenomedullary catecholamines in man. Circulation. 1979;59:637–643. doi: 10.1161/01.cir.59.4.637. [DOI] [PubMed] [Google Scholar]
- 264.Robinson BJ, Johnson RH. Why does vasodilatation occur during syncope? Clin Sci. 1988;74:347–350. doi: 10.1042/cs0740347. [DOI] [PubMed] [Google Scholar]
- 265.Rodriguez M, Sabate M, Troncoso E. Time and frequency domain analysis for the assessment of heart autonomic control in Parkinson’s disease. J Neural Transm. 1996;103:447–454. doi: 10.1007/BF01276420. [DOI] [PubMed] [Google Scholar]
- 266.Romero-Bermejo FJ, Ruiz-Bailen M, Gil-Cebrian J, Huertos-Ranchal MJ. Sepsis-induced cardiomyopathy. Curr Cardiol Rev. 2011;7:163–183. doi: 10.2174/157340311798220494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Rona G. Catecholamine cardiotoxicity. J Mol Cell Cardiol. 1985;17:291–306. doi: 10.1016/s0022-2828(85)80130-9. [DOI] [PubMed] [Google Scholar]
- 268.Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, Tomiyama H, Nakashima K, Hasegawa K, Obata F, Yoshikawa T, Kawakami H, Sakoda S, Yamamoto M, Hattori N, Murata M, Nakamura Y, Toda T. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41:1303–1307. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- 269.Schachter S, Singer J. Cognitive, social, and physiological determinants of emotional state. Psychol Rev. 1962;69:379–399. doi: 10.1037/h0046234. [DOI] [PubMed] [Google Scholar]
- 270.Scherrer U, Vissing S, Morgan BJ, Hanson P, Victor RG. Vasovagal syncope after infusion of a vasodilator in a heart-transplant recipient. N Engl J Med. 1990;322:602–604. doi: 10.1056/NEJM199003013220906. [DOI] [PubMed] [Google Scholar]
- 271.Schlaich MP, Sobotka PA, Krum H, Lambert E, Esler MD. Renal sympathetic-nerve ablation for uncontrolled hypertension. N Engl J Med. 2009;361:932–934. doi: 10.1056/NEJMc0904179. [DOI] [PubMed] [Google Scholar]
- 272.Schommer NC, Hellhammer DH, Kirschbaum C. Dissociation between reactivity of the hypothalamus-pituitary-adrenal axis and the sympathetic-adrenal-medullary system to repeated psychosocial stress. Psychosom Med. 2003;65:450–460. doi: 10.1097/01.psy.0000035721.12441.17. [DOI] [PubMed] [Google Scholar]
- 273.Schultz W. Behavioral dopamine signals. Trends Neurosci. 2007;30:203–210. doi: 10.1016/j.tins.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 274.Scott EM, Greenwood JP, Stoker JB, Mary DA, Gilbey SG. Sympathetic nerve hyperactivity is associated with increased peripheral vascular resistance in hypopituitary patients with growth hormone deficiency. Clin Endocrinol (Oxf) 2002;56:759–763. doi: 10.1046/j.1365-2265.2002.01546.x. [DOI] [PubMed] [Google Scholar]
- 275.Seals DR, Esler MD. Human ageing and the sympathoadrenal system. J Physiol. 2000;528:407–417. doi: 10.1111/j.1469-7793.2000.00407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Seller H, Illert M. The localization of the first synapse in the carotid sinus baroreceptor reflex pathway and its alteration of the afferent input. Pflugers Arch. 1969;306:1–19. doi: 10.1007/BF00586608. [DOI] [PubMed] [Google Scholar]
- 277.Selye H. The Physiology and Pathology of Exposure to Stress. A Treatise Based on the Concepts of the General-Adaptation Syndrome and the Diseses of Adaptation. Montreal, Canada: Acta, Inc; 1950. [Google Scholar]
