

Abstract
The dopamine transporter (DAT) plays an important homeostatic role in the control of both the extracellular and intraneuronal concentrations of dopamine, thereby providing effective control over activity of dopaminergic transmission. Since brain dopamine is known to be involved in numerous neuropsychiatric disorders, investigations using mice with genetically altered DAT function and thus intensity of dopamine-mediated signaling have provided numerous insights into the pathology of these disorders and highlight novel pathological mechanisms that could be targeted to provide new therapeutic approaches for these disorders. In this brief overview we discuss recent investigations involving animals with genetically altered DAT function, particularly focusing on translational studies providing new insights into pathology and pharmacology of dopamine-related disorders. Perspective applications of these and newly developed models of DAT dysfunction are also discussed.
Keywords: Dopamine transporter (DAT), Knockout, Schizophrenia, Parkinson’s disease, Addiction, Attention Deficit Hyperactivity Disorder (ADHD)
The dopamine (DA) system plays an important role in numerous physiological, behavioral and endocrine functions (Carlsson, 1987; Gainetdinov, 2008; Greengard, 2001). Dopamine system abnormalities are involved in a number of pathological conditions, including Parkinson’s disease (PD), attention deficit hyperactivity disorder (ADHD), depression, addiction, schizophrenia and Tourette syndrome (Miller et al., 1999; Wilson et al., 1996). DA neurons selectively express DAT, a member of a family of Na+/Cl− - dependent neurotransmitter transporters (Reith et al., 1997). Termination of extracellular DA action is primarily achieved by its rapid DAT-mediated re-uptake from the synaptic cleft into presynaptic terminals making DAT an important determinant of functionality of the dopamine system (Amara & Kuhar, 1993; Giros et al., 1996). In the central nervous system DAT is mainly expressed within nigrostriatal and mesolimbic dopaminergic neurons with the highest level detected in the striatum and nucleus accumbens. In the periphery it is present in retina, lung, kidney and pancreas (Ciliax et al., 1995). At the cellular level DAT is mostly localized perisynaptically on the presynaptic membrane (Nirenberg et al., 1997; Sesack et al., 1998).
Several transgenic animal models with altered DAT expression were created including strains with deficient DAT function, knock-in of modified DAT gene and over-expressing DAT gene (Gainetdinov, 2008; Giros et al., 1996). Studies on these strains have provided important information not only on DAT functioning, but also on the mechanisms of action of psychotropic drugs, interaction between neurotransmitter systems, and pathological mechanisms involved in brain disorders (Gainetdinov & Caron, 2003; Gainetdinov, 2008).
DAT knockout (DAT-KO) mice
General phenotype of DAT-KO mice
Strains with a disrupted DAT gene were created using in vivo homologous recombination (Giros et al., 1996; Sora et al., 1998). Despite lacking a major protein of the DA system, mice appeared to be viable, yet they showed pronounced alterations in neurochemistry, phenotype and behavior. Most importantly, mice without DAT were viable (Giros et al., 1996; Sora et al., 1998), however only about 70% of animals survied after 10 weeks (Giros et al., 1996). Mice are fertile and mate, but female knockout mice showed inability to lactate and impaired maternal behavior and therefore could not take care of the litter (Bossé et al., 1997; Giros et al., 1996). DAT-KO mice showed normal development in general, yet they gained weight more slowly compared to wild type animals and their weight remained lower by 30% in adults (Bossé et al., 1997). Importantly, a significant number of DAT-KO mice (30–40%) sporadically develop a DA-dependent progressive locomotor disorder characterized by the loss of hyperactivity, occurrence of dyskenetic movements, paralysis and eventual death several days after appearance of this phenotype. Morphological investigation of the nigrostriatal system of the affected population showed reduction in populations of dopamine-receptive medium spiny GABA neurons (Cyr et al., 2003). Degenerating spiny neurons were TUNEL positive and contained a detectable level of caspase 3 indicating activation of apoptotic pathways. The same neurons also showed accumulation of the hyperphosphorylated microtubule associated protein tau that has been reported to be associated with Alzheimer’s disease and also other degenerative diseases (Lee, 2001). It has been demonstrated that this striatal neurodegeneration is caused by overactivity of D1 dopamine receptor–mediated signaling as evidenced by up-regulation of DeltaFosB, cyclin-dependent kinase 5, and p35 (Cyr et al., 2003) and this phenotype can be potentiated if DAT-KO mice are crossed with a mouse model of Huntington’s disease (Cyr et al., 2005).
As for physiological functions, DAT-KO mice have significantly slower breathing rate with prolonged inspiration time. The reaction to the hypoxia condition is reduced in DAT-KO mice, compared to wild-type (WT) mice while CO2 production does not differ in mutants (Vincent et al., 2007). Also, an alteration in body temperature of DAT-KO mice was found. Circadian measurement showed that they have lower body temperature during resting time (daylight time) (Vincent et al., 2007). The involvement of DAT in thermoregulation is also supported by studies of methylone-induced lethality that is thought to be related to body hyperthermia. In this experimental paradigm, DAT-KO mice showed reduced lethality compared to WT and serotonin transporter (SERT) KO mice (Piao et al., 2015).
Changes in DA system in DAT-KO mice
Lack of dopamine transporter in DAT-KO animals leads to greatly reduced clearance of DA from the synaptic cleft (Giros et al., 1996). Indeed, basal extracellular dopamine level measured by microdialysis is five-fold higher in KO mice compared to wild-type animals (Jones et al., 1998a). Unlike WT animals, DAT-KO mice lack diurnal oscillation of extracellular DA levels (Ferris et al., 2014). Clearance of extracellular DA measured by cyclic voltammetry after single pulse stimulation was up to 300 times slower in knockout mice (Giros et al., 1996). The extracellular level of dopamine was still depolarization-dependent vesicular exocytosis – infusion of tetrodotoxin or Ca2+ media in in vivo microdialysis measurements gradually decreased the level of DA to undetectable levels (Gainetdinov et al., 1999a). DA clearance time remained unaltered by application of inhibitors of other monoamine transporters – SERT and norepinephrine transporter (Budygin et al., 2002; Jones et al., 1998a). When measured by carbon fiber amperometry with electrical stimulation of medial forebrain bundle, catechol-o-methyl transferase inhibitor had no effect on DA clearance, however the monoamine oxidase inhibitor pargyline slightly increased the lifetime of DA, showing that monoamine oxidase is somehow involved in DA clearance from synaptic cleft (Benoit-Marand et al., 2000).
Despite persistent elevated levels of extracellular DA it was found that the amount of dopamine released in response to a single pulse stimulation was dramatically reduced – up to 75% less when measured by fast-scan voltammetry and up to 93% if measured by amperometry (Benoit-Marand et al., 2000; Jones et al., 1998b). Total content of DA that represents storage vesicular pool of DA in brain tissue is reduced up to 20-fold in comparison to wild type animals (Giros et al., 1996). The level of DA metabolite 3,4-dihydroxyphenylacetic acid (DOPAC) is unaltered and the level of homovanillic acid (HVA) is elevated (Jones et al., 1998b). As there is no reuptake of DA from the synaptic cleft, all of the present DA should be newly synthesized by tyrosine hydroxylase (TH) - the key enzyme of DA synthesis. Indeed, striatal DA in DAT-KO mice is extremely sensitive to inhibition of TH – when an inhibitor of TH alpha-methyl-para-tyrosine is injected, dopamine decreases to undetectable levels (Jones et al., 1998b). Such an approach provided an easy way to develop an acute model of absolute DA deficiency that is used as a Parkinson’s model to study effects of antiparkinsonian drugs (Sotnikova et al., 2005).
Striatal level of TH is decreased, yet the amount of TH-mRNA per neuron is not altered in mutants (Jaber et al., 1999; Jones et al., 1998b). The number of TH-positive neurons is just slightly lower. Levels of another DA synthesis enzyme aromatic L-amino acid decarboxylase(L-AADC) is not changed, however functional activity of TH in striatum as measured by L-DOPA accumulation is increased in DAT-KO mice (Jones et al., 1998b). Also, the level of VMAT2, a vesicular transporter that moves DA to vesicles, is just slightly lower (Gainetdinov et al., 1998; Jaber et al., 1999), however, the level of VMAT2 mRNA is unaltered (Jaber et al., 1999).
