

Abstract
Here, we report a supramolecular strategy for detecting specific proteins in complex media using hyperpolarized 129Xe NMR. A cucurbit[6]uril (CB[6]) based molecular relay was programmed for three sequential equilibrium conditions by designing a two-faced guest (TFG) that initially binds CB[6] and blocks CB[6]-Xe interaction. Protein analyte recruits the TFG and frees CB[6] for Xe binding. TFGs containing CB[6]- and carbonic anhydrase II (CAII)-binding domains were synthesized in one or two steps. X-ray crystallography confirmed TFG binding to Zn2+ in the deep, active-site CAII cleft, which precludes simultaneous CB[6] binding. The molecular relay was reprogrammed to detect avidin using a different TFG. Finally, CB[6]-Xe binding was detected in buffer and in E. coli cultures expressing CAII via ultrasensitive 129Xe NMR spectroscopy.
Keywords: Host-guest systems, Sensors, NMR spectroscopy, Molecular recognition, Bioinorganic chemistry
Graphical Abstract
A cucurbit[6]uril (CB[6]) based molecular relay enables detection of proteins using 129Xe NMR. A two-faced guest (TFG) initially binds CB[6], TFG is sequestered by cognate protein target thereby freeing CB[6], and, lastly, xenon binds CB[6]. The TFG is engineered such that CB[6]-129Xe NMR signal is absent until addition of target protein.
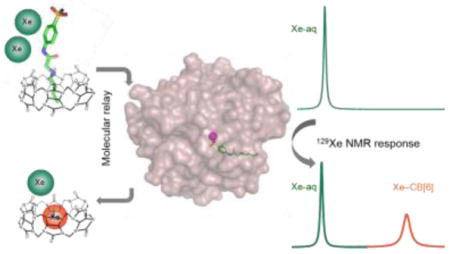
Methods for controlling the sequential interaction of molecules in solution can yield new functionality and complexity, as evidenced by many enzyme- and small-molecule-mediated tandem reactions, drug delivery strategies, and nanoscale and mesoscale architectures and working ‘machines’.[1] Less investigated are supramolecular strategies for improving in situ sample analysis, e.g., for generating high contrast in bio-assays and molecular imaging. Our lab and Schröder’s recently showed that commercially available cucurbit[6]uril (CB[6], Scheme 1) provides exceptional contrast at pM concentration via hyperpolarized (hp) 129Xe chemical exchange saturation transfer (Hyper-CEST) NMR.[2] In Hyper-CEST, encapsulated hp 129Xe is selectively depolarized by radiofrequency pulses, and through rapid exchange, the depolarized 129Xe accumulates in the solvent pool where decrease of signal can be readily monitored (Supporting Information, Scheme S1).[3] CB[6] is not readily functionalized for biosensing applications. Thus, we exploited the versatile host-guest chemistry of CB[6] to develop a de novo “molecular relay” that reports on specific proteins in solution.
Scheme 1.

Molecular relay produces 129Xe NMR signal upon analyte detection.
129Xe NMR/MRI has enabled investigations of many complex porous systems, from inorganic and organic materials[4] to bacterial spores[5] and mammalian lungs.[6] For detection of specific proteins in solution, however, a specific encapsulating agent is required as Xe has low affinity for endogenous biomolecules. Cryptophane-A and its derivatives are the most studied Xe-binding cages and these exhibit the largest association constants.[7] However, cryptophane biosensors require multi-step synthesis that yields only milligram quantities.[8] This has motivated the search for new 129Xe-binding scaffolds.[9]
The rigid structure and unique molecular recognition properties of cucurbit[n]uril (CB[n]) compounds make these useful as drug delivery vehicles, chemical reaction chambers, components of enzyme assays and building blocks in stimuli-responsive supramolecular architectures.[10][11] Importantly, CB[6] has a suitable cavity size to bind Xe (D ≈ 4.4 Å), and CB[6] and a water-soluble derivative were reported to bind Xe with useful affinities (KA = 500–3000 M−1 at rt).[12],[2a] Here, we describe CB[6]-129Xe NMR biosensors programmed for a molecular relay with three sequential recognition events (Scheme 1): a two-faced guest (TFG) is engineered to control this relay, such that CB[6]-129Xe NMR signal is absent until addition of analyte.