- 278.Selye H. The Stress of Life. New York: McGraw-Hill; 1956. [Google Scholar]
- 279.Selye H. Stress without Distress. New York: New American Library; 1974. [Google Scholar]
- 280.Sevoz-Couche C, Comet MA, Bernard JF, Hamon M, Laguzzi R. Cardiac baroreflex facilitation evoked by hypothalamus and prefrontal cortex stimulation: Role of the nucleus tractus solitarius 5-HT2A receptors. Am J Physiol. 2006;291:R1007–R1015. doi: 10.1152/ajpregu.00052.2006. [DOI] [PubMed] [Google Scholar]
- 281.Sevoz-Couche C, Hamon M, Laguzzi R. Antinociceptive effect of cardiopulmonary chemoreceptor and baroreceptor reflex activation in the rat. Pain. 2002;99:71–81. doi: 10.1016/s0304-3959(02)00055-6. [DOI] [PubMed] [Google Scholar]
- 282.Shah SD, Tse TF, Clutter WE, Cryer PE. The human sympathochromaffin system. Am J Physiol. 1984;247:E380–E384. doi: 10.1152/ajpendo.1984.247.3.E380. [DOI] [PubMed] [Google Scholar]
- 283.Sharabi Y, Dendi R, Holmes C, Goldstein DS. Baroreflex failure as a late sequela of neck irradiation. Hypertension. 2003;42:110–116. doi: 10.1161/01.HYP.0000077441.45309.08. [DOI] [PubMed] [Google Scholar]
- 284.Sharabi Y, Imrich R, Holmes C, Pechnik S, Goldstein DS. Generalized and neurotransmitter-selective noradrenergic denervation in Parkinson’s disease with orthostatic hypotension. Mov Disord. 2008;23:1725–1732. doi: 10.1002/mds.22226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Siegel S. Conditioning of insulin effects. J Comp Physiol Psychol. 1972;78:233–241. doi: 10.1037/h0032180. [DOI] [PubMed] [Google Scholar]
- 286.Simpson A, Maynard V. A longitudinal study of the effect of Antarctic residence on energy dynamics and aerobic fitness. Int J Circumpolar Health. 2012;71:17227. doi: 10.3402/ijch.v71i0.17227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Singal PK, Dhillon KS, Beamish RE, Kapur N, Dhalla NS. Myocardial cell damage and cardiovascular changes due to iv infusion of adrenochrome in rats. Br J Pathol. 1982;63:167–176. [PMC free article] [PubMed] [Google Scholar]
- 288.Singleton A, Gwinn-Hardy K, Sharabi Y, Li ST, Holmes C, Dendi R, Hardy J, Crawley A, Goldstein DS. Association between cardiac denervation and parkinsonism caused by alpha-synuclein gene triplication. Brain. 2004;127:768–772. doi: 10.1093/brain/awh081. [DOI] [PubMed] [Google Scholar]
- 289.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 290.Sofuoglu M, Nelson D, Babb DA, Hatsukami DK. Intravenous cocaine increases plasma epinephrine and norepinephrine in humans. Pharmacol Biochem Behav. 2001;68:455–459. doi: 10.1016/s0091-3057(01)00482-8. [DOI] [PubMed] [Google Scholar]
- 291.Sossi V, de la Fuente-Fernandez R, Schulzer M, Troiano AR, Ruth TJ, Stoessl AJ. Dopamine transporter relation to dopamine turnover in Parkinson’s disease: A positron emission tomography study. Ann Neurol. 2007;62:468–474. doi: 10.1002/ana.21204. [DOI] [PubMed] [Google Scholar]
- 292.Southwick SM, Bremner JD, Rasmusson A, Morgan CA, III, Arnsten A, Charney DS. Role of norepinephrine in the pathophysiology and treatment of posttraumatic stress disorder. Biological Psychiatry. 1999;46:1192–1204. doi: 10.1016/s0006-3223(99)00219-x. [DOI] [PubMed] [Google Scholar]
