

Abstract
Muraymycins are a promising class of antimicrobial natural products. These uridine-derived nucleoside-peptide antibiotics inhibit the bacterial membrane protein translocase I (MraY), a key enzyme in the intracellular part of peptidoglycan biosynthesis. This review describes the structures of naturally occurring muraymycins, their mode of action, synthetic access to muraymycins and their analogues, some structure–activity relationship (SAR) studies and first insights into muraymycin biosynthesis. It therefore provides an overview on the current state of research, as well as an outlook on possible future developments in this field.
Keywords: antibiotics, natural products, nucleosides, peptides, structure–activity relationship
Introduction
The treatment of infectious diseases caused by bacteria is a severe issue. With multiresistant bacterial strains rendering well-established therapeutic procedures ineffective, the exploration of novel antimicrobial agents is of growing significance. The discovery of penicillin [1] and the proof of its in vivo efficacy [2] marked the starting point for the research on antibacterial drugs during the so-called "golden age" of antibiotics. Despite the early occurrence of first resistances [3–5], an innovation gap followed from the 1960s onwards, during which only few antibiotics were introduced into the market. Most of them were modifications of established substances already in clinical use. Current and future developments will have to consider these improved 2nd and 3rd generation antibiotics [6] alongside the search for completely unknown structures. For such novel agents, natural products appear to be a promising source [7–9].
Bacteria deploy different mechanisms to escape the toxic effect of an antibacterial drug [10–12]. These include the structural modification and degradation of a drug, as it is reported for aminoglycoside-modifying proteins [13], and alteration of the drug target, as can be found in macrolide-resistant bacteria that contain mutations in the bacterial ribosome [14]. Further mechanisms are an increased efflux [15] and a change in permeability of the cell wall [16–17]. Due to the evolutionary pressure exerted by antibiotics, bacteria featuring the aforementioned mutations survive, proliferate and may even develop resistances against multiple drug classes. Excessive application of antibiotics fuels the emergence of multiresistant strains such as hospital and community-associated methicillin-resistant Staphylococcus aureus (MRSA) [18–19] and vancomycin-resistant Enterococcus (VRE) [20]. This development raises the demand for antibiotics exploiting yet unused modes of action. Potential targets within bacteria include peptidoglycan biosynthesis, protein biosynthesis, DNA and RNA replication and folate metabolism [21].
Promising candidates meeting the requirements for new drugs are nucleoside antibiotics, i.e., uridine-derived compounds that address the enzyme translocase I (MraY) as a novel target, thereby interfering with a membrane-associated intracellular step of peptidoglycan biosynthesis. This review will focus on muraymycins as a subclass of nucleoside antibiotics, covering their mode of action, synthetic approaches as well as SAR studies on several derivatives. Furthermore, first insights into the biosynthesis of these Streptomyces-produced secondary metabolites will be discussed.
Review
Structures of naturally occurring muraymycins
The muraymycins were first isolated in 2002 from a broth of a Streptomyces sp. [22]. McDonald et al. discovered and characterised 19 naturally occurring muraymycins (Figure 1). These compounds belong to the family of nucleoside antibiotics which have a uridine-derived core structure in common. Their antibiotic potency is based on the inhibition of MraY, thereby blocking a membrane-associated intracellular step of bacterial cell-wall biosynthesis. The structure elucidation was carried out using one- and two-dimensional NMR experiments as well as FT mass spectrometry [22].
Figure 1.

Structures of the naturally occurring muraymycins isolated by McDonald et al. [22].
Muraymycins have a glycyl-uridine motif, which is connected via an aminopropyl linker to a urea peptide moiety consisting of L-leucine or L-hydroxyleucine, L-epicapreomycidine (a non-proteinogenic cyclic arginine derivative) and L-valine. The uridine structure is glycosylated in its 5'-position with an aminoribose unit and in some cases a lipophilic side chain is attached to the hydroxyleucine residue. The 19 compounds are divided into four different series (A–D) which mainly vary in the leucine residue and the lipophilic side chain or the amino sugar (Figure 1). The aminoribose is missing in muraymycins A5 and C4, which may eventually be hydrolysis products. The series A and B have lipophilic side chains with varying chain lengths, which are either ω-functionalised with a guanidino or hydroxyguanidino-function in case of series A or unfunctionalised but terminally branched in case of series B. Muraymycins of series C contain unfunctionalised L-hydroxyleucine while in series D proteinogenic L-leucine occurs instead.
Muraymycin A1 is one of the most active members of this family and shows good activity mainly against Gram-positive (Staphylococcus MIC: 2–16 μg/mL, Enterococcus MIC: 16–64 µg/mL) but also a few Gram-negative bacteria (E. coli MIC: down to 0.03 μg/mL). Since the activity against wild-type E. coli is clearly lower (MIC > 128 μg/mL) [22], it is assumed that this might be an effect resulting from low membrane permeability.
There are other naturally occurring nucleoside antibiotics which address the same biological target, thereby inhibiting peptidoglycan biosynthesis. Figure 2 shows the structures of selected other classes of nucleoside antibiotics, with structural similarities being highlighted. A broad overview of antimicrobial nucleoside antibiotics blocking peptidoglycan biosynthesis is given by Bugg et al. in two review articles [23–24] and by Ichikawa et al. in a recent review [25].
Figure 2.

Structures of selected classes of nucleoside antibiotics. Similarities to the muraymycins are highlighted in different colours.
Representing the first discovered nucleoside antibiotics, the tunicamycins were isolated in 1971 from Streptomyces lysosuperficus nov. sp. by Takatsuki and Tamura et al. [26–28]. They contain a uridine moiety, two O-glycosidically linked sugars, the so-called tunicamine and a fatty acid moiety, which typically is terminally branched and unsaturated. Two closely related nucleoside antibiotics were isolated later on and named streptoviridins (isolated in 1975 from Streptomyces griseoflavus subsp. thuringiensis [29–31]) and corynetoxins (isolated in 1981 from Corynebacterium rathayi [32]). These classes have merely the uracil nucleoside core structure in common with the muraymycins and the terminally branched lipophilic side chain resembles the acyl moiety in muraymycins of group B.
Capuramycin, a nucleoside antibiotic isolated in 1986 from Streptomyces griseus, shares the uracil-derived nucleoside moiety with the muraymycins [33–34]. The antibiotic FR-900493, which is structurally closely related to muraymycins, was isolated from Bacillus cereus and characterised in 1990 [35]. In comparison to the muraymycins, only the urea peptide moiety and the lipopeptidyl motif are absent.
The mureidomycins [36–38] and pacidamycins [39–41], both reported in 1989, the napsamycins (1994) [42] and the sansanmycins (2007) [43–44] are structurally closely related. They consist of a 3'-deoxyuridine unit with a unique enamide linkage and the non-proteinogenic N-methyl-2,3-diaminobutyric acid, which branches into two peptide moieties. They differ in the amino acid residues AA2, AA4 and AA5, with AA2 and AA5 being aromatic in all four classes. The amino acid residue AA4 is either methionine for mureidomycins, napsamycins and sansanmycins or alanine in case of pacidamycins. Remarkably, these natural products share a urea peptide motif with the muraymycins. They are mainly active against Gram-negative bacteria, which is a noteworthy difference to the muraymycins and other related nucleoside antibiotics.
The liposidomycins (isolated in 1985) [45] and the related caprazamycins (isolated in 2003) [46–47] have a unique diazepanone ring, and in case of the caprazamycins a permethylated rhamnose residue. They resemble the muraymycins in their uridine-derived core structure, which is also glycosylated in 5'-position with an aminoribose unit, and they contain a fatty acid moiety as well. Caprazamycins also display noteworthy antimicrobial activity against M. tuberculosis as well as most Gram-positive bacteria (Table 1) [46,48].
Table 1.
Comparison of the antimicrobial activities of selected representative compounds of different classes of nucleoside antibiotics against selected bacterial species.a
| Gram-positive | Gram-negative | ||||
| S. aureus | B. subtilis | M. smegmatis | E. coli | P. aeruginosa | |
| Muraymycin A1 | ++ | n.r. | n.r. | ++/+b | +/− |
| Tunicamycin | − | ++ | − | − | − |
| Capuramycin | − | − | ++ | − | − |
| FR-900493 | ++ | ++ | n.r. | n.r. | n.r. |
| Mureidomycin C | − | n.r. | n.r. | − | ++ |
| Caprazamycin B | ++ | ++/+ | ++ | − | ++ |
| Liposidomycin A | − | − | n.r.c | − | n.r. |
a++: good activity (MIC < 10 μg/mL), +: moderately active (10 μg/mL < MIC < 32 μg/mL), −: no notable activity (MIC > 32 μg/mL), n.r.: not reported. bNot active against wild-type E. coli. cActive against M. phlei.
All aforementioned nucleoside antibiotics address the same biological target and most likely have the same mode of action by inhibiting MraY (see below), but their in vitro activity differs significantly. It is important to notice that a comprehensive comparison of minimum inhibitory concentrations (MIC values) is difficult because naturally occurring nucleoside antibiotics have been tested against different bacterial strains. However, synthetic analogues of the nucleoside antibiotics listed in Table 1 have been tested against some of the listed bacterial species. It can therefore be assumed that the parent natural products display similar activities even though there are no data available. Furthermore, the activity of a compound against different strains of a bacterial species can vary. Nonetheless, there are certain trends and differences that can be observed. Muraymycin A1 is mainly active against Gram-positive bacteria such as S. aureus or E. faecalis, but also against some Gram-negative E. coli strains [49]. Tunicamycin, capuramycin and FR-900493 only show antimicrobial activity against Gram-positive strains. For mureidomycin C (R5 = Gly, AA2 = AA5 = m-Tyr, AA4 = Met, B = uracil, see Figure 2) as a representative compound, no activity against Gram-positive bacteria was observed, but it displayed pronounced antibacterial activity against P. aeruginosa. This remarkable finding distinguishes the mureidomycins, pacidamycins, sansanmycins and napsamycins from other nucleoside antibiotics. On the other hand, caprazamycin B shows good activity against Gram-positive bacteria, Pseudomonas and M. tuberculosis [48]. The related liposidomycins display good activity against M. phlei, while they are not active against a range of other bacteria [45].
Mode of action
To develop an effective antibiotic one needs to choose a target that is essential for bacterial survival or growth and offers selectivity to strike only bacterial cells (without cytotoxicity to human cells). There are mainly four classical target processes for antibiotics: bacterial cell wall biosynthesis, bacterial protein biosynthesis, DNA replication and folate metabolism [21]. Novel approaches that differ from these established modes of action are under investigation, but many new compounds in development still address bacterial cell wall biosynthesis. They are accompanied by a rich variety of prominent antibiotics in clinical use such as the penicillins [23,50–51]. All bacteria, i.e., Gram-positive and Gram-negative congeners, have a cell wall as part of their cell envelope. While its thickness differs among bacteria – Gram-positive strains usually have a thicker cell wall relative to Gram-negative ones – the principle molecular structure remains identical: Bacterial cell walls consist of peptidoglycan, a heteropolymer with long chains of alternating units of N-acetylmuramic acid (MurNAc) and N-acetylglucosamine (GlcNAc) that are cross-linked through peptide chains attached to the muramic acid sugar (Figure 3) [52].
Figure 3.

