

Abstract
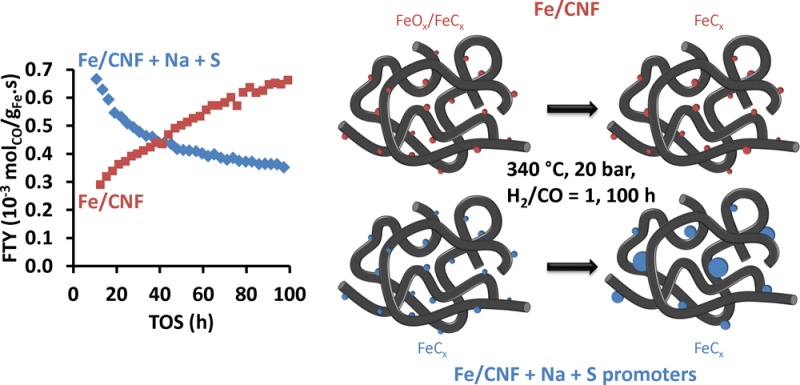
The Fischer–Tropsch Synthesis converts synthesis gas from alternative carbon resources, including natural gas, coal, and biomass, to hydrocarbons used as fuels or chemicals. In particular, iron-based catalysts at elevated temperatures favor the selective production of C2–C4 olefins, which are important building blocks for the chemical industry. Bulk iron catalysts (with promoters) were conventionally used, but these deactivate due to either phase transformation or carbon deposition resulting in disintegration of the catalyst particles. For supported iron catalysts, iron particle growth may result in loss of catalytic activity over time. In this work, the effects of promoters and particle size on the stability of supported iron nanoparticles (initial sizes of 3–9 nm) were investigated at industrially relevant conditions (340 °C, 20 bar, H2/CO = 1). Upon addition of sodium and sulfur promoters to iron nanoparticles supported on carbon nanofibers, initial catalytic activities were high, but substantial deactivation was observed over a period of 100 h. In situ Mössbauer spectroscopy revealed that after 20 h time-on-stream, promoted catalysts attained 100% carbidization, whereas for unpromoted catalysts, this was around 25%. In situ carbon deposition studies were carried out using a tapered element oscillating microbalance (TEOM). No carbon laydown was detected for the unpromoted catalysts, whereas for promoted catalysts, carbon deposition occurred mainly over the first 4 h and thus did not play a pivotal role in deactivation over 100 h. Instead, the loss of catalytic activity coincided with the increase in Fe particle size to 20–50 nm, thereby supporting the proposal that the loss of active Fe surface area was the main cause of deactivation.
Keywords: Fischer−Tropsch, FTO, iron, lower olefins, synthesis gas, stability, sintering
Introduction
The Fischer–Tropsch synthesis (FTS) is a catalytic surface polymerization reaction which converts synthesis gas (CO and H2) into valuable hydrocarbons, such as lower olefins.1 Synthesis gas can be produced from a wide array of carbon sources including natural gas, coal, and biomass, and product selectivity toward chemicals and fuels can be adjusted by catalyst design, which makes this a flexible option to the industry.2−5
Iron-based FT catalysts are favored due to their low cost, high abundance, low methane formation, and useful water–gas shift (WGS) activity.6−9 Bulk Fe catalysts (often modified with promoters and synthesized via coprecipitation or sintering) were extensively studied and displayed promising results, but these suffered from poor mechanical stability.10 Carbon formation which occurs via the Boudouard reaction (2 CO → C + CO2), leads to blocking of active sites and disintegration of catalyst particles. To improve on the mechanical properties of Fe-based catalysts, dispersing Fe nanoparticles on supports was attempted. A concern to the use of oxidic supports, such as silica and high surface area alumina, is the inhibition of critical phase transformation to active Fe carbide species due to strong support–metal interactions.11 Thus, weakly interacting supports such as nanostructured carbon materials and low surface area alumina were preferred for supported Fe FT catalysts.12−15
Earlier research showed that sodium and sulfur promoters, as well as the particle size of iron nanoparticles supported on the carbon nanofibers (CNF), affect the activity and selectivity of lower olefins.16 This breakthrough in increasing the lower olefins selectivity made the direct production of lower olefins from synthesis gas (Fischer–Tropsch to olefins, FTO) a more attractive option. However, the stability of the improved Fe FT catalysts remained a challenge.
