

Abstract
Background
Following severe traumatic injury, critically ill patients have a prolonged hypercatacholamine state that is associated with bone marrow (BM) dysfunction and persistent anemia. However, current animal models of injury and shock result in a transient anemia. Daily restraint stress (CS) has been shown to increase catecholamines. We hypothesize that adding CS following injury or injury and shock in rats will prolong the hypercatecholaminemia, and prolong the initial anemia, despite elevated erythropoietin levels.
Methods
Male Sprague-Dawley Rats (N=6–8/group) underwent lung contusion (LC) or combined lung contusion/hemorrhagic shock (LCHS) followed by six days of chronic stress (CS). CS consisted of a two hour restraint period interrupted with repositioning and alarms every 30 minutes. At seven days, urine was assessed for norepinephrine (NE) levels, blood for erythropoietin (EPO) and hemoglobin (Hgb), and BM for erythroid progenitor growth.
Results
Animals undergoing LC or combined LCHS predictably recovered by day seven; urine NE, EPO and Hgb levels were normal. The addition of CS to LC and LCHS models was associated with a significant elevation in NE on day six. The addition of CS to LC led to a persistent 20–25% decrease in the growth of BM HPCs. These findings were further exaggerated when CS was added following LCHS, resulting in a 20–40% reduction in BM erythroid progenitor colony growth and a 20% decrease in Hgb when compared to LCHS alone.
Conclusions
Exposing injured animals to CS results in prolonged elevation of norepinephrine and erythropoietin which is associated with worsening BM erythroid function and persistent anemia. Chronic restraint stress following injury and shock provides a clinically relevant model to further evaluate persistent injury-associated anemia seen in critically ill trauma patients. Furthermore, alleviating chronic stress after severe injury is a potential therapeutic target to improve BM dysfunction and anemia.
INTRODUCTION
Severe traumatic injury is associated with bone marrow (BM) dysfunction manifest by increased susceptibility to infection and persistent injury-associated anemia lasting more than 14 days following admission (1, 2). This severe injury-associated anemia persists regardless of blood transfusion (3). In 45 critically ill trauma patients, Livingston et al. (2), demonstrated a decrease in BM progenitor cell growth, an increase of erythroid progenitors into the circulation, and despite elevated erythropoietin levels, there was a persistent anemia. In a rodent model of lung contusion and hemorrhagic shock (LCHS) acute BM dysfunction is manifest by increased mobilization of hematopoietic progenitor cells (HPCs) to peripheral blood, reduction of BM cellularity and HPC colony growth, and decreased hemoglobin (Hgb) (4–6). However, in this acute rodent injury model, BM function is nearly recovered at seven days and Hgb levels have returned to normal (6). This recovery of BM function is not seen in patients following severe traumatic injury. In these critically ill patients there is an elevation of catecholamine levels two to ten times normal lasting beyond a week following initial injury (6). We have shown that supraphysiologic levels of norepinephrine (NE) suppress rat BM HPC colony growth both in vitro and in vivo (7, 8).
The factors responsible for BM dysfunction after trauma remain unknown. Specifically, the role of erythropoietin (EPO) in BM dysfunction and injury-associated persistent anemia is not well defined. In ICU patients following traumatic injury, EPO levels have been shown to be variably elevated but there is no corresponding reticulocytosis which indicates ongoing BM dysfunction (2). EPO levels have been shown to be disproportionately low as compared to degree of anemia in chronic medical illnesses including chronic kidney disease, chronic obstructive pulmonary disease, and heart failure (9). This entity has become known as EPO-resistant anemia, and may be secondary to antagonistic actions of proinflammatory cytokines, direct inhibition of erythroid progenitor cells, or disruption of iron metabolism (9). The relationship between EPO levels and persistent anemia has not been evaluated over time.
Critically ill patients in the intensive care unit (ICU) continue to have numerous physiologic stressors following their initial injury including additional operations, repeated bouts of sepsis, and disturbed sleep-wake cycles (10, 11). This repeated exposure to daily stress may contribute to the prolonged hypercatecholaminemia seen in ICU patients and ongoing exposure to high levels of catecholamines may impede BM recovery, contributing to persistent injury-associated anemia (6, 11–12). This ongoing stress may be what is missing in current acute injury models in rodents. Animal models of chronic restraint stress (CS) have been shown to be associated with continuously elevated levels of stress hormones, including catecholamines (13–16). CS has been shown to negatively affect immune surveillance, gastrointestinal integrity, coronary artery disease, and wound healing (17–21). Using the addition of CS following injury and hemorrhagic shock, we hypothesize that the daily stress prolongs BM dysfunction and leads to persistent injury-associated anemia despite elevated erythropoietin levels.