Structure of peptidoglycan. Long chains of glycosides (alternating GlcNAc (green) and MurNAc (blue)) are cross-linked through the MurNAc peptide chain. The exact composition of the peptide chain varies among different bacterial species.
The biosynthesis of peptidoglycan is illustrated in Figure 4 and has been described in detail in several reviews (e.g., [51,53–57]). It can be divided into three parts: first, the formation of the monomeric building blocks in the cytosol (Figure 4, step A); second, the membrane-bound steps with the attachment to the lipid linker, transformation to a disaccharide and transport to the extracellular side of the membrane (Figure 4, steps B, C); finally, polymerisation to long oligosaccharide chains and cross-linking occur (Figure 4, steps D, F).
Figure 4.

Schematic representation of bacterial cell wall biosynthesis.
In the cytosol, uridine diphosphate-N-acetylglucosamine (UDP-GlcNAc), that is formed from fructose-6-phosphate in four steps, is transformed into UDP-MurNAc-pentapeptide in a number of enzyme-catalysed reactions (Figure 4, step A). The exact composition of the peptide chain varies in different organisms. Examples given in Figure 3 are frequently occurring ones and a more comprehensive list has been reported elsewhere [52].
The membrane-associated steps commence with the transfer of UDP-MurNAc-pentapeptide to the lipid carrier undecaprenyl phosphate, catalysed by translocase I (MraY), to give lipid I (Figure 4, product of step B). The glycosyltransferase MurG attaches a GlcNAc sugar to furnish lipid II (Figure 4, product of step C). This building block is then transported to the extracellular side of the membrane. It is speculated that there might be some kind of 'flippase' involved but this particular step is still unclear and requires further investigation [55]. On the extracellular side of the membrane, the building blocks are connected by transglycosylases to form long chains (Figure 4, step D) and then are cross-linked by transpeptidases (Figure 4, step E). Both enzymes are members of the family of penicillin-binding proteins [23].
As mentioned above, there are many antibiotics in clinical use that target at least one step of bacterial cell wall biosynthesis. Prominent examples besides penicillins are cephalosporins, cycloserine, vancomycin, fosfomycin and daptomycin [9]. All of them (except fosfomycin and cycloserine) inhibit late, extracellular steps of cell wall formation. Thus, there are still many steps not addressed by clinically used drugs, which implies that cell wall biosynthesis still offers promising novel targets for the development of antibiotics with new modes of action. Muraymycins and other nucleoside antibiotics target translocase I (MraY) that represents such a potential novel molecular target [22].
Overexpression of the mraY gene, identified in an mra (murein region A) cluster, led to an increase of UDP-N-acetylmuramoyl-pentapeptide: undecaprenyl phosphate phospho-N-acetylmuramoyl-pentapeptide transferase activity [58]. Gene knockout experiments revealed the MraY-catalysed reaction in cell wall biosynthesis to be an essential process for bacterial viability and growth [59–63].
The chemical transformation catalysed by MraY is shown in Figure 5. The cytosolic precursor UDP-MurNAc-pentapeptide is linked to undecaprenyl phosphate, a C55-isoprenoid lipid carrier that is located in the cellular membrane. With concomitant release of uridine monophosphate (UMP), this furnishes a diphosphate linkage between the two substrates. The reaction is reversible and MraY accelerates the adjustment of the equilibrium state. Whereas this reaction was known for a long time [64–65], the structure of the MraY protein remained unclear. The mechanism of the MraY-catalysed reaction was investigated by kinetic studies by Heydanek, Neuhaus et al. in the 1960s. They proposed a two-step mechanism for lipid I formation that was later revised (Figure 6A) [55,66–69].
Figure 5.

Translocase I (MraY) catalyses the reaction of UDP-MurNAc-pentapeptide with undecaprenyl phosphate towards lipid I.
Figure 6.

Proposed mechanisms for the MraY-catalysed reaction. A: Two-step mechanism postulated by Heydanek et al. [66]; B: one-step mechanism postulated by Bouhss et al. [69].
The identification of the mraY gene [58] facilitated the alignment of MraY homologue sequences by van Heijenoort et al. and resulted in a two-dimensional topology model of MraY from E. coli, among others [70]. Bugg et al. identified three conserved residues with nucleophilic side chains within the superfamily of polyisoprenyl-phosphate N-acetyl hexosamine 1-phosphate transferases (PNPT). Mutation of these three aspartate residues (D115, D116 and D267 in the E. coli protein) resulted in a complete loss of catalytic activity. This led to a proposed model for the active site of MraY in accordance with previous findings [66]: D115 and D116 bind a Mg2+-cofactor, UDP-MurNAc-pentapeptide also binds the Mg2+-cofactor and D267 acts as a nucleophile within the proposed two-step mechanism (Figure 6) [68]. In a study with purified MraY from B. subtilis, Bouhss et al. found small remaining activity in the D231N mutant (corresponding to D267 in MraY from E. coli). They assumed that this would contradict the two-step mechanism as a nucleophilic residue is essential for the previously proposed mechanism. They found D98 to be crucial for activity and proposed its role to deprotonate undecaprenyl phosphate. This was speculated to be followed by a one-step nucleophilic attack of the C55-alkyl phosphate at the UDP-MurNAc-pentapeptide (Figure 6B) [69].
In 2013, Lee et al. reported an X-ray crystal structure (3.3 Å resolution) of MraY from Aquifex aeolicus (MraYAA) as the first structure of a member of the PNPT superfamily. MraYAA crystallised as a dimer and additional experiments showed that it also exists as a dimer in detergent micelles and membranes [71]. The previously proposed models are in agreement with the solved structure showing ten transmembrane helices and five cytoplasmic loops. The authors identified a cleft at the cytoplasmic side of the membrane that showed the highest conservation in sequence mapping. Furthermore, it is also the region where most of the previously identified, functionally important residues [69] are located [71]. The location and binding mode of the Mg2+ ion in the crystal does not support the proposed model for a two-step mechanism [68]. In experiments with Mn2+ exchange no interaction of the metal with D117 and D118 could be detected. Surface calculation of MraYAA showed an inverted U-shaped groove that could harbour the undecaprenyl phosphate co-substrate. The locations of this groove, the Mg2+ and D265 do at least not contradict the proposed one-step mechanism. Nevertheless, there is still a need for further studies to fully understand the MraY-catalysed reaction at the molecular level [71].
In the context of a different MraY inhibitor, i.e., lysis protein E from bacteriophage 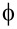 X174, Bugg et al. reported a different site of inhibition in pronounced distance to the proposed active site. It has been demonstrated before that mutation of phenylalanine 288 (F288L) in helix 9 of MraY caused resistance against lysis protein E [72–73]. An interaction between F288 and glutamic acid 287 (E287) with the peptide motif arginine-tryptophan-x-x-tryptophan (RWxxW, x represents an arbitrary amino acid) was found. Mutants F288L and E287A showed reduced or no detectable enzyme inhibition, thus indicating a secondary binding site for potential MraY inhibitors. Nevertheless, it remains unclear how binding at helix 9 can inhibit MraY function and further studies are probably inevitable [74].
X174, Bugg et al. reported a different site of inhibition in pronounced distance to the proposed active site. It has been demonstrated before that mutation of phenylalanine 288 (F288L) in helix 9 of MraY caused resistance against lysis protein E [72–73]. An interaction between F288 and glutamic acid 287 (E287) with the peptide motif arginine-tryptophan-x-x-tryptophan (RWxxW, x represents an arbitrary amino acid) was found. Mutants F288L and E287A showed reduced or no detectable enzyme inhibition, thus indicating a secondary binding site for potential MraY inhibitors. Nevertheless, it remains unclear how binding at helix 9 can inhibit MraY function and further studies are probably inevitable [74].
In order to investigate the biological potencies of MraY inhibitors such as the muraymycins, in vitro assay systems are needed. A widely used and universal method to evaluate the in vitro activity of potential agents against certain bacteria is the determination of minimum inhibitory concentrations (MIC). MICs are defined as the lowest concentration at which a potential antimicrobial agent inhibits the visible growth of a microorganism [75]. They are easily determined and reflect several effects such as target interaction, cellular uptake and potential resistance mechanisms of the microorganism. MIC values are therefore widely used, also in studies on muraymycin analogues (e.g., [22,76–78]) and have been the basis of many structure–activity relationship studies (see below).
This bacterial growth assay, however, does not elucidate the inhibitory potency of the potential antimicrobial solely against the target protein MraY. Thus, another assay system that is not based on the interaction with whole cells but only with the target protein is required. For MraY, there are three different assays available that provide such inhibition data: i) a fluorescence-based and ii) a radioactivity-based assay as well as iii) a relatively new Förster resonance energy transfer (FRET)-based method.
The fluorescence-based assay was developed by Bugg et al. [79–80] and uses a fluorescently labelled (dansylated) analogue of the MraY substrate UDP-MurNAc-pentapeptide. The reaction of this substrate analogue with undecaprenyl phosphate leads to an increase in fluorescence intensity that can be used as a measure for enzymatic activity (e.g., [74,78]). The assay reported by Bouhss et al. [81] uses a radioactively labelled UDP-MurNAc-pentapeptide and thin layer chromatography (TLC) separation of undecaprenyl-linked MurNAc-pentapeptide from unreacted substrate (e.g., [77,82]). The third assay was introduced in 2012 by Shapiro et al. and uses a FRET system with the FRET donor attached to the UDP-MurNAc-pentapeptide and the FRET acceptor in a detergent or detergent/lipid micelle that also hosts the MraY protein [83].
The overexpression and purification of the transmembrane protein MraY is challenging. MraY from different bacterial strains was heterologous overexpressed in E. coli and was used in assays mentioned above as a crude cellular membrane preparation or as a detergent-solubilised membrane protein mixture [79,84]. A purification to homogeneity was reported for MraY from B. subtilis by Bouhss et al. in 2004 [81] and for the congener from Aquifex aeolicus by Lee et al. in 2013 [71]. Wang, Bernhard et al. achieved a cell-free production of MraY from B. subtilis and E. coli, also experiencing the need of pronounced adjustments in expression conditions [85].
Synthetic access
Following the isolation of muraymycins [22], a group of scientists from Wyeth reported the semisynthetic access towards 16 derivatives of muraymycin C1 for structure–activity relationship (SAR) studies [86]. At the same time, a first set of fully synthetic structurally simplified muraymycin analogues was described [76]. Starting from uridine (1), protected uridine-5'-aldehyde 2 was prepared in four steps (Scheme 1) [87–88]. This was followed by an aldol reaction of aldehyde 2 with N,N-dibenzylglycine tert-butyl ester (3) [89] and LDA as a key step of the synthesis (Scheme 1). The resultant products were the two 5'-epimers 4 (5'R,6'S) in 31% yield and 5 (5'S,6'S) in 14% yield, which could be separated by column chromatography. After debenzylation, the resultant primary amines were connected with amido aldehydes 6 substituted with different moieties R and R' by reductive amination with R being either a hydroxy group or a hydrogen and R' representing an alkyl, allyl, ester or a protected amino moiety. This led to many truncated muraymycin analogues based on the structures 7 and 8 [76]. Cbz deprotection and subsequent peptide coupling with the L-arginine-L-valine-derived urea dipeptide 9 gave various full-length muraymycin analogues 10 and 11 [76]. Some of the truncated and the full-length compounds were able to inhibit lipid II formation. These active compounds are discussed in the section on structure–activity relationship (SAR) studies.
Scheme 1.