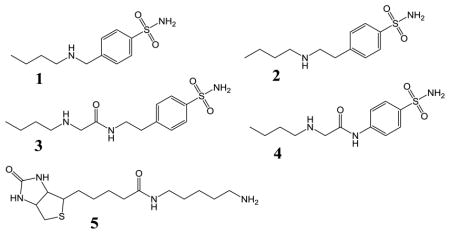
CB[6]-guest binding affinities first measured by Mock and coworkers[13] guided our design of TFGs that bind CB[6] with intermediate affinity (displacing Xe), and are readily sequestered upon binding the opposite TFG face to a higher-affinity protein target. Butylamine was measured by ITC to exhibit intermediate affinity for CB[6], KA = 2.85 × 105 M−1 in pH 7.2, PBS solution at 300 K (Supporting Information, Figure S1a). We chose human carbonic anhydrase II (CAII, EC 4.2.1.1) as the initial target, as this is a model single-site enzyme with biomedical significance[14] and its inhibitors have been well studied.[15] The active site of CAII resides at the base of a ~15-Å deep conical pocket that can differentiate between TFGs even if they share the same Zn2+-targeting moiety, as previously demonstrated for many CA inhibitors.[16] We therefore designed TFGs 1–4 (Table 1) with a CAII-binding p-benzenesulfonamide moiety and CB[6]-binding butylamine tail, while varying the length and chemical structure of the linker.
Table 1.
Two-faced guests (TFGs) used in this study and thermodynamic data for binding to CB[6], CAII, or avidin.
| |||||
|---|---|---|---|---|---|
| TFG | Host | KD (μM) | ΔH (kcal/mol) | TΔS (kcal/mol) | ΔG (kcal/mol) |
| 1 | CB[6] | 4.07 ± 0.55 | −1.17 ± 0.02 | 6.24 | −7.41 |
| CAII | 1.11 ± 0.29 | 1.23 ± 0.03 | 9.39 | −8.16 | |
| 2 | CB[6] | 6.58 ± 0.89 | −0.91 ± 0.03 | 6.21 | −7.12 |
| CAII | 2.29 ± 0.46 | −2.19 ± 0.08 | 5.55 | −7.74 | |
| 3 | CB[6] | 5.55 ± 0.72 | −2.33 ± 0.05 | 4.89 | −7.22 |
| CAII | 1.00 ± 0.14 | −3.86 ± 0.06 | 4.38 | −8.24 | |
| 4 | CB[6] | 3.61 ± 0.90 | −2.08 ± 0.10 | 5.40 | −7.48 |
| CAII | 0.48 ± 0.07 | −2.22 ± 0.02 | 6.45 | −8.67 | |
| 5 | CB[6] | 236 ± 30 | −4.79 ± 0.66 | 0.20 | −4.99 |
| Avidin [a] | ≈ 10−9 | N.D. | N.D. | ≈ −20 | |
Estimated values shown are based on measurements from ref. 21.
Synthetic details for TFGs 1–4 are provided in Scheme S2. Briefly, following procedures modified from Salvatore et al.,[17] 1 and 2 were synthesized from the primary amine and alkyl bromide in a single step using cesium hydroxide monohydrate as the base in 30% and 43% yield. TFGs 3 and 4 were synthesized in two steps. In the first step, 2-chloroacetyl chloride was reacted with 4-(2-aminoethyl)benzenesulfonamide or 4-aminobezenesulfonamide in 68% or 70% yield and subsequently reacted with 1-butylamine to deliver 3 or 4 in 56% or 50% yield.
To test whether the TFGs function as desired in the prototypal CB[6] molecular relay, binding affinities were measured by ITC separately for CB[6] and CAII (Table 1, Figure S1). The association constants for CB[6] titrated with 1, 2, 3, or 4 were all in the range of 1–3 × 105 M−1 at 300 K in pH 7.2, PBS buffer with 1% DMSO. We then performed ITC to test binding of 1–4 to CAII in the same buffer, and 4 displayed the highest affinity. The likely origins of the enthalpic and entropic contributions towards 1 binding to CAII are observed in the crystal structure of the enzyme-TFG complex (Figure S2). The benzenesulfonamide moiety of 1 forms the interactions typically observed for arylsulfonamide inhibitors: the sulfonamidate (N) coordinates to the catalytic Zn2+ and donates a hydrogen bond to the hydroxyl oxygen of T199, and a sulfonamide oxygen accepts a hydrogen bond from the backbone amide nitrogen of T199. The n-butyl tail of 1 occupies a hydrophobic pocket lined by residues L198, P202, L204, F131, V135, and L141. Other sulfonamide inhibitors with flexible, hydrophobic tails have also been observed to occupy this pocket.[18] The amine nitrogen in the tail of 1 is positioned near residues L198 and P202 but does not form hydrogen bonds with the protein or solvent. This limits enthalpic contributions to TFG binding, whereas solvent displacement contributes significant entropy to ΔG.