- 293.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 294.Sterling P, Eyer J. Allostasis: A new paradigm to explain arousal pathology. In: Fisher J, Reason J, editors. Handbook of Life Stress, Cognition, and Health. New York: Johns Wiley & Sons, Inc; 1988. pp. 629–649. [Google Scholar]
- 295.Sverrisdottir YB, Elam M, Herlitz H, Bengtsson BA, Johannsson G. Intense sympathetic nerve activity in adults with hypopituitarism and untreated growth hormone deficiency. J Clin Endocrinol Metab. 1998;83:1881–1885. doi: 10.1210/jcem.83.6.4895. [DOI] [PubMed] [Google Scholar]
- 296.Szatalowicz VL, Arnold PE, Chaimovitz C, Bichet D, Berl T, Schrier RW. Radioimmunoassay of plasma arginine vasopressin in hyponatremic patients with congestive heart failure. N Engl J Med. 1981;305:263–266. doi: 10.1056/NEJM198107303050506. [DOI] [PubMed] [Google Scholar]
- 297.Tatar P, Bulas J, Kvetnansky R, Strec V. Venous plasma adrenaline response to orthostatic syncope during tilting in healthy men. Clin Physiol. 1986;6:303–309. doi: 10.1111/j.1475-097x.1986.tb00627.x. [DOI] [PubMed] [Google Scholar]
- 298.Taylor TN, Caudle WM, Shepherd KR, Noorian A, Jackson CR, Iuvone PM, Weinshenker D, Greene JG, Miller GW. Nonmotor symptoms of Parkinson’s disease revealed in an animal model with reduced monoamine storage capacity. J Neurosci. 2009;29:8103–8113. doi: 10.1523/JNEUROSCI.1495-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Theopistou A, Gatzoulis K, Economou E, Sideris S, Hantzos K, Stefanadis C, Toutouzas P. Biochemical changes involved in the mechanism of vasovagal syncope. Am J Cardiol. 2001;88:376–381. doi: 10.1016/s0002-9149(01)01682-4. [DOI] [PubMed] [Google Scholar]
- 300.Thomas CB. Experimental hypertension from section of moderator nerves. BullJohnsHopkinsHosp. 1944;74:335–377. [Google Scholar]
- 301.Thomas JA, Marks BH. Plasma norepinephrine in congestive heart failure. Am J Cardiol. 1978;41:233–243. doi: 10.1016/0002-9149(78)90162-5. [DOI] [PubMed] [Google Scholar]
- 302.Thoren P, Skarphendinsson JO, Carlsson S. Sympathetic inhibition from vagal afferents during severe hemorrhage in rats. Acta Physiol Scand Suppl. 1988;571:97–105. [PubMed] [Google Scholar]
- 303.Tsutamoto T, Nishiyama K, Sakai H, Tanaka T, Fujii M, Yamamoto T, Yamaji M, Horie M. Transcardiac increase in norepinephrine and prognosis in patients with chronic heart failure. Eur J Heart Fail. 2008;10:1208–1214. doi: 10.1016/j.ejheart.2008.09.011. [DOI] [PubMed] [Google Scholar]
- 304.Turkka JT, Tolonen U, Myllyla VV. Cardiovascular reflexes in Parkinson’s disease. Eur Neurol. 1987;26:104–112. doi: 10.1159/000116319. [DOI] [PubMed] [Google Scholar]
- 305.Uchiyama M, Otsuka T, Sakai K. Response of plasma renin activity to postural change in vasovagal syncope in children, with observations on syncope. Horm Res. 1986;23:147–150. doi: 10.1159/000180310. [DOI] [PubMed] [Google Scholar]
- 306.Udelsman R, Goldstein DS, Loriaux DL, Chrousos GP. Catecholamine-glucocorticoid interactions during surgical stress. J Surg Res. 1987;43:539–545. doi: 10.1016/0022-4804(87)90128-4. [DOI] [PubMed] [Google Scholar]
- 307.Uschold-Schmidt N, Nyuyki KD, Fuchsl AM, Neumann ID, Reber SO. Chronic psychosocial stress results in sensitization of the HPA axis to acute heterotypic stressors despite a reduction of adrenal in vitro ACTH responsiveness. Psychoneuroendocrinology. 2012;37:1676–1687. doi: 10.1016/j.psyneuen.2012.02.015. [DOI] [PubMed] [Google Scholar]