First synthetic access towards simplified muraymycin analogues as reported by Yamashita et al. [76].
In 2005, Ichikawa, Matsuda et al. reported the synthesis of (+)-caprazol [90–92] which contains the same uridine-derived core structure as the muraymycins. The latest and optimised synthesis is shown in Scheme 2 [92]. Oxidation of the isopropylidene-protected uridine 12 to the 5'-aldehyde and a Wittig reaction [93] gave olefin 13. The key step was a subsequent asymmetric Sharpless aminohydroxylation [94] furnishing (5'S,6'S)-nucleosyl amino acid 14 in 96% yield (98% de) [92,95]. A novel β-selective glycosylation of the 5'-hydroxy group was also established. Thus, 14 was reacted with the ribosyl fluoride 15 and BF3·Et2O, which afforded the glycosylated product 16 in 77% yield and with a β/α-selectivity of 24:1 [91–92]. This reaction was followed by an azide reduction, Boc protection, saponification of the ester, peptide coupling with the amino acid 17, oxidative cleavage of the double bond to give 18 and an intramolecular reductive amination in order to construct the seven-membered ring. Methylation with subsequent acidic global deprotection led to the target compound (+)-caprazol (19) [90,92].
Scheme 2.

Synthesis of (+)-caprazol (19) reported by Ichikawa, Matsuda et al. [92].
For the synthesis of muraymycins, Ichikawa, Matsuda et al. furthermore developed a new route towards the epicapreomycidine-containing urea dipeptide unit via C–H activation (Scheme 3) [96–97]. For this purpose, the commercially available δ-N-Boc-α-N-Cbz-L-ornithine (20) was transformed into sulfamate 21. Subsequently, the C–H insertion representing the key step of this synthesis was examined with two different catalysts and different reaction conditions. Despite different ratios in the outcome of the C–H insertion in favour of the unwanted diastereomer 22, the synthesis was finished with the desired minor component 23. Boc deprotection followed by reaction with guanidinylation reagent 24 gave bicyclic compound 25. The next steps included a desulfonylation and the reaction with 26 leading to protected epicapreomycidine-containing urea dipeptide 27 [96–97].
Scheme 3.

Synthesis of the epicapreomycidine-containing urea dipeptide via C–H activation [96–97].
Starting from the uridine derivative 28 used in the synthesis of (+)-caprazol, Ichikawa and Matsuda built up muraymycin D2 and its epimer (Scheme 4). They used an Ugi four-component reaction with an isonitrile derivative 29 obtained from the uridine-derived core structure 28, aldehyde 30, amine 31 and the urea dipeptide building block 27. A two-step global deprotection then gave the desired muraymycin D2 and its epimer which could be separated by HPLC [96–97].
Scheme 4.

Synthesis of muraymycin D2 and its epimer reported by Ichikawa, Matsuda et al. [96–97].
In 2012, Kurosu et al. also reported the synthesis of potential key intermediates for the total synthesis of muraymycins (Scheme 5) [98]. A fully protected ureidomuraymycidine tripeptide was prepared through lactone opening followed by urea formation and a final Mitsunobu ring closure as key steps. A Strecker reaction of the benzylimine 34 followed by several steps afforded the alcohol 35. A thermal lactonisation as a first key step of the synthesis led to a 1:1 mixture of the two epimers 36 and 37, and the undesired lactone 37 could be epimerised and converted into 36 by treatment with DBU [98]. Epimerisation and simultaneous lactone opening could be achieved in another key step using L-valine tert-butyl ester. Acetylation of the thus formed primary alcohol resulted in compound 38. This was followed by benzyl and Cbz deprotection and the subsequent urea formation with the imidazolium salt 39 to furnish tripeptide 40. After Boc deprotection, the resultant amine was guanidinylated using isothiourea 41. The thus obtained precursor 42 was treated with DIAD and PPh3 in a final step for an intramolecular Mitsunobu ring closure to finish the synthesis of the fully protected ureidomuraymycidine 43 (Scheme 5) [98].
Scheme 5.

Synthesis of the urea tripeptide unit as a building block for muraymycins reported by Kurosu et al. [98].
In 2010, Ducho et al. reported an alternative synthesis of the naturally occurring uridine-derived muraymycin core structure (Scheme 6) [78,99]. The key step of their route was a sulfur-ylide reaction with high substrate-controlled diastereoselectivity [100–102]. This epoxide-forming sulfur-ylide reaction had been established before by Sarabia et al. [103–104]. After some initial confusion regarding the stereochemical configuration of the epoxide product, it could be unambiguously proven that the transformation of uridine-5'-aldehyde 44 with sulfonium salt 45 under basic conditions furnished epoxide 46 with high diastereoselectivity (Scheme 6). Subsequent ring opening of this epoxide with Bu4NBr resulted in bromohydrine 47, followed by levulinyl (Lev) protection of the hydroxy group (product 48). Nucleophilic substitution at the 6'-position with Bu4NN3 gave the naturally occurring (5'S,6'S)-stereochemistry of the uridine core structure in a double inversion manner [78,99]. DDQ oxidation then provided indolamide 49. Hydrolysis of the amide, formation of the synthetically more versatile tert-butyl ester, azide reduction and final Cbz protection resulted in the uridine-derived building block 50 for the synthesis of naturally occurring muraymycins (Scheme 6). Furthermore, 5'- and 6'-epi analogues of muraymycins were also synthesised via suitable epoxide precursors by Ducho et al. [105].
Scheme 6.

Synthesis of the uridine-derived core structure of naturally occuring muraymycins reported by Ducho et al. [78,99].
Ducho's synthesis of epicapreomycidine (Scheme 7) started from the (R)-configured Boc-protected Garner aldehyde 51 [106], which was transformed into the N-benzylimine 52. The latter was then diastereoselectively converted with a Grignard reagent into the amine 53 as a key step of the synthesis [78]. Cbz protection followed by ozonolysis with subsequent reductive amination and hydrogenolysis led to the 1,3-diamine 54. The cyclisation to the guanidine functionality was achieved with the novel guanidinylation reagent 55. With the protected epicapreomycidine precursor 56 in hand, the Boc and acetonide protecting groups were removed. Urea formation with the valine derivative 57 with final oxidation of the primary hydroxy function afforded the desired dipeptide 58 [78] (Scheme 7).
Scheme 7.

Synthesis of the epicapreomycidine-containing urea dipeptide from Garner's aldehyde reported by Ducho et al. [78].
Furthermore, Ducho et al. synthesised the hydroxyleucine moiety found in naturally occurring muraymycins of classes A to C (Scheme 8) [107]. Adapting a strategy developed by Zhu et al., D-serine (59) was stereoselectively converted into the protected amino alcohol 60 [108]. Key intermediate 60 was then Cbz- and acetonide protected to give 61. A sequence of desilylation and oxidation furnished the acid 62. Peptide coupling with amine 63 and acidic deprotection then afforded the desired aldehyde 64, which already contained the muraymycin linker unit (Scheme 8) [107]. Together with the uridine core structure 50 and the urea dipeptide 58, the aldehyde 64 was the third building block of Ducho's envisioned stereocontrolled tripartite route towards muraymycins, in contrast to Ichikawa's and Matsuda's modular multicomponent, but non-stereocontrolled approach (see above).
Scheme 8.

Synthesis of a hydroxyleucine-derived aldehyde building block reported by Ducho et al. [107].
This novel tripartite approach was then used by Ducho et al. to synthesise the structurally simplified natural product analogue 5'-deoxy muraymycin C4 (65), which formally differs from the parent natural product only by absence of one oxygen atom (Scheme 9) [78,109–110]. Starting from protected uridine-5'-aldehyde 44, the first key step of the synthesis was a (Z)-selective Wittig–Horner reaction with phosphonate 66 [111] in order to obtain the didehydro amino acid 67. The next important step of this route was an asymmetric catalytic hydrogenation [112–113] with the chiral Rh(I)–DuPHOS catalyst 68 to prepare the (6'S)-configured product 69 [109–110]. Subsequent hydrogenolytic cleavage of the Cbz group gave the nucleosyl amino acid 70. To complete the tripartite approach, the reductive amination with the aldehyde 64 furnished 71, and Cbz deprotection and peptide coupling with the epicapreomycidine-containing urea dipeptide 58, followed by acidic global deprotection, gave the desired 5'-deoxy muraymycin C4 (65) (Scheme 9) [78].
Scheme 9.