A crystal structure of CAII bound to the longest TFG 3 (Figure 1) shows that the amine nitrogen is only 4.8, 5.1 and 5.4 Å from three neighboring residues, respectively. Based on these distances, TFG 3 is not available to bind simultaneously to CB[6], with outer diameter of 6.9 Å at its portal and 14.4 Å at its widest point.[19] Molecular modeling (Figure S3) helps to illustrate that a stable ternary structure cannot form between CAII, TFG 3 and CB[6] due to steric clashes. Finally, a crystal structure of CAII with TFG 4 (lacking an ethyl linker), confirms that the butyl tail is even less solvent accessible in this complex (Figure S4).
Figure 1.

X-ray crystal structure of TFG 3 bound to CAII. (Left) Simulated annealing omit map (contoured at 3σ) shows 3 bound to Zn2+ (green sphere) in the active site of CAII. Interatomic distances (solid purple lines) are labeled in Å. Water molecules are omitted for clarity. (Right) CAII active-site channel is represented as a surface model (hydrophobes in red; hydrophilic residues in white).
The CB[6]-TFG complexes were incubated with CAII for 20 min, and the hydrolysis rates of p-nitrophenyl acetate (pNPA) enzymatically catalyzed by CAII were determined by monitoring an increase in absorbance at 400 nm (Figure 2). Compounds 3 and 4 displayed stronger CAII inhibition compared to the other TFGs, which is consistent with their higher CAII affinity. alone did not show any CAII inhibition, which suggested lack of direct interaction between CAII and CB[6]. All TFGs when complexed with CB[6] inhibited CAII activity only slightly less than TFG alone, which provides further evidence that the TFGs shuttle from CB[6] to CAII, as designed. Furthermore, titration of CB[6] led to recovery of CAII activity (Figure S5), proving that the shuttling of TFG is under thermodynamic control. This is also consistent with butylamine undergoing fast exchange (τexch < 10 ms) with CB[6].[20]
Figure 2.

Comparison of CAII esterase activity with different TFGs and CB[6]-TFG complexes monitored using pNPA as substrate. All assays were performed after 20 min incubation. Standard errors were determined from three or more replicates for each condition.
Based on its highest affinity for CAII and intermediate affinity with CB[6] (Table 1), TFG 4 was selected for testing our “turn-on” 129Xe NMR biosensing strategy. First, 1 μM CB[6] was dissolved in pH 7.2 PBS solution, and multiple selective Dsnob-shaped saturation pulses were scanned over the chemical shift range of 90–230 ppm in 5-ppm steps. Two saturation responses were observed (Figure 3), centered at 193 ppm (129Xe-aq) and 122 ppm (129Xe-CB[6]). Upon addition of 4 μM TFG 4, the 129Xe-CB[6] Hyper-CEST signal at 122 ppm was greatly reduced as a result of less free CB[6] (~ 0.5 μM) in the sample solution. Finally, when 4 μM CAII was added to the solution, 129Xe-CB[6] Hyper-CEST signal was mostly restored, confirming that TFG 4 was sequestered by CAII.
Figure 3.

Frequency-dependent 129Xe NMR saturation spectra in pH 7.2 PBS at 300 K showed CAII detection via CB[6]-4 molecular relay.
To apply the molecular relay to a system with different thermodynamic properties, we employed a commercially available pentylamine-biotin (pAB) TFG that binds CB[6] much less tightly than its target protein avidin. TFG 5 was determined by ITC (Table 1) to bind to CB[6] with modest affinity, KA = 4240 M−1, whereas avidin affinity was estimated to be much higher, KA ≈ 1014 M−1.[21] In order to block xenon access initially, we added 50 μM TFG 5 to 1 μM CB[6] solution, and as expected, the 129Xe-CB[6] Hyper-CEST signal was completely turned off. Subsequently, administration of 50 μM avidin fully recovered 129Xe-CB[6] signal (Figure 4). These data highlight the ease of reprogramming the CB[6]-TFG relay for assaying a wide range of biomolecules in solution.
Figure 4.

Frequency-dependent 129Xe NMR saturation spectra in pH 7.2 PBS at 300 K showed avidin detection via CB[6]-pAB molecular relay.