METHODS
Animals
Male Sprague-Dawley rats (Charles River, Wilmington, MA) weighing 300–400g were housed at 25°C under barrier-sustained conditions with 12-hour light/dark cycles. Animals were provided ad lib access to water and chow (Teklad22/5 Rodent Diet W-8640; Harlan Teklad, Madison, WI). The animal facility environment and animals were maintained in accordance with the regulations detailed in the Guide for the Care and Use of Laboratory Animals. The protocols were approved by the Institutional Animal Care and Use Committee.
Reagents
Bovine serum albumin (BSA) and 2-mercaptoethanol were purchased from Sigma-Aldrich (St. Louis, MO). Methylcellulose was purchased from Stemcell Technologies (Vancouver, Canada). Fetal bovine serum (FBS), Iscove’s Modified Dulbecco’s Medium (IMDM), glutamine, penicillin/streptomycin, and trypan blue were obtained from Invitrogen (Carlsbad, CA). All cytokines rhEpo, rhIL-3, rhGM-CSF were purchased from R&D Systems (Minneapolis, MN). Sodium pentobarbital was purchased from Lundbeck Inc. (Deerfield, IL) and heparin was obtained from Hospira Inc. (Lakefront, IL).
Experimental Groups
Animals (N=6–9 animals/group) were assigned to the following experimental groups. Control groups consisted of uninjured animals that underwent daily handling. Experimental groups were defined as: lung contusion (LC) alone, lung contusion followed by six days of chronic restraint stress (LC/CS), lung contusion followed by hemorrhagic shock (LCHS), or LCHS followed by six days of chronic restraint stress (LCHS/CS). Rodents in all groups were euthanized following rodent cocktail, followed by cardiac puncture and tissue removal. On day seven BM and blood were harvested and urine was collected on post injury days one, three, and six.
Lung Contusion Model
Experimental animals were anesthetized with intraperitoneal injections of sodium pentobarbital (50 mg/kg). Unilateral LC was inflicted using a blast wave percussive nail gun (Craftsman 968514 Stapler, Sears Brands Chicago, IL) applied to a 12mm metal plate adherent to the right axilla of the rat. This LC model has previously been shown to produce a clinically significant lung injury as demonstrated by histology and radiography (22).
Hemorrhagic Shock Model
Immediately following LC, rats in the LCHS and LCHS/CS groups underwent hemorrhagic shock for 45 minutes by removal of blood and maintaining a MAP of 30–35mmHg for 45 minutes. This well established hemorrhagic shock model has been previously described by Baranski et al (4). Following the completion of the shock period, shed blood was re-infused at a rate of 1mL/min.
Chronic Stress Model
Following LC or LCHS, every day between 8am-noon, animals underwent restraint stress. Animals were placed in restraint containers, which measured 16.5cm in length and 7.5cm in diameter, for two hours daily. To prevent acclimation to containers, animals were stimulated at 30, 60, and 90 minutes. Stimulation consisted of two minutes of continuous alarms (80–85 decibels) transmitted via speakers placed adjacent to restraint containers as well as repositioning. Animals were removed from their restraint containers at two hours and returned to normal housing in 12-hour light/dark cycles. Since animals undergoing CS were not permitted feed or water, animals in all other experimental groups were not given access to feed or water during this two hour period.
Bone Marrow Cellularity
BM cells were obtained by removing the femoral epiphysis and aspirating BM with an 18 gauge needle on a 5cc syringe filled with of 1mL IMDM supplemented with 10% FBS. A suspension was prepared by passing cells through a 40μm sterile nylon strainer to remove particulate matter. Total viable cell counts were then determined using a hemocytometer after staining with 0.4% Trypan blue.
Bone Marrow Progenitor Cell Cultures
Colony-forming unit-granulocyte-, erythrocyte-, monocyte-, megakaryocyte (CFU-GEMM) were used to assess growth of early BM progenitor cells. To specifically explore the effects on the erythroid cell lines, burst-forming unit-erythroid (BFU-E) and colony-forming unit-erythroid (CFU-E) were evaluated. Based on the cellularity, a stock solution of BM mononuclear cells was prepared to grow CFU-GEMM, BFU-E and CFU-E cultures as previously described.5 CFU-E colonies were counted at day 7, BFU-E colonies at day 14, and CFU-GEMM colonies at day 17 by an observer blinded to the origin of the samples.
Hemoglobin Measurements
Blood was obtained by via cardiac puncture using a 10mL heparinized syringe. Three hundred microliter aliquots were then sampled and analyzed within 10 minutes of collection using a Coulter Hmx Hematology Analyzer (Beckman Coulter; Brea, CA).
Erythropoietin Levels
Plasma EPO levels were determined by a commercial ELISA kit (R&D systems, Minneapolis, MN) run in duplicate. Briefly, 50μL of assay diluent was added to each well of the ELISA plate followed by 50μL of standard, control, or rat plasma. Wells were precoated with murine monoclonal antimouse EPO antibodies. Plates were incubated at room temperature for two hours on a microplate horizontal orbital shaker. Following incubation, the plate was washed five times and 100μL Mouse EPO conjugate was then added to each well and incubated for two hours. Following five additional washings, 100μL of substrate solution was then added to each well followed by a 30 minute dark incubation. Then 100μL stop solution was added to each well and the plate was then read with wavelength correction of 450–540nm.