Synthesis of 5'-deoxy muraymycin C4 (65) as a closely related natural product analogue [78,109–110].
In addition to the described synthetic routes, a range of other muraymycin analogues has been prepared. In the interest of conciseness, this synthetic work is not discussed here, but the biological properties of such analogues will be summarised in the following section on SAR studies.
Structure–activity relationship studies
With various structurally diverse compounds at hand, the stage has been set for SAR studies on muraymycins. The antimicrobial activities found by McDonald et al. introduced muraymycins as a promising subject of study [22]. The naturally occurring muraymycins isolated from Streptomyces guided first insights into the structural features essential for MraY inhibition. For the most active member of the family, i.e., muraymycin A1, antibiotic activity could be found against various bacteria ranging from Staphylococci with MIC values of 2 to 16 μg/mL, Enterococci with 16 μg/mL and higher to some Gram-negative bacteria (8 μg/mL). Against an E. coli mutant with increased membrane permeability, an MIC value below 0.03 μg/mL was obtained, suggesting that inhibition is a matter of cellular uptake of the compound. In vivo efficacy was demonstrated for muraymycin A1 with an ED50 of 1.1 mg/kg in Staphylococcus aureus-infected mice.
Five of the 19 naturally occurring compounds (i.e., muraymycins A1, A5, B6, C2 and C3) were capable of inhibiting both MraY and peptidoglycan synthesis at the lowest concentration tested (IC50 = 0.027 μg/mL), which represented activities comparable to those of liposidomycin C (0.05 μg/mL) and mureidomycin A (0.03 μg/mL). As a general trend, higher antimicrobial activities were found for acylated compounds, in particular with longer and functionalised fatty acid side chains.
Lin et al. employed a semisynthetic approach for modifications of muraymycin C1 as starting point of their SAR studies (Figure 7) [86]. In accordance with the results reported by McDonald et al., their work was based on the assumption that the cellular uptake required for MraY inhibition is mainly dependent on fatty acids connected to the hydroxyleucine moiety. The attachment of lipophilic groups on either the primary or both the primary and secondary amino function was supposed to have similar effects. The muraymycin derivatives 72–86 were thus evaluated against the target in a coupled MraY–MurG in vitro assay employing radiolabelled UDP-N-acetylglucosamine. Disubstituted analogues were not active at the concentrations tested, suggesting that one free amino group is vital for activity. Hydantoin-derived compounds 79 with C12H25 and 80 with PhCH2 as residues R at the hydantoin moiety gave the best results with inhibition of lipid II formation at 6.25 μg/mL, which is comparable to muraymycin C1. Good activity was also found for hydantoin derivative 77 with the 4-FC6H4 substituent, showing inhibition of lipid II formation at 25 μg/mL. The only N-alkylated derivative inhibiting in the same order of magnitude was 83 with n-C11H23 substitution. However, activities of the other compounds within this group also coincided with the previous observation that lipophilic compounds were more active. Overall, the tested monosubstituted hydantoin derivatives confirmed the assumed correlation between inhibitory activities and lipophilicity of the substituent.
Figure 7.

Summary of modifications on semisynthetic muraymycin analogues tested by Lin et al. [86]. Most active compounds are highlighted (in vitro inhibition of lipid II formation at 6.25 μg/mL: orange; 25–50 μg/mL: green).
Yamashita et al. studied truncated muraymycin analogues lacking the lipophilic side chain as described in the section on synthetic access (compounds of type 7, 8 and 10) [76]. The activities measured in a soluble peptidoglycan assay indicated a stereochemical preference for the (5'S)-configuration, contrary to the results of MIC value determination. Further studies were then carried out with (5'R)-derivatives only, i.e., with 5'-epimers of the parent natural products. The influence of protecting groups was examined applying a strategy of stepwise deprotection. This led to the observation that fully protected compounds were not active at all, as well as the completely deprotected analogues. Remarkably, some partially protected congeners 87–90 with the free terminal amino group were found to show good inhibition (MIC = 1–16 μg/mL) of the growth of Gram-positive bacteria including S. aureus and E. faecalis strains, with best results obtained for 88 (Figure 8). Evaluation of the inhibition of lipid II formation revealed the importance of the substitution pattern of the terminal amino acid.
Figure 8.

Bioactive muraymycin analogues identified by Yamashita et al. [76].
In 2010 and 2011, Ichikawa, Matsuda et al. published SAR studies with a range of synthetic muraymycin analogues [77,114]. The IC50 values were measured in an in vitro assay mentioned above to examine the inhibitory activity of the prepared analogues against the target enzyme. MIC values were determined against several bacterial strains. The inhibitory activities of the synthesised muraymycin D2 33 (with an L-leucine unit) and its epimer (with a D-leucine unit) on the purified MraY enzyme from B. subtilis were determined. Both compounds showed good inhibitory activities with IC50 values of 0.01 μM and 0.09 μM, respectively. However, their antibacterial activities against several Gram-positive bacteria (S. aureus, E. faecalis, E. faecium) were low (MIC values up to 64 μg/mL). In comparison to the analogues of the A and B series, which showed good antibacterial activities (see above), muraymycin D2 (33) and its epimer lack the hydrophobic side chain at the leucine moiety [22]. It was postulated that this lipophilic side chain may not be necessary for target inhibition, but for cellular uptake through the lipid bilayer of the cytoplasmic membrane, as an increased lipophilicity is advantageous for this [77,114].
Consequently, several lipophilic derivatives 91a–d were prepared (Figure 9). Long-chain lipophilic amino acids were incorporated into the muraymycin core structure as a simplified replacement of the O-acylated hydroxyleucine moiety. Compound 91a (highlighted in orange) with the pentadecyl side chain showed the best activity as an MraY inhibitor (IC50 = 0.33 μM (with L-leucine moiety), IC50 = 0.74 μM (with D-leucine moiety)), but relative to muraymycin D2 and its epimer, this implied a 33-fold and 8-fold, respectively, decrease of inhibitory activity. In bacterial growth assays, the analogue 91a exhibited the best MIC values ranging between 0.25 μg/mL and 4 μg/mL (see Table 2). These values were comparable to those of the naturally occurring congeners of the A and B series [22]. Generally, derivatives with the naturally occurring L-configuration in the leucine moiety showed slightly better activities. These lipophilic analogues were also tested for cytotoxicity towards Hep G2 cells and showed no cytotoxicity (IC50 > 100 μg/mL) [114].
Figure 9.

Muraymycin D2 and several non-natural lipidated analogues 91a–d [77,114].
Table 2.
Inhibitory (against MraY) and antibacterial activities of non-natural lipophilic muraymycin analogues [77,114].
| Compound | (L-Leu) (D-Leu) |
IC50 (μM)a | MIC (μg/mL)b |
| muraymycin D2 (33) | 0.01 0.09 |
>64 >64 |
|
| 91a | 0.33 0.74 |
2–4 0.25–4 |
|
| 91b | n.d. | 4–16 4–16 |
|
| 91c | n.d. | 16–64 4–64 |
|
| 91d | n.d. | 4–8 4–16 |
|
| 92a | n.d. | 2–4 2–4 |
|
| 92b | n.d. | 1–2 2–4 |
|
| 92c | n.d. | 2–8 4–8 |
|
| 92d | n.d. | 4–8 4–8 |
|
| 92e | n.d. | 4–8 4–8 |
|
| Compound (L-Leu) | IC50 (μM)a | MIC (μg/mL)b | |
| 92f | n.d. | 4–8 | |
| 92g | n.d. | 4 | |
| 92h | n.d. | 4–8 | |
| 93 | 5 | 32 to ≥64 | |
In another series of analogues with different peptide units, the pentadecyl side chain of 91a was kept. The L-epicapreomycidine (L-epi-Cpm) unit of 91a was replaced by L-capreomycidine (L-Cpm, 92a), L-arginine (L-Arg, 92b) and L-ornithine (L-Orn, 92c) in order to investigate the role of the cyclic guanidine functionality (Figure 10) [77].
Figure 10.

Non-natural muraymycin analogues with varying peptide structures [77,114].
These compounds were all active against MRSA and VRE with varying MIC values (Table 2). The most active analogues of this series were 92a and 92b (Figure 10, highlighted in orange) with MIC values between 1 μg/mL and 4 μg/mL. Derivatives with unnatural D-stereochemistry in the pentadecyl glycine motif possessed a similar antibacterial activity (potency within factor 2). Truncated analogues lacking the L-valine urea terminus (Cbz-protected 92d and N-terminally unprotected 92e) showed only a minor loss of activity (MIC = 4–8 μg/mL) (Table 2). These results indicated that the guanidine motif of analogues 91a, 92a and 92b (MICs between 0.25 μg/mL and 4 μg/mL) is preferred, but that amino analogues 92c and 92f still show good activity (MICs between 2 μg/mL to 8 μg/mL). The different stereochemistry at the central leucine unit and the terminal truncation had no crucial effects on the antibacterial activity (Table 2). Truncated derivatives 92f–h (Figure 10) without the L-valine urea terminus contained L-ornithine (L-Orn, 92f), L-arginine (L-Arg, 92g) and L-methionine (L-Met, 92h), respectively. They were also tested and showed reasonable activity against some bacterial strains (MIC = 4–8 μg/mL), which further indicated that significant variations in the peptide moiety are tolerated. The truncated analogue 93 (Figure 10) only consisted of the N-alkylated nucleoside core structure. Its inhibitory activity was 6 to 12-fold reduced (IC50 = 5 μM) and the antibacterial activity decreased with MIC values between 32 μg/mL and 64 μg/mL. In summary, these systematic SAR studies demonstrated the importance of the lipophilic side chain for the antibacterial activity. The urea dipeptide motif is important for antibacterial activity as well, but it could be diversified with simpler amino acids as well as being truncated in order to provide bioactive analogues. A graphical summary of these results is provided in Figure 11.
Figure 11.