Deactivation of Fe FT catalysts could be due to Fe particle growth and/or carbon deposition, and/or transformation of active Fe carbide species into inactive Fe carbide/oxide/metallic species.17−20 Sintering, either via Ostwald ripening or particle migration and coalescence, results in loss of active Fe carbide surface area, and thereby loss of catalytic activity. Phase transformation and carbon deposition are speculated to be the main causes of deactivation for bulk Fe catalysts,21−28 while the surface chemistry of support material dictates the deactivation mechanism for supported Fe catalysts. Fe nanoparticles supported on oxidic supports were stable due to strong support–metal interactions, hence sintering and phase transformations were not expected, and C deposition was likely to be the dominant cause for activity loss.29,30 Conversely, Fe nanoparticles supported on weakly interacting supports were prone to Fe particle growth.31−33 Because the mode of deactivation is affected by the support material, there are different strategies to improve catalytic stability by means of proper support selection. Carbon supports are widely used, and to improve on the catalytic stability, catalyst design has been focused on encapsulating Fe nanoparticles to prevent Fe particle growth.34,35
In this work, the aim is to comprehend the effect of iron particle size and the presence or absence of promoters on catalyst stability. Various causes of deactivation, such as Fe phase transformations, carbon deposition, and Fe particle growth will be assessed. To eliminate the contribution from oxidic supports, carbon nanofibers were used to support the iron nanoparticles. Iron nanoparticles supported on CNF with/without Na and S promoters were prepared via incipient wetness impregnation. By varying the loading of iron between 2 to 20 wt %, iron oxide nanoparticles between 3–9 nm were obtained. Catalytic tests were performed to determine the catalytic activity, selectivity, and stability. The as-synthesized and spent catalysts were characterized ex situ by transmission electron microscopy (TEM). In situ Mössbauer spectroscopy was used to characterize the iron phases under FTO conditions. A tapered element oscillating microbalance (TEOM) was used to monitor the rate of carbon deposited during the FTO process.36
Experimental Methods
Catalyst Preparation and Characterization
Growth of CNF Support
A 5 wt % Ni/SiO2 catalyst (sieve fraction of 425–825 μm) was synthesized via homogeneous deposition precipitation as reported previously.37 Five grams of the catalyst was reduced under the flow of 190 mL/min H2 and 625 mL/min N2 at a pressure of 2.8 bar and 700 °C for 2 h (5 °C/min). Temperature was lowered to 550 °C (3.5 °C/min) after reduction, and carbon nanofibers with fishbone structure were grown by flowing diluted syngas with the composition of 102 mL/min H2, 266 mL/min CO, and 450 mL/min N2 for 24 h. To remove SiO2, the CNF grown was refluxed thrice in 400 mL of 1 M KOH followed by washing with demineralized water to pH 7. To remove Ni and to introduce oxygen-containing groups on the surface, the purified CNF was refluxed in 400 mL of 65% HNO3 for 1.5 h followed by washing with demineralized water to pH 7.
Preparation of Unpromoted Supported Catalysts
Four unpromoted catalysts with different iron loadings (2, 5, 10, 20 wt % Fe) were prepared using incipient wetness impregnation as described previously.16 In the initial step, 7.014 g ammonium iron citrate (Fluka, purum p.a., 14.5–16 wt % Fe) was dissolved in 25 mL of demineralized water to form a stock solution. Depending on the iron loading, different volumes of this stock solution were impregnated onto CNF to achieve the desired loading. Except for the 2 wt % Fe-loaded catalyst, every catalyst required successive impregnation steps. The samples were dried under static air at 120 °C between impregnation steps and after the final impregnation step for 1 and 2 h, respectively. Heat treatment was performed at 500 °C for 2 h (5 °C/min; 100 mL/min for 2 g catalyst) under the nitrogen flow. After it was cooled to room temperature, the catalyst was passivated by controlled surface oxidation. The oxygen concentration was increased stepwise (2% v/v increase every 30 min) until reaching 20% v/v. The number in the sample code indicates the surface area-average particle size of iron nanoparticles measured by TEM.
Preparation of Promoted Supported Catalysts
Four promoted catalysts with different iron loadings (2, 5, 10, 20 wt % Fe) were prepared using incipient wetness impregnation as described above. The determined promoter loading is shown in Table 1. Initially, 6.954 g of ammonium iron citrate (Fluka, purum p.a., 14.5–16 wt % Fe), 0.199 g of sodium citrate tribasic dihydrate (Sigma-Aldrich, > 99.0%), and 0.056 g of iron(II) sulfate heptahydrate (Merck) were dissolved in 25 mL of demineralized water to form a stock solution. Subsequent steps were performed as described above. In addition to the number in the sample code which indicates the surface area-average particle size of Fe2O3 nanoparticles measured by TEM, the letter P was included to identify promoted catalysts.
Table 1. Properties of As-Synthesized Promoted and Unpromoted CNF-Supported Fe Catalysts.
| wt % loadinga |
average particle size (nm) | |||
|---|---|---|---|---|
| Fe | Na | S | Fe2O3b | |
| 3Fe | 2 | 0.03 | <0.005 | 3 |
| 4Fe | 5 | 0.03 | <0.006 | 4 |
| 7Fe | 9 | 0.02 | <0.004 | 7 |
| 9Fe | 15 | 0.03 | 0.007 | 9 |
| 3FeP | 2 | 0.10 | 0.02 | 3 |
| 4FeP | 5 | 0.24 | 0.04 | 4 |
| 7FeP | 9 | 0.41 | 0.07 | 7 |
| 9FeP | 16 | 0.68 | 0.11 | 9 |
wt % loading determined using ICP-AES.
Surface area average determined by TEM.
Characterization
TEM was used to determine the iron particle size distribution and the spatial distribution of iron nanoparticles on the support. At least 300 iron nanoparticles per catalyst were measured to obtain an average particle size. The images were attained with a Philips Tecnai-20 FEG (200 kV) microscope equipped with an EDX and HAADF detector. Temperature-programmed reduction (TPR) measurements were carried out with a Micromeritics AutoChem II equipped with a TCD detector. Relevant reduction conditions were used (i.e., 350 °C (5 °C/min), 5% H2 in Ar, 2 h). The composition of the Fe phases before reaction, after reduction, and after FTO reaction was determined in situ with transmission 57Fe Mössbauer spectroscopy. Transmission 57Fe Mössbauer spectra were collected at 300 and 4.2 K with a sinusoidal velocity spectrometer using a 57Co(Rh) source. Velocity calibration was carried out using an α-Fe foil. The source and the absorbing samples were kept at the same temperature during the measurements. The Mössbauer spectra were fitted using the Mosswinn 4.0 program.38 The experiments were performed in a state-of-the-art high-pressure Mössbauer in situ cell—recently developed at Reactor Institute Delft.39 The high-pressure beryllium windows used in this cell contain 0.08% Fe impurity, and its spectral contribution was fitted and removed from the final spectra. The Mössbauer transmission cell has a tubular reaction chamber with an internal diameter of 15 mm, and the catalyst bed lengths were 1.5–3 mm (catalyst loading of 100–300 mg). Although the reactant gases pass through the catalyst bed, the Mössbauer cell is not a plug-flow reactor due to a large dead volume (∼7 cm3) before the catalyst bed. A total flow rate of 100 mL/min was used during treatments, corresponding to a gas hourly space velocity (GHSV) of about 12 000–24 000 h–1. The reaction conditions were as described in the catalytic tests below.