Urinary Norepinephrine Levels
To reduce the stress of urine collection, the animals not allocated to CS were stroked while lifting their tails to induce passive urination. Those animals allocated to CS had spontaneous urine collection done during the restraint period on days one, three, and six. The collected urine was acidified with 5microliters of 6N HCl and frozen at −80° Celsius. Urine samples were analyzed for NE using a competitive ELISA (2-CAT ELISA, IBL-America, Minneapolis, MN). All samples were assayed in duplicate and preformed according to the manufacturer’s instruction.
Statistical Analysis
All data are expressed as mean ± SD. Statistical analyses were performed using one-way analysis of variance (ANOVA) followed by Tukey-Kramer’s multiple comparison post-test or Student’s t-test with GraphPad Prism (Version 4.0, San Diego, CA). Results were considered significant if *p <0.05 vs. LC or **p <0.05 vs. LCHS.
RESULTS
Effects of CS following injury and shock on urinary NE levels
To determine the relationship between the injury models and the NE response, serial urine analysis was performed. NE levels in control animals average 39±15 ng/mL. There is a significant increase in urinary NE levels on day 1 in all models as compared to control animals (Figure 1). The addition of CS to LC results in a mild increase in urinary NE on day three but the increase in urinary NE on day six is statistically significant as compared to LC alone (52±21* vs. 10±1). The LCHS model keeps urinary NE levels elevated but not significantly as compared to control animals. But the addition of CS to the combined LCHS persistently elevates urinary NE at both three and six days after the initial injury. The use of CS following LCHS keeps urinary NE levels elevated three times that of LCHS alone (85±20** vs. 31±8).
Figure 1. Urinary NE levels one, three, and six days after injury, shock, and chronic stress.
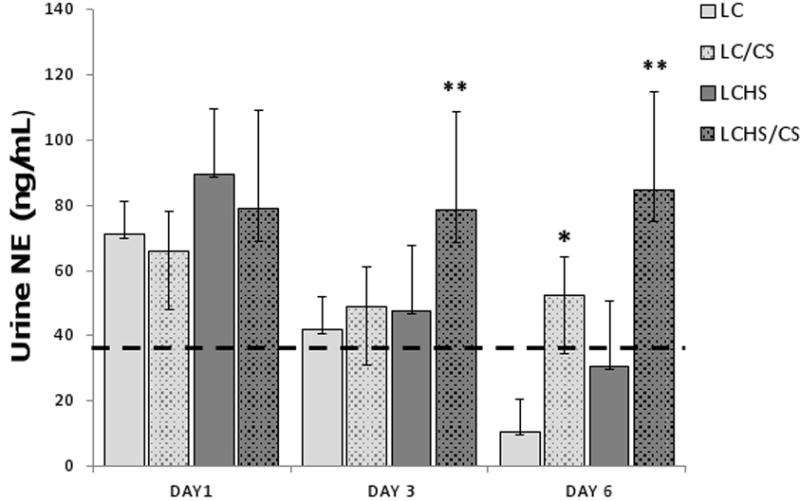
On day 1 all groups have elevated unnary NE levels compared to control animals (represented by the dashed line). The addition of CS to LC and LCHS results in significantly increased urinary NE levels on day 6. (LC=lung contusion alone, LC/CS=lung contusion followed by CS; LCHS=lung contusion followed by hemorrhagic shock; LCHS/CS=LCHS followed by CS). * p<0 05 vs. LC alone, ** p<0.05 vs. LCHS alone. Dashed line represents control level.
Effects of CS on BM cellularity
Seven days following LC alone there is no significant difference in BM cellularity as compared to controls (213±12 vs. 225±6). The addition of CS to LC resulted in a significant reduction in BM cellularity when compared to LC alone (163.3±8* vs. 213±12) (Figure 2A). In the combined injured and shock model, LCHS alone results in a significant reduction of BM cellularity at seven days as compared to control. However, there is further 28% reduction in BM cellularity with the addition of CS to LCHS. (163±8** vs. 197±7) (Figure 2A).
Figure 2. A–D. BM cellularity and HPC colony growth seven days after injury, shock, and chronic stress.