SAR results for several structural variations of the muraymycin scaffold.
In 2014, Ichikawa, Matsuda et al. continued their SAR studies with respect to urgently needed anti-Pseudomonas agents [115]. These Gram-negative bacteria possess an outer membrane which acts as an additional permeability barrier, making them generally less sensitive to antibacterial agents. In this context, the aforementioned muraymycin analogues (91a, 92a–h) were tested for MraY inhibitory activity again, with MraY enzyme from S. aureus (Table 3). However, antibacterial activities against several Pseudomonas strains were moderate to low with MICs between 8 μg/mL and >64 μg/mL. Analogue 92g was the most active congener in this series with MIC values between 8 μg/mL and 32 μg/mL. Compounds 92e and 92f showed nearly no activity (MIC = 32 to >64 μg/mL). More lipophilic truncated analogues 94 without the urea dipeptide unit (Figure 12) were synthesised and tested, but they all showed nearly no activity.
Table 3.
Inhibitory (against MraY) and antibacterial activities of non-natural muraymycin analogues against Pseudomonas aeruginosa [115].
| Compound | IC50 (nM)a | MIC (μg/mL)b |
| 91a, 92a–d | 0.7–4.2 | ≥ 64 |
| 92e,f | 2.4–3.8 | 32 to ≥ 64 |
| 92g | 2.2 | 8–32 |
| 92h | 8.5 | 16 to ≥ 64 |
| 94 (R1 = -H or -COCH3) | 2.6–2.7 | 32 to ≥ 64 |
| 94 (R1 = -CO(het)aryl ) | 6.4–105 | ≥ 64 |
| 95 | 1.6 | 8–32 |
| 96 | 0.14 | 8–16 |
| 97 | 12.2 | 16–32 |
| 98 | 0.60 | 4–8 |
Figure 12.

Muraymycin analogues designed for potential anti-Pseudomonas activity (most active analogues are highlighted in orange) [115].
These results indicated the importance of the presence of a guanidine residue and a lipophilic side chain for potential antibacterial activity against Pseudomonas strains. Hence, several derivatives were prepared in which the positions and numbers of the guanidine groups and the lipophilic side chains were varied in order to optimise their relative orientation for best biological activity. This strategy resulted in the bioactive analogues 95–98 (Figure 12). Analogue 95 with an interconversion of the lipid side chain and the guanidine group had a slightly reduced activity compared to lipidated analogue 92g. Analogue 96 showed an increased antibacterial activity towards some of the tested Pseudomonas strains. Analogue 97 is an interconverted version of 96 and displayed a comparatively poor activity. The most active analogue was compound 98 which is a hybrid type of the aforementioned analogues 95–97. The results indicate that a lipophilic side chain and guanidine groups are necessary for antibacterial potency. Compounds 95–98 showed antibacterial activity, with the branched-type compound 96 (MIC values between 8 μg/mL and 16 μg/mL) and the hybrid-type compound 98 (MIC between 4 μg/mL and 8 μg/mL) being the most active congeners. A limitation of both analogues 96 and 98 is their increased cytotoxicity against HepG2 cells with IC50 values of 4.5 μg/mL and 34 μg/mL, respectively. Further, the metabolic stability was studied in vitro for the analogues 95, 96 and 98 using human or rat liver microsomes and all of them proved to be reasonably stable [115].
In 2014, Ducho et al. reported the synthesis of 5'-deoxy muraymycin C4 (65, see above) [78]. Biological assays revealed that 65 inhibited the MraY enzymes of E. coli and S. aureus with potencies in the range of tunicamycins. The antibacterial activity of 65 was tested against some selected E. coli and S. aureus strains although the lack of a lipophilic moiety indicated that the compound should not be a potent antibiotic. However, an unexpected moderate activity against E. coli DH5 alpha was observed, whereas 65 was weakly active against E. coli strain ΔtolC but not active against the S. aureus Newman strain. Further studies indicated excellent plasma and metabolic stability and no cytotoxicity. Overall, the structurally simplified 5'-deoxy muraymycin scaffold 65 may therefore be useful for further antibacterial development. It should also be noticed that it has inspired the design of a novel oligonucleotide backbone modification [116–117].
Biosynthesis
So far, there are only limited insights into muraymycin biosynthesis. The biosynthetic gene cluster for the formation of muraymycins in Streptomyces sp. NRRL 30471 has been identified by Chen, Deng et al. in 2011 [118]. The sequence analysis revealed the cluster to contain 33 open reading frames (ORFs) with 26 of them being involved in muraymycin formation. Based on their elucidation of the gene cluster and sequence homologies, Chen, Deng et al. proposed an outline pathway for muraymycin biosynthesis (Scheme 10).
Scheme 10.

Proposed outline pathway for muraymycin biosynthesis based on the analysis of the biosynthetic gene cluster by Chen, Deng et al. [118]. MTA = 5'-deoxy-5'-(methylthio)adenosine.
According to this biosynthetic proposal, uridine (1) is enzymatically oxidised to give uridine-5'-aldehyde 99. Aldehyde 99 then supposedly undergoes an aldol addition with glycine 100 as the enol(ate) component, thus furnishing the amino acid–nucleoside hybrid 5'-C-glycyluridine (GlyU, 101). Alkylation of the 6'-amino group is then achieved by reaction with S-adenosyl methionine (SAM), and the resultant intermediate 102 is decarboxylated to provide diamine 103. Attachment of the aminoribosyl moiety (which is supposedly also derived from uridine (1) over several enzymatic steps) finally affords the aminopropyl-substituted 5'-O-aminoribosylated GlyU core structure 104. Transformation of 104 with the thioester-activated peptide moiety 105 then gives muraymycin C2 (Scheme 10), which is speculated to serve as an intermediate en route to other muraymycins, in particular towards O-lipidated congeners of the A and B series (see Figure 2).
A fragmented non-ribosomal peptide synthetase (NRPS) system appears to be responsible for the assembly of the urea tripeptide building block 105. However, the non-proteinogenic amino acids need to be formed first. It has been proposed that L-arginine (106) undergoes 3-hydroxylation (giving 3-hydroxy-L-arginine (107)) and subsequent ring closure to furnish L-epicapreomycidine ((2S,3S)-capreomycidine, 108), that is then activated as thioester 109 (Scheme 10). This proposal is based on the elucidated formation of the epimeric amino acid L-capreomycidine ((2S,3R)-capreomycidine) as part of viomycin biosynthesis in Streptomyces vinaceus. In this producing organism, L-arginine is diastereoselectively hydroxylated to afford (3S)-3-hydroxy-L-arginine. The ring-closure reaction then occurs with formal inversion of the β-stereocenter (but quite likely through an aza-Michael addition to the α,β-unsaturated intermediate) [119–121]. The exact stereochemical course of epicapreomycidine formation in muraymycin biosynthesis is unclear though as the stereochemical configuration at C-3 of the intermediate 3-hydroxy-L-arginine (107) has not been identified yet. It cannot be ruled out that an epimerisation reaction might be involved in the biosynthesis of 108, in particular with respect to other epimerisation steps in bacterial biosynthetic pathways [122]. Consequently, synthetic routes towards both 3-epimers of 3-hydroxy-L-arginine have been developed which would also enable the preparation of isotopically labelled congeners for biosynthetic studies [123–124]. It should also be noted that a biomimetic domino guanidinylation–aza-Michael-addition reaction for the synthesis of the capreomycidine scaffold has been developed, which only furnished the target structures as stereoisomeric mixtures though [125].
The epicapreomycidine-derived thioester 109 is proposed to be converted into the urea dipeptide motif with valine derivative 110 and possibly hydrogen carbonate as a C1-building block for urea formation, thus furnishing 111. The 3-hydroxy-L-leucine moiety might be obtained by stereoselective enzymatic β-hydroxylation of thioester-activated L-leucine 112, which leads to the formation of 113. Finally, peptide formation by condensation of 111 with 113 affords the complete thioester-activated urea tripeptide unit 105 (Scheme 10). One interesting aspect of this biosynthetic proposal by Chen, Deng et al. is that they assume the putative dioxygenase Mur16 to catalyse β-hydroxylations of two structurally distinct amino acid substrates, i.e., L-arginine (106) and thioester-activated L-leucine 112.
As pointed out, there is a lack of experimental insights into muraymycin biosynthesis beyond the elucidation of its gene cluster. However, Van Lanen et al. have studied the early steps of the biosynthesis of A-90289 nucleoside antibiotics in detail (Scheme 11) [126]. The A-90289 subclass is structurally closely related to caprazamycins and liposidomycins, and its aminoribosylated nucleoside core is identical to that of muraymycins (Figure 2). This supports the assumption that the early steps of the biosynthesis of all these subclasses are probably highly similar, if not identical. For the A-90289 nucleoside antibiotics, Van Lanen et al. have demonstrated that uridine monophosphate (UMP, 114) is the actual source of uridine-5'-aldehyde 99, which is furnished in an oxidative transformation of UMP 114 with the 2-oxoglutarate (2-OG)-dependent non-haem Fe(II)-oxygenase LipL [127]. This result challenges the proposal by Chen, Deng et al. that aldehyde 99 might be formed by oxidation of uridine (1) in muraymycin biosynthesis. Aldehyde 99 then undergoes the aforementioned aldol-type transformation to GlyU 101, catalysed by the enzyme LipK. However, aldehyde 99 also serves as a source of the aminoribosyl moiety. Thus, it is converted into 5'-amino-5'-deoxyuridine (115) in a transamination reaction mediated by LipO. This is followed by the LipP-catalysed displacement of the uracil with a phosphate moiety to afford 5-amino-5-deoxyribose-1-phosphate (116). The LipM-mediated reaction of ribosyl phosphate 116 with a nucleoside triphosphate (NTP) then yields nucleoside diphosphate (NDP)-aminoribose 117. Finally, aminoribosylation of 101 with glycosyl donor 117, catalysed by glycosyltransferase LipN, furnishes the complete nucleoside core structure 118 (Scheme 11). The order of 6'-N-(3-aminopropyl) attachment and 5'-O-aminoribosylation is not fully clear yet, i.e., it is not elucidated if 101 or 6'-N-aminoalkyl intermediate 103 (see Scheme 10) act as the glycosyl acceptor in the aminoribosylation step.
Scheme 11.

Biosynthesis of the nucleoside core structure of A-90289 antibiotics (which is identical to the muraymycin nucleoside core) according to the studies of Van Lanen et al. [126]. 2-OG = 2-oxoglutarate.
Van Lanen et al. then studied the LipK-catalysed aldol-type formation of GlyU 101 in more detail [128]. Surprisingly and in contrast to Chen's and Deng's proposal, L-threonine (119) turned out to be the source of the enol(ate) component instead of glycine (100). Hence, LipK was revealed to be a transaldolase mediating a retro-aldol reaction of L-threonine (119) towards the enol(ate) and acetaldehyde (120), followed by a stereoselective aldol addition of the former to uridine-5'-aldehyde 99 (Scheme 12). Using synthetic reference compounds, it could be proven that (5'S,6'S)-GlyU 101 is the stereoisomer furnished in this reaction, so that no epimerisation at a later stage of the biosynthetic route is required for the formation of the A-90289 nucleoside antibiotics.
Scheme 12.

