

Abstract
This article addresses three inter-related subjects: the development of the Human Plasma Proteome Peptide Atlas, the launch of the Human Proteome Project, and the emergence of alternative splice variant transcripts and proteins as important features of evolution and pathogenesis. The current Plasma Peptide Atlas provides evidence on which peptides have been detected for every protein confidently identified in plasma; there are links to their spectra and their estimated abundance, facilitating the planning of targeted proteomics for biomarker studies. The Human Proteome Project (HPP) combines a chromosome-centric C-HPP with a biology and disease-driven B/D-HPP, upon a foundation of mass spectrometry, antibody, and knowledgebase resource pillars. The HPP aims to identify the approximately 7000 “missing proteins” and to characterize all proteins and their many isoforms. Success will enable the larger research community to utilize newly-available peptides, spectra, informative MS transitions, and databases for targeted analyses of priority proteins for each organ and disease. Among the isoforms of proteins, splice variants have the special feature of greatly enlarging protein diversity without enlarging the genome; evidence is accumulating of striking differential expression of splice variants in cancers. In this era of RNA-sequencing and advanced mass spectrometry, it is no longer sufficient to speak simply of increased or decreased expression of genes or proteins without carefully examining the splice variants in the protein mixture produced from each multi-exon gene. This article is part of a Special Issue entitled: Biomarkers: A Proteomic Challenge.
Keywords: Human Plasma Peptide Atlas, Human Proteome Peptide Atlas, Human Proteome Project, Splice variant protein, Splice variant transcript, Biomarker
1. Introduction to the Hupo Human Plasma Proteome Project (Hpp) and the Human Peptide Atlas
Five days after the publication 15–16 February 2001 of the full issues of Nature [1] and Science [2] about the Human Genome Sequence and its implications, The Financial Times of London ran a feature story about “The Next Holy Grail, Deciphering the Whole Protein Set” (the proteome). The accompanying cartoon showed a dancing globular protein on center stage, with the double-helix of DNA in the shadows offstage! Indeed, proteomics is needed to link knowledge of the genome to the functions of the cell and the whole organism or patient [3].
The Human Proteome Organization (HUPO) was formed in 2001 and soon launched an array of excellent projects to identify and characterize the proteomes of plasma, brain, liver, kidney and urine, and later other organs, along with the Human Antibody Initiative and the Protein Standards Initiative (www.hupo.org). After much discussion and planning, a decade later the Human Proteome Project was initiated [4], as will be described below.
1.1. The Human Plasma Protein Project
A total of 55 laboratories around the world in 2003 requested reference samples of human serum, EDTA-anticoagulated plasma, heparin anticoagulated plasma, and citrate-anticoagulated plasma from Caucasian, African-American, Asian-American, and Chinese donors from the Human Plasma Proteome Project (HPPP). A great variety of technology platforms was employed by the various laboratories, most using only a few of the many requested reference samples. In all, 18 laboratories reported LC-MS/MS results, which served as the backbone for the publication in Proteomics [5] of an overview of the many aspects of the Project and a tabulation of 3020 proteins identified with two or more high-confidence peptides. Many features of the protein sets were analyzed and presented. The Project concluded that the most reliable results came from the EDTA-anticoagulated plasma samples and recommended this sample choice for future studies.
All data and meta-data were made available from the University of Michigan and the European Bioinformatics Institute/PRIDE. Subsets of the data were used to create the first version of the Plasma Peptide Atlas. Data from four sources using antibodies to quantitate selected proteins were compared with the spectral counts of all the proteins to generate an estimate of abundance for the entire protein set. The HPPP special issue of Proteomics included a total of 28 articles, half from lab-specific studies and half from multi-lab collaborative analyses. An alternative analysis of the same data with much more stringent criteria for high-confidence identification and with adjustment for multiple comparisons gave a list of 889 proteins [6]. These studies demonstrated that different proteomics measurements using different sample preparation and analysis techniques identify significantly different sets of proteins and that a comprehensive plasma proteome can be compiled only by combining data from many different experiments and specimens, preferably using a standardized analytical pipeline.
1.2. The Human Plasma Peptide Atlas
Over the years the Plasma Peptide Atlas at the Institute for Systems Biology has collected raw datasets from numerous investigators in academe and in industry and re-analyzed the spectra with the Trans-Proteomic Pipeline [7] to generate a standardized data resource readily utilized by the larger community. Any investigator wanting to know whether a protein has been detected by mass spectrometry, and which peptides were detected, can benefit from such information in the Peptide Atlas. In 2011, Farrah et al. [8] published a complex and useful framework for the Plasma Peptide Atlas, with 1929 unambiguous, unreplicated, canonical plasma proteins at a protein false-discovery rate (FDR) of 1 percent. The stringency corresponds to approximately 0.2% FDR for the peptide level and 0.02% FDR for the peptide spectrum match (PSM), based on 20,433 distinct peptides from 91 LC-MS/MS datasets. The layered scheme in Fig. 1 shows the results for several sets with increasingly relaxed criteria (arrow).
Fig. 1.
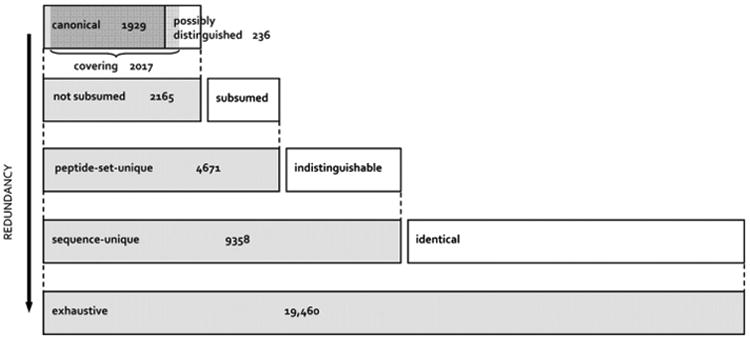
The scheme of Farrah et al. [8], for six levels of stringency or redundancy in generating the Human Plasma Peptide Atlas, with six shaded bars (two of which overlap). Beginning at the bottom: Exhaustive set: contains any protein sequence in the atlas' combined protein sequence database (Swiss-Prot 2010–04 + IPI v3.71 + Ensembl v57.37) that includes at least one identified peptide. Sequence-unique set: exhaustive set with exact duplicates removed. Peptide-set-unique set: a subset of the sequence-unique set within which no two protein sequences include the exact same set of identified peptides. Not subsumed set: peptide-set-unique set with subsumed protein sequences removed (those for which the identified peptides form a proper subset of the identified peptides for another protein sequence). Canonical set: a subset of the not subsumed set within which no protein sequence includes more than 80% of the peptides of any other member of the set. Protein sequences that are not subsumed, but are not canonical are called possibly distinguished, because each has a peptide set that is close, but not identical, to that of a canonical protein sequence. Covering set: a minimal set of protein sequences that can explain all of the identified peptides.