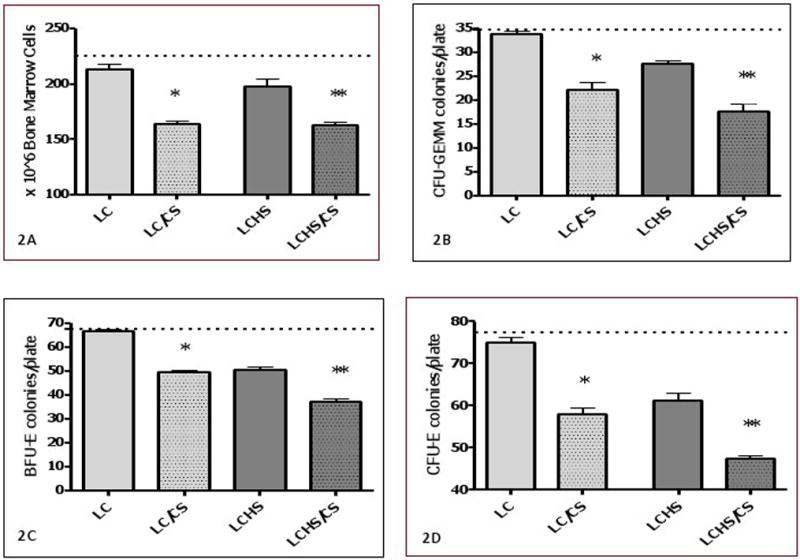
In 2A, the addition of CS after LC or LCHS causes a significant reduction in BM cellularity compared to LC or LCHS alone In Figures 2B–D, The addition of CS after LC or LCHS causes a further significant reduction in BM CFU-GEMM, BFU-E and CFU-E colony growth compared to LC or LCHS alone. (BM = bone marrow; LC = lung contusion alone; LC/CS = lung contusion followed by CS; LCHS=lung contusion followed by hemorrhagic shock, LCHS/CS=LCHS followed by CS). * p<0.05 vs. LC and **p<0.05 vs. LCHS. Dashed line represents control levels.
Effects of CS on BM HPC cultures
To determine the effects of CS following injury and shock models on BM erythroid function, BM HPC growth was evaluated. Seven days following LC alone, there is no significant difference in BM CFU-GEMM, BFU-E, or CFU-E growth as compared to control animals (Figure 2B–D). However, the addition of CS to LC results in significant reduction in BM CFU-GEMM colony growth as compared to LC alone (22±4* vs. 34±1)(Figure 2B) as well as BM BFU-E colony growth as compared to LC alone (49±3* vs. 66±2)(Figure 2C). The effects of CS persist when examining late erythroid progenitor cells, CFU-E (Figure 2D). There is marked reduction of BM CFU-E colony growth in LC/CS seven days following injury.
Seven days following LCHS alone, there remains approximately a 20% decrease in BM CFU-GEMM, BFU-E and CFU-E growth as compared to control animals (Figure 2B–D). The addition of CS to LCHS further worsens the persistent reduction of BM CFU-GEMM growth as compared to LCHS alone (18±4** vs. 28±1) (Figure 2B). Similar to CFU-GEMM, the addition of CS to LCHS significantly reduces both BM BFU-E and CFU-E colony growth as compared to LCHS alone (Figure 2C–D).
Effects of CS following injury and shock on hemoglobin
The average Hgb of control rodents are 13.3±0.8 g/dL. Seven days after LC alone and LCHS, there is no significant decrease in Hgb (14.2±0.3 and 13.2±0.8). The addition of CS to LC and LCHS results in a significant decrease in Hgb as compared to LC and LCHS alone (Figure 3). LCHS/CS results in significant persistent injury anemia at 7 days as compared to controls (11.1±0.7** vs. 13.3±0.8).
Figure 3. Hemoglobin seven days after injury, shock, and chronic stress.
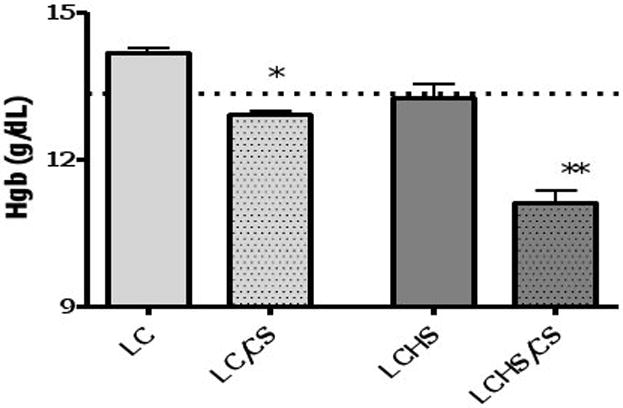
Hgb after seven days of CS is also only slightly decreased from control. Hgb at seven days after LCHS approaches control levels. The addition of CS after LCHS causes a significant reduction in Hgb at seven days. (Hgb=Hemoglobin; LC = lung contusion alone; LC/CS = lung contusion followed by CS; LCHS=lung contusion followed by hemorrhagic shock; LCHS/CS=LCHS followed by CS). * p<0 01 vs LC alone **p<0.001 vs. LCHS. Dashed line represents control levels.
Effects of CS following injury and shock on erythropoietin
To further assess the factors involved in the persistent anemia seen with LCHS/CS, EPO levels were determined. EPO levels in control animals average 16.4±6 pg/mL. There is no significant increase in EPO levels following LC alone seven days following injury (Figure 4). The addition of CS to LC results in an increase in EPO but does not reach statistical significance (46.1±55 vs. 18.6±10).