Transaldolase-catalysed formation of the key intermediate GlyU 101 in the biosynthesis of muraymycin-related A-90289 antibiotics [128].
Based on the elucidation of the LipK-mediated reaction, Van Lanen et al. then performed a PCR-based screening of a collection of ≈2500 actinomycete strains for similar transaldolase-encoding genes [129]. They could identify the gene sphJ from a Sphaerisporangium sp., which encoded the transaldolase SphJ having 51% amino acid sequence identity with LipK. Following detailed characterisation of this enzyme, the sphJ gene was employed as a probe to clone the entire genetic locus consisting of 34 putative ORFs. The expression of three selected genes (including sphJ) was monitored under different growth conditions. Under the thereby identified optimal conditions, the actinomycete produced a set of four unprecedented MraY-inhibiting nucleoside antibiotics named sphaerimicin A to D [129]. Hence, detailed studies on LipK-like transaldolases led to the discovery of novel antimicrobially active secondary metabolites.
It remains to be proven that the results obtained for the early steps of A-90289 and sphaerimicin biosynthesis are also valid for the biosynthetic formation of muraymycins. Bioinformatic analyses of the biosynthetic gene clusters of A-90289 antibiotics, caprazamycins and muraymycins revealed six shared ORFs overall [128]. A sequence comparison of a range of transaldolases gave 47% identity and 78% similarity of Mur17 with LipK [129]. Overall, these insights suggest that the formation of the GlyU intermediate 101 and very likely also of the whole aminoribosylated nucleoside core structure occur in a conserved manner. Further studies on muraymycin biosynthesis are still pending.
Conclusion
In summary, this review describes a promising class of antimicrobially active natural products, the uridine-derived muraymycins. Muraymycins are one subclass of nucleoside antibiotics inhibiting the membrane protein translocase I (MraY), a key enzyme in the intracellular part of peptidoglycan formation. Synthetic methodology for the preparation of muraymycins and their analogues has been established, and first SAR insights revealed that the design of structurally simplified, biologically active muraymycin analogues is an auspicious approach. However, further SAR studies as well as investigations on the interplay of target inhibition and cellular uptake for the antibiotic activity are surely desirable. Studies on muraymycin biosynthesis may not only be of academic interest, but could also lead to semi- or mutasynthetic methodology for the preparation of novel muraymycin analogues. Several laboratories around the world currently perform research on muraymycins and other uridine-derived nucleoside antibiotics. Hopefully, this work will contribute to the development of urgently needed novel antimicrobial drugs.
Acknowledgments
We thank the Deutsche Forschungsgemeinschaft (DFG, SFB 803 "Functionality controlled by organization in and between membranes") and the Fonds der Chemischen Industrie (FCI, Sachkostenzuschuss) for financial support of our research on muraymycin antibiotics. G. N. is grateful for a doctoral fellowship of the Fonds der Chemischen Industrie, and D. W. is grateful for a doctoral fellowship of the Konrad-Adenauer-Stiftung.
This article is part of the Thematic Series "Natural products in synthesis and biosynthesis II" and is dedicated to Professor Wittko Francke on the occasion of his 75th birthday.
References
- 1.Fleming A. Bull W H O. 2001;79:780–790. [PMC free article] [PubMed] [Google Scholar]
- 2.Chain E, Florey H W, Gardner A D, Heatley N G, Jennings M A, Orr-Ewing J, Sanders A G. Lancet. 1940;236:226–228. doi: 10.1016/S0140-6736(01)08728-1. [DOI] [Google Scholar]
- 3.Abraham E P, Chain E. Nature. 1940;146:837. doi: 10.1038/146837a0. [DOI] [Google Scholar]
- 4.Kirby W M M. Science. 1944;99:452–453. doi: 10.1126/science.99.2579.452. [DOI] [PubMed] [Google Scholar]
- 5.Levy S B, Marshall B. Nat Med. 2004;10:S122–S129. doi: 10.1038/nm1145. [DOI] [PubMed] [Google Scholar]
- 6.Bush K. Curr Opin Pharmacol. 2012;12:527–534. doi: 10.1016/j.coph.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 7.O'Connell K M G, Hodgkinson J T, Sore H F, Welch M, Salmond G P C, Spring D R. Angew Chem, Int Ed. 2013;52:10706–10733. doi: 10.1002/anie.201209979. Angew. Chem.2013,125, 10904–10932. doi:10.1002/ange.201209979. [DOI] [PubMed] [Google Scholar]
- 8.von Nussbaum F, Brands M, Hinzen B, Weigand S, Häbich D. Angew Chem, Int Ed. 2006;45:5072–5129. doi: 10.1002/anie.200600350. Angew. Chem.2006,118, 5194–5254. doi:10.1002/ange.200600350. [DOI] [PubMed] [Google Scholar]
- 9.Butler M S, Buss A D. Biochem Pharmacol. 2006;71:919–929. doi: 10.1016/j.bcp.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 10.Alekshun M N, Levy S B. Cell. 2007;128:1037–1050. doi: 10.1016/j.cell.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 11.Walsh C. Nature. 2000;406:775–781. doi: 10.1038/35021219. [DOI] [PubMed] [Google Scholar]
- 12.Wright G D. Curr Opin Chem Biol. 2003;7:563–569. doi: 10.1016/j.cbpa.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 13.Wright G D. Curr Opin Microbiol. 1999;2:499–503. doi: 10.1016/S1369-5274(99)00007-7. [DOI] [PubMed] [Google Scholar]
- 14.Katz L, Ashley G W. Chem Rev. 2005;105:499–528. doi: 10.1021/cr030107f. [DOI] [PubMed] [Google Scholar]
- 15.Li X-Z, Nikaido H. Drugs. 2009;69:1555–1623. doi: 10.2165/11317030-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Denyer S P, Maillard J-Y. J Appl Microbiol. 2002;92:35S–45S. doi: 10.1046/j.1365-2672.92.5s1.19.x. [DOI] [PubMed] [Google Scholar]
- 17.Lambert P A. J Appl Microbiol. 2002;92:46S–54S. doi: 10.1046/j.1365-2672.92.5s1.7.x. [DOI] [PubMed] [Google Scholar]
- 18.Barrett J F. Expert Opin Ther Targets. 2004;8:515–519. doi: 10.1517/14728222.8.6.515. [DOI] [PubMed] [Google Scholar]
- 19.Otto M. Int J Med Microbiol. 2013;303:324–330. doi: 10.1016/j.ijmm.2013.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bonten M J M, Willems R, Weinstein R A. Lancet Infect Dis. 2001;1:314–325. doi: 10.1016/S1473-3099(01)00145-1. [DOI] [PubMed] [Google Scholar]
- 21.Walsh C. Nat Rev Microbiol. 2003;1:65–70. doi: 10.1038/nrmicro727. [DOI] [PubMed] [Google Scholar]
- 22.McDonald L A, Barbieri L R, Carter G T, Lenoy E, Lotvin J, Petersen P J, Siegel M M, Singh G, Williamson R T. J Am Chem Soc. 2002;124:10260–10261. doi: 10.1021/ja017748h. [DOI] [PubMed] [Google Scholar]
- 23.Kimura K, Bugg T D H. Nat Prod Rep. 2003;20:252–273. doi: 10.1039/b202149h. [DOI] [PubMed] [Google Scholar]
- 24.Winn M, Goss R J M, Kimura K, Bugg T D H. Nat Prod Rep. 2010;27:279–304. doi: 10.1039/B816215H. [DOI] [PubMed] [Google Scholar]
- 25.Ichikawa S, Yamaguchi M, Matsuda A. Curr Med Chem. 2015;22:3951–3979. doi: 10.2174/0929867322666150818103502. [DOI] [PubMed] [Google Scholar]
- 26.Takatsuki A, Arima K, Tamura G. J Antibiot. 1971;24:215–223. doi: 10.7164/antibiotics.24.215. [DOI] [PubMed] [Google Scholar]
- 27.Takatsuki A, Tamura G. J Antibiot. 1971;24:224–231. doi: 10.7164/antibiotics.24.224. [DOI] [PubMed] [Google Scholar]
- 28.Takatsuki A, Tamura G. J Antibiot. 1971;24:785–794. doi: 10.7164/antibiotics.24.785. [DOI] [PubMed] [Google Scholar]
- 29.Eckardt K, Thrum H, Bradler G, Tonew E, Tonew M. J Antibiot. 1975;28:274–279. doi: 10.7164/antibiotics.28.274. [DOI] [PubMed] [Google Scholar]
- 30.Thrum H, Eckardt K, Bradler G, Fügner R, Tonew E, Tonew M. J Antibiot. 1975;28:514–521. doi: 10.7164/antibiotics.28.514. [DOI] [PubMed] [Google Scholar]
- 31.Eckardt K, Ihn W, Tresselt D, Krebs D. J Antibiot. 1981;34:1631–1632. doi: 10.7164/antibiotics.34.1631. [DOI] [PubMed] [Google Scholar]