Catalyst Performance
Catalytic experiments were performed using high throughput fixed-bed reactors as described elsewhere.40 The catalysts were first reduced in situ at 340 °C (5 °C/min), 3 bar, He/H2 = 2, GHSV = 7200 h–1 for 2 h. Synthesis gas mixture (H2/CO/He = 45/45/10) was introduced at 280 °C and 3 bar, and temperature and pressure were subsequently increased over 0.5 h to 340 °C and 20 bar. Two different gas hourly space velocities (GHSV) were employed, specifically 7200 h–1 and 54 000 h–1 for mimicking industrially relevant conditions and C deposition test conditions, respectively. A blank experiment using CNF support showed zero activity under relevant FTO conditions. Catalytic activity, expressed as iron time yield (FTY), was expressed as moles of CO converted per gram of Fe per second. CO conversion (%) was calculated as
| 1 |
Where CCO, R and CHe,R correspond to concentration of CO and He at the reactor outlet, respectively. CHe,blk and CCO, blk correspond to concentrations of CO and He at the outlet of the blank reactor. The product selectivity to hydrocarbons up to C9 was determined with online gas chromatography (GC) and was calculated on a carbon atom basis. Selectivity toward CO2 was also measured. This analysis method is consistent with previous literature.16
C Deposition
Carbon deposition was measured using a TEOM (TEOM series 1500 Pulse Mass Analyzer, Rupprecht & Patashnick Co.,Inc.).36 The procedure started with flushing the tapered element with N2 at room temperature and pressure. The pressure and temperature were increased to 2 bar and 340 °C (10 °C/min) respectively. The resulting decrease in gas density was reflected by a decrease in the mass signal. Upon stabilization of the mass signal which took ∼4 h, the gas feed was switched to a mixture of N2 and H2, resulting in a sharp decrease in the mass signal. Each catalyst was reduced at 2 bar, 340 °C, N2/H2 = 2, and GHSV = 54 000 h–1 for 2 h, and this reduction step was apparent from the gradual decrease in the mass signal. After reduction, the gas feed was switched from N2 and H2 mixture to pure N2 to determine the mass loss during reduction. Stabilization of the mass signal was again needed (∼0.5 h) before synthesis gas feed was introduced. C deposition was monitored for 4 h at 340 °C, 20 bar, CO/H2 = 1, and GHSV = 54 000 h–1. Finally, the feed was switched back to pure N2 gas feed after 4 h of synthesis gas exposure. The CNF support was also tested in a blank experiment and no mass difference was observed during reduction and Fischer–Tropsch reaction. To prevent accumulation of hydrocarbon products in the catalyst bed, a high GHSV was required. As the quartz element may break due to an increase in the catalyst volume from coking, time-on-stream (TOS) was limited to 4 h.
Results and Discussion
An overview of the fresh catalysts and their properties is presented in Table 1. More information on elemental loadings and the particle size distributions can be found in Supporting Information, SI 1 and 2, respectively.
Tables 2 and 3 summarize the activity and product selectivity of these supported Fe catalysts at low conversions during the initial period (TOS = 12 h) and steady state (TOS = 100 h), respectively. Activity and product selectivity of these supported Fe catalysts at high conversions are included in SI 3. Data in Table 2 show that the addition of promoters resulted in increased activity and product selectivity for CO2, C2–C4 olefins, and C5+ hydrocarbons,16 and these promoted catalysts exhibited stable product selectivities after 12 h on-stream (Table 3). Product selectivities were influenced by various factors, for example, CO conversion and Fe particle size at different times on-stream.
Table 2. Catalytic Performance of CNF-Supported Fe Catalysts under FTO Conditionsa.
| product
selectivity (% Cat, CO2 free) |
|||||||
|---|---|---|---|---|---|---|---|
| CO conv. (%) | FTY (10–3 molCO/gFe·s) | CO2 sel. (%) | CH4 | C2–C4 olefins | C2–C4 paraffins | C5+ | |
| 3Fe | 4 | 0.8 | 12 | 52 | 26 | 22 | 0 |
| 4Fe | 3 | 0.3 | 18 | 50 | 26 | 24 | 1 |
| 7Fe | 4 | 0.2 | 20 | 47 | 28 | 22 | 3 |
| 9Fe | 4 | 0.1 | 23 | 46 | 36 | 14 | 4 |
| 3FeP | 7 | 1.7 | 35 | 20 | 54 | 14 | 12 |
| 4FeP | 7 | 0.7 | 38 | 11 | 61 | 6 | 22 |
| 7FeP | 10 | 0.5 | 40 | 8 | 60 | 6 | 25 |
| 9FeP | 16 | 0.3 | 42 | 8 | 60 | 5 | 27 |
Conditions: 340 °C, 20 bar, H2/CO/He = 45/45/10, GHSV = 54 000 h–1, TOS = 12 h.