Major postsynaptic DA receptors, D1 and D2, are downregulated by approximately 50% in the striatum (Giros et al., 1996). In quantitative in situ hybridization studies, the level of mRNA of D1 and D2 was decreased. Phenotypical organization of D1 and D2 expressing neurons in striatum in KO mice is similar to WT mice but expression level of D3 receptors is increased as well as the density of D3 expressing neurons (Fauchey et al., 2000).
Thus, DAT-KO animals show major changes in DA system, including altered synthesis, total and extracellular levels of DA and metabolites and DA receptors density. As DA system plays an important role in multiple brain functions, lack of DAT causes significant alterations in behavioral phenotype of animals and thus DAT-KO mice are considered as a valuable animal model of different neuropathologies related to DA system.
DAT-KO mice as ADHD model
DAT-KO mice have a very pronounced behavioral phenotype. As a result of permanent elevation of extracellular DA, DAT-KO mice show hyperlocomotion, disrupted sensorimotor gating, increased impulsivity and impaired ability to perform learning tasks. When exposed to a novel environment, DAT-KO mice show highly elevated spontaneous locomotion (Giros et al., 1996; Sora et al., 1998). No habituation to a novel enviroment occurs for over 240 min, whereas for WT mice active exploratory time is about 30–40 min. Under the condition of repetitive exposure to an open field, KO animals also show no habituation (Gainetdinov et al., 1999a). Antagonists of both D1 and D2 dopamine receptors are able to block this hyperactivity (Gainetdinov et al., 1999a; Ralph et al., 2001).
Several lines of evidence suggest that DAT dysfunction may contribute to the pathogenesis of Attention Deficit Hyperactivity Disorder (ADHD). Multiple genetic studies in patients have demonstrated association of DAT gene variants with ADHD (Faraone et al., 2014). Apart from that, it is known that in patients with ADHD psychostimulants interacting with the DAT, such as amphetamine and methylphenidate, can improve behavioral deficits. Hyperactivity and cognitive deficits found in DAT-KO mice suggest that these mice can be a valuable model for ADHD. Furthermore, the antihyperkinetic effect of amphetamine (AMPH) and methylphenidate (MPH) which was observed in DAT-KO mice further supporting face validity and predictive validity of DAT-KO mice as a model of ADHD (Gainetdinov et al., 1999b; Leo & Gainetdinov, 2013).
MPH is widely used in the treatment of ADHD in patients. In DAT-KO mice it showed its ability to reduce spontaneous hyperactivity (Gainetdinov et al., 1999b) and increase the extracellular level of DA in the prefrontal cortex, but not in the striatum (Takamatsu et al., 2015). Rewarding properties of MPH are also preserved in DAT-KO mice in conditional place preference test (Sora et al., 1998).
Symptoms of ADHD do not include only hyperactivity, but also learning deficits and increased impulsivity. Increased impulsivity of mutants was shown in a cliff avoidance test (Yamashita et al., 2013). When tested in an 8-arm labyrinth, DAT-KO mice showed a notable deficit in spatial learning making more preservative errors when performing the test (Dzirasa et al., 2009; Gainetdinov et al., 1999a). Also, in contrast to hyperactivity in the open field, during learning DAT-KO animals were less active compared to WT animals, yet were involved in other extraneous activities (sniffing, rearing, grooming, etc.) (Gainetdinov et al., 1999b). In another learning task, Morris water maze cued version, DAT-KO animals also were unable to performed the task successfully (Morice et al., 2007). In the water maze, following haloperidol administration, DAT-KOs became as successful as WT mice in finding the platform, and their performance markedly improved with training (Morice et al., 2007). Finally, DAT-KO mice showed impairment of avoidance behavior in shuttle box, which could be reversed by methylphenidate (Takamatsu et al., 2015).
Apart from altered learning abilities, significant changes in postsynaptic plasticity do occur reflecting altered dopamine-glutamate interplay in mutants. Thus, DAT-KO mice exhibit enhanced long-term potentiation (LTP) of the frontocortico-accumbal glutamatergic synapses due to PSD-95 reduction (Yao et al., 2004). Intriguingly, similar reduction of PSD-95 was observed in chronically cocaine-sensitized mice indicating an important role of PSD-95 in psychostimulant action (Yao et al., 2004) and D1 dopamine receptor function (Zhang et al., 2007, 2009). Such modulatory influence of PSD-95 may provide a common molecular and cellular mechanism shared between drug-related plasticity and learning (Mohn et al., 2004) that might be involved also in striatal neurodegeration (Zhang et al., 2014).
Hyperdopaminergic DAT-KO mice were also instrumental to uncover molecular mechanisms of dopamine-dependent plasticity in the frontal cortical neurons. It has been observed that in DAT-KO mice LTP in layer V pyramidal neurons cannot be established in the PFC unless D2 dopamine receptors were blocked. Furthermore, suppression of postsynaptic protein phosphatase 1 (PP1) also restored LTP indicating that coupling of D2 dopamine receptors to the PP1 is critical for this deficit (Xu et al., 2009). Intriguingly, amphetamine treatment restored the loss of LTP in DAT-KO mice primarily as a result of activation of the noradrenergic system by this drug (Xu et al., 2010)
At the same time an impaired long-term depression (LTD) in the hippocampal areas that is correlated with cognitive inflexibility was observed in mutants (Morice et al., 2007). In general, glutamate neurotransmission in hippocampal neurons was not remarkably changed in mutants, as evidenced by an analysis of N-methyl-d-aspartate (NMDA) and non-NMDA receptor-mediated synaptic currents (Morice et al., 2007). D2 receptor antagonist, haloperidol, but not methylphenidate, is able to facilitate LTD in DAT-KO mice, bringing it to levels observed in WT mice (Morice et al., 2007). It was also found that blockade of NMDA receptors by MK-801 remarkably enhances hyperactivity of DAT-KO mice (Gainetdinov et al., 2001a). Enhancement of glutamatergic transmission through positive modulation of AMPA receptors by AMPAkines or increase of glycine concentration leads to inhibition of hyperactivity (Gainetdinov et al., 2001a). Inhibition of NMDA neurotransmission prevents effects of psychostimulants and 5-HT-based drugs suggesting that inhibitory effect of 5-HT on hyperactivity might be mediated via modulation of glutamate neurotransmission (Gainetdinov et al., 2001a).
Altogether, DAT-KO mice proved to be a good model for ADHD studies, showing hyperactivity, deficit in learning abilities and positive effect of MPH, a major drug for ADHD treatment.
Action of drugs of abuse on DAT-KO mice
This strain of mice also became very useful in determining the involvement of the dopaminergic system in action mechanisms of different drugs of abuse, including psychostimulants and alcohol. It is now well established that DAT is a principal target for many psychostimulants such as cocaine, amphetamine, methamphetamine, methylphenidate, etc. (Fumagalli et al., 1998; Gainetdinov et al., 2002; Gainetdinov et al., 1999b; Jones et al., 1998a; Rocha et al., 1998). It is known that cocaine acts as an inhibitor of DAT, increasing the extracellular level of DA (Giros et al., 1996). Indeed, in DAT-KO mice cocaine does not change clearance rate of DA from the synaptic cleft as measured by fast-scan cyclic voltammetry in striatal slices (Budygin et al., 2002; Giros et al., 1996; Jones et al., 1998b).
Similarly, extracellular levels of DA in the striatum and nucleus accumbens measured by microdialysis following cocaine remain unaffected in DAT-KO mice (Rocha et al., 1998; Shen et al., 2004). Cocaine-induced hyperlocomotion is eliminated in DAT-KO mice; instead, like amphetamine, it reduces locomotor hyperactivity (Gainetdinov et al., 1999b; Giros et al., 1996), bringing evidence that DA plays a major role in cocaine-induced hyperactivity. However, it seems that DAT is not the only target for cocaine action. In DAT-KO mice cocaine still possesses rewarding properties that can be seen in a conditional place preference test (Sora et al., 1998). Also, DAT-KO mice are able to develop long-term expression of sensitization to cocaine (Morice et al., 2009). Importantly, when self-administration of cocaine was tested, DAT-KO mice showed the ability to perform the task (Rocha et al., 1998), albeit much reduced, whereas performance to food reinforcement remained normal (Thomsen et al., 2009a).The mechanism of action of AMPH still remains a question of investigation. It is well established that AMPH enters presynaptic neurons through the DAT, however, it also can enter into the neuron via diffusion (Seiden et al., 1993). Inside the terminal it can also interact with the vesicular transporter resulting in massive efflux of DA into the extracellular space (Sulzer et al., 1995). In DAT-KO mice injection of AMPH has no influence on the extracellular level of DA in striatum, yet AMPH retains its intraneuronal actions (Jones et al., 1998b). However, in the nucleus accumbens, AMPH still increases extracellular levels of DA, likely via heteroreceptor mechanisms (Budygin et al., 2004). Importantly, despite the lack of DAT, AMPH retains its rewarding properties in mutant mice (Budygin et al., 2004).