The canonical set is a highly non-redundant set of protein sequences explaining nearly all of the identified peptides; it serves as a reference set. It includes the highest probability protein sequence from each ProteinProphet protein group, which is the group representative. No protein sequence shares more than 80% of its observed peptides with any other member of the set, which obviously excludes closely related protein family members and most isoforms. Thus, highly non-redundant sets are example lists, each entry of which may represent several proteins that are as likely, or almost as likely, to exist in the sample. In contrast, the exhaustive set includes any entry from the combined Swiss-Prot + IPI (International Protein Index) + Ensembl database to which any identified peptide maps; this highly redundant set includes multiple copies of identical sequences. The covering set is somewhat larger than the canonical set because it contains some of the possibly-distinguished protein sequences; it is better for estimating FDR using the Mayu adjustment for large datasets and proteome coverage [9], see ref. [8].
The previous Plasma Peptide Atlas Build in 2007 had 2738 proteins identified, despite many fewer datasets. The 2010 Build had four methodologic improvements: spectral library searching of real spectra; iProphet to increase discrimination between true and false identifications; PSM FDR cutoff instead of a probability cutoff of 0.9 which admitted more false PSMs for poor experiments than for high quality experiments; and a new decoy-estimated protein FDR, using Mayu. There are many interesting details in this paper, including quantitation (down to 0.5 ng/mL) using spectral counting, analysis of N-glyco-motifs and N-glycoproteins, sequence variants from polymorphic mutations or splicing, and discussion of whether to include 124 immunoglobulins and 36 keratins as examples of large families of proteins. We also compared 1% vs 5% protein FDR, which captured about 410 (20 percent) more proteins. Manual curation was employed for the highest-quality single spectrum peptides. Comparing proteins identified with more sensitive immunological methods with those in MS/MS datasets, we found that many of the additional proteins could be found in the exhaustive set. Finally, we calculated an empirical observability score (EOS) for peptides as a guide for targeted proteomics studies.
One of the special features of the Peptide Atlases is the facilitation of comparative proteomic analysis, as is ongoing between plasma, kidney, and urine proteomes (Yamamoto, Farrah, Deutsch, Omenn, unpublished). Our recommended scheme is to compare the canonical set of one proteome (say, plasma) against the exhaustive set of the other proteome (kidney or urine); this approach captures the many ambiguous identifications, including homologous proteins, that are excluded in the generation of the canonical list of proteins. Then the reverse analysis should be done, using the canonical set for kidney to compare with the exhaustive set for plasma.
1.3. The Human Proteome Peptide Atlas
The Human Proteome Peptide Atlas has been expanded to be a compendium of LC-MS/MS proteomics data from 470 experiments with human specimens from many laboratories, all processed consistently through the Trans-Proteomic Pipeline and subjected to the 1% protein FDR filter. The 2012 Build [10] contains results for many organ and biofluid compartments, mapped to Swiss-Prot, the protein sequence database curated to contain one entry per human protein coding gene. The newly added datasets include vitreous humor, seminal plasma, integral membrane proteins, signaling proteins, mitotic spindle, nucleosome, and colorectal tissue to broaden the sampling of the proteome. This latest Peptide Atlas (PA) contains at least one peptide for each of 12,509 Swiss-Prot entries, leaving about 7500 gene products yet to be confidently identified and catalogued. With each Build there will be additional datasets incorporated, such as Cao et al. [11], noted by a reviewer.
Farrah et al. [10] then compared the “PA-unseen proteins” with those identified so far in terms of tissue localization, transcript abundance, and Gene Ontology enrichment and proposed reasons for their absence from Peptide Atlas and strategies for detecting them. Some of the PA-unseen proteins have no tryptic peptides in the detectable range (7–30 residues); 35% of these proteins are embedded in transmembrane regions; 24% are very hydrophobic and insoluble in trypsin digestion protocols; 2% are very basic and might be detectable with ETD (electron transfer dissociation), but not with the standard CID (collision-induced dissociation) for mass spectrometry. Comparison with the antibody-based immunohistochemical findings for 46 cell types led to recognition that skeletal muscle myocytes, liver hepatocytes, and kidney glomerular cells had highest percentages of PA-unseen proteins; not surprisingly, samples from skeletal muscle, liver, and kidney cells have not yet been included in Peptide Atlas, nor have the various organs/regions of the gastrointestinal tract. Gene Ontology analyses showed deficiency of membrane proteins, including olfactory receptor proteins. Of course, low abundance proteins and transiently expressed proteins are likely not to have been detected.
Finally, the requirement for high-confidence and very low error rates means that excluding large numbers of false-positives will always result in discarding valuable true positives. Many of the PA-unseen proteins are probably in the data, but were not detected with sufficient confidence to be included. The first eight human experiments with SRM submitted to PASSEL (The Peptide Atlas SRM Experiment Library) have yielded some additional proteins; of 917 Swiss-Prot entries represented so far in PASSEL, 83% are in PA-seen and 17% (153) are in the PA-unseen set [10].
2. The Human Proteome Project (HPP)
About a decade after the Human Genome Sequencing Project (HGP) was reported to be (nearly) complete, the goal of characterization and annotation of proteins encoded by each gene remains elusive with regard to presence, abundance, function, structural modifications through mutations or splicing or proteolysis, and post-transcriptional side-chain modifications. HUPO announced the plans and strategy for a Human Proteome Project (HPP) at the annual World Congress in Sydney, Australia, in September 2010 and launched the HPP in Geneva a year later. By September 2012 at the Boston World Congress there was an extensive program of presentations and workshops, leading up to a special issue of the Journal of Proteome Research in January 2013 organized by the leaders of the C-HPP with a total of 33 papers from or related to the Chromosome-centric HPP, including papers from chromosome teams 1, 4, 7, 8,11,13,16,17,18,19, 20, X, and Y and multiple database articles (http://pubs.acs.org/toc/jprobs/12/1) An additional 15 papers for the 2013 C-HPP special issue appeared in the Journal in June 2013 (http://pubs.acs.org/toc/jprobs/12/6).