Table 3. Catalytic Performance of CNF-Supported Fe Catalysts under FTO Conditionsa.
| product
selectivity (% Cat, CO2 free) |
|||||||
|---|---|---|---|---|---|---|---|
| CO conv. (%) | FTY (10–3 molCO/gFe·s) | CO2 sel. (%) | CH4 | C2–C4 olefins | C2–C4 paraffins | C5+ | |
| 3Fe | 6 | 1.3 | 15 | 49 | 25 | 21 | 5 |
| 4Fe | 8 | 0.7 | 21 | 40 | 31 | 20 | 9 |
| 7Fe | 13 | 0.5 | 30 | 38 | 36 | 14 | 12 |
| 9Fe | 18 | 0.4 | 33 | 35 | 36 | 15 | 14 |
| 3FeP | 2 | 0.8 | 33 | 14 | 72 | 7 | 7 |
| 4FeP | 4 | 0.3 | 37 | 11 | 66 | 7 | 16 |
| 7FeP | 7 | 0.3 | 41 | 10 | 62 | 6 | 22 |
| 9FeP | 8 | 0.2 | 40 | 9 | 62 | 5 | 24 |
Conditions: 340 °C, 20 bar, H2/CO/He = 45/45/10, GHSV = 54 000 h–1, TOS = 100 h.
Figure 1 shows the catalytic activity as a function of time, thereby providing insights in the stability of these catalysts at high temperature and pressure. The unpromoted catalysts had the lowest catalytic activity during the initial period. On the other hand, the promoted catalysts had the highest catalytic activity during the initial period, but catalytic activity decreased over time (Figure 1b). It is noted that the catalytic activity after 100 h on-stream of promoted and unpromoted catalysts was similar, except for 3Fe(P). Formation of different Fe species, carbon accumulation, and Fe particle growth are possible causes of deactivation, and these were further investigated.
Figure 1.

Iron time yield (FTY) of (a) unpromoted catalysts and (b) promoted catalysts (340 °C, 20 bar, H2/CO/He = 45/45/10, and GHSV = 54 000 h–1).
In situ Mössbauer spectroscopy determined quantitatively the various Fe species present during reduction and FTO conditions. Upon reduction, 25 mol % Fe carbide species were measured for both 4FeP and 4Fe (Figures 2a,c and SI 4). It is thus suggested that the addition of promoters did not have a significant influence on the reduction step. This observation was confirmed by the similar TPR profiles obtained for 4PFe and 4Fe (Figure S10). To further investigate the reduction behavior of a promoted catalyst (7PFe), Mössbauer spectra were measured after 2 and 24 h reduction (Ar/H2 = 2, 350 °C, 2 bar). Despite the longer reduction duration, incomplete reduction was observed and similar content of Fe carbide phase was measured (Table S8 and Figure S11). However, after 20 h of FTO conditions, 4FeP was fully carburized and 4Fe had only 23 mol % Fe carbide species (Figures 2b,d and SI 4). The remaining Fe atoms were present as Fe2+ species having Mössbauer spectra similar to those of Wüstite and its nonstoichiometric equivalent (Fe1–xO).41 The promoters induced formation of more Fe carbide species in the initial period, which gave higher catalytic activity. Alkali promoters such as Na are known to increase Fe carburization rate,42 but the addition of S showed the opposite effect.43 It is noted that 4FeP had four times more Fe carbide species, but its catalytic activity was only twice higher than 4Fe.
Figure 2.
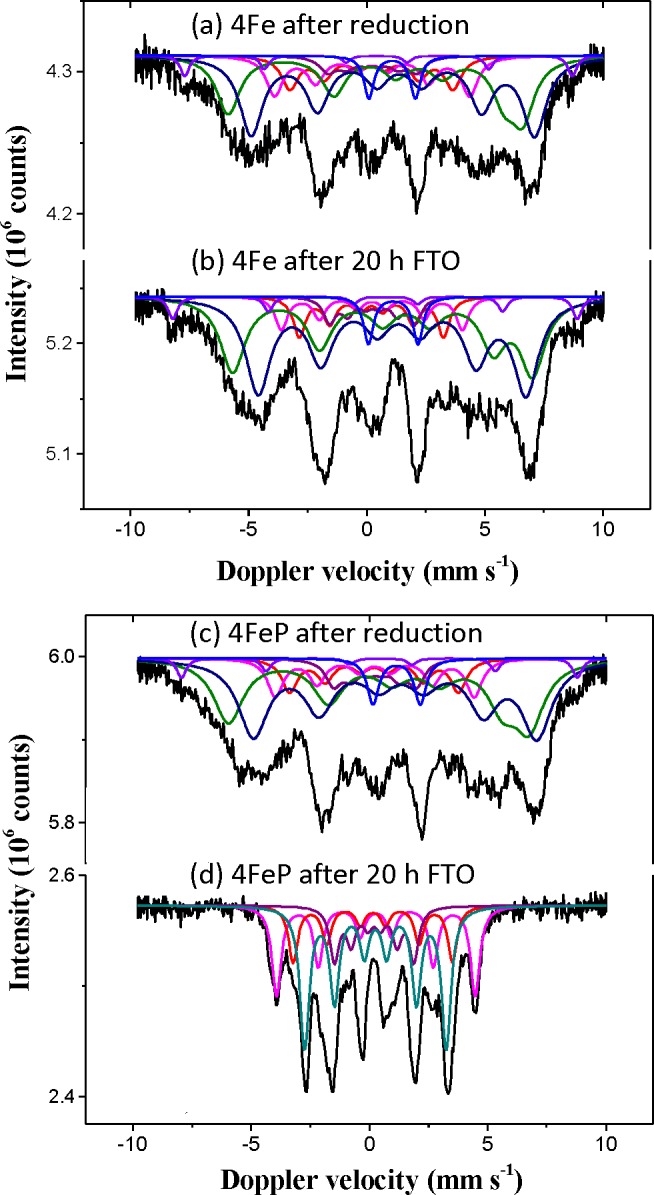
In situ Mossbauer spectra of (a) 4Fe after reduction, (b) 4Fe after 20 h TOS, (c) 4FeP after reduction, and (d) 4FeP after 20 h TOS. Reduction: 350 °C, 2 bar, Ar/H2 = 2, 2 h. FTO: 340 °C, 20 bar, H2/CO = 1, TOS = 20 h. (3 Fe sites for Hägg carbide: magenta, red, and purple; 1 Fe site for ε′ carbide: dark cyan; 3–4 Fe sites for Fe1–xO: violet, olive, navy, blue).