AMPH has a pronounced effect on the behavior of DAT-KO mice, eliminating their pronounced hyperactivity. It was proposed that AMPH, as well as other psychostimulants, can act by modulating the DA system via indirect action of the serotonin (5-HT) system. Indeed, several serotonergic drugs including SERT inhibitor fluoxetine, 5-HT receptor agonist quipazine and 5-HT precursors tryptophan and 5-hydroxytryptophan can influence behavior of DAT-KO mice by decreasing locomotor activity (Gainetdinov et al., 1999b). Recent findings further supported the idea that 5-HT receptors play an important role in modulation of locomotor activity, in DAT-KO animals either acute chemical or permanent genetic reduction of 5-HT1B receptor leads to lower locomotor activity in DAT-KO mice (Hall et al., 2014a). Reduction of 5-HT1B receptor function in absence of DAT leads to restoration of cocaine-induced locomotor activation. Interestingly, an antagonist of 5-HT2A receptor (M100907) also reversed the behavioral deficit of DAT-KO mice such as hyperlocomotor activity and pre-pulse inhibition deficit (Barr et al., 2004).
DAT-KO mice have also provided an opportunity to directly evaluate whether alcohol is altering DA dynamics directly via the DAT or by another mechanism. In fact, there was much debate on the acute effects of alcohol on striatal DA uptake. Acute ethanol administration has been reported to increase (Sabeti et al., 2003), decrease (Robinson et al., 2005), or to have no effect on DA uptake (Budygin et al., 2001a, 2001b; Jones et al., 2006). This is an important question, since revealing how alcohol modulates striatal DA transmission aids in understanding alcohol-mediated reinforcement and, ultimately, alcoholism. The acute effects of ethanol on DA release and uptake in the caudate-putamen of mice with and without the DAT were evaluated using microdialysis and voltammetry (Mathews et al., 2006). The fact that alcohol had identical effects on striatal DA in WT and DAT-KO mice proved the idea that ethanol does not directly interact with the DAT to produce its effects on DA transmission.
DA plays an important role in the development of addiction (Franken et al., 2005; Volkow et al., 2004) and many studies have been devoted to understand its role in the reward circuitry associated with mesolimbic and mesocortical pathways (Bonci et al., 2003; Laakso et al., 2002; Robbins & Everitt, 2002). The mesocortical and mesolimbic projections are part of the brain “reward” circuitry and are a target of psychostimulant drugs of abuse (Nestler, 2005). Functional studies of reward circuitry showed that in DAT-KO animals the connectivity of prefrontal- ventral striatopallidal part of this circuitry remains unaltered, whereas they have diminished robustness in reward circuitry distal to the thalamus (Zhang et al., 2010). DAT-KO mice with a two-bottle choice had stronger preference toward sucrose solution, than WT mice (Costa et al., 2007). Furthermore, DAT-KO animals have an altered licking pattern – they had slower licking frequency and longer licking durations when drinking sucrose solution and were much more sensitive to feedback function, with no difference in licking pattern with water solution (Rossi & Yin, 2015). For aversive stimuli (quinine water) there was decreased licking rate for WT, but not for KO animals (Costa et al., 2007). It is suggested that DA plays an important role in appetitive and consummatory behavior (Salamone & Correa, 2012; Szczypka et al., 2001).
DA system is critically involved in the action of psychostimulants and other drugs of abuse. Hyperdopaminergic DAT-KO mice proved to be a valuable model for determining mechanism of action of several abused substances and deciphering the particularities of the involvement of DA in rewarding properties of them.
DAT-KO mice as a model for other psychiatric disorders
DAT-KO animals show some behavior considered to be schizophrenia–related such as deficit of prepulse inhibition (PPI) (Ralph et al., 2001). DA hypothesis of schizophrenia is one of the most established theories of schizophrenia to date. The involvement of DA is supported by the clinical observations of amphetamine-induced psychosis and effectiveness of D2 dopamine receptor-mediated treatment (Seeman, 1987; Snyder, 1973). D2 dopamine receptor antagonist antipsychotics are successfully used to treat positive symptoms of schizophrenia (Lewis & Lieberman, 2000; Ross et al., 2006). Another theory suggests that hypofunction of glutamate NMDA receptors is an underlying cause of schizophrenia (Coyle, 2006; Harrison & Weinberger, 2005; Mohn et al., 1999). DAT-KO mice, much like amphetamine-treated animals, show behaviors that mimic positive symptoms of schizophrenia making it an interesting model to study schizophrenia (Gainetdinov et al., 2001b; Ralph et al., 2001). DAT-KO mice were instrumental in uncovering a novel model of D2 dopamine receptor signaling relevant to schizophrenia and bipolar disorder (Beaulieu et al., 2004, 2009). By analyzing effects of the antimaniac drug lithium in DAT-KO mice it has been demonstrated that alongside the classical PKA-cAMP-DARPP32 signaling cascade, D2 DA receptors can engage in arrestin-mediated AKT-GSK3 signaling with significant behavioral consequences (Beaulieu et al., 2005). Furthermore, the antimaniac drug lithium and antipsychotics seem to have a potent effect on this pathway known to be dysregulated in schizophrenia and bipolar disorder (Beaulieu & Gainetdinov, 2011; Beaulieu et al., 2008).
Neurosteroids, that are used as an alternative treatment for schizophrenia patients were tested on DAT-KO mice. Pregnenolone showed its effectiveness to rescue schizophrenia-like behavior of mutants (Wong et al., 2012). Its soluble form, pregnenolone sulfate, also known as a positive allosteric modulator of NMDA receptors, was also able to rescue PPI, cognitive deficit and normalize hyperlocomotion (Wong et al., 2015).
One of the morphological abnormalities shown in postmortem studies of schizophrenia patients’ brains are changes of dendritic spines of pyramidal neurons (Rajkowska et al., 1998). Intriguingly, DAT-KO mice have decreased density of dendritic spines of pyramidal neurons in the medial prefrontal cortex, yet no changes in motor cortex or amygdala were observed. That may suggest that abnormalities in DA transmission might be involved in dendritic changes occurring in schizophrenia (Kasahara et al., 2015). Furthermore, DAT-KOs have slightly altered communication between hemispheres (Zhang et al., 2010), as well as decreased volume of anterior striatum, where the number of neurons was found to be less (Cyr et al., 2005). In the striatal medium spiny neurons of DAT-KO mice a loss of dendritic spines on the proximal portion of dendrite was also reported (Berlanga et al., 2011).
Altogether, DAT-KO mice can be used to understand dopamine-related pathologies such as ADHD, addiction, schizophrenia and bipolar disorder. At present, DAT-KO mice are one of the best available models of ADHD, yet cognitive aspects are not well investigated in these mice due to technical difficulties in assessing complex cognitive function in hyperactive mice. DAT-KO mice recapitulate only a part of endophenotypes relevant to positive symptoms of schizophrenia and bipolar disorder. Nevertheless, studies in DAT-KO mice brought important insights on the pathogenesis of schizophrenia and bipolar disorder by answering a number of questions regarding circuitry involved, uncovering mechanisms of drug action and finding new targets for treatment of these disorders.
Other DAT mutant mice
Apart from full knockout mice, several models with partially reduced level of DAT have been created.
DAT-heterozygous mice
In these animals, the level of DAT is reduced two-fold leading to proportionally increased extracellular DA tone (Giros et al., 1996; Jones et al., 1998a). However, despite elevated dopamine, no change in locomotion was observed in heterozygous animals (Giros et al., 1996). DAT heterozygous animals are much less studied in comparison to KO mice, yet they can also bring important information about the role and function of the DA system. Locomotor activity of DAT-het mice does not differ from WT in youth, yet age-related changes progress differently in these two genotypes. Effect of aging on habituated locomotor activity and injection-stimulated locomotor activity is diminished in heterozygous mice, as well as the effect of aging on cocaine-induced locomotion (Hall et al., 2014b). Also it was found that DAT-het mice were protected from age-related decline of the DA system (Dluzen et al., 2010).
In DAT heterozygous mice, sex of the animal influenced the level of striatal dopamine: in females DA tissue content level was significantly lower compared to WT, whereas in males no difference was found (Dluzen & McDermott, 2008).