In parallel, the Biology and Disease-driven HPP has emerged, as envisioned by Legrain et al. [4] in Fig. 2, which also shows the mass spectrometry (MS), protein capture (antibody, AB), and knowledge base (KB) resource pillars for the HPP. At the present time there are 25 chromosome-centric teams (Fig. 3A) [C-HPP newsletter on c-hpp.org] and 16 biology and disease-driven teams (Fig. 3B) [12].
Fig. 2.
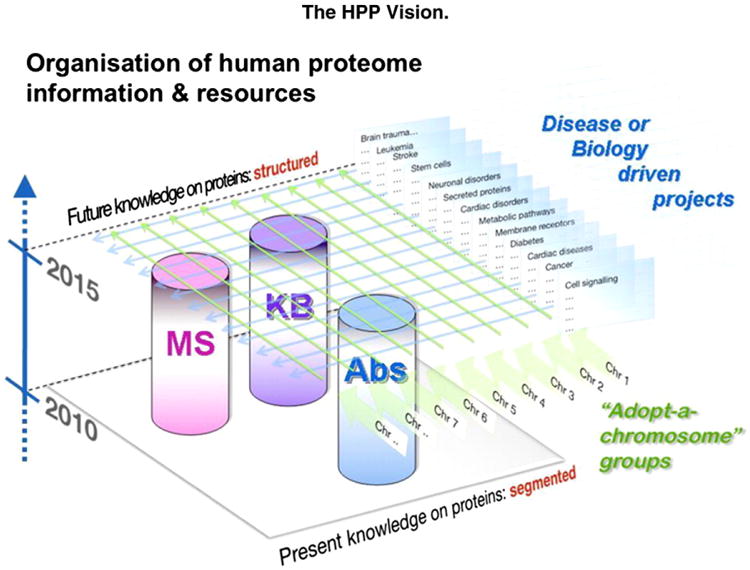
Scheme for the organization of the Human Proteome Project (HPP), showing the mass spectrometry (MS), antibody-based protein capture (AB), and knowledgebase (KB) resource pillars, the chromosome-centric “adopt-a-chromosome” approach, and the complementary biology and disease-driven approach. From Legrain et al. [4] with permission.
Fig. 3.
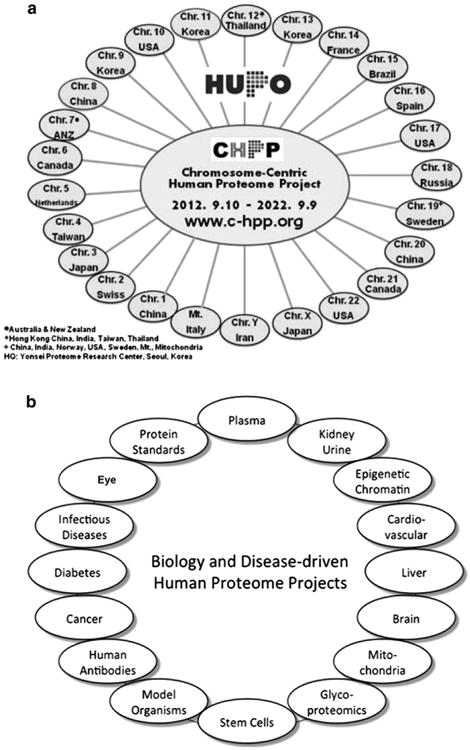
The Chromosome-centric Human Proteome Project teams by chromosome and national leadership (a) and the Biology and the Disease-driven Human Proteome Project teams (b) by organ, biofluid, biological processes, or disease. Details about all the teams can be found at www.thehpp.org, as well as www.c-hpp.org.
The MS resource committee is addressing advances in instrumentation, including development of robust moderate-cost instruments for clinical laboratories; the AB committee addresses emerging methods and the regular updates of the huge Human Protein Atlas project, a major input to the HPP; and the KB committee has greatly facilitated the linkages across neXtProt, Peptide Atlas, GPMdb, and Human Protein Atlas, the emergence of ProteomeXchange, and the assistance with data analysis by the many C-HPP and B/D-HPP teams.
We have created a Master Table as a baseline for the HPP and specifically for the C-HPP using five standard metrics for each chromosome [13]: Ensembl (v69) provides the number of protein-coding genes; neXtProt (gold), Peptide Atlas (canonical), and GPMdb (green) provide numbers for confidently identified proteins from mass spectrometry studies, with special features for each; and the Human Protein Atlas gives us the number of proteins for which polyclonal antibodies generated against one or two different epitopes along the protein sequence have been used to characterize protein expression across 46 cell types, intracellular organelles, and selected cancer cell lines (with evidence scored at the medium or high levels). As of December 2012, the numbers across those five resources were 20,059 for Ensembl, 13,664 for neXtProt, 12,509 for Human Peptide Atlas, 14,300 for GPMdb, and 10,794 for Human Protein Atlas. We estimated that 32.7 percent were “missing”. The article explains in considerable depth the special features of these complementary resources [12].
There are many reasons for missing proteins. First, we must be systematically missing proteins expressed significantly only in unusual organs or cell types; promising sites include brain (with extreme histologic and functional heterogeneity), nasal epithelium/olfactory cortex (with the very large family of olfactory receptor genes) testis, and placenta. However, the Human Protein Atlas [14], multiple cultured cells from different cell lineages [15-17], and results for placenta from the Chr 13 team [18] have not revealed many truly tissue-specific proteins as “missing proteins”. A second approach is developmental, pointing to analysis of embryonic and fetal stages, but the embryonic and induced pluripotent cell lines studied by Heck and collaborators found relatively few unknown proteins [19]. Third, there are large families of proteins that we miss systematically due to sample preparation challenges or inability to distinguish highly homologous families of proteins through peptide matches; these include cytokeratins, immunoglobulins, histocompatibility antigens, and olfactory receptors, plus such large classes as kinases, phosphatases, G-protein coupled receptors (GPCR), and many other membrane-embedded proteins. Finally, there are many proteins whose abundance is below our present limits of detection or have short half-lives. Alternative technology platforms are likely to be complementary in extending the limits of detection, as in the case of ETD mass spectrometry [18]. Of course, there remain very significant sample collection and pre-analytical variables, which are exacerbated by the wide dynamic range and extreme complexity of serum and plasma samples [11,20].