Carbon deposition over time was monitored in situ using the TEOM. No carbon deposition was measured during 4 h for the unpromoted catalysts with different particle sizes (Figure S12), and it is proposed to be due to the lack of active χ-Fe5C2 phase. This proposal was supported by in situ Mössbauer spectroscopy data (Figure 2) and catalyst performance data. The promoted catalysts showed carbon accumulation over time (Figure S13). During the early stages (first 30 min approximately), there were two opposite occurrences for mass changes which resulted in a negligible net mass change. Further reduction of Fe oxide species to Fe carbide species caused a decrease in mass, while carbon laydown due to the Fe carbide species already present after reduction caused an increase in mass. Thus, the net effect was a lower mass change rate than when the catalyst reached a state where no further reduction took place. Subsequently, the coking rate decreased over 4 h. It is believed that the measured mass change was caused directly by carbon build-up and not hydrocarbons because of the reaction conditions used.36 Carbon laydown rate decreased with increasing particle size initially (1st hour), but the rate increased with increasing initial particle size at steady state (4th hour). This indicates that the effect of particle size on carbon deposition rate was different when the surfaces were relatively clean and after reaction had proceeded for a longer period. Possible reasons for this decrease in coking rate include blocking of active sites (surface covered with amorphous or graphitic carbon), growth of Fe carbide particles, and phase transformations that do not convert CO to carbon. Thus, it appeared that the carbon deposition was most severe in the initial period and was not the main reason for deactivation over a longer period (10–100 h TOS).
The spent catalysts after carbon deposition studies (TOS = 4 h) and after catalytic tests (TOS = 100 h) were characterized with TEM, and images are shown in Figure S2 and Figure 3 respectively. It was mentioned earlier that the addition of promoters did not lead to a significant change in Fe particle size distribution of the fresh catalysts. However, the Fe particle size distributions of spent promoted and unpromoted catalysts were strikingly different. The promoted catalysts displayed a higher degree of sintering compared to the unpromoted catalysts. In the extreme case (9FeP) depicted in Figure 3h, Fe particles of approximately 100 nm were observed. In addition, the promoted Fe nanoparticles seemed to have a core–shell structure (Figure 3e–h). TEM-EDX maps (Figure 3i–m) showed that the core was rich in Fe and the shell was mainly Fe oxide. The formation of Fe oxide was most likely due to exposure to air after reaction.
Figure 3.

TEM images of spent (a–d) unpromoted catalysts and (e–h) promoted catalysts after carbon deposition studies (340 °C, 20 bar, H2/CO = 1, GHSV = 54 000 h–1, TOS = 100 h). TEM-EDX maps of spent 7FeP catalyst (i–m).
In Figure 4, the average iron particle sizes of fresh and spent catalysts are compared. The particle size distributions of spent catalysts can be found in SI 1. It was thus shown that the promoted Fe nanoparticles produced more carbon and displayed more sintering while the unpromoted catalysts showed limited sintering even after 100 h TOS.
Figure 4.
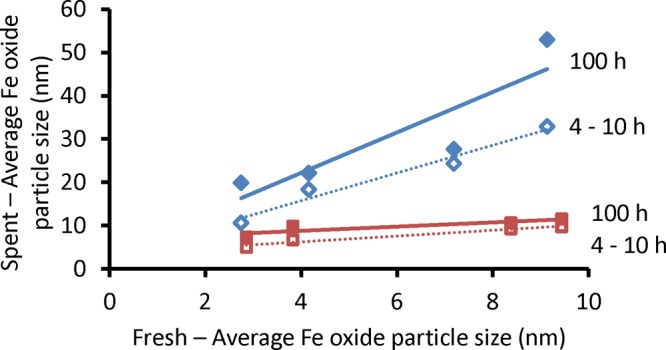
Iron particle size of (blue-outline ◇) promoted catalysts and (red-outline □) unpromoted catalysts after carbon deposition studies, TOS = 4 h. Iron particle size of (blue ◆) promoted catalysts and (red ■) unpromoted catalysts after catalytic tests, TOS = 100 h (340 °C, 20 bar, H2/CO = 1, GHSV = 54 000 h–1).
Figure 5 reveals the effects of the particle size on the stability of unpromoted Fe FT catalysts. The unpromoted catalysts showed increased catalytic activity and limited sintering over time, and it is proposed tentatively that the increased catalytic activity is related to increased Fe carbidization.
Figure 5.
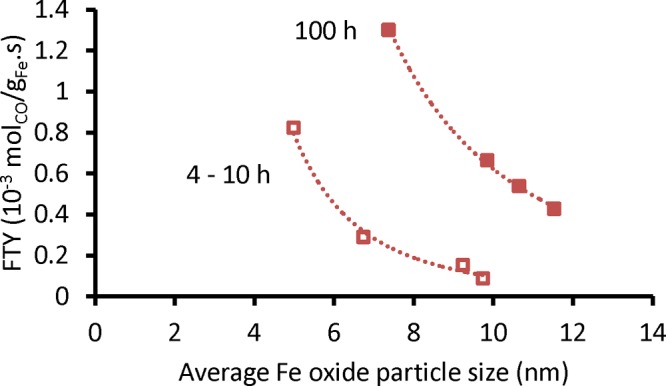
Catalytic activity as a function of average iron particle size of spent unpromoted catalysts (red-outline □) at the initial period, TOS = 4–10 h, and (red ■) at steady state, TOS = 100 h (340 °C, 20 bar, H2/CO = 1, GHSV = 54 000 h–1). Lines were added to guide the eye.