DAT-knock-down (DAT-KD) mice
Another model of DAT dysfunction is the DAT knockdown (DAT-KD) mouse which has reduced DAT expression down to 10% of normal level (Zhuang et al., 2001). In these mice, DA clearance rate is significantly lower, resulting in 2-fold higher extracellular level of DA (Cagniard et al., 2014; Ralph-Williams et al., 2003). AMPH increases DA level, just to a much lower extent compared to WT. Cocaine also increased the extracellular level of DA, but with earlier onset of action (Cagniard et al., 2014). DAT-KDs have higher food and water intake compared to WTs (Pecina et al., 2003) and unlike DAT-KO mice are normal size, have normal lactation and do not show increased lethality in population (Zhuang et al., 2001). In a novel environment DAT-KD animals display hyperlocomotion due to impaired response habituation that could be inhibited by AMPH (Zhuang et al., 2001). DAT-KD mice have increased stereotypy and an instinctive behavioral sequence, such as grooming chain patterns, that are stronger and more resistant to interruption (Berridge et al., 2005). PPI in DAT-KOs is not changed (Ralph-Williams et al., 2003). DAT-KD mice show changes in temporally controlled responses showing earlier reaction in two peak procedures, that can be normalized by the D2 antagonist raclopride, supporting the idea that the DA system is involved in internal timing (Balci et al., 2010). Elevated DA enhances motivation to work for food reward, but does not improve learning abilities (Cagniard et al., 2006a). Also, studies suggest that elevated DA tone reduces selectivity of stimulus control over conditional behavior and does not affect instrumental learning (Yin et al., 2006). In case of change of task DAT-KD animals perform as well as WT animals, showing no learning deficit (Beeler et al., 2010). Alongside with hyperlocomotion, DAT-KD animals show increased preservative behaviors that can be attenuated by the effective antimania drug valproate (Ralph-Williams et al., 2003; van Enkhuizen et al., 2013), making DAT-KD mice a good model for manic behavior of bipolar disorder (BD) patients. Indeed, some genetic studies showed relevance of DAT variation with bipolar disorders (Greenwood et al., 2001, 2006; Kelsoe et al., 1996) with lower DAT binding in BD patients and reduced DAT expression on the cell surface (Amsterdam & Newberg, 2007; Horschitz et al., 2005). BD patients are known to exhibit increased risk taking behavior and DAT-KD mice also demonstrate riskier patterns of behavior (Young et al., 2011). Also mania-like behavior disappears in DAT-KD mice in a familiar environment and the DAT inhibitor GRB 12909 is able to reinstate mania-like behavior (Young et al., 2010). Alpha-methyl-p-tyrosine (AMPT), a competitive inhibitor of TH, also attenuates some mania like behavior such as activity level and movement organization(van Enkhuizen et al., 2014), however these effects could be also be relevant to other catecholamine systems, as AMPT lacks specificity for the DA system. Yet this model of BD has its limitations, as, apart from hyperactivity, valproate does not affect other aspects of mania-like behavior: increased specific exploration and more linear movement (van Enkhuizen et al., 2013).
DAT – Cocaine Insensitive (DAT-CI) mice
To determine the effect of cocaine’s action, a knock-in mouse strain with change in the DAT-cocaine binding site was created. As a result of knock-in mutated DAT mice not only is there a 68-fold less sensitivity to cocaine but also a 50% reduction of DAT activity leading to spontaneous hyperactivity, thereby creating an interesting model to study cocaine action and drug addiction. By using this model it has been demonstrated that cocaine-induced increase of dendritic spine density requires DAT inhibition (Martin et al., 2011). In these mice, injection of cocaine suppressed locomotor activity did not change DA level in nucleus accumbens and had no rewarding properties in conditional place preference, indicating that DAT blockade is necessary for cocaine’s rewarding action (Chen et al., 2006; Tilley et al., 2007, 2009). However, DAT-CI mice were able to perform tasks for a food reward and would self-administer AMPH and the direct DA agonist SKF 82958 (Thomsen et al., 2009b). This mutation in DAT also leads to 47-fold reduced MPH inhibition suggesting that it has the same site of action as cocaine. At the same time, AMPH and MPH retained their action (Chen et al., 2005). Injection of AMPH reduced locomotor activity potentially via selective aberrant phasic activation of D1R/сAMP/PKA/DARPP32 signaling in response to increased striatal extracellular DA level (Napolitano et al., 2010). The behavioral effect of amphetamine was not dependent on genetic background and retained in mice after backcrossing (O’Neill & Gu, 2013). At the same time, cocaine’s effect is dependent of genetic background: C57Bl/6J strain does not show preference or aversion to cocaine, whereas 129S1/SvImJ × C57Bl/6J (129B6) background has strong conditional place aversion to cocaine (O’Neill et al., 2013). Viral-mediated expression of DAT wild type in DAT-KD mice in rostrolateral striatum results in restoration of cocaine-induced hyperactivity and rewarding properties. Restoration of DAT in DAT-KD mice in the nucleus accumbens and dorsal striatum did not affect cocaine action (O’Neill et al., 2014).
Inducible DAT-knockdown (iDAT-KD) mice
A line with tetracycline inducible DAT knockdown was created (Cagniard et al., 2006b). Inhibition of DAT expression leads to increase of extracellular DA clearance time in nucleus accumbens. DAT suppression leads to chronically elevated DA tone without changes in phasic DA activity (Cagniard et al., 2006b). DAT knockdown leads to increased motivation for preferable reward, but not enhanced feeding, in general, and influenced task performance only under the condition of pronounced motivation drive (Cagniard et al., 2006b).
Another way to create inducible DAT depletion (35–40% lower) is to use non-viral short interfering RNA (si-RNA). Infusion of DAT-siRNA in adult mice leads to increase in locomotion (Hoyer et al., 2006; Salahpour et al., 2007; Thakker et al., 2004) that could be decreased by injection of MPH (Chen & Lai, 2010). DAT-siRNA does not affect anxiety of animals and PPI, however these animals show stress-induced immobility in a tail-suspension test indicating increased depressive-like behavior (Chen & Lai, 2010). In a learning task (Morris water maze) DAT-siRNA treated animals show no difference from control animals (Chen & Lai, 2010).
DAT-overexpressing (DAT-OE) mice
Mice carrying additional copies of the DAT gene were created by the BAC transgenic approach (BAC DAT-tg) which creates a several-fold increase in the level of DAT, and, as a result, a reduced extracellular DA level. Accordingly, BAC DAT-tg mice have an increased level of D1 and D2 dopamine receptors (Ghisi et al., 2009). Locomotor activity in BAC DAT-tg mice does not differ from WT mice and also effects of DAT blockade by cocaine, MPH or GBR12909 were not different from WT. AMPH had, however, a markedly increased locomotor response in mutants compared to WT mice. Amount of DA released by AMPH injection also increased by 3-fold. BAC DAT-tg mice display impaired operant responding to natural reward and appear to be much more sensitive to amphetamine, displaying place preference with much lower doses of AMPH. These results indicate that up-regulation of DAT leads to increase of sensitivity to psychomotor and rewarding properties of AMPH (Salahpour et al., 2008). Another application of BAC-Tg approach resulted in the creation of mouse line [Dat1-enhanced green fluorescent protein (eGFP)] that expresses eGFP under control of the dopamine transporter (DAT) promoter. These mice provided new valuable tool for identification and purification of midbrain dopaminergic neurons (Apuschkin et al., 2015).
Other DAT knock-in (DAT-KI) mice
DAT-KO mice are proven to be a valuable model to understand ADHD and other psychiatric disorders, jet one of disadvantages of this model is that it does not replicate directly known genetic alterations. One of the variations of DAT gene related to bipolar affective disorder and ADHD is Val559 mutation (Grünhage et al., 2000; Mergy et al., 2014a). In vitro studies on transfected cells with Val559 variant showed normal total and surface DAT protein expression, and normal DA uptake rates (Mazei-Robison & Blakely, 2005). Further studies revealed that these cells however have abnormal outward “leak” of cytoplasmic DA and AMPH application on transfected cells did not change efflux of DA and its effect was similar to cocaine action(Mazei-Robison et al., 2008). DAT-KI mouse line with Val559 mutation in DAT gene was created (Mergy et al., 2014b). These mice have normal levels of DA and its metabolites in the brain tissue. At the same time they have elevated level of extracellular DA compared to wild type animals (Mergy et al., 2014a). No changes in locomotion was observed in Val559 DAT-KI mice, yet pronounced decrease in vertical activity was found (Mergy et al., 2014a). Injection of AMPH to DAT-KI animals had much less behavioral impact compared to wild type animals (Mergy et al., 2014a).