The HPP investigators have committed to open and timely sharing of datasets and metadata. Fortunately, a repository coordination system has emerged from efforts of the past five years to create the ProteomeXchange, based at the European Bioinformatics Institute in England [Vizcaino et al., submitted]. Datasets registered with ProteomeXchange (PX) are made available through curation at EBI/PRIDE and Swiss Institute for Bioinformatics (SIB)/neXtProt and are downloaded and re-analyzed by Peptide Atlas (using Trans-Proteomic Pipeline) and by the Global Proteome Machine Database (GPMDB, using X!Tandem). Results from targeted proteomics with selected reaction monitoring/multiple reaction monitoring (SRM/MRM) now are shared through the SRMAtlas at the Institute for Systems Biology and PASSEL, the Peptide Atlas SRM Experiment Library. The SRM peptide reagents, spectral libraries, and most informative transitions from data-independent acquisition (DIA) MS are now public for proteins from nearly all human protein–coding genes. Polyclonal antibodies are available for 13,985 proteins from the Human Protein Atlas [21], and monoclonal antibodies are being made. The B/D-HPP and the MS pillar committee are working with manufacturers to encourage robust, simple-to-operate mass spectrometers for use by the non-expert community in clinical laboratories; this aim will be highlighted at the 2013 HUPO World Congress in Yokohama, Japan, alongside many workshop sessions presented by the HPP.
The overriding goal for the HPP, beyond building the parts list of proteins and their isoforms, interactions, and functions, is to enable the much broader scientific community, including clinical investigators, to use proteomics methods as part of an integrated omics strategy to link genome and phenotype through pathways, modules, networks, and regulatory mechanisms [12]. Quantification of protein abundances and of the myriad products of protein enzymes is necessary to drive that strategy. The B/D-HPP will provide to all researchers priority lists of proteins and the attendant reagents and data for studies of functional modules in particular organs and diseases. This approach should help researchers study a much broader array of proteins and protein families [22].
2.1. The phenomenon of alternative splicing
Multi-cellular organisms have evolved remarkable splicing mechanisms to convert heterogeneous nuclear RNA transcribed from multi-exon genes to mRNAs. There may be several or numerous alternative transcripts. What was first thought to be highly unusual when reported by Philip Sharp and by Richard Roberts in 1977 for adenovirus-infected cells has now been recognized to be ubiquitous in multi-cellular organisms and their multi-exon genes.
Alternative splicing generates protein diversity without increasing genome size. This phenomenon helps explain how humans generate high complexity with fewer genes than some other species, with “only” 20,000 protein-coding genes, compared with predictions that there would be 50,000 to 100,000 or more protein-coding genes when the Human Genome Sequencing Project was initiated and estimates of 35,000 when the Human Genome Project results were published in 2001. Splicing events that affect the protein-coding region of the mRNA produce proteins differing in sequence and functions; splicing within the non-coding regions can alter regulatory elements for protein expression such as translational enhancers or RNA stability domains.
The implications are huge: When we speak or write about “up-” or “down” expression of genes, we must recognize we are dealing with a mixture of gene products whose actions cannot reliably be related to the combined protein concentration. The relative expression of the splice isoforms must be quantitated, and the structure/function relationships of each variant must be elucidated.
As shown in Fig. 4, pre-mRNA molecules transcribed from DNA have a sequence of 5′UTR, exon, intron, exon, intron, and 3′UTR regions from which the introns must be spliced out to generate the mRNA coding for protein. There is an elaborate molecular process to perform the removal of introns. At each site, a spliceosome, composed of five small nuclear RNAs (<200 nucleotides in length) and up to 100 proteins, forms an RNA–protein complex. ATP hydrolysis is required for the assembly and complex rearrangements of the spliceosome. The intron sequence is characterized by 5′-GU and 3′AG splice sites, with an adenine (A) branch point and a polypyrimidine tract, which is recognized by certain splicing proteins, near the 3′ end. The branchpoint adenine in the intron attacks the 5′-splice site, the sugar-phosphate backbone there is cut, and the released free 3′-OH end of the exon sequences reacts with the start of the next exon, releasing the intron in the shape of a lariat, which is degraded. Introns can range in length from 10 to > 100,000 nucleotides [24].
Fig. 4.
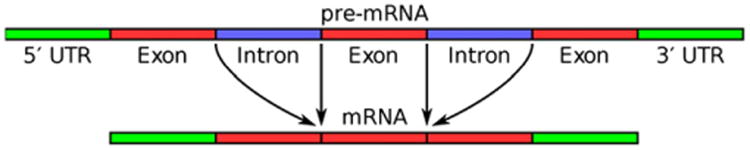
Scheme of a multi-exon gene with pre-mRNA to be spliced to produce mRNA. [See text for details.]
The mechanisms for splicing include mutually exclusive exons (exon-swaps), cassette exons (used or skipped), intron retention (with read-through and inclusion in the protein sequence if in-frame), alternative 5′ start sites, alternative 3′ start sites, alternative promoters, and alternative polyadenylation [23]. Alberts et al. noted that the exon– intron arrangement facilitates the emergence of new and useful proteins over evolutionary time scales [24]. Many modern proteins resemble patchworks composed from a common set of protein domains.
In a 2001 review, Gravely [25] noted three striking cases of functional splice variants: (a) inner ear cochlear hair cells are tuned from 20 to 20,000 Hz as a gradient for sound frequencies along the length of the basilar membrane, with >500 variants of the calcium-activated potassium channel gene slo and functional differences mediated by the protein STREX; (b) >1000 neurexin mRNAs from 3 genes create a code for neural connectivity in synaptogenesis and neuronal signaling, as studied in the rat and cow; and (c) Dscam (the Down syndrome cell adhesion molecule) yields 38,000 mRNA isoforms for distinct receptors and cues for axon migration and connections in Drosophila. In addition, protein diversity is increased through RNA editing.
It is feasible to detect splice variant sequences at the peptide level through mass spectrometry, ideally the peptide with the actual splice junction. Nesvizhskii et al. [26] sought to identify the high percentage (about 85%) of mass spectra that do not match protein sequences with standard database search algorithms. Besides post-translational modifications, sequence polymorphisms, and metabolites, they noted novel peptides that may be due to splicing. Subsequently, Ning and Nesvizhskii [27] reported that about 5% of the RNA splice variants detected in mitochondria from brain stem, liver, and skeletal muscle of the mouse could be identified as translated into peptide sequences by MS/MS with LTQ-Orbitrap; the yield rose sharply with log10 RKPM (abundance) of the transcripts, as would be expected.
Weatheritt et al. [28] used protein structural annotation methods to demonstrate that protein isoform diversity is coupled to modulation of short linear motifs in intrinsically disordered regions of proteins. For example, different phenotypes of TP53 isoforms are attributed to removal or inclusion of short linear motif-containing exons. Loss of the MDM2-binding site increases half-life, due to reduced degradation by attachment of ubiquitin. Absence of a nuclear export signal in another variant explains its exclusive nuclear localization. Yet another variant has an added exon with a putative KEN-box degron, indicating degradation by the APC/C complex during anaphase.