Figure 4. Erythropoietin levels seven days aits injury, shock, and chronic stress.
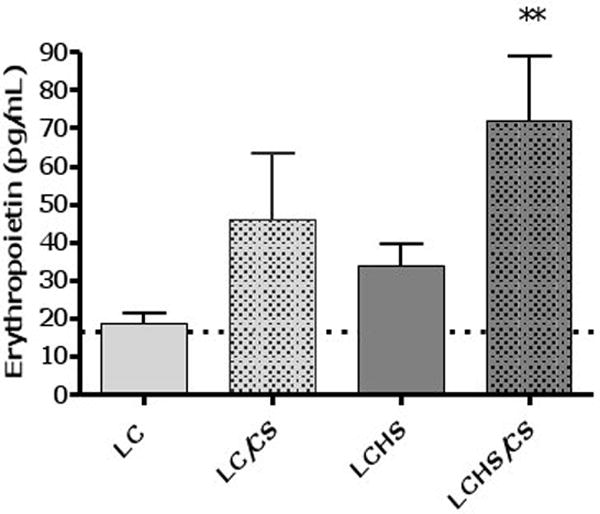
The addition of CS to LC and LCHS results in increased EPO levels. (LC=lung contusion alone, LC/CS=lung contusion followed by CS; LCHS=lung contusion followed by hemorrhagic shock; LCHS/CS=LCHS followed by CS). ** p<0.05 vs. LCHS alone. Dashed line represents control level.
Seven days following LCHS, EPO levels are twice that of controls (33.9±22 vs. 16.4±6). With the addition of CS to LCHS, EPO levels are significantly increased to twice that of LCHS alone (72.1±66** vs. 33.9±22) (Figure 4).
DISCUSSION
BM dysfunction that manifests as an injury-associated anemia has been established following extensive soft tissue injury, burns, hemorrhagic shock and severe traumatic injury (2, 5, 23–25). Injury-associated anemia is multifactorial in nature and likely secondary to a combination of acute blood loss, multiple phlebotomies, nutritional abnormalities, and BM dysfunction resulting in inadequate production of erythrocytes (1–3). While this anemia appears early following traumatic injury, it often persists for months regardless of blood transfusions (1–3). In the current study, we describe for the first time a model of chronic restraint stress that mimics the persistent stress seen in critical illness. The addition of CS to LC and LCHS is synergistic resulting in significant decreases in BM cellularity as well as HPC colony growth that persists seven days following injury. These changes are associated with a persistent anemia despite elevations in EPO levels.
Previous studies have demonstrated the acute effects of LC alone and LCHS alone on BM dysfunction (4–6). In this study, the addition of CS to LC and LCHS models results in a significant reduction in BM cellularity and BM colony growth at seven days which is likely due to the persistent hypercatecholaminemia that accompanies both CS models. This study demonstrates significantly elevated urinary NE levels in both LC/CS and LCHS/CS models six days after injury. Because of the pulsatility in the secretion of catecholamines, urine rather than plasma was utilized to allow for a more accurate basal assessment of sympathetic activity. The use of restraint stress is a well-established model known to activate the sympathetic nervous system (26). To prevent the phenomena of habituation, the use of alarms and repositioning during the restraint period was performed (26). Our findings confirm the presence of a hypercatecholamine state after injury with the use of CS. This study supports the notion that erythropoiesis is impacted by NE. Fonseca et al. (7) established a dose-dependent reduction in erythroid progenitor colony growth in response to increasing levels of NE in vitro. Penn et al. (8) further determined that while a physiologic level of NE is necessary in vivo for erythroid growth, this growth is inhibited at higher catecholamine levels. Similarly, Schraml et al. (27) further demonstrated that treatment with α- and β- adrenergic agonists in vitro leads to significantly decreased HPC colony formation capacity and that this decline was related to the formation of reactive oxygen species as well as activation of the p38/mitogen-activated protein kinase (MAPK) pathway, suggesting that α- and β- adrenergic agonists cause cell-cycle arrest that leads to hematopoietic failure.
Previous animal models demonstrated that LC with or without HS resulted in a significant decrease in BM colony growth of CFU-GM, CFU-E, and BFU-E, however, the BM recovers after seven days (6). This study confirms these findings in both the LC and LCHS groups as well as demonstrates that the addition of chronic restraint stress results in prolonged BM dysfunction with markedly decreased BM colony growth and BM cellularity seven days after injury and shock. This combined injury model with chronic restraint stress creates persistent BM dysfunction which is similar to what is found in critically ill trauma patients (2). In addition, the use of beta blockade has been shown to improve BM function in injury and hemorrhagic shock models which further implicates the role of catecholamines in BM dysfunction (4–5).