Figure 6 depicts the effects of the particle size on the stability of promoted Fe FT catalysts. The decrease in catalytic activity appeared to be in agreement with the increase in Fe particle size over time. The loss of surface area per gram Fe follows the relation: surface area/volume α 1/Fe particle diameter; thus, FTY was fitted to be inversely proportional to particle size (trend line in Figure 6). The first data point (smallest average particle size) appeared to be an anomaly, and that is attributed to the presence of small particles around 3 and 4 nm. These small Fe particles were shown previously to be highly active and produced mainly methane.16
Figure 6.
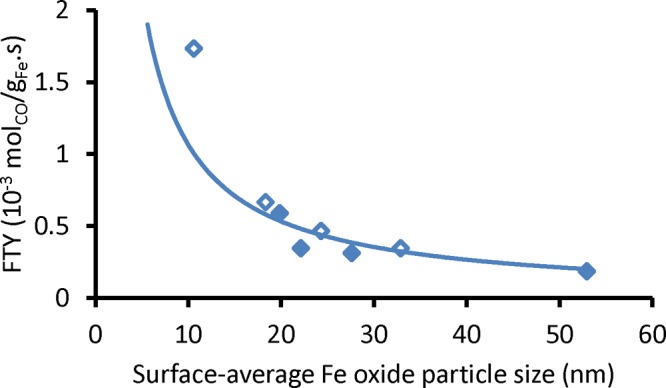
Catalytic activity as a function of average iron particle size of spent promoted catalysts (blue-outline ◇) at the initial period, TOS = 4–10 h, and (blue ◆) at TOS = 100 h (340 °C, 20 bar, H2/CO = 1, GHSV = 54 000 h–1). The trend line is fitted to y α x–1 for all data points except the first point.
For the promoted catalysts, Fe particle growth was concluded to be the main cause of deactivation. Phase transformations, which occurred when water was produced but not removed from the catalyst bed, were reported previously to be a reason for deactivation;25−27 however, it is not expected to be relevant here because of the use of high space velocities and low conversion conditions. Carbon deposition rates decreased over the initial hours and were not expected to play a significant role over a longer period. The use of sulfur, in the absence of sodium, to increase the resistance against carbon deposition was demonstrated earlier,29 and this may be a possibility to explain the relatively low carbon deposition rates when compared to other Fe-based catalysts. The sintering of Fe nanoparticles supported on O-functionalized CNTs was concluded previously to be more severe due to weak support–metal interactions and a low concentration of surface defects for anchoring of Fe nanoparticles.33 Although a weakly interacting support CNF was used in this study, very different extent of particle growth was observed for the promoted and unpromoted Fe nanoparticles. This proves that the severe sintering of promoted Fe nanoparticles was not solely due to weak metal–support interaction. Intrinsically, the FTO process would be a high temperature FT route, thus sintering is expected to be more prominent than at the less severe conditions of low-temperature FT, which is typical for coal-to-liquids (CTL).
Conclusion
The activity, product selectivity and stability of CNF-supported Fe catalysts under industrially relevant Fischer–Tropsch to olefins (FTO) conditions were investigated. It was observed that the activity of unpromoted catalysts increased over time, regardless of particle size. With addition of promoters, maximum activity was attained in the initial period and deactivation was prominent.
In situ Mössbauer spectroscopy revealed that both promoted and unpromoted catalysts attained similar Fe carbidization levels after reduction. However, after 20 h of synthesis gas treatment, the promoted catalyst was fully carbided, and the carbidization level of the unpromoted catalyst did not increase beyond 25%. This difference in phase transformation upon exposure to synthesis gas resulted in a higher initial activity of the promoted catalyst. As the correlation of activity and Fe carbide species was not linear, deactivation via carbon deposition and/or Fe particle growth is proposed to occur simultaneously with phase transformations.
A tapered element oscillating microbalance (TEOM) was utilized to measure the rate of carbon deposition under industrially relevant FTO conditions. No carbon laydown was detected for the unpromoted catalysts, and this was rationalized by the lack of active Fe carbide phases. In contrast, the presence of promoters facilitated Fe activation which resulted in significant carbon deposition over the first hours of operation. While different particle sizes resulted in different coking rates, coking rates decreased over time. This suggests that carbon deposition is not the leading cause of deactivation over longer periods.
Limited sintering was observed for the unpromoted catalysts while severe sintering was seen for the promoted catalysts. This indicated that the promoters led to formation of mobile and active Fe phases which resulted in a higher degree of particle growth.
For the unpromoted catalysts, phase transformation was considered to be the leading cause for the increase in catalytic activity over time. For the promoted catalysts, catalytic activity was shown to be inversely proportional to Fe particle diameter of spent catalysts which leads to the conclusion that here Fe particle growth is the main reason for deactivation over time.
In this work, the catalytic performance of CNF-supported Fe catalysts under industrially relevant FTO conditions were thoroughly and critically assessed. Although the activity and C2–C4 olefins selectivity of the promoted Fe catalysts were highly encouraging, the stability needs improvement. Carbon deposition for Fe nanoparticles was less significant compared to bulk Fe catalysts and is proposed not to play a pivotal role in the deactivation. Sintering was, however, the major cause of deactivation, and it is hence believed that sintering is a vital factor affecting stability of these highly active and selective promoted Fe catalysts. Thus, the direction of future research is on designing highly active and selective Fe catalysts which are more resistant to sintering. It is of interest for future studies to unravel the mechanism and details of particle growth of promoted and unpromoted Fe nanoparticles.