SpliceMap is a Python computational method developed to handle long reads (50–100 nt) from RNA-Seq with Illumina, especially those that span across exon–exon junctions, and to exploit half-reads and paired-reads to improve sensitivity, accuracy, and specificity of mapping splice junctions compared with TopHat [29]. Its use was illustrated with human brain data.
Finally, Merkin et al. [30] reported thousands of previously unknown, lineage-specific, conserved alternative exons from nine tissues from rat, mouse, macaque, cow and chicken. They had signatures of binding by MBNL, PTB, RBFOX, STAR, and ITA family splicing factors, suggesting that these are ancestral mammalian splicing regulators. They concluded that alternative splicing of a core of about 500 highly conserved phosphorylated exons may delimit the scope of kinase/phosphatase signaling networks. Moreover, exons undergoing extensive variation between species that exceeds intra-species differences between tissues may contribute to evolutionary rewiring of signaling networks. These findings were contrasted with splice isoforms that differ only in untranslated regions, potentially affecting mRNA stability, localization, translation, or degradation.
3. Splice variants as a new class of protein biomarker candidates
As summarized in detail elsewhere [31], our group has published a series of papers identifying splice variant peptides and proteins and quantifying the expression of alternate isoforms in mouse models of human KrasG12D activation/Ink4a/Arf deletion-caused pancreatic cancer [32] and human Her2/neu (ERBB2)-induced breast cancer [33], as well as human ERBB2+ cancer cell lines [34]. In each study, we started with deep proteomic analyses and mapped peptides against our modified ECgene database with all potential protein sequences, totaling 10.4 million entries for the mouse and 14.2 million entries for the human [32]. We reported 420 splice isoforms in plasma for the pancreatic cancer model and 608 splice isoforms in mammary tissue for the breast cancer model, with striking differential expression between the tumor-bearing and normal mice in each case.
We used qRT-PCR to demonstrate the presence of mRNAs corresponding to sets of known and novel peptide sequences we had identified as splice variants. To annotate the splice variant proteins, we used InterProScan, MotifScan, Gene Ontology, and FuncAssociate and displayed protein-protein interactions with the Cytoscape plug-in for Michigan Molecular Interactions (MiMI), a consolidated resource of six different databases for protein-protein interactions [35].
We have utilized I-TASSER and TM-align computational protein folding methods to deduce the conformational and functional differences between pairs of splice isoforms [36]. This bridge between structural biology of individual proteins and proteomics of large numbers of potentially interacting proteins has great potential. Different splice isoforms of the same gene can have different, even opposing biological functions and have different functional interactions.
Now we have the benefit of deep RNA-Seq datasets to identify and quantify splice variant transcripts. Proteomic analyses are still needed to show that the transcripts are actually translated into proteins and then to deduce the functional characteristics of the protein isoforms. We are collaborating with Guan and colleagues, who have built models of functional relationship networks for genes [37], identifying disease-gene associations. Li et al. have created an isoform-level high-resolution map of functional networks with a Bayesian network-based multiple instance learning approach that integrates public RNA-seq data resolved at the transcript level and gene-level functional annotations. This model predicts the active splice isoform of each multi-isoform gene; the results gave significantly higher accuracies in predicting functional relationships than did a comparable gene-level network. These results are supported with proteomic data from normal mouse liver cells and breast cells [Li et al., submitted].
As part of the Chromosome-centric Human Proteome Project (C-HPP) chromosome 17 team, we have analyzed splice variant transcripts and proteins in ERBB2+ cancer cell lines [34]. We noted 4, 15, 9, and 6 splice variants in the Chr 17 oncogenes ERBB2, IKZF3, TP53, and BRCA1, respectively.
Fig. 5 shows variation in quantitative expression of splice variant transcripts of Septin 9, Erbb2, Fbxl20, cdk12, Grb7, and Ppp1r1b in the six ERBB2+ cell lines from colon (LIM2405, LIM1899), gastric (KATOIII, SNU16), and breast cancers (SUM149, SUM190) [34]. Across these six cell lines, we found 195 distinct alternative splice transcripts from 144 genes, of which 46 had more than one alternative transcript expressed. The seven splice transcripts of Sept9 had strikingly different expression levels (Fig. 5a), with sept9 epsilon by far the highest expressed. The shorter protein variant of Erbb2 (ENSP000000385185) had remarkably high expression in SUM190 (Fig. 5b). SUM190 also had extremely high expression of the long splice transcript for Grb7, which is frequently co-expressed with Erbb2asnear neighbors in the ERBB2 amplicon (Fig. 5c). Finally, the short variant of Ppp1r1b was strikingly over-expressed in the KATOIII gastric cancer cell line (Fig. 5d). We are currently completing extensive additional splice isoform studies of SUM149, SUM190, and SKBR3 breast cancer cell lines [36], the same cell lines characterized for pathways related to ERBB2 and EGFR (ERBB1) by Zhang et al. [38].
Fig. 5.
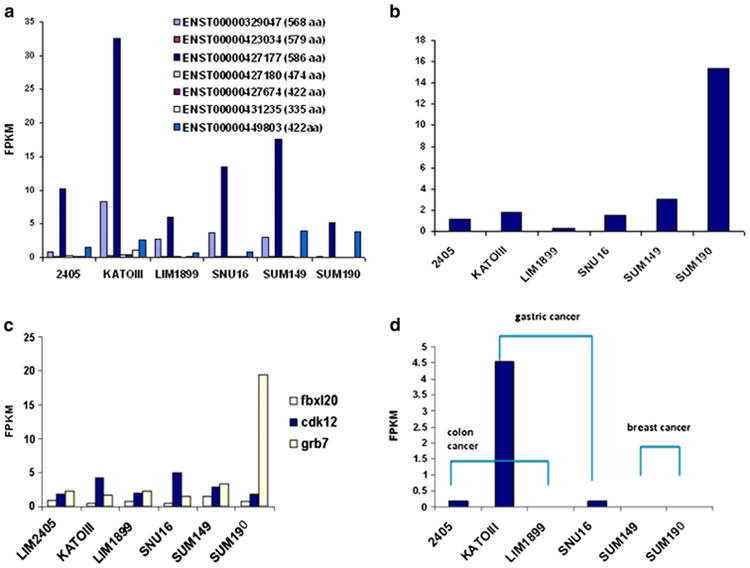
Variation in splice transcript expression across six ERBB2+ cell lines, two each from colon, stomach and breast (see text). (a) Septin 9 (SEPT9) transcript expression for seven different transcripts from this gene. (b) Relative abundance (in fragments per kilobase of transcript per million fragments, FPKM) of the shorter variant of ERBB2 (ENSP00000385185). (c) Relative expression levels of splice transcripts of three genes around ERBB2 on chromosome 17. (d) Relative expression levels of the shorter transcript variant of Ppp1r1b. From [34].