- 32.Vogel P, Petterson D S, Berry P H, Frahn J L, Anderton N, Cockrum P A, Edgar J A, Jago M V, Lanigan G W, Payne A L, et al. Aust J Exp Biol Med Sci. 1981;59:455–467. doi: 10.1038/icb.1981.39. [DOI] [PubMed] [Google Scholar]
- 33.Seto H, Otake N, Sato S, Yamaguchi H, Takada K, Itoh M, Lu H S M, Clardy J. Tetrahedron Lett. 1988;29:2343–2346. doi: 10.1016/S0040-4039(00)86055-4. [DOI] [Google Scholar]
- 34.Yamaguchi H, Sato S, Yoshida S, Takada K, Itoh M, Seto H, Otake N. J Antibiot. 1986;39:1047–1053. doi: 10.7164/antibiotics.39.1047. [DOI] [PubMed] [Google Scholar]
- 35.Ochi K, Ezaki M, Iwami M, Komori T, Kohsaka M, inventors. FR-900493 substance, a process for its production and a pharmaceutical composition containing the same. US4950605 A. U.S. Patent. 1990 Aug 21;
- 36.Inukai M, Isono F, Takahashi S, Enokita R, Sakaida Y, Haneishi T. J Antibiot. 1989;42:662–666. doi: 10.7164/antibiotics.42.662. [DOI] [PubMed] [Google Scholar]
- 37.Isono F, Inukai M, Takahashi S, Haneishi T, Kinoshita T, Kuwano H. J Antibiot. 1989;42:667–673. doi: 10.7164/antibiotics.42.667. [DOI] [PubMed] [Google Scholar]
- 38.Isono F, Katayama T, Inukai M, Haneishi T. J Antibiot. 1989;42:674–679. doi: 10.7164/antibiotics.42.674. [DOI] [PubMed] [Google Scholar]
- 39.Karwowski J P, Jackson M, Theriault R J, Chen R H, Barlow G J, Maus M L. J Antibiot. 1989;42:506–511. doi: 10.7164/antibiotics.42.506. [DOI] [PubMed] [Google Scholar]
- 40.Chen R H, Buko A M, Whittern D N, McAlpine J B. J Antibiot. 1989;42:512–520. doi: 10.7164/antibiotics.42.512. [DOI] [PubMed] [Google Scholar]
- 41.Fernandes P B, Swanson R N, Hardy D J, Hanson C W, Coen L, Rasmussen R R, Chen R H. J Antibiot. 1989;42:521–526. doi: 10.7164/antibiotics.42.521. [DOI] [PubMed] [Google Scholar]
- 42.Chatterjee S, Nadkarni S R, Vijayakumar E K S, Patel M V, Ganguli B N, Fehlhaber H-W, Vertesy L. J Antibiot. 1994;47:595–598. doi: 10.7164/antibiotics.47.595. [DOI] [PubMed] [Google Scholar]
- 43.Xie Y, Chen R, Si S, Sun C, Xu H. J Antibiot. 2007;60:158–161. doi: 10.1038/ja.2007.16. [DOI] [PubMed] [Google Scholar]
- 44.Xie Y, Xu H, Si S, Sun C, Chen R. J Antibiot. 2008;61:237–240. doi: 10.1038/ja.2008.34. [DOI] [PubMed] [Google Scholar]
- 45.Isono K, Uramoto M, Kusakabe H, Kimura K-I, Izaki K, Nelson C C, McCloskey J A. J Antibiot. 1985;38:1617–1621. doi: 10.7164/antibiotics.38.1617. [DOI] [PubMed] [Google Scholar]
- 46.Igarashi M, Nakagawa N, Doi N, Hattori S, Naganawa H, Hamada M. J Antibiot. 2003;56:580–583. doi: 10.7164/antibiotics.56.580. [DOI] [PubMed] [Google Scholar]
- 47.Igarashi M, Takahashi Y, Shitara T, Nakamura H, Naganawa H, Miyake T, Akamatsu Y. J Antibiot. 2005;58:327–337. doi: 10.1038/ja.2005.41. [DOI] [PubMed] [Google Scholar]
- 48.Naganawa H, Hamada M, Igarashi M, Takeuchi T, inventors. Antibiotic caprazamycins and process for producing the same. CA2388050 A1. Canadian Patent. 2001 Feb 22;
- 49.Carter G T, Lotvin J A, McDonald L A, inventors. Antibiotics AA-896. WO2002085310 A3. WO Patent. 2003 Nov 20;
- 50.Bugg T D H, Walsh C T. Nat Prod Rep. 1992;9:199–215. doi: 10.1039/np9920900199. [DOI] [PubMed] [Google Scholar]
- 51.van Heijenoort J. Nat Prod Rep. 2001;18:503–519. doi: 10.1039/a804532a. [DOI] [PubMed] [Google Scholar]
- 52.Vollmer W, Blanot D, De Pedro M A. FEMS Microbiol Rev. 2008;32:149–167. doi: 10.1111/j.1574-6976.2007.00094.x. [DOI] [PubMed] [Google Scholar]
- 53.Osborn M J. Annu Rev Biochem. 1969;38:501–538. doi: 10.1146/annurev.bi.38.070169.002441. [DOI] [PubMed] [Google Scholar]
- 54.Barreteau H, Kovač A, Boniface A, Sova M, Gobec S, Blanot D. FEMS Microbiol Rev. 2008;32:168–207. doi: 10.1111/j.1574-6976.2008.00104.x. [DOI] [PubMed] [Google Scholar]
- 55.Bouhss A, Trunkfield A E, Bugg T D H, Mengin-Lecreulx D. FEMS Microbiol Rev. 2008;32:208–233. doi: 10.1111/j.1574-6976.2007.00089.x. [DOI] [PubMed] [Google Scholar]
- 56.Gautam A, Vyas R, Tewari R. Crit Rev Biotechnol. 2011;31:295–336. doi: 10.3109/07388551.2010.525498. [DOI] [PubMed] [Google Scholar]
- 57.Egan A J F, Vollmer W. Ann N Y Acad Sci. 2013;1277:8–28. doi: 10.1111/j.1749-6632.2012.06818.x. [DOI] [PubMed] [Google Scholar]
- 58.Ikeda M, Wachi M, Jung H K, Ishino F, Matsuhashi M. J Bacteriol. 1991;173:1021–1026. doi: 10.1128/jb.173.3.1021-1026.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boyle D S, Donachie W D. J Bacteriol. 1998;180:6429–6432. doi: 10.1128/jb.180.23.6429-6432.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Branstrom A A, Midha S, Longley C B, Han K, Baizman E R, Axelrod H R. Anal Biochem. 2000;280:315–319. doi: 10.1006/abio.2000.4530. [DOI] [PubMed] [Google Scholar]
- 61.Barbosa M D F S, Ross H O, Hillman M C, Meade R P, Kurilla M G, Pompliano D L. Anal Biochem. 2002;306:17–22. doi: 10.1006/abio.2001.5691. [DOI] [PubMed] [Google Scholar]
- 62.Thanassi J A, Hartman-Neumann S L, Dougherty T J, Dougherty B A, Pucci M J. Nucleic Acids Res. 2002;30:3152–3162. doi: 10.1093/nar/gkf418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lara B, Mengin-Lecreulx D, Ayala J A, van Heijenoort J. FEMS Microbiol Lett. 2005;250:195–200. doi: 10.1016/j.femsle.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 64.Struve W G, Neuhaus F C. Biochem Biophys Res Commun. 1965;18:6–12. doi: 10.1016/0006-291X(65)90873-9. [DOI] [PubMed] [Google Scholar]
- 65.Anderson J S, Matsuhashi M, Haskin M A, Strominger J L. Proc Natl Acad Sci U S A. 1965;53:881–889. doi: 10.1073/pnas.53.4.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Heydanek M G, Jr, Struve W G, Neuhaus F C. Biochemistry. 1969;8:1214–1221. doi: 10.1021/bi00831a056. [DOI] [PubMed] [Google Scholar]
- 67.Pless D D, Neuhaus F C. J Biol Chem. 1973;248:1568–1576. [PubMed] [Google Scholar]
- 68.Lloyd A J, Brandish P E, Gilbey A M, Bugg T D H. J Bacteriol. 2004;186:1747–1757. doi: 10.1128/JB.186.6.1747-1757.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Al-Dabbagh B, Henry X, Ghachi M E, Auger G, Blanot D, Parquet C, Mengin-Lecreulx D, Bouhss A. Biochemistry. 2008;47:8919–8928. doi: 10.1021/bi8006274. [DOI] [PubMed] [Google Scholar]
- 70.Bouhss A, Mengin-Lecreulx D, Le Beller D, van Heijenoort J. Mol Microbiol. 1999;34:576–585. doi: 10.1046/j.1365-2958.1999.01623.x. [DOI] [PubMed] [Google Scholar]
- 71.Chung B C, Zhao J, Gillespie R A, Kwon D-Y, Guan Z, Hong J, Zhou P, Lee S-Y. Science. 2013;341:1012–1016. doi: 10.1126/science.1236501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bernhardt T G, Roof W D, Young R. Proc Natl Acad Sci U S A. 2000;97:4297–4302. doi: 10.1073/pnas.97.8.4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bernhardt T G, Struck D K, Young R. J Biol Chem. 2001;276:6093–6097. doi: 10.1074/jbc.M007638200. [DOI] [PubMed] [Google Scholar]
- 74.Rodolis M T, Mihalyi A, O'Reilly A, Slikas J, Roper D I, Hancock R E W, Bugg T D H. ChemBioChem. 2014;15:1300–1308. doi: 10.1002/cbic.201402064. [DOI] [PubMed] [Google Scholar]
- 75.Andrews J M. J Antimicrob Chemother. 2001;48:5–16. doi: 10.1093/jac/48.suppl_1.5. [DOI] [PubMed] [Google Scholar]
- 76.Yamashita A, Norton E, Petersen P J, Rasmussen B A, Singh G, Yang Y, Mansour T S, Ho D M. Bioorg Med Chem Lett. 2003;13:3345–3350. doi: 10.1016/S0960-894X(03)00671-1. [DOI] [PubMed] [Google Scholar]
- 77.Tanino T, Al-Dabbagh B, Mengin-Lecreulx D, Bouhss A, Oyama H, Ichikawa S, Matsuda A. J Med Chem. 2011;54:8421–8439. doi: 10.1021/jm200906r. [DOI] [PubMed] [Google Scholar]
- 78.Spork A P, Büschleb M, Ries O, Wiegmann D, Boettcher S, Mihalyi A, Bugg T D H, Ducho C. Chem – Eur J. 2014;20:15292–15297. doi: 10.1002/chem.201404775. [DOI] [PubMed] [Google Scholar]
- 79.Brandish P E, Burnham M K, Lonsdale J T, Southgate R, Inukai M, Bugg T D H. J Biol Chem. 1996;271:7609–7614. doi: 10.1074/jbc.271.13.7609. [DOI] [PubMed] [Google Scholar]