Recently, two different dopamine transporter knock-in mice with disrupted PDZ-binding motifs (DAT-AAA and DAT+Ala) were created (Rickhag et al., 2013). These mutants are characterized by dramatic loss of dopamine transporter expression in the striatum, spontaneous hyperlocomotion and attenuated response to amphetamine thereby validating a critical role of PDZ-domain interactions for synaptic distribution of DAT (Rickhag et al., 2013).
Conclusions
Mice with dysregulated DA transmission due to genetic alterations of DAT function have provided numerous advances in understanding the pathology and pharmacology of dopamine-related brain disorders, such as schizophrenia, ADHD, addiction, Parkinson’s disease and bipolar disorder. The use of these animals has significantly advanced our knowledge of the mechanism of action of psychostimulants, antipsychotics, antimaniac and antiparkinsonian drugs. Diversity of DAT models, from full DAT knockouts to DAT overexpressing and knock-in mice show an importance of DAT in normal brain functioning and its involvement in a number of pathological processes.
While there is little doubt that these models will continue to provide new exciting advances in numerous fields, other models are being created. In particular, recent progress in the development of gene editing approaches such as TALEN or CRISPR-CAS nuclease technologies made it possible to perform targeted genetic manipulations in rats, a preferred species for behavioral neuroscience. DAT mutant rats may become a model of a new generation to focus on cognitive aspects of DA-related abnormalities, a topic. Furthermore, these rats could provide an opportunity to investigate the role of small nuclei and brain areas involved in dopaminergic dysfunction. Further studies in DAT mutant rats and already existing and emerging mouse lines will be instrumental for future developments in this rapidly growing field.
Figure 1.
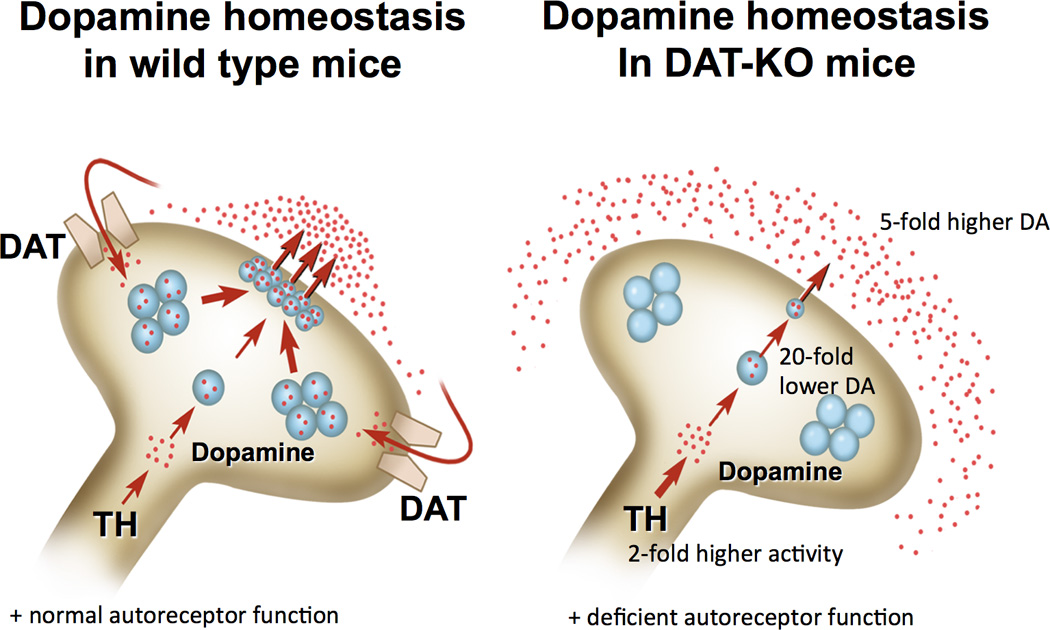
Schematic diagram representing the status of presynaptic dopamine homeostasis in striatal terminals of normal mice and mice lacking the dopamine transporter (DAT). In DAT knockout (DAT-KO) mice synthesis of dopamine (DA) is doubled despite dramatic reduction of tyrosine hydroxylase levels, intarneuronal DA is dramatically (20-fold) decreased, whereas extracellular DA is 5-fold increased. In part due to excessive dopaminergic tone, an autoreceptor function is almost absent in the striatal neurons of DAT-KO mice.
Acknowledgments
Funded by NIH grant AA022449 (EAB) and by the Russian Science Foundation grant 14-15-00131 (TDS, EVE and RRG).
References
- Amara SG, Kuhar MJ. Neurotransmitter transporters: recent progress. Annual Review of Neuroscience. 1993;16:73–93. doi: 10.1146/annurev.ne.16.030193.000445. [DOI] [PubMed] [Google Scholar]
- Amsterdam JD, Newberg AB. A preliminary study of dopamine transporter binding in bipolar and unipolar depressed patients and healthy controls. Neuropsychobiology. 2007;55(3–4):167–170. doi: 10.1159/000106476. [DOI] [PubMed] [Google Scholar]
- Apuschkin M, Stilling S, Rahbek-Clemmensen T, Sørensen G, Fortin G, Herborg Hansen F, Rickhag M. A novel dopamine transporter transgenic mouse line for identification and purification of midbrain dopaminergic neurons reveals midbrain heterogeneity. The European Journal of Neuroscience. 2015;42(7):2438–2454. doi: 10.1111/ejn.13046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balci F, Ludvig EA, Abner R, Zhuang X, Poon P, Brunner D. Motivational effects on interval timing in dopamine transporter (DAT) knockdown mice. Brain Research. 2010;1325:89–99. doi: 10.1016/j.brainres.2010.02.034. [DOI] [PubMed] [Google Scholar]
- Barr AM, Lehmann-Masten V, Paulus M, Gainetdinov RR, Caron MG, Geyer MA. The selective serotonin-2A receptor antagonist M100907 reverses behavioral deficits in dopamine transporter knockout mice. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology. 2004;29(2):221–228. doi: 10.1038/sj.npp.1300343. [DOI] [PubMed] [Google Scholar]
- Beaulieu J-M, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacological Reviews. 2011;63(1):182–217. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- Beaulieu J-M, Gainetdinov RR, Caron MG. Akt/GSK3 signaling in the action of psychotropic drugs. Annual Review of Pharmacology and Toxicology. 2009;49:327–347. doi: 10.1146/annurev.pharmtox.011008.145634. [DOI] [PubMed] [Google Scholar]
- Beaulieu J-M, Marion S, Rodriguiz RM, Medvedev IO, Sotnikova TD, Ghisi V, Caron MG. A beta-arrestin 2 signaling complex mediates lithium action on behavior. Cell. 2008;132(1):125–136. doi: 10.1016/j.cell.2007.11.041. [DOI] [PubMed] [Google Scholar]
- Beaulieu J-M, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell. 2005;122(2):261–273. doi: 10.1016/j.cell.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Beaulieu J-M, Sotnikova TD, Yao W-D, Kockeritz L, Woodgett JR, Gainetdinov RR, Caron MG. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(14):5099–5104. doi: 10.1073/pnas.0307921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeler JA, Daw N, Frazier CRM, Zhuang X. Tonic dopamine modulates exploitation of reward learning. Frontiers in Behavioral Neuroscience. 2010;4:170. doi: 10.3389/fnbeh.2010.00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit-Marand M, Jaber M, Gonon F. Release and elimination of dopamine in vivo in mice lacking the dopamine transporter: functional consequences. The European Journal of Neuroscience. 2000;12(8):2985–2992. doi: 10.1046/j.1460-9568.2000.00155.x. [DOI] [PubMed] [Google Scholar]
- Berlanga ML, Price DL, Phung BS, Giuly R, Terada M, Yamada N, Ellisman MH. Multiscale imaging characterization of dopamine transporter knockout mice reveals regional alterations in spine density of medium spiny neurons. Brain Research. 2011;1390:41–49. doi: 10.1016/j.brainres.2011.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge KC, Aldridge JW, Houchard KR, Zhuang X. Sequential super-stereotypy of an instinctive fixed action pattern in hyper-dopaminergic mutant mice: a model of obsessive compulsive disorder and Tourette’s. BMC Biology. 2005;3:4. doi: 10.1186/1741-7007-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonci A, Bernardi G, Grillner P, Mercuri NB. The dopamine-containing neuron: maestro or simple musician in the orchestra of addiction? Trends in Pharmacological Sciences. 2003;24(4):172–177. doi: 10.1016/S0165-6147(03)00068-3. [DOI] [PubMed] [Google Scholar]
- Bossé R, Fumagalli F, Jaber M, Giros B, Gainetdinov RR, Wetsel WC, Caron MG. Anterior pituitary hypoplasia and dwarfism in mice lacking the dopamine transporter. Neuron. 1997;19(1):127–138. doi: 10.1016/s0896-6273(00)80353-0. [DOI] [PubMed] [Google Scholar]
- Budygin EA, Brodie MS, Sotnikova TD, Mateo Y, John CE, Cyr M, Jones SR. Dissociation of rewarding and dopamine transporter-mediated properties of amphetamine. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(20):7781–7786. doi: 10.1073/pnas.0401418101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budygin EA, John CE, Mateo Y, Jones SR. Lack of cocaine effect on dopamine clearance in the core and shell of the nucleus accumbens of dopamine transporter knock-out mice. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience. 2002;22(10):RC222. doi: 10.1523/JNEUROSCI.22-10-j0002.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budygin EA, Phillips PE, Robinson DL, Kennedy AP, Gainetdinov RR, Wightman RM. Effect of acute ethanol on striatal dopamine neurotransmission in ambulatory rats. The Journal of Pharmacology and Experimental Therapeutics. 2001a;297(1):27–34. [PubMed] [Google Scholar]