In myelodysplastic syndromes and other malignant hematological disorders, Visconte et al. [39] reported mutations in the spliceosome machinery. The splicing factors mutated include splicing factor 3 sub-unit b1 (SF3b1), U2 small nuclear RNA auxillary factor 1 (U2AF1), and serine arginine-rich splicing factor 2 (SRSF2), occurring in different frequencies across disease subtypes with differences in survival. Mutations may lead to alternative splicing or may even cause splice isoforms of splicing factors, as we have found for carcinomas, amplifying the changes in splicing compared with splice variants of other classes of proteins. Spliceosome proteins are potential targets in early-stage drug development [39].
4. Conclusion
There is currently a broad array of advances in proteomics technologies and informatics, with numerous applications. Enhanced by the Human Proteome Project, we are completing the parts list of the overall human proteome and organ and biofluid proteomes; creating peptide and antibody reagents, spectral libraries, databases, and priority protein sets for targeted proteomics of various diseases; and learning how to identify, quantitate, and characterize splice variants, post-translational modifications, polymorphic mutations, copy number variants, fusions, and protein-protein interactions, pathways, modules, and networks [12,21]. Comprehensive knowledge of proteins is essential to connect the knowledge of the genome to biological processes and phenotypes [3,40].
Acknowledgments
GSO acknowledges support from NIH grants RM-08-029; P30U54ES017885; and UL1RR24986.
Footnotes
This article is part of a Special Issue entitled: Biomarkers: A Proteomic Challenge.
Edited by Professor Kai Stuehler, kai.stuehler@uni-duesseldorf.de.
References
- 1.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itoh T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Smith DR, Doucette-Stamm L, Rubenfield M, Weinstock K, Lee HM, Dubois J, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A, Yang H, Yu J, Wang J, Huang G, Gu J, Hood L, Rowen L, Madan A, Qin S, Davis RW, Federspiel NA, Abola AP, Proctor MJ, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Shimizu N, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Roe BA, Chen F, Pan H, Ramser J, Lehrach H, Reinhardt R, McCombie WR, de la Bastide M, Dedhia N, Blocker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen HC, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JG, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang W, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz J, Slater G, Smit AF, Stupka E, Szustakowski J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang SP, Yeh RF, Collins F, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Patrinos A, Morgan MJ, de Jong P, Catanese JJ, Osoegawa K, Shizuya H, Choi S, Chen YJ. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 2.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amanatides P, Ballew RM, Huson DH, Wortman JR, Zhang Q, Kodira CD, Zheng XH, Chen L, Skupski M, Subramanian G, Thomas PD, Zhang J, Gabor Miklos GL, Nelson C, Broder S, Clark AG, Nadeau J, Mc Kusick VA, Zinder N, Levine AJ, Roberts RJ, Simon M, Slayman C, Hunkapiller M, Bolanos R, Delcher A, Dew I, Fasulo D, Flanigan M, Florea L, Halpern A, Hannenhalli S, Kravitz S, Levy S, Mobarry C, Reinert K, Remington K, Abu-Threideh J, Beasley E, Biddick K, Bonazzi V, Brandon R, Cargill M, Chandramouliswaran I, Charlab R, Chaturvedi K, Deng Z, Di Francesco V, Dunn P, Eilbeck K, Evangelista C, Gabrielian AE, Gan W, Ge W, Gong F, Gu Z, Guan P, Heiman TJ, Higgins ME, Ji RR, Ke Z, Ketchum KA, Lai Z, Lei Y, Li Z, Li J, Liang Y, Lin X, Lu F, Merkulov GV, Milshina N, Moore HM, Naik AK, Narayan VA, Neelam B, Nusskern D, Rusch DB, Salzberg S, Shao W, Shue B, Sun J, Wang Z, Wang A, Wang X, Wang J, Wei M, Wides R, Xiao C, Yan C, Yao A, Ye J, Zhan M, Zhang W, Zhang H, Zhao Q, Zheng L, Zhong F, Zhong W, Zhu S, Zhao S, Gilbert D, Baumhueter S, Spier G, Carter C, Cravchik A, Woodage T, Ali F, An H, Awe A, Baldwin D, Baden H, Barnstead M, Barrow I, Beeson K, Busam D, Carver A, Center A, Cheng ML, Curry L, Danaher S, Davenport L, Desilets R, Dietz S, Dodson K, Doup L, Ferriera S, Garg N, Gluecksmann A, Hart B, Haynes J, Haynes C, Heiner C, Hladun S, Hostin D, Houck J, Howland T, Ibegwam C, Johnson J, Kalush F, Kline L, Koduru S, Love A, Mann F, May D, McCawley S, McIntosh T, McMullen I, Moy M, Moy L, Murphy B, Nelson K, Pfannkoch C, Pratts E, Puri V, Qureshi H, Reardon M, Rodriguez R, Rogers YH, Romblad D, Ruhfel B, Scott R, Sitter C, Smallwood M, Stewart E, Strong R, Suh E, Thomas R, Tint NN, Tse S, Vech C, Wang G, Wetter J, Williams S, Williams M, Windsor S, Winn-Deen E, Wolfe K, Zaveri J, Zaveri K, Abril JF, Guigo R, Campbell MJ, Sjolander KV, Karlak B, Kejariwal A, Mi H, Lazareva B, Hatton T, Narechania A, Diemer K, Muruganujan A, Guo N, Sato S, Bafna V, Istrail S, Lippert R, Schwartz R, Walenz B, Yooseph S, Allen D, Basu A, Baxendale J, Blick L, Caminha M, Carnes-Stine J, Caulk P, Chiang YH, Coyne M, Dahlke C, Mays A, Dombroski M, Donnelly M, Ely D, Esparham S, Fosler C, Gire H, Glanowski S, Glasser K, Glodek A, Gorokhov M, Graham K, Gropman B, Harris M, Heil J, Henderson S, Hoover J, Jennings D, Jordan C, Jordan J, Kasha J, Kagan L, Kraft C, Levitsky A, Lewis M, Liu X, Lopez J, Ma D, Majoros W, McDaniel J, Murphy S, Newman M, Nguyen T, Nguyen N, Nodell M, Pan S, Peck J, Peterson M, Rowe W, Sanders R, Scott J, Simpson M, Smith T, Sprague A, Stockwell T, Turner R, Venter E, Wang M, Wen M, Wu D, Wu M, Xia A, Zandieh A, Zhu X. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 3.Hood LE, Omenn GS, Moritz RL, Aebersold R, Yamamoto KR, Amos M, Hunter-Cevera J, Locascio L. New and improved proteomics technologies for understanding complex biological systems: addressing a grand challenge in the life sciences. Proteomics. 2012;12:2773–2783. doi: 10.1002/pmic.201270086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Legrain P, Aebersold R, Archakov A, Bairoch A, Bala K, Beretta L, Bergeron J, Borchers CH, Corthals GL, Costello CE, Deutsch EW, Domon B, Hancock W, He F, Hochstrasser D, Marko-Varga G, Salekdeh GH, Sechi S, Snyder M, Srivastava S, Uhlen M, Wu CH, Yamamoto T, Paik YK, Omenn GS. The human proteome project: current state and future direction. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M111.009993. M111 009993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Omenn GS, States DJ, Adamski M, Blackwell TW, Menon R, Hermjakob H, Apweiler R, Haab BB, Simpson RJ, Eddes JS, Kapp EA, Moritz RL, Chan DW, Rai AJ, Admon A, Aebersold R, Eng J, Hancock WS, Hefta SA, Meyer H, Paik YK, Yoo JS, Ping P, Pounds J, Adkins J, Qian X, Wang R, Wasinger V, Wu CY, Zhao X, Zeng R, Archakov A, Tsugita A, Beer I, Pandey A, Pisano M, Andrews P, Tammen H, Speicher DW, Hanash SM. Overview of the HUPO Plasma Proteome Project: results from the pilot phase with 35 collaborating laboratories and multiple analytical groups, generating a core dataset of 3020 proteins and a publicly-available database. Proteomics. 2005;5:3226–3245. doi: 10.1002/pmic.200500358. [DOI] [PubMed] [Google Scholar]
- 6.States DJ, Omenn GS, Blackwell TW, Fermin D, Eng J, Speicher DW, Hanash SM. Challenges in deriving high-confidence protein identifications from data gathered by a HUPO plasma proteome collaborative study. Nat Biotechnol. 2006;24:333–338. doi: 10.1038/nbt1183. [DOI] [PubMed] [Google Scholar]
- 7.Keller A, Eng J, Zhang N, Li XJ, Aebersold R. A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol Syst Biol. 2005;1:2005 0017. doi: 10.1038/msb4100024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Farrah T, Deutsch EW, Omenn GS, Campbell DS, Sun Z, Bletz JA, Mallick P, Katz JE, Malmstrom J, Ossola R, Watts JD, Lin B, Zhang H, Moritz RL, Aebersold R. A high-confidence human plasma proteome reference set with estimated concentrations in Peptide Atlas. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M110.006353. M110 006353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reiter L, Claassen M, Schrimpf SP, Jovanovic M, Schmidt A, Buhmann JM, Hengartner MO, Aebersold R. Protein identification false discovery rates for very large proteomics data sets generated by tandem mass spectrometry. Mol Cell Proteomics. 2009;8:2405–2417. doi: 10.1074/mcp.M900317-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farrah T, Deutsch EW, Hoopmann MR, Hallows JL, Sun Z, Huang CY, Moritz RL. The state of the human proteome in 2012 as viewed through Peptide Atlas. J Proteome Res. 2013;12:162–171. doi: 10.1021/pr301012j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao Z, Yende S, Kellum JA, Robinson RA. Additions to the Human Plasma Proteome via a Tandem MARS Depletion iTRAQ-Based Workflow. Int J Proteomics. 2013:654356. doi: 10.1155/2013/654356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aebersold R, Bader GD, Edwards AM, van Eyk JE, Kussmann M, Qin J, Omenn GS. The biology/disease-driven human proteome project (B/D-HPP): enabling protein research for the life sciences community. J Proteome Res. 2013;12:23–27. doi: 10.1021/pr301151m. [DOI] [PubMed] [Google Scholar]
- 13.Marko-Varga G, Omenn GS, Paik YK, Hancock WS. A first step toward completion of a genome-wide characterization of the human proteome. J Proteome Res. 2013;12:1–5. doi: 10.1021/pr301183a. [DOI] [PubMed] [Google Scholar]
- 14.Fagerberg L, Oksvold P, Skogs M, Algenas C, Lundberg E, Ponten F, Sivertsson A, Odeberg J, Klevebring D, Kampf C, Asplund A, Sjostedt E, Al-Khalili Szigyarto C, Edqvist PH, Olsson I, Rydberg U, Hudson P, Ottosson Takanen J, Berling H, Bjorling L, Tegel H, Rockberg J, Nilsson P, Navani S, Jirstrom K, Mulder J, Schwenk JM, Zwahlen M, Hober S, Forsberg M, von Feilitzen K, Uhlen M. Contribution of antibody-based protein profiling to the Human Chromosome-Centric Proteome Project (C-HPP) J Proteome Res. 2013;12:2439–2448. doi: 10.1021/pr300924j. [DOI] [PubMed] [Google Scholar]
- 15.Beck M, Schmidt A, Malmstroem J, Claassen M, Ori A, Szymborska A, Herzog F, Rinner O, Ellenberg J, Aebersold R. The quantitative proteome of a human cell line. Mol Syst Biol. 2011;7:549. doi: 10.1038/msb.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagaraj N, Wisniewski JR, Geiger T, Cox J, Kircher M, Kelso J, Paabo S, Mann M. Deep proteome and transcriptome mapping of a human cancer cell line. Mol Syst Biol. 2011;7:548. doi: 10.1038/msb.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geiger T, Wehner A, Schaab C, Cox J, Mann M. Comparative proteomic analysis of eleven common cell lines reveals ubiquitous but varying expression of most proteins. Mol Cell Proteomics. 2012;11 doi: 10.1074/mcp.M111.014050. M111 014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee HJ, Jeong SK, Na K, Lee MJ, Lee SH, Lim JS, Cha HJ, Cho JY, Kwon JY, Kim H, Song SY, Yoo JS, Park YM, Hancock WS, Paik YK. Comprehensive genome-wide proteomic analysis of human placental tissue for the chromosome-centric human proteome project. J Proteome Res. 2013;12:2458–2466. doi: 10.1021/pr301040g. [DOI] [PubMed] [Google Scholar]
- 19.Munoz J, Low TY, Kok YJ, Chin A, Frese CK, Ding V, Choo A, Heck AJ. The quantitative proteomes of human-induced pluripotent stem cells and embryonic stem cells. Mol Syst Biol. 2011;7:550. doi: 10.1038/msb.2011.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gelfand CA, Omenn GS. Preanalytical variables for plasma and serum proteome analyses. In: Ivanov AR, Lazarev AV, editors. Sample Preparation in Biological Mass Spectrometry. Springer; New York: 2011. pp. 269–289. [Google Scholar]