- 308.Vaddadi G, Esler MD, Dawood T, Lambert E. Persistence of muscle sympathetic nerve activity during vasovagal syncope. Eur Heart J. 2010;31:2027–2033. doi: 10.1093/eurheartj/ehq071. [DOI] [PubMed] [Google Scholar]
- 309.Vaddadi G, Esler MD, Dawood T, Lambert E. Persistence of muscle sympathetic nerve activity during vasovagal syncope. European Heart J. 2010;31:2027–2033. doi: 10.1093/eurheartj/ehq071. [DOI] [PubMed] [Google Scholar]
- 310.Valappil RA, Black JE, Broderick MJ, Carrillo O, Frenette E, Sullivan SS, Goldman SM, Tanner CM, Langston JW. Exploring the electrocardiogram as a potential tool to screen for premotor Parkinson’s disease. Movement Disorders. 2010;25:2296–2303. doi: 10.1002/mds.23348. [DOI] [PubMed] [Google Scholar]
- 311.Virtanen R, Jula A, Salminen JK, Voipio-Pulkki LM, Helenius H, Kuusela T, Airaksinen J. Anxiety and hostility are associated with reduced baroreflex sensitivity and increased beat-to-beat blood pressure variability. Psychosom Med. 2003;65:751–756. doi: 10.1097/01.psy.0000088760.65046.cf. [DOI] [PubMed] [Google Scholar]
- 312.Vissing SF. Differential activation of sympathetic discharge to skin and skeletal muscle in humans. Acta Physiol Scand Suppl. 1997;639:1–32. [PubMed] [Google Scholar]
- 313.von Euler US. A specific sympathomimetic ergone in adrenergic nerve fibres (sympathin) and its relations to adrenaline and nor-adrenaline. Acta Physiol Scand. 1946;12:73–96. [Google Scholar]
- 314.Wallin BG, Sundlof G. Sympathetic outflow to muscles during vaso-vagal syncope. J Autonom Nerv Sys. 1982;6:287–291. doi: 10.1016/0165-1838(82)90001-7. [DOI] [PubMed] [Google Scholar]
- 315.Watabe T, Tanaka K, Kumagae M, Itoh S, Takeda F, Morio K, Hasegawa M, Horiuchi T, Miyabe S, Shimizu N. Hormonal responses to insulin-induced hypoglycemia in man. J Clin Endocrinol Metab. 1987;65:1187–1191. doi: 10.1210/jcem-65-6-1187. [DOI] [PubMed] [Google Scholar]
- 316.Weil-Malherbe H, Axelrod J, Tomchick R. Blood-brain barrier for adrenaline. Science. 1959;129:1226–1227. doi: 10.1126/science.129.3357.1226. [DOI] [PubMed] [Google Scholar]
- 317.Wey M, Fernandez E, Martinez PA, Sullivan P, Goldstein DS, Strong R. Neurodegeneration and motor dysfunction in mice lacking cytosolic and mitochondrial aldehyde dehydrogenases: Implications for Parkinson’s disease. PLoS ONE. 2012;7:e31522. doi: 10.1371/journal.pone.0031522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Williams GC. Pleiotropy, natural selection, and the evolution of senescence. Evolution. 1957;11:398–411. [Google Scholar]
- 319.Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, Hetzer C, Loher T, Vilar M, Campioni S, Tzitzilonis C, Soragni A, Jessberger S, Mira H, Consiglio A, Pham E, Masliah E, Gage FH, Riek R. In vivo demonstration that {alpha}-synuclein oligomers are toxic. Proc Natl Acad Sci U S A. 2011;108:4194–4199. doi: 10.1073/pnas.1100976108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Wittstein IS, Thiemann DR, Lima JA, Baughman KL, Schulman SP, Gerstenblith G, Wu KC, Rade JJ, Bivalacqua TJ, Champion HC. Neurohumoral features of myocardial stunning due to sudden emotional stress. N Engl J Med. 2005;352:539–548. doi: 10.1056/NEJMoa043046. [DOI] [PubMed] [Google Scholar]