- Budygin EA, Phillips PE, Wightman RM, Jones SR. Terminal effects of ethanol on dopamine dynamics in rat nucleus accumbens: an in vitro voltammetric study. Synapse (New York, N.Y.) 2001b;42(2):77–79. doi: 10.1002/syn.1101. [DOI] [PubMed] [Google Scholar]
- Cagniard B, Balsam PD, Brunner D, Zhuang X. Mice with chronically elevated dopamine exhibit enhanced motivation, but not learning, for a food reward. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology. 2006a;31(7):1362–1370. doi: 10.1038/sj.npp.1300966. [DOI] [PubMed] [Google Scholar]
- Cagniard B, Beeler JA, Britt JP, McGehee DS, Marinelli M, Zhuang X. Dopamine scales performance in the absence of new learning. Neuron. 2006b;51(5):541–547. doi: 10.1016/j.neuron.2006.07.026. [DOI] [PubMed] [Google Scholar]
- Cagniard B, Sotnikova TD, Gainetdinov RR, Zhuang X. The dopamine transporter expression level differentially affects responses to cocaine and amphetamine. Journal of Neurogenetics. 2014;28(1–2):112–121. doi: 10.3109/01677063.2014.908191. [DOI] [PubMed] [Google Scholar]
- Carlsson A. Perspectives on the Discovery of Central Monoaminergic Neurotransmission. Annual Review of Neuroscience. 1987;10(1):19–40. doi: 10.1146/annurev.ne.10.030187.000315. [DOI] [PubMed] [Google Scholar]
- Chen R, Han DD, Gu HH. A triple mutation in the second transmembrane domain of mouse dopamine transporter markedly decreases sensitivity to cocaine and methylphenidate. Journal of Neurochemistry. 2005;94(2):352–359. doi: 10.1111/j.1471-4159.2005.03199.x. [DOI] [PubMed] [Google Scholar]
- Chen R, Tilley MR, Wei H, Zhou F, Zhou F-M, Ching S, Gu HH. Abolished cocaine reward in mice with a cocaine-insensitive dopamine transporter. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(24):9333–9338. doi: 10.1073/pnas.0600905103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-C, Lai W-S. Behavioural phenotyping of dopamine transporter knockdown mice using local small interference RNA. Behavioural Brain Research. 2010;214(2):475–479. doi: 10.1016/j.bbr.2010.06.008. [DOI] [PubMed] [Google Scholar]
- Ciliax BJ, Heilman C, Demchyshyn LL, Pristupa ZB, Ince E, Hersch SM, Levey AI. The dopamine transporter: immunochemical characterization and localization in brain. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 1995;15(3 Pt 1):1714–1723. doi: 10.1523/JNEUROSCI.15-03-01714.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa RM, Gutierrez R, de Araujo IE, Coelho MRP, Kloth AD, Gainetdinov RR, Simon SA. Dopamine levels modulate the updating of tastant values. Genes, Brain, and Behavior. 2007;6(4):314–320. doi: 10.1111/j.1601-183X.2006.00257.x. [DOI] [PubMed] [Google Scholar]
- Coyle JT. Glutamate and schizophrenia: beyond the dopamine hypothesis. Cellular and Molecular Neurobiology. 2006;26(4–6):365–384. doi: 10.1007/s10571-006-9062-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyr M, Beaulieu J-M, Laakso A, Sotnikova TD, Yao W-D, Bohn LM, Caron MG. Sustained elevation of extracellular dopamine causes motor dysfunction and selective degeneration of striatal GABAergic neurons. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(19):11035–11040. doi: 10.1073/pnas.1831768100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyr M, Caron MG, Johnson GA, Laakso A. Magnetic resonance imaging at microscopic resolution reveals subtle morphological changes in a mouse model of dopaminergic hyperfunction. NeuroImage. 2005;26(1):83–90. doi: 10.1016/j.neuroimage.2005.01.039. [DOI] [PubMed] [Google Scholar]
- Dluzen DE, Ji J, McDermott JL. Age-related changes in nigrostriatal dopaminergic function in heterozygous mutant dopamine transporter knock-out mice. Neuroscience Letters. 2010;476(2):66–69. doi: 10.1016/j.neulet.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Dluzen DE, McDermott JL. Sex differences in dopamine- and vesicular monoamine-transporter functions. Annals of the New York Academy of Sciences. 2008;1139:140–150. doi: 10.1196/annals.1432.010. [DOI] [PubMed] [Google Scholar]
- Dzirasa K, Santos LM, Ribeiro S, Stapleton J, Gainetdinov RR, Caron MG, Nicolelis MAL. Persistent hyperdopaminergia decreases the peak frequency of hippocampal theta oscillations during quiet waking and REM sleep. PloS One. 2009;4(4):e5238. doi: 10.1371/journal.pone.0005238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraone SV, Bonvicini C, Scassellati C. Biomarkers in the diagnosis of ADHD--promising directions. Current Psychiatry Reports. 2014;16(11):497. doi: 10.1007/s11920-014-0497-1. [DOI] [PubMed] [Google Scholar]
- Fauchey V, Jaber M, Caron MG, Bloch B, Le Moine C. Differential regulation of the dopamine D1, D2 and D3 receptor gene expression and changes in the phenotype of the striatal neurons in mice lacking the dopamine transporter. European Journal of Neuroscience. 2000;12(1):19–26. doi: 10.1046/j.1460-9568.2000.00876.x. [DOI] [PubMed] [Google Scholar]
- Ferris MJ, España RA, Locke JL, Konstantopoulos JK, Rose JH, Chen R, Jones SR. Dopamine transporters govern diurnal variation in extracellular dopamine tone. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(26):E2751–E2759. doi: 10.1073/pnas.1407935111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franken IHA, Booij J, van den Brink W. The role of dopamine in human addiction: from reward to motivated attention. European Journal of Pharmacology. 2005;526(1–3):199–206. doi: 10.1016/j.ejphar.2005.09.025. [DOI] [PubMed] [Google Scholar]
- Fumagalli F, Gainetdinov RR, Valenzano KJ, Caron MG. Role of dopamine transporter in methamphetamine-induced neurotoxicity: evidence from mice lacking the transporter. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience. 1998;18(13):4861–4869. doi: 10.1523/JNEUROSCI.18-13-04861.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainetdinov RR. Dopamine transporter mutant mice in experimental neuropharmacology. Naunyn-Schmiedeberg’s Archives of Pharmacology. 2008;377(4–6):301–313. doi: 10.1007/s00210-007-0216-0. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Caron MG. Monoamine transporters: from genes to behavior. Annual Review of Pharmacology and Toxicology. 2003;43:261–284. doi: 10.1146/annurev.pharmtox.43.050802.112309. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Jones SR, Caron MG. Functional hyperdopaminergia in dopamine transporter knock-out mice. Biological Psychiatry. 1999a;46(3):303–311. doi: 10.1016/s0006-3223(99)00122-5. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Jones SR, Fumagalli F, Wightman RM, Caron MG. Re-evaluation of the role of the dopamine transporter in dopamine system homeostasis. Brain Research Brain Research Reviews. 1998;26(2–3):148–153. doi: 10.1016/s0165-0173(97)00063-5. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Mohn AR, Bohn LM, Caron MG. Glutamatergic modulation of hyperactivity in mice lacking the dopamine transporter. Proceedings of the National Academy of Sciences of the United States of America. 2001a;98(20):11047–11054. doi: 10.1073/pnas.191353298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainetdinov RR, Mohn AR, Caron MG. Genetic animal models: focus on schizophrenia. Trends in Neurosciences. 2001b;24(9):527–533. doi: 10.1016/s0166-2236(00)01886-5. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Sotnikova TD, Caron MG. Monoamine transporter pharmacology and mutant mice. Trends in Pharmacological Sciences. 2002;23(8):367–373. doi: 10.1016/s0165-6147(02)02044-8. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Wetsel WC, Jones SR, Levin ED, Jaber M, Caron MG. Role of serotonin in the paradoxical calming effect of psychostimulants on hyperactivity. Science (New York, N.Y.) 1999b;283(5400):397–401. doi: 10.1126/science.283.5400.397. [DOI] [PubMed] [Google Scholar]