Finally, we investigated our molecular relay in cellular environments. BL21(DE3) E. coli expressing recombinant CAII was cultured in LB medium and induced with 1 mM IPTG and 1 mM ZnCl2. Cells were then pelleted, washed, resuspended in PBS buffer, and lysed with two freeze-thaw cycles. Quantitative SDS gel indicated expression level of 12 nmol CAII (3 mL of 4 μM sample solution) from cell lysate equivalent to OD600nm = 2 (Figure S6). Non-transformed E. coli were grown and lysed in the same manner to serve as a negative control. Screening experiments revealed that 16 μM CB[6] gave useful residual 129Xe Hyper-CEST NMR signal, despite CB[6] binding to many components of the bacterial lysate. Addition of 4 μM TFG 4 to control sample with 16 μM CB[6] greatly reduced the Hyper-CEST signal, whereas in lysate from CAII-overexpressing E. coli, the original NMR signal intensity was observed (Figure 5). These experiments highlight the ability of the CB[6] detection scheme to identify a specific protein target within a complex mixture. Thus, the molecular relay extends the utility of CB[6] for ultrasensitive 129Xe NMR biosensing.
Figure 5.

Frequency-dependent 129Xe NMR saturation spectra showed CAII detection via CB[6]-4 relay in bacterial lysate (OD600nm = 2) at 300 K.
In summary, by exploiting versatile CB[6] host-guest chemistry and facile attachment of a CB[6]-binding domain to a protein-binding moiety, we were able to generate novel 129Xe NMR biosensors. The TFG programs the molecular relay for exquisite control over chemical equilibria between several interacting components CB[6], TFG, target molecule and Xe. This strategy produces CB[6]-129Xe NMR signal and makes it possible to “target” CB[6] for specific biomolecule detection. In the Hyper-CEST setup, because Xe (~136 μM, all isotopes) is more abundant than other components, analyte detection sensitivity should be maximized when the TFG is engineered to achieve well discriminated binding affinities: analyte-TFG ≫ CB[6]-TFG ≫ CB[6]-Xe. The thermodynamic and kinetic stability of the starting CB[6]-TFG complex will hold additional significance in future in vivo applications for molecular imaging.
Supplementary Material
Acknowledgments
This work was supported by NIH R01-GM097478 and CDMRP-LCRP Concept Award #LC130824. The ITC was purchased via NIH S10-DO016260. We thank Drs. George Furst and Jun Gu for assistance with NMR spectroscopy. We thank Dr. David Christianson for the CAII plasmid and assistance with crystallography. We thank the Stanford Synchrotron Radiation Lightsource (SSRL) and Advanced Photon Source (APS) for access to their respective beamlines for X-ray crystallographic data collection.
References
- 1.a) Cheng CY, McGonigal PR, Schneebeli ST, Li H, Vermeulen NA, Ke CF, Stoddart JF. Nat Nanotechnol. 2015;10:547–553. doi: 10.1038/nnano.2015.96. [DOI] [PubMed] [Google Scholar]; b) Whitesides GM, Grzybowski B. Science. 2002;295:2418–2421. doi: 10.1126/science.1070821. [DOI] [PubMed] [Google Scholar]; c) Browne WR, Feringa BL. Nat Nanotechnol. 2006;1:25–35. doi: 10.1038/nnano.2006.45. [DOI] [PubMed] [Google Scholar]
- 2.a) Wang Y, Dmochowski IJ. Chem Commun. 2015;51:8982–8985. doi: 10.1039/c5cc01826a. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kunth M, Witte C, Hennig A, Schröder L. Chem Sci. 2015 doi: 10.1039/c5sc01400j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schröder L, Lowery TJ, Hilty C, Wemmer DE, Pines A. Science. 2006;314:446–449. doi: 10.1126/science.1131847. [DOI] [PubMed] [Google Scholar]
- 4.a) Demarquay J, Fraissard J. Chem Phys Lett. 1987;136:314–318. [Google Scholar]; b) Sozzani P, Bracco S, Comotti A, Mauri M, Simonutti R, Valsesia P. Chem Commun. 2006:1921–1923. doi: 10.1039/b602040b. [DOI] [PubMed] [Google Scholar]