Multiple studies have shown persistent anemia in patients with critical illness, with approximately 95% of patients admitted to an ICU for more than three days developing anemia (3, 28–29). Despite higher transfusion rates in trauma patients there is a persistence of anemia (28). Approximately 80% of ICU patients remain anemic at discharge when using a restrictive transfusion protocol (30). The current study implicates chronic stress as a contributing factor in the persistence of anemia following injury and hemorrhagic shock.
Persistent anemia has been shown in animal models developed to mimic diseases of chronic inflammation, such as arthritis or autoimmune disorders as well as chronic infection, such as osteomyelitis and endocarditis (31). Carter et al. (32) recently demonstrated persistent anemia with associated elevation in plasma EPO levels in two mouse models of colitis. In these mouse models, one an acute ingestion-mediated colitis and the other a more chronic, T-cell transfer model, both induced a 20–35% decrease in hematocrit as well as a 10-fold increase in EPO levels (32). The increase in EPO was attributed to the effect of a decreased hematocrit and the presence of tissue hypoxia induced by colonic inflammation (32). EPO is a hormone that control erythropoiesis and is required for the terminal differentiation of CFU-E to erythroblasts and erythrocytes. Our study demonstrates a persistent anemia at seven days despite an elevation in EPO levels. Our persistent anemia may be caused by the hypercatecholaminergic state induced by CS following a pro-inflammatory state caused by injury and ischemia-reperfusion. Interestingly, we demonstrate a persistent anemia despite EPO levels that are increased four-fold as compared to controls. Anemia despite increased circulating levels of EPO in chronic medical diseases is known as erythropoietin-resistant anemia and this entity is thought to be mediated by several pro-inflammatory factors and cytokines, including tumor necrosis factor-alpha, transforming growth factor-beta, interferon-gamma, c-reactive protein, and interleukin-1 (9, 31). It is also possible that while the amount of EPO may be adequate following severe trauma, the EPO receptor in the BM may be dysfunctional. BM dysfunction is not unique to trauma. It is also seen in patients with congestive heart failure (CHF)(33). In patients with CHF, there is impaired clonogenic potential of HPCs, differentiated erythroid cells have markedly increased apoptosis, as well as a blunted EPO response (33). In addition, exogenous EPO administration did not rescue the dysfunction of erythroid lineage in animal models of heart failure (34). Therefore, further study into the mechanism of anemia seen with LC/CS and LCHS/CS may implicate these or other mediators of persistent anemia in the face of elevated circulating EPO.
In summary, the results of this study implicate chronic stress as a cause of persistent NE elevation that is associated with prolonged BM dysfunction and anemia seen following severe traumatic injury. The addition of CS to either LC or LCHS results in a prolonged decrease in BM cellularity and BM HPC growth. This BM dysfunction is associated with a prolonged anemia despite significant elevation in circulating EPO levels. Chronic restraint stress following injury and shock provides a clinically relevant model to further evaluate persistent injury-associated anemia, which mimics critical illness seen in trauma ICU patients.
Acknowledgments
This research was supported by the National Institutes of Health grants R01 NIH GM105893-01A1 and T32 GM069330.
Footnotes
The authors have nothing to disclose or have any conflicts to report.
This was presented as an oral presentation at the Twenty-eighth Annual Meeting of the Eastern Association for the Surgery of Trauma to be held in Orlando, FL January 14–17, 2015
Author Contribution:
Literature search: Bible, Pasupuleti, Gore, Mohr
Study design: Sifri, Mohr
Data collection: Bible, Pasupuleti, Gore, Kannan
Data analysis: Bible, Pasupuleti, Gore, Kannan
Data interpretation: Bible, Pasupuleti, Sifri, Mohr
Writing: Bible, Pasupuleti, Mohr
Critical revision: Bible, Pasupuleti, Sifri, Kannan, Mohr
Contributor Information
Letitia E. Bible, Email: lebible@gmail.com.
Latha V. Pasupuleti, Email: lath413@gmail.com.
Amy V. Gore, Email: goreav@njms.rutgers.edu.
Ziad C. Sifri, Email: sifrizi@njms.rutgers.edu.
Kolenkode B. Kannan, Email: Kolenkode.Kannan@surgery.ufl.edu.
Alicia M. Mohr, Email: alicia.mohr@surgery.ufl.edu.