- Ghisi V, Ramsey AJ, Masri B, Gainetdinov RR, Caron MG, Salahpour A. Reduced D2-mediated signaling activity and trans-synaptic upregulation of D1 and D2 dopamine receptors in mice overexpressing the dopamine transporter. Cellular Signalling. 2009;21(1):87–94. doi: 10.1016/j.cellsig.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379(6566):606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- Greengard P. The neurobiology of slow synaptic transmission. Science (New York, N.Y.) 2001;294(5544):1024–1230. doi: 10.1126/science.294.5544.1024. [DOI] [PubMed] [Google Scholar]
- Greenwood TA, Alexander M, Keck PE, McElroy S, Sadovnick AD, Remick RA, Kelsoe JR. Evidence for linkage disequilibrium between the dopamine transporter and bipolar disorder. American Journal of Medical Genetics. 2001;105(2):145–151. doi: 10.1002/1096-8628(2001)9999:9999<::aid-ajmg1161>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Greenwood TA, Schork NJ, Eskin E, Kelsoe JR. Identification of additional variants within the human dopamine transporter gene provides further evidence for an association with bipolar disorder in two independent samples. Molecular Psychiatry. 2006;11(2):125–133. 115. doi: 10.1038/sj.mp.4001764. [DOI] [PubMed] [Google Scholar]
- Grünhage F, Schulze TG, Müller DJ, Lanczik M, Franzek E, Albus M, Nöthen MM. Systematic screening for DNA sequence variation in the coding region of the human dopamine transporter gene (DAT1) Molecular Psychiatry. 2000;5(3):275–282. doi: 10.1038/sj.mp.4000711. [DOI] [PubMed] [Google Scholar]
- Hall FS, Sora I, Hen R, Uhl GR. Serotonin/dopamine interactions in a hyperactive mouse: reduced serotonin receptor 1B activity reverses effects of dopamine transporter knockout. PloS One. 2014a;9(12):e115009. doi: 10.1371/journal.pone.0115009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall FS, Itokawa K, Schmitt A, Moessner R, Sora I, Lesch KP, Uhl GR. Decreased vesicular monoamine transporter 2 (VMAT2) and dopamine transporter (DAT) function in knockout mice affects aging of dopaminergic systems. Neuropharmacology. 2014b;76 Pt A:146–155. doi: 10.1016/j.neuropharm.2013.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Molecular Psychiatry. 2005;10(1):40–68. doi: 10.1038/sj.mp.4001558. image 5. [DOI] [PubMed] [Google Scholar]
- Horschitz S, Hummerich R, Lau T, Rietschel M, Schloss P. A dopamine transporter mutation associated with bipolar affective disorder causes inhibition of transporter cell surface expression. Molecular Psychiatry. 2005;10(12):1104–1109. doi: 10.1038/sj.mp.4001730. [DOI] [PubMed] [Google Scholar]
- Hoyer D, Thakker DR, Natt F, Maier R, Huesken D, Müller M, Cryan JF. Global down-regulation of gene expression in the brain using RNA interference, with emphasis on monoamine transporters and GPCRs: implications for target characterization in psychiatric and neurological disorders. Journal of Receptor and Signal Transduction Research. 2006;26(5–6):527–547. doi: 10.1080/10799890600929663. [DOI] [PubMed] [Google Scholar]
- Jaber M, Dumartin B, Sagne C, Haycock JW, Roubert C, Giros B, Caron MG. Differential regulation of tyrosine hydroxylase in the basal ganglia of mice lacking the dopamine transporter. European Journal of Neuroscience. 1999;11(10):3499–3511. doi: 10.1046/j.1460-9568.1999.00764.x. [DOI] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Jaber M, Giros B, Wightman RM, Caron MG. Profound neuronal plasticity in response to inactivation of the dopamine transporter. Proceedings of the National Academy of Sciences of the United States of America. 1998a;95(7):4029–4034. doi: 10.1073/pnas.95.7.4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Wightman RM, Caron MG. Mechanisms of amphetamine action revealed in mice lacking the dopamine transporter. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience. 1998b;18(6):1979–1986. doi: 10.1523/JNEUROSCI.18-06-01979.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SR, Mathews TA, Budygin EA. Effect of moderate ethanol dose on dopamine uptake in rat nucleus accumbens in vivo. Synapse (New York, N.Y.) 2006;60(3):251–255. doi: 10.1002/syn.20294. [DOI] [PubMed] [Google Scholar]
- Kasahara Y, Arime Y, Hall FS, Uhl GR, Sora I. Region-specific dendritic spine loss of pyramidal neurons in dopamine transporter knockout mice. Current Molecular Medicine. 2015;15(3):237–244. doi: 10.2174/1566524015666150330143613. [DOI] [PubMed] [Google Scholar]
- Kelsoe JR, Sadovnick AD, Kristbjarnarson H, Bergesch P, Mroczkowski-Parker Z, Drennan M, Remick RA. Possible locus for bipolar disorder near the dopamine transporter on chromosome 5. American Journal of Medical Genetics. 1996;67(6):533–540. doi: 10.1002/(SICI)1096-8628(19961122)67:6<533::AID-AJMG4>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Laakso A, Mohn AR, Gainetdinov RR, Caron MG. Experimental genetic approaches to addiction. Neuron. 2002;36(2):213–228. doi: 10.1016/s0896-6273(02)00972-8. [DOI] [PubMed] [Google Scholar]
- Lee VM. Biomedicine. Tauists and beta-aptists united--well almost! Science (New York, N.Y.) 2001;293(5534):1446–1447. doi: 10.1126/science.1064684. [DOI] [PubMed] [Google Scholar]
- Leo D, Gainetdinov RR. Transgenic mouse models for ADHD. Cell and Tissue Research. 2013;354(1):259–271. doi: 10.1007/s00441-013-1639-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Lieberman JA. Catching Up on Schizophrenia. Neuron. 2000;28(2):325–334. doi: 10.1016/s0896-6273(00)00111-2. [DOI] [PubMed] [Google Scholar]
- Martin BJ, Naughton BJ, Thirtamara-Rajamani K, Yoon DJ, Han DD, Devries AC, Gu HH. Dopamine transporter inhibition is necessary for cocaine-induced increases in dendritic spine density in the nucleus accumbens. Synapse (New York, N.Y.) 2011;65(6):490–496. doi: 10.1002/syn.20865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews TA, John CE, Lapa GB, Budygin EA, Jones SR. No role of the dopamine transporter in acute ethanol effects on striatal dopamine dynamics. Synapse (New York, N.Y.) 2006;60(4):288–294. doi: 10.1002/syn.20301. [DOI] [PubMed] [Google Scholar]