- 80.Brandish P E, Kimura K, Inukai M, Southgate R, Lonsdale J T, Bugg T D. Antimicrob Agents Chemother. 1996;40:1640–1644. doi: 10.1128/aac.40.7.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bouhss A, Crouvoisier M, Blanot D, Mengin-Lecreulx D. J Biol Chem. 2004;279:29974–29980. doi: 10.1074/jbc.M314165200. [DOI] [PubMed] [Google Scholar]
- 82.Fer M J, Bouhss A, Patrão M, Le Corre L, Pietrancosta N, Amoroso A, Joris B, Mengin-Lecreulx D, Calvet-Vitale S, Gravier-Pelletier C. Org Biomol Chem. 2015;13:7193–7222. doi: 10.1039/C5OB00707K. [DOI] [PubMed] [Google Scholar]
- 83.Shapiro A B, Jahić H, Gao N, Hajec L, Rivin O. J Biomol Screening. 2012;17:662–672. doi: 10.1177/1087057112436885. [DOI] [PubMed] [Google Scholar]
- 84.Mengin-Lecreulx D, Parquet C, Desviat L R, Plá J, Flouret B, Ayala J A, van Heijenoort J. J Bacteriol. 1989;171:6126–6134. doi: 10.1128/jb.171.11.6126-6134.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ma Y, Münch D, Schneider T, Sahl H-G, Bouhss A, Ghoshdastider U, Wang J, Dötsch V, Wang X, Bernhard F. J Biol Chem. 2011;286:38844–38853. doi: 10.1074/jbc.M111.301085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lin Y-I, Li Z, Francisco G D, McDonald L A, Davis R A, Singh G, Yang Y, Mansour T S. Bioorg Med Chem Lett. 2002;12:2341–2344. doi: 10.1016/S0960-894X(02)00469-9. [DOI] [PubMed] [Google Scholar]
- 87.Zhu X-F, Williams H J, Scott A I. J Chem Soc, Perkin Trans 1. 2000:2305–2306. doi: 10.1039/B003562I. [DOI] [Google Scholar]
- 88.Myers A G, Gin D Y, Rogers D H. J Am Chem Soc. 1994;116:4697–4718. doi: 10.1021/ja00090a018. [DOI] [Google Scholar]
- 89.Banfi L, Cardani S, Potenza D, Scolastico C. Tetrahedron. 1987;43:2317–2322. doi: 10.1016/S0040-4020(01)86816-2. [DOI] [Google Scholar]
- 90.Hirano S, Ichikawa S, Matsuda A. Angew Chem, Int Ed. 2005;44:1854–1856. doi: 10.1002/anie.200462439. Angew. Chem.2005,117, 1888–1890. doi:10.1002/ange.200462439. [DOI] [PubMed] [Google Scholar]
- 91.Hirano S, Ichikawa S, Matsuda A. J Org Chem. 2007;72:9936–9946. doi: 10.1021/jo701699h. [DOI] [PubMed] [Google Scholar]
- 92.Hirano S, Ichikawa S, Matsuda A. J Org Chem. 2008;73:569–577. doi: 10.1021/jo702264e. [DOI] [PubMed] [Google Scholar]
- 93.Kimura J, Kobayashi H, Miyahara O, Mitsunobu O. Bull Chem Soc Jpn. 1986;59:869–874. doi: 10.1246/bcsj.59.869. [DOI] [Google Scholar]
- 94.Tao B, Schlingloff G, Sharpless K B. Tetrahedron Lett. 1998;39:2507–2510. doi: 10.1016/S0040-4039(98)00350-5. [DOI] [Google Scholar]
- 95.Ichikawa S, Hayashi R, Hirano S, Matsuda A. Org Lett. 2008;10:5107–5110. doi: 10.1021/ol8018743. [DOI] [PubMed] [Google Scholar]
- 96.Tanino T, Hirano S, Ichikawa S, Matsuda A. Nucleic Acids Symp Ser. 2008;52:557–558. doi: 10.1093/nass/nrn282. [DOI] [PubMed] [Google Scholar]
- 97.Tanino T, Ichikawa S, Shiro M, Matsuda A. J Org Chem. 2010;75:1366–1377. doi: 10.1021/jo9027193. [DOI] [PubMed] [Google Scholar]
- 98.Aleiwi B A, Schneider C M, Kurosu M. J Org Chem. 2012;77:3859–3867. doi: 10.1021/jo300205b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Spork A P, Koppermann S, Dittrich B, Herbst-Irmer R, Ducho C. Tetrahedron: Asymmetry. 2010;21:763–766. doi: 10.1016/j.tetasy.2010.03.037. [DOI] [Google Scholar]
- 100.Aggarwal V K, Ford J G, Thompson A, Jones R V H, Standen M C H. J Am Chem Soc. 1996;118:7004–7005. doi: 10.1021/ja961144+. [DOI] [Google Scholar]
- 101.Aggarwal V K, Harvey J N, Richardson J. J Am Chem Soc. 2002;124:5747–5756. doi: 10.1021/ja025633n. [DOI] [PubMed] [Google Scholar]
- 102.Spork A P, Koppermann S, Ducho C. Synlett. 2009;15:2503–2507. doi: 10.1055/s-0029-1217742. [DOI] [Google Scholar]
- 103.Sarabia F, Martín-Ortiz L, López-Herrera F J. Org Lett. 2003;5:3927–3930. doi: 10.1021/ol0355074. [DOI] [PubMed] [Google Scholar]
- 104.Sarabia F, Martín-Ortiz L. Tetrahedron. 2005;61:11850–11865. doi: 10.1016/j.tet.2005.09.086. [DOI] [Google Scholar]
- 105.Spork A P, Ducho C. Synlett. 2013;24:343–346. doi: 10.1055/s-0032-1318117. [DOI] [Google Scholar]
- 106.Dondoni A, Perrone D. Org Synth. 2000;77:64–70. doi: 10.15227/orgsyn.077.0064. [DOI] [Google Scholar]
- 107.Ries O, Büschleb M, Granitzka M, Stalke D, Ducho C. Beilstein J Org Chem. 2014;10:1135–1142. doi: 10.3762/bjoc.10.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Laïb T, Chastanet J, Zhu J. J Org Chem. 1998;63:1709–1713. doi: 10.1021/jo971468w. [DOI] [Google Scholar]
- 109.Spork A P, Ducho C. Org Biomol Chem. 2010;8:2323–2326. doi: 10.1039/c003092a. [DOI] [PubMed] [Google Scholar]
- 110.Spork A P, Wiegmann D, Granitzka M, Stalke D, Ducho C. J Org Chem. 2011;76:10083–10098. doi: 10.1021/jo201935w. [DOI] [PubMed] [Google Scholar]
- 111.Schmidt U, Lieberknecht A, Schanbacher U, Beuttler T, Wild J. Angew Chem, Int Ed. 1982;21:776–777. doi: 10.1002/anie.198207761. Angew. Chem.1982,94, 797–798. doi:10.1002/ange.19820941027. [DOI] [Google Scholar]
- 112.Burk M J. J Am Chem Soc. 1991;113:8518–8519. doi: 10.1021/ja00022a047. [DOI] [Google Scholar]
- 113.Masquelin T, Broger E, Müller K, Schmid R, Obrecht D. Helv Chim Acta. 1994;77:1395–1411. doi: 10.1002/hlca.19940770518. [DOI] [Google Scholar]
- 114.Tanino T, Ichikawa S, Al-Dabbagh B, Bouhss A, Oyama H, Matsuda A. ACS Med Chem Lett. 2010;1:258–262. doi: 10.1021/ml100057z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Takeoka Y, Tanino T, Sekiguchi M, Yonezawa S, Sakagami M, Takahashi F, Togame H, Tanaka Y, Takemoto H, Ichikawa S, et al. ACS Med Chem Lett. 2014;5:556–560. doi: 10.1021/ml5000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schmidtgall B, Spork A P, Wachowius F, Höbartner C, Ducho C. Chem Commun. 2014;50:13742–13745. doi: 10.1039/C4CC06371F. [DOI] [PubMed] [Google Scholar]
- 117.Schmidtgall B, Höbartner C, Ducho C. Beilstein J Org Chem. 2015;11:50–60. doi: 10.3762/bjoc.11.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cheng L, Chen W, Zhai L, Xu D, Huang T, Lin S, Zhou X, Deng Z. Mol BioSyst. 2011;7:920–927. doi: 10.1039/C0MB00237B. [DOI] [PubMed] [Google Scholar]
- 119.Yin X, Zabriskie T M. ChemBioChem. 2004;5:1274–1277. doi: 10.1002/cbic.200400082. [DOI] [PubMed] [Google Scholar]
- 120.Yin X, McPhail K L, Kim K, Zabriskie T M. ChemBioChem. 2004;5:1278–1281. doi: 10.1002/cbic.200400187. [DOI] [PubMed] [Google Scholar]
- 121.Ju J, Ozanick S G, Shen B, Thomas M G. ChemBioChem. 2004;5:1281–1285. doi: 10.1002/cbic.200400136. [DOI] [PubMed] [Google Scholar]
- 122.Hamed R B, Gomez-Castellanos J R, Henry L, Ducho C, McDonough M A, Schofield C J. Nat Prod Rep. 2013;30:21–107. doi: 10.1039/C2NP20065A. [DOI] [PubMed] [Google Scholar]
- 123.Lemke A, Büschleb M, Ducho C. Tetrahedron. 2010;66:208–214. doi: 10.1016/j.tet.2009.10.102. [DOI] [Google Scholar]
- 124.Lemke A, Ducho C. Eur J Org Chem. 2016;2016:87–98. doi: 10.1002/ejoc.201501109. [DOI] [Google Scholar]
- 125.Büschleb M, Granitzka M, Stalke D, Ducho C. Amino Acids. 2012;43:2313–2328. doi: 10.1007/s00726-012-1309-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chi X, Pahari P, Nonaka K, Van Lanen S G. J Am Chem Soc. 2011;133:14452–14459. doi: 10.1021/ja206304k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yang Z, Chi X, Funabashi M, Baba S, Nonaka K, Pahari P, Unrine J, Jacobsen J M, Elliott G I, Rohr J, et al. J Biol Chem. 2011;286:7885–7892. doi: 10.1074/jbc.M110.203562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Barnard-Britson S, Chi X, Nonaka K, Spork A P, Tibrewal N, Goswami A, Pahari P, Ducho C, Rohr J, Van Lanen S G. J Am Chem Soc. 2012;134:18514–18517. doi: 10.1021/ja308185q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Funabashi M, Baba S, Takatsu T, Kizuka M, Ohata Y, Tanaka M, Nonaka K, Spork A P, Ducho C, Chen W-C L, et al. Angew Chem, Int Ed. 2013;52:11607–11611. doi: 10.1002/anie.201305546. Angew. Chem.2013,125, 11821–11825. doi:10.1002/ange.201305546. [DOI] [PMC free article] [PubMed] [Google Scholar]