- 21.Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M, Zwahlen M, Kampf C, Wester K, Hober S, Wernerus H, Bjorling L, Ponten F. Towards a knowledge-based Human Protein Atlas. Nat Biotechnol. 2010;28:1248–1250. doi: 10.1038/nbt1210-1248. [DOI] [PubMed] [Google Scholar]
- 22.Edwards AM, Isserlin R, Bader GD, Frye SV, Willson TM, Yu FH. Too many roads not taken. Nature. 2011;470:163–165. doi: 10.1038/470163a. [DOI] [PubMed] [Google Scholar]
- 23.Rajan P, Elliott DJ, Robson CN, Leung HY. Alternative splicing and biological heterogeneity in prostate cancer. Nat Rev Urol. 2009;6:454–460. doi: 10.1038/nrurol.2009.125. [DOI] [PubMed] [Google Scholar]
- 24.Alberts B. Molecular Biology of the Cell. 5th. Garland Science; New York: 2008. [Google Scholar]
- 25.Graveley BR. Alternative splicing: increasing diversity in the proteomic world. Trends Genet. 2001;17:100–107. doi: 10.1016/s0168-9525(00)02176-4. [DOI] [PubMed] [Google Scholar]
- 26.Nesvizhskii AI, Roos FF, Grossmann J, Vogelzang M, Eddes JS, Gruissem W, Baginsky S, Aebersold R. Dynamic spectrum quality assessment and iterative computational analysis of shotgun proteomic data: toward more efficient identification of post-translational modifications, sequence polymorphisms, and novel peptides. Mol Cell Proteomics. 2006;5:652–670. doi: 10.1074/mcp.M500319-MCP200. [DOI] [PubMed] [Google Scholar]
- 27.Ning K, Nesvizhskii AI. The utility of mass spectrometry-based proteomic data for validation of novel alternative splice forms reconstructed from RNA-Seq data: a preliminary assessment. BMC Bioinf. 2010;11(Suppl. 11):S14. doi: 10.1186/1471-2105-11-S11-S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weatheritt RJ, Davey NE, Gibson TJ. Linear motifs confer functional diversity onto splice variants. Nucleic Acids Res. 2012;40:7123–7131. doi: 10.1093/nar/gks442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Au KF, Jiang H, Lin L, Xing Y, Wong WH. Detection of splice junctions from paired-end RNA-seq data by SpliceMap. Nucleic Acids Res. 2010;38:4570–4578. doi: 10.1093/nar/gkq211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Merkin J, Russell C, Chen P, Burge CB. Evolutionary dynamics of gene and isoform regulation in mammalian tissues. Science. 2012;338:1593–1599. doi: 10.1126/science.1228186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Omenn GS, Menon R, Zhang Y. Innovations in proteomic profiling of cancers: alternative splice variants as a new class of cancer biomarker candidates and bridging of proteomics with structural biology. J Proteomics. 2013;90:28–37. doi: 10.1016/j.jprot.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Menon R, Zhang Q, Zhang Y, Fermin D, Bardeesy N, DePinho RA, Lu C, Hanash SM, Omenn GS, States DJ. Identification of novel alternative splice isoforms of circulating proteins in a mouse model of human pancreatic cancer. Cancer Res. 2009;69:300–309. doi: 10.1158/0008-5472.CAN-08-2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Menon R, Omenn GS. Proteomic characterization of novel alternative splice variant proteins in human epidermal growth factor receptor 2/neu-induced breast cancers. Cancer Res. 2010;70:3440–3449. doi: 10.1158/0008-5472.CAN-09-2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu S, Im H, Bairoch A, Cristofanilli M, Chen R, Dalton S, Deutsch EW, Fanayan S, Gates C, Gaudet P, Hincapie M, Hanash S, Kim H, Jeong SK, Lundberg E, Mias G, Menon R, Mu Z, Nice E, Paik YK, Uhlén M, Wells L, Wu SL, Yan F, Zhang F, Zhang Y, Snyder M, Omenn GS, Beavis R, Hancock WS. A chromosome-centric human proteome project (C-HPP) to characterize the sets of proteins encoded in chromosome 17. J Proteome Res. 2013;12:45–57. doi: 10.1021/pr300985j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao J, Ade AS, Tarcea VG, Weymouth TE, Mirel BR, Jagadish HV, States DJ. Integrating and annotating the interactome using the MiMI plugin for Cytoscape. Bioinformatics. 2009;25:137–138. doi: 10.1093/bioinformatics/btn501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Menon R, Roy A, Mukherjee S, Belkin S, Zhang Y, Omenn GS. Functional implications of structural predictions for alternative splice proteins expressed in Her2/neu-induced breast cancers. J Proteome Res. 2011;10:5503–5511. doi: 10.1021/pr200772w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guan Y, Ackert-Bicknell CL, Kell B, Troyanskaya OG, Hibbs MA. Functional genomics complements quantitative genetics in identifying disease-gene associations. PLoS Comput Biol. 2010;6:e1000991. doi: 10.1371/journal.pcbi.1000991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang EY, Cristofanilli M, Robertson F, Reuben JM, Mu Z, Beavis RC, Im H, Snyder M, Hofree M, Ideker T, Omenn GS, Fanayan S, Jeong SK, Paik YK, Zhang AF, Wu SL, Hancock WS. Genome wide proteomics of ERBB2 and EGFR and other oncogenic pathways in inflammatory breast cancer. J Proteome Res. 2013;12:2805–2817. doi: 10.1021/pr4001527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Visconte V, Makishima H, Maciejewski JP, Tiu RV. Emerging roles of the spliceosomal machinery in myelodysplastic syndromes and other hematological disorders. Leukemia. 2012;26:2447–2454. doi: 10.1038/leu.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vidal M, Chan DW, Gerstein M, Mann M, Omenn GS, Tagle D, Sechi S. The human proteome – a scientific opportunity for transforming diagnostics, therapeutics, and healthcare. Clin Proteomics. 2012;9:6. doi: 10.1186/1559-0275-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]