- 5.Bai Y, Wang Y, Goulian M, Driks A, Dmochowski IJ. Chem Sci. 2014;5:3197–3203. doi: 10.1039/c4sc01190b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mugler JP, 3rd, Altes TA. J Magn Reson Imaging. 2013;37:313–331. doi: 10.1002/jmri.23844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Brotin T, Dutasta JP. Chem Rev. 2009;109:88–130. doi: 10.1021/cr0680437. [DOI] [PubMed] [Google Scholar]; b) Taratula O, Hill PA, Khan NS, Carroll PJ, Dmochowski IJ. Nat Commun. 2010;1:148. doi: 10.1038/ncomms1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taratula O, Hill PA, Bai Y, Khan NS, Dmochowski IJ. Org Lett. 2011;13:1414–1417. doi: 10.1021/ol200088f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Stevens TK, Ramirez RM, Pines A. J Am Chem Soc. 2013;135:9576–9579. doi: 10.1021/ja402885q. [DOI] [PubMed] [Google Scholar]; b) Klippel S, Freund C, Schröder L. Nano Lett. 2014;14:5721–5726. doi: 10.1021/nl502498w. [DOI] [PubMed] [Google Scholar]; c) Shapiro MG, Ramirez RM, Sperling LJ, Sun G, Sun J, Pines A, Schaffer DV, Bajaj VS. Nat Chem. 2014;6:629–634. doi: 10.1038/nchem.1934. [DOI] [PubMed] [Google Scholar]
- 10.a) Isaacs L. Chem Commun. 2009:619–629. doi: 10.1039/b814897j. [DOI] [PubMed] [Google Scholar]; b) Kim K, Selvapalam N, Ko YH, Park KM, Kim D. J Kim, Chem Soc Rev. 2007;36:267–279. doi: 10.1039/b603088m. [DOI] [PubMed] [Google Scholar]; c) Walker S, Oun R, McInnes FJ, Wheate NJ. Isr J Chem. 2011;51:616–624. [Google Scholar]; d) Hennig A, Bakirci H, Nau WM. Nat Methods. 2007;4:629–632. doi: 10.1038/nmeth1064. [DOI] [PubMed] [Google Scholar]; e) Chinai JM, Taylor AB, Ryno LM, Hargreaves ND, Morris CA, Hart PJ, Urbach AR. J Am Chem Soc. 2011;133:8810–8813. doi: 10.1021/ja201581x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Bhasikuttan AC, Mohanty J, Nau WM, Pal H. Angew Chem Int Ed. 2007;46:4120–4122. doi: 10.1002/anie.200604757. [DOI] [PubMed] [Google Scholar]; b) Berdnikova DV, Aliyeu TM, Paululat T, Fedorov YV, Fedorova OA, Ihmels H. Chem Commun. 2015;51:4906–4909. doi: 10.1039/c5cc01025j. [DOI] [PubMed] [Google Scholar]; c) Ghosh S, Isaacs L. J Am Chem Soc. 2010;132:4445–4454. doi: 10.1021/ja910915k. [DOI] [PubMed] [Google Scholar]
- 12.Kim BS, Ko YH, Kim Y, Lee HJ, Selvapalam N, Lee HC, Kim K. Chem Commun. 2008:2756–2758. doi: 10.1039/b805724a. [DOI] [PubMed] [Google Scholar]
- 13.Mock WL, Shih NY. J Org Chem. 1986;51:4440–4446. [Google Scholar]
- 14.Chambers JM, Hill PA, Aaron JA, Han ZH, Christianson DW, Kuzma NN, Dmochowski IJ. J Am Chem Soc. 2009;131:563–569. doi: 10.1021/ja806092w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Supuran CT. Nat Rev Drug Discovery. 2008;7:168–181. doi: 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- 16.Gao JM, Qiao S, Whitesides GM. J Med Chem. 1995;38:2292–2301. doi: 10.1021/jm00013a005. [DOI] [PubMed] [Google Scholar]
- 17.Salvatore RN, Nagle AS, Jung KW. J Org Chem. 2002;67:674–683. doi: 10.1021/jo010643c. [DOI] [PubMed] [Google Scholar]
- 18.a) Elbaum D, Nair SK, Patchan MW, Thompson RB, Christianson DW. J Am Chem Soc. 1996;118:8381–8387. [Google Scholar]; b) Bozdag M, Pinard M, Carta F, Masini E, Scozzafava A, McKenna R, Supuran CT. J Med Chem. 2014;57:9673–9686. doi: 10.1021/jm501497m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a) Bardelang D, Udachin KA, Leek DM, Ripmeester JA. CrystEngComm. 2007;9:973–975. [Google Scholar]; b) Lee JW, Samal S, Selvapalam N, Kim HJ, Kim K. Acc Chem Res. 2003;36:621–630. doi: 10.1021/ar020254k. [DOI] [PubMed] [Google Scholar]
- 20.Mock WL, Shih NY. J Am Chem Soc. 1989;111:2697–2699. [Google Scholar]
- 21.Green NM. Biochem J. 1963;89:585–591. doi: 10.1042/bj0890585. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.