References
- 1.Livingston DH, Gentile PS, Malangoni MA. Bone marrow failure after hemorrhagic shock. Circ Shock. 1990;30:255–263. [PubMed] [Google Scholar]
- 2.Livingston DH, Anjaria D, Wu J, Hauser CJ, Chang V, Deitch EA, Rameshwar P. Bone marrow failure following severe injury in humans. Ann Surg. 2003;238:748–753. doi: 10.1097/01.sla.0000094441.38807.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Corwin HL, Surgenor SD, Gettinger A. Transfusion practice in the critically ill. Crit Care Med. 2003;31:S668–S671. doi: 10.1097/01.CCM.0000099348.99451.84. [DOI] [PubMed] [Google Scholar]
- 4.Baranski GM, Offin MD, Sifri ZC, Elhassan IO, Hannoush EJ, Alzate WD, Rameshwar P, Livingston DH, Mohr AM. Beta-blockade protection of bone marrow following trauma: the role of G-CSF. J Surg Res. 2011;170:325–331. doi: 10.1016/j.jss.2011.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mohr AM, Elhassan IO, Hannoush EJ, Sifri ZC, Offin MD, Alzate WD, Rameshwar P, Livingston DH. Does beta blockade postinjury prevent bone marrow suppression? J Trauma. 2011;70:1043–1050. doi: 10.1097/TA.0b013e3182169326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pasupuleti LV, Cook KM, Sifri ZC, Kotamarti S, Calderon GM, Alzate WD, Livingston DH, Mohr AM. Does selective beta-1 blockade provide bone marrow protection after trauma/hemorrhagic shock? Surgery. 2012;152:322–330. doi: 10.1016/j.surg.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fonseca RB, Mohr A, Wang L, Sifri ZC, Rameshwar P, Livingston DH. The impact of a hypercatecholamine state on erythropoiesis following severe injury and the role of IL-6. J Trauma. 2005;59:884–890. doi: 10.1097/01.ta.0000187653.64300.f5. [DOI] [PubMed] [Google Scholar]
- 8.Penn A, Mohr AM, Shah SG, Sifri ZC, Kaiser VL, Rameshwar P, Livingston DH. Dose-Response relationship between norepinephrine and erythropoiesis: evidence for a critical threshold. J Surg Res. 2010;163:e85–e90. doi: 10.1016/j.jss.2010.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van der Putten K, Braam B, Jie K, Gaillard C. Mechanisms of disease: erythropoietin resistance in patients with both heart and kidney failure. Nat Clin Pract Nephrol. 2008;4:47–57. doi: 10.1038/ncpneph0655. [DOI] [PubMed] [Google Scholar]
- 10.Friese RS. Sleep and recovery from critical illness and injury: a review of theory, current practice, and future directions. Crit Care Med. 2008;36:697–705. doi: 10.1097/CCM.0B013E3181643F29. [DOI] [PubMed] [Google Scholar]
- 11.Epstein J, Breslow MJ. The stress response of critical illness. Crit Care Clin. 1999;15:17–33. doi: 10.1016/s0749-0704(05)70037-3. [DOI] [PubMed] [Google Scholar]
- 12.Dunser MW, Hasibeder WR. Sympathetic overstimulation during critical illness: adverse effects of adrenergic stress. J Intensive Care Med. 2009;24:293–316. doi: 10.1177/0885066609340519. [DOI] [PubMed] [Google Scholar]
- 13.Hunzeker J, Padgett DA, Sheridan PA, Dhabhar FS, Sheridan JF. Modulation of natural killer cell activity by restraint stress during an influenza A/PR8 infection in mice. Brain Behav Immun. 2004;18:526–535. doi: 10.1016/j.bbi.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 14.Kiank C, Holtfreter B, Starke A, Mundt A, Wilke C, Schutt C. Stress susceptibility predicts the severity of immune depression and the failure to combat bacterial infections in chronically stressed mice. Brain Behav Immun. 2006;20:359–368. doi: 10.1016/j.bbi.2005.10.151. [DOI] [PubMed] [Google Scholar]
- 15.Jin J, Wang X, Wang Q, Guo X, Cao J, Zhang X, Zhu T, Zhang D, Wang W, Wang J, et al. Chronic Psychological Stress induces the accumulation of myeloid-derived suppressor cells in mice. PLoS One. 2013;8:e74497. doi: 10.1371/journal.pone.0074497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng Z, Liu L, Zhang C, Zheng T, Wang J, Lin M, Zhao Y, Wang X, Levine AJ, Hu W. Chronic restraint stress attenuates p53 function and promotes tumorigenesis. Proc Natl Acad Sci USA. 2012;109:7013–7018. doi: 10.1073/pnas.1203930109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meddings JB, Swain MG. Environmental stress-induced gastrointestinal permeability is mediated by endogenous glucocorticoids in the rat. Gastroenterology. 2000;119:1019–1028. doi: 10.1053/gast.2000.18152. [DOI] [PubMed] [Google Scholar]