Acknowledgments
This research received funding from The Netherlands Organisation for Scientific Research (NWO) in the framework of the TASC Technology Area “Syngas, a Switch to Flexible New Feedstock for the Chemical Industry (TA-Syngas)”. Dow and Johnson Matthey are also acknowledged for the funding received. K.P.d.-J. acknowledges the European Research Council, EU FP7 ERC Advanced Grant no. 338846. H. Meeldijk and W. Lamme are thanked for the TEM images. Peter Bramwell is acknowledged for the TPR measurements. H. C. de Waard is acknowledged for the ICP-AES measurements. G. Bonte (Dow Benelux) is thanked for catalyst performance testing. M. Watson and L. van de Water (Johnson Matthey) are acknowledged for fruitful discussion.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.6b00321.
TEM characterization of fresh and spent catalysts, catalytic performance, in situ Mössbauer spectroscopy, and in situ carbon deposition studies (TEOM) (PDF)
Author Present Address
∥ (T.G.) Albemarle Catalysts Company B.V., Nieuwendammerkade 1–3, 1022 AB, Amsterdam
The authors declare no competing financial interest.
Supplementary Material
References
- van Santen R. A. Acc. Chem. Res. 2009, 42, 57–66. 10.1021/ar800022m. [DOI] [PubMed] [Google Scholar]
- Wender I. Fuel Process. Technol. 1996, 48, 189–297. 10.1016/S0378-3820(96)01048-X. [DOI] [Google Scholar]
- Dry M. E. Catal. Today 2002, 71, 227–241. 10.1016/S0920-5861(01)00453-9. [DOI] [Google Scholar]
- van Steen E.; Claeys M. Chem. Eng. Technol. 2008, 31, 655–666. 10.1002/ceat.200800067. [DOI] [Google Scholar]
- Zhang Q.; Kang J.; Wang Y. ChemCatChem 2010, 2, 1030–1058. 10.1002/cctc.201000071. [DOI] [Google Scholar]
- Davis B. H. Catal. Today 2003, 84, 83–98. 10.1016/S0920-5861(03)00304-3. [DOI] [Google Scholar]
- Li S.; Krishnamoorthy S.; Li A.; Meitzner G. D.; Iglesia E. J. Catal. 2002, 206, 202–217. 10.1006/jcat.2001.3506. [DOI] [Google Scholar]
- Torres Galvis H. M.; de Jong K. P. ACS Catal. 2013, 3, 2130–2149. 10.1021/cs4003436. [DOI] [Google Scholar]
- Khodakov A. Y.; Chu W.; Fongarland P. Chem. Rev. 2007, 107, 1692–1744. 10.1021/cr050972v. [DOI] [PubMed] [Google Scholar]
- Shroff M. D.; Kalakkad D. S.; Coulter K. E.; Köhler S. D.; Harrington M. S.; Jackson N. B.; Sault A. G.; Datye A. K. J. Catal. 1995, 156, 185–207. 10.1006/jcat.1995.1247. [DOI] [Google Scholar]
- Yang Y.; Xiang H.-W.; Tian L.; Wang H.; Zhang C.-H.; Tao Z.-C.; Xu Y.-Y.; Zhong B.; Li Y.-W. Appl. Catal., A 2005, 284, 105–122. 10.1016/j.apcata.2005.01.025. [DOI] [Google Scholar]
- Torres Galvis H. M.; Bitter J. H.; Khare C. B.; Ruitenbeek M.; Dugulan A. I.; de Jong K. P. Science 2012, 335, 835–838. 10.1126/science.1215614. [DOI] [PubMed] [Google Scholar]
- Bahome M. C.; Jewell L. L.; Hildebrandt D.; Glasser D.; Coville N. J. Appl. Catal., A 2005, 287, 60–67. 10.1016/j.apcata.2005.03.029. [DOI] [Google Scholar]
- Moussa S. O.; Panchakarla L. S.; Ho M. Q.; El-shall M. S. ACS Catal. 2014, 4, 535–545. 10.1021/cs4010198. [DOI] [Google Scholar]
- Keyvanloo K.; Mardkhe M. K.; Alam T. M.; Bartholomew C. H.; Woodfield B. F.; Hecker W. C. ACS Catal. 2014, 4, 1071–1077. 10.1021/cs401242d. [DOI] [Google Scholar]
- Torres Galvis H. M.; Bitter J. H.; Davidian T.; Ruitenbeek M.; Dugulan A. I.; de Jong K. P. J. Am. Chem. Soc. 2012, 134, 16207–16215. 10.1021/ja304958u. [DOI] [PubMed] [Google Scholar]
- Bartholomew C. H. Appl. Catal., A 2001, 212, 17–60. 10.1016/S0926-860X(00)00843-7. [DOI] [Google Scholar]
- Moulijn J. A.; van Diepen A. E.; Kapteijn F. Appl. Catal., A 2001, 212, 3–16. 10.1016/S0926-860X(00)00842-5. [DOI] [Google Scholar]
- de Smit E.; Weckhuysen B. M. Chem. Soc. Rev. 2008, 37, 2758–2781. 10.1039/b805427d. [DOI] [PubMed] [Google Scholar]
- de Smit E.; Cinquini F.; Beale A. M.; Safonova O. V.; van Beek W.; Sautet P.; Weckhuysen B. M. J. Am. Chem. Soc. 2010, 132, 14928–14941. 10.1021/ja105853q. [DOI] [PubMed] [Google Scholar]