- 321.Wolfovitz E, Grossman E, Folio CJ, Keiser HR, Kopin IJ, Goldstein DS. Derivation of urinary dopamine from plasma dihydroxyphenylalanine in humans. Clin Sci. 1993;84:549–557. doi: 10.1042/cs0840549. [DOI] [PubMed] [Google Scholar]
- 322.Wong ML, Kling MA, Munson PJ, Listwak S, Licinio J, Prolo P, Karp B, McCutcheon IE, Geracioti TD, Jr, DeBellis MD, Rice KC, Goldstein DS, Veldhuis JD, Chrousos GP, Oldfield EH, McCann SM, Gold PW. Pronounced and sustained central hypernoradrenergic function in major depression with melancholic features: Relation to hypercortisolism and corticotropin-releasing hormone. Proc Natl Acad Sci U S A. 2000;97:325–330. doi: 10.1073/pnas.97.1.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Xu Q, Park Y, Huang X, Hollenbeck A, Blair A, Schatzkin A, Chen H. Physical activities and future risk of Parkinson disease. Neurology. 2010;75:341–348. doi: 10.1212/WNL.0b013e3181ea1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Zakowski SG, Hall MH, Klein LC, Baum A. Appraised control, coping, and stress in a community sample: A test of the goodness-of-fit hypothesis. Ann Behav Med. 2001;23:158–165. doi: 10.1207/S15324796ABM2303_3. [DOI] [PubMed] [Google Scholar]
- 325.Zeng C, Zhu Z, Liu G, Hu W, Wang X, Yang C, Wang H, He D, Tan J. Randomized, double-blind, placebo-controlled trial of oral enalapril in patients with neurally mediated syncope. Am Heart J. 1998;136:852–858. doi: 10.1016/s0002-8703(98)70131-0. [DOI] [PubMed] [Google Scholar]
- 326.Ziegler D, Weise F, Langen KJ, Piolot R, Boy C, Hubinger A, Muller-Gartner HW, Gries FA. Effect of glycaemic control on myocardial sympathetic innervation assessed by [123I]metaiodobenzylguanidine scintigraphy: A 4-year prospective study in IDDM patients. Diabetologia. 1998;41:443–451. doi: 10.1007/s001250050928. [DOI] [PubMed] [Google Scholar]
- 327.Zigmond MJ, Abercrombie ED, Berger TW, Grace AA, Stricker EM. Compensations after lesions of central dopaminergic neurons: Some clinical and basic implications. Trends Neurosci. 1990;13:290–296. doi: 10.1016/0166-2236(90)90112-n. [DOI] [PubMed] [Google Scholar]
- 328.Zimlichman R, Goldstein DS, Zimlichman S, Keiser HR. Angiotensin II increases cytosolic calcium and stimulates catecholamine release in cultured bovine adrenomedullary cells. Cell Calcium. 1987;8:315–325. doi: 10.1016/0143-4160(87)90006-6. [DOI] [PubMed] [Google Scholar]
- 329.Zimlichman R, Levinson PD, Kelly G, Stull R, Keiser HR, Goldstein DS. Derivation of urinary dopamine from plasma dopa. Clin Sci. 1988;75:515–520. doi: 10.1042/cs0750515. [DOI] [PubMed] [Google Scholar]
- 330.Zohar J, Yahalom H, Kozlovsky N, Cwikel-Hamzany S, Matar MA, Kaplan Z, Yehuda R, Cohen H. High dose hydrocortisone immediately after trauma may alter the trajectory of PTSD: Interplay between clinical and animal studies. Eur Neuropsychopharmacol. 2011;21:796–809. doi: 10.1016/j.euroneuro.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 331.Zuckerman-Levin N, Tiosano D, Eisenhofer G, Bornstein S, Hochberg Z. The importance of adrenocortical glucocorticoids for adrenomedullary and physiological response to stress: A study in isolated glucocorticoid deficiency. J Clin Endocrinol Metab. 2001;86:5920–5924. doi: 10.1210/jcem.86.12.8106. [DOI] [PubMed] [Google Scholar]