- 18.Marucha PT, Kiecolt-Glaser JK, Favagehi M. Mucosal wound healing is impaired by examination stress. Psychosom Med. 1998;60:362–365. doi: 10.1097/00006842-199805000-00025. [DOI] [PubMed] [Google Scholar]
- 19.Tymen SD, Rojas IG, Zhou X, Fang ZJ, Zhao Y, Marucha PT. Restraint stress alters neutrophil and acrophage phenotypes during wound healing. Brain Behav Immun. 2013;28:207–217. doi: 10.1016/j.bbi.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin Q, Wang F, Yang R, Zheng X, Gao H, Zhang P. Effect of chronic restraint stress of human colorectal carcinoma growth in mice. PLoS One. 2013;8:e61435. doi: 10.1371/journal.pone.0061435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Romana-Souza B, Porto LC, Monte-Alto-Costa A. Cutaneous wound healing of chronically stressed mice is improved through catecholamine blockade. Exp Dermatol. 2010;19:821–829. doi: 10.1111/j.1600-0625.2010.01113.x. [DOI] [PubMed] [Google Scholar]
- 22.Badami CD, Livingston DH, Sifri ZC, Caputo FJ, Bonilla L, Mohr AM, Deitch EA. Hematopoietic progenitor cells mobilize to the site of injury after trauma and hemorrhagic shock in rats. J Trauma. 2007;63:596–602. doi: 10.1097/TA.0b013e318142d231. [DOI] [PubMed] [Google Scholar]
- 23.Gamelli RL, He LK, Liu H. Marrow granulocyte-macrophage progenitor cell response to burn injury as modified by endotoxin and indomethacin. J Trauma. 1994;37:339–346. doi: 10.1097/00005373-199409000-00002. [DOI] [PubMed] [Google Scholar]
- 24.Raff G, Livingston DH, Wang MT, Rameshwar P. Hemorrhagic shock abolishes the myelopoietic response to turpentine-induced soft tissue injury. J Surg Res. 1995;59:75–79. doi: 10.1006/jsre.1995.1134. [DOI] [PubMed] [Google Scholar]
- 25.Fontes B, Moore FA, Moore EE, Koike K, Kim F, Trew CE, Perterson VM. Gut ischemia induces bone marrow failure and increases risk of infection. J Surg Res. 1994;57:505–509. doi: 10.1006/jsre.1994.1176. [DOI] [PubMed] [Google Scholar]
- 26.Hucklebridge FH, Nowell NW. Plasma catecholamine response to physical and psychological aspects of fighting in mice. Physiol Beh. 1974;13:35–40. doi: 10.1016/0031-9384(74)90303-5. [DOI] [PubMed] [Google Scholar]
- 27.Schraml E, Fuchs R, Kotzbeck P, Grillari J, Schauenstein K. Acute adrenergic stress inhibits proliferation of murine hematopoietic progenitor cells via p38/MAPK signaling. Stem Cells Dev. 2009;18:215–227. doi: 10.1089/scd.2008.0072. [DOI] [PubMed] [Google Scholar]
- 28.Hobisch-Hagen P, Wiedermann F, Mayr A, Fries D, Jelkmann W, Fuchs D, Hasibeder W, Mutz N, Klingler A, Schobersberger W. Blunted erythropoietic response to anemia in multiply traumatized patients. Crit Care Med. 2000;29:743–747. doi: 10.1097/00003246-200104000-00009. [DOI] [PubMed] [Google Scholar]
- 29.Corwin HL, Gettinger A, Pearl RG, Fink MP, Levy MM, Abraham E, MacIntyre NR, Shabot MM, Duh MS, Shapiro MJ. The CRIT study: anemia and blood transfusion in the critically ill—current clinical practice in the United States. Crit Care Med. 2004;32:39–52. doi: 10.1097/01.CCM.0000104112.34142.79. [DOI] [PubMed] [Google Scholar]
- 30.Walsh TS, Lee RJ, Maciver CR, Garrioch M, Mackirdy F, Binning AR, Cole S, McClelland DB. Anemia during and at discharge from intensive care: the impact of restrictive blood transfusion practice. Intensive Care Med. 2006;32:100–109. doi: 10.1007/s00134-005-2855-2. [DOI] [PubMed] [Google Scholar]
- 31.Similowski T, Agusti A, MacNee W, Schonhofer B. The potential impact of anemia of chronic disease in COPD. Eur Respir J. 2006;27:390–396. doi: 10.1183/09031936.06.00143704. [DOI] [PubMed] [Google Scholar]
- 32.Carter PR, Watts MN, Kosloski-Davidson M, Prasai K, Grisham MB, Harris NR. Iron status, anemia, and plasma erythropoietin levels in acute and chronic mouse models of colitis. Inflamm Bowel Dis. 2013;19:1260–1265. doi: 10.1097/MIB.0b013e3182813466. [DOI] [PubMed] [Google Scholar]
- 33.Westenbrink BD, Voors AA, deBoer RA, Schuringa JJ, Klinkenberg T, van der Harst P, Vellenga E, van Veldhuisen DJ, van Gilst WH. Bone marrow dysfunction in chronic heart failure patients. Eur J Heart Fail. 2010;12:676–684. doi: 10.1093/eurjhf/hfq061. [DOI] [PubMed] [Google Scholar]
- 34.Ruifrok WP, Qian C, Sillje HH, van Goor H, van Veldhuisen DJ, van Gilst WH, de Boer RA. Heart failure-associated anemia: bone marrow dysfunction and response to erythropoietin. J Mol Med. 2011;89:377–387. doi: 10.1007/s00109-010-0710-6. [DOI] [PMC free article] [PubMed] [Google Scholar]