- Niemantsverdriet J. W.; van der Kraan A. M. J. Catal. 1981, 72, 385–388. 10.1016/0021-9517(81)90025-7. [DOI] [Google Scholar]
- Eliason S. A.; Bartholomew C. H. Appl. Catal., A 1999, 186, 229–243. 10.1016/S0926-860X(99)00146-5. [DOI] [Google Scholar]
- Nakhaei Pour A.; Housaindokht M. R.; Zarkesh J.; Tayyari S. F. J. Ind. Eng. Chem. 2010, 16, 1025–1032. 10.1016/j.jiec.2010.09.003. [DOI] [Google Scholar]
- Pendyala V. R. R.; Graham U. M.; Jacobs G.; Hamdeh H. H.; Davis B. H. ChemCatChem 2014, 6, 1952–1960. 10.1002/cctc.201402073. [DOI] [Google Scholar]
- Pendyala V. R. R.; Graham U. M.; Jacobs G.; Hamdeh H. H.; Davis B. H. Catal. Lett. 2014, 144, 1704–1716. 10.1007/s10562-014-1336-z. [DOI] [Google Scholar]
- Ning W.; Koizumi N.; Chang H.; Mochizuki T.; Itoh T.; Yamada M. Appl. Catal., A 2006, 312, 35–44. 10.1016/j.apcata.2006.06.025. [DOI] [Google Scholar]
- Li S.; O'Brien R. J.; Meitzner G. D.; Hamdeh H.; Davis B. H.; Iglesia E. Appl. Catal., A 2001, 219, 215–222. 10.1016/S0926-860X(01)00694-9. [DOI] [Google Scholar]
- Niemantsverdriet J. W.; van der Kraan A. M.; van Dijk W. L.; van der Baan H. S. J. Phys. Chem. 1980, 84, 3363–3370. 10.1021/j100462a011. [DOI] [Google Scholar]
- Zhou X.; Ji J.; Wang D.; Duan X.; Qian G.; Chen D.; Zhou X. Chem. Commun. 2015, 51, 8853–8856. 10.1039/C5CC00786K. [DOI] [PubMed] [Google Scholar]
- Galuszka J.; Sano T.; Sawicki J. A. J. Catal. 1992, 136, 96–109. 10.1016/0021-9517(92)90109-U. [DOI] [Google Scholar]
- Abbaslou R. M. M.; Tavasoli A.; Dalai A. K. Appl. Catal., A 2009, 355, 33–41. 10.1016/j.apcata.2008.11.023. [DOI] [Google Scholar]
- Abbaslou R. M. M.; Tavassoli A.; Soltan J.; Dalai A. K. Appl. Catal., A 2009, 367, 47–52. 10.1016/j.apcata.2009.07.025. [DOI] [Google Scholar]
- Schulte H. J.; Graf B.; Xia W.; Muhler M. ChemCatChem 2012, 4, 350–355. 10.1002/cctc.201100275. [DOI] [Google Scholar]
- Yu G.; Sun B.; Pei Y.; Xie S.; Yan S.; Qiao M.; Fan K.; Zhang X.; Zong B. J. Am. Chem. Soc. 2010, 132, 935–937. 10.1021/ja906370b. [DOI] [PubMed] [Google Scholar]
- Santos V. P.; Wezendonk T. a.; Jaén J. J. D.; Dugulan a. I.; Nasalevich M. a.; Islam H.-U.; Chojecki A.; Sartipi S.; Sun X.; Hakeem A. a.; Koeken A. C. J.; Ruitenbeek M.; Davidian T.; Meima G. R.; Sankar G.; Kapteijn F.; Makkee M.; Gascon J. Nat. Commun. 2015, 6, 6451. 10.1038/ncomms7451. [DOI] [PubMed] [Google Scholar]
- Koeken A. C. J.; Torres Galvis H. M.; Davidian T.; Ruitenbeek M.; de Jong K. P. Angew. Chem., Int. Ed. 2012, 51, 7190–7193. 10.1002/anie.201200280. [DOI] [PubMed] [Google Scholar]
- van der Lee M. K.; van Dillen A. J.; Geus J. W.; de Jong K. P.; Bitter J. H. Carbon 2006, 44, 629–637. 10.1016/j.carbon.2005.09.031. [DOI] [Google Scholar]
- Klencsar Z. Nucl. Instrum. Methods Phys. Res., Sect. B 1997, 129, 527–533. 10.1016/S0168-583X(97)00314-5. [DOI] [Google Scholar]
- Dugulan A. I.Final report of the ACTS-programme ASPECT; The Netherlands Organisation for Scientific Research: The Hague, Netherlands, 2012. [Google Scholar]
- Shibata H.; McAdon M.; Schroden R.; Meima G.; Chojecki A.; Catry P.; Bardin B. In Modern Applications of High Throughput R&D in Heterogenous Catalysis; Hagemeyer A., Volpe A. F., Eds.; Bentham Science Publishers: United Arab Emirates, 2014; Vol. 48, pp 173–196. [Google Scholar]
- Schweika W.; Hoser A.; Martin M.; Carlsson A. E. Phys. Rev. B: Condens. Matter Mater. Phys. 1995, 51, 15771–15788. 10.1103/PhysRevB.51.15771. [DOI] [PubMed] [Google Scholar]
- Ribeiro M. C.; Jacobs G.; Davis B. H.; Cronauer D. C.; Kropf A. J.; Marshall C. L. J. Phys. Chem. C 2010, 114, 7895–7903. 10.1021/jp911856q. [DOI] [Google Scholar]
- Xu J.; Chang Z.; Zhu K.; Weng X.; Weng W.; Zheng Y.; Huang C.; Wan H. Appl. Catal., A 2016, 514, 103–113. 10.1016/j.apcata.2015.12.023. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.