- Mazei-Robison MS, Blakely RD. Expression studies of naturally occurring human dopamine transporter variants identifies a novel state of transporter inactivation associated with Val382Ala. Neuropharmacology. 2005;49(6):737–749. doi: 10.1016/j.neuropharm.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Mazei-Robison MS, Bowton E, Holy M, Schmudermaier M, Freissmuth M, Sitte HH, Blakely RD. Anomalous dopamine release associated with a human dopamine transporter coding variant. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience. 2008;28(28):7040–7046. doi: 10.1523/JNEUROSCI.0473-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mergy MA, Gowrishankar R, Gresch PJ, Gantz SC, Williams J, Davis GL, Blakely RD. The rare DAT coding variant Val559 perturbs DA neuron function, changes behavior, and alters in vivo responses to psychostimulants. Proceedings of the National Academy of Sciences of the United States of America. 2014a;111(44):E4779–E4788. doi: 10.1073/pnas.1417294111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mergy MA, Gowrishankar R, Davis GL, Jessen TN, Wright J, Stanwood GD, Blakely RD. Genetic targeting of the amphetamine and methylphenidate-sensitive dopamine transporter: On the path to an animal model of attention-deficit hyperactivity disorder. Neurochemistry International. 2014b;73:56–70. doi: 10.1016/j.neuint.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GW, Gainetdinov RR, Levey AI, Caron MG. Dopamine transporters and neuronal injury. Trends in Pharmacological Sciences. 1999;20(10):424–429. doi: 10.1016/s0165-6147(99)01379-6. [DOI] [PubMed] [Google Scholar]
- Mohn AR, Gainetdinov RR, Caron MG, Koller BH. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell. 1999;98(4):427–436. doi: 10.1016/s0092-8674(00)81972-8. [DOI] [PubMed] [Google Scholar]
- Mohn AR, Yao W-D, Caron MG. Genetic and genomic approaches to reward and addiction. Neuropharmacology. 2004;47(Suppl 1):101–110. doi: 10.1016/j.neuropharm.2004.07.025. [DOI] [PubMed] [Google Scholar]
- Morice E, Billard J-M, Denis C, Mathieu F, Betancur C, Epelbaum J, Nosten-Bertrand M. Parallel loss of hippocampal LTD and cognitive flexibility in a genetic model of hyperdopaminergia. Neuropsychopharmacology : Official Publication of the American College of Neuropsychopharmacology. 2007;32(10):2108–2116. doi: 10.1038/sj.npp.1301354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morice E, Denis C, Giros B, Nosten-Bertrand M. Evidence of long-term expression of behavioral sensitization to both cocaine and ethanol in dopamine transporter knockout mice. Psychopharmacology. 2009;208(1):57–66. doi: 10.1007/s00213-009-1707-0. [DOI] [PubMed] [Google Scholar]
- Napolitano F, Bonito-Oliva A, Federici M, Carta M, Errico F, Magara S, Usiello A. Role of aberrant striatal dopamine D1 receptor/cAMP/protein kinase A/DARPP32 signaling in the paradoxical calming effect of amphetamine. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience. 2010;30(33):11043–11056. doi: 10.1523/JNEUROSCI.1682-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ. Is there a common molecular pathway for addiction? Nature Neuroscience. 2005;8(11):1445–1449. doi: 10.1038/nn1578. [DOI] [PubMed] [Google Scholar]
- Nirenberg MJ, Chan J, Pohorille A, Vaughan RA, Uhl GR, Kuhar MJ, Pickel VM. The Dopamine Transporter: Comparative Ultrastructure of Dopaminergic Axons in Limbic and Motor Compartments of the Nucleus Accumbens. J Neurosci. 1997;17(18):6899–6907. doi: 10.1523/JNEUROSCI.17-18-06899.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill B, Gu HH. Amphetamine-induced locomotion in a hyperdopaminergic ADHD mouse model depends on genetic background. Pharmacology, Biochemistry, and Behavior. 2013;103(3):455–459. doi: 10.1016/j.pbb.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill B, Tilley MR, Gu HH. Cocaine produces conditioned place aversion in mice with a cocaine-insensitive dopamine transporter. Genes, Brain, and Behavior. 2013;12(1):34–38. doi: 10.1111/j.1601-183X.2012.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill B, Tilley MR, Han DD, Thirtamara-Rajamani K, Hill ER, Bishop GA, Gu HH. Behavior of knock-in mice with a cocaine-insensitive dopamine transporter after virogenetic restoration of cocaine sensitivity in the striatum. Neuropharmacology. 2014;79:626–633. doi: 10.1016/j.neuropharm.2013.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecina S, Cagniard B, Berridge KC, Aldridge JW, Zhuang X. Hyperdopaminergic Mutant Mice Have Higher “Wanting” But Not “Liking” for Sweet Rewards. J Neurosci. 2003;23(28):9395–9402. doi: 10.1523/JNEUROSCI.23-28-09395.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piao Y-S, Hall FS, Moriya Y, Ito M, Ohara A, Kikura-Hanajiri R, Sora I. Methylone-induced hyperthermia and lethal toxicity: role of the dopamine and serotonin transporters. Behavioural Pharmacology. 2015;26(4):345–352. doi: 10.1097/FBP.0000000000000135. [DOI] [PubMed] [Google Scholar]
- Rajkowska G, Selemon LD, Goldman-Rakic PS. Neuronal and glial somal size in the prefrontal cortex: a postmortem morphometric study of schizophrenia and Huntington disease. Archives of General Psychiatry. 1998;55(3):215–224. doi: 10.1001/archpsyc.55.3.215. [DOI] [PubMed] [Google Scholar]
- Ralph RJ, Paulus MP, Fumagalli F, Caron MG, Geyer MA. Prepulse inhibition deficits and perseverative motor patterns in dopamine transporter knock-out mice: differential effects of D1 and D2 receptor antagonists. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience. 2001;21(1):305–313. doi: 10.1523/JNEUROSCI.21-01-00305.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralph-Williams RJ, Paulus MP, Zhuang X, Hen R, Geyer MA. Valproate attenuates hyperactive and perseverative behaviors in mutant mice with a dysregulated dopamine system. Biological Psychiatry. 2003;53(4):352–359. doi: 10.1016/s0006-3223(02)01489-0. [DOI] [PubMed] [Google Scholar]
- Reith ME, Xu C, Chen N-H. Pharmacology and regulation of the neuronal dopamine transporter. European Journal of Pharmacology. 1997;324(1):1–10. doi: 10.1016/s0014-2999(97)00065-4. [DOI] [PubMed] [Google Scholar]
- Rickhag M, Owens WA, Winkler M-T, Strandfelt KN, Rathje M, Sørensen G, Gether U. Membrane-permeable C-terminal dopamine transporter peptides attenuate amphetamine-evoked dopamine release. The Journal of Biological Chemistry. 2013;288(38):27534–27544. doi: 10.1074/jbc.M112.441295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins TW, Everitt BJ. Limbic-striatal memory systems and drug addiction. Neurobiology of Learning and Memory. 2002;78(3):625–636. doi: 10.1006/nlme.2002.4103. [DOI] [PubMed] [Google Scholar]
- Robinson DL, Volz TJ, Schenk JO, Wightman RM. Acute ethanol decreases dopamine transporter velocity in rat striatum: in vivo and in vitro electrochemical measurements. Alcoholism, Clinical and Experimental Research. 2005;29(5):746–755. doi: 10.1097/01.alc.0000164362.21484.14. [DOI] [PubMed] [Google Scholar]
- Rocha BA, Fumagalli F, Gainetdinov RR, Jones SR, Ator R, Giros B, Caron MG. Cocaine self-administration in dopamine-transporter knockout mice. Nature Neuroscience. 1998;1(2):132–137. doi: 10.1038/381. [DOI] [PubMed] [Google Scholar]
- Ross CA, Margolis RL, Reading SAJ, Pletnikov M, Coyle JT. Neurobiology of schizophrenia. Neuron. 2006;52(1):139–153. doi: 10.1016/j.neuron.2006.09.015. [DOI] [PubMed] [Google Scholar]
- Rossi MA, Yin HH. Elevated dopamine alters consummatory pattern generation and increases behavioral variability during learning. Frontiers in Integrative Neuroscience. 2015;9:37. doi: 10.3389/fnint.2015.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeti J, Gerhardt GA, Zahniser NR. Chloral hydrate and ethanol, but not urethane, alter the clearance of exogenous dopamine recorded by chronoamperometry in striatum of unrestrained rats. Neuroscience Letters. 2003;343(1):9–12. doi: 10.1016/s0304-3940(03)00301-x. [DOI] [PubMed] [Google Scholar]
- Salahpour A, Medvedev IO, Beaulieu J-M, Gainetdinov RR, Caron MG. Local knockdown of genes in the brain using small interfering RNA: a phenotypic comparison with knockout animals. Biological Psychiatry. 2007;61(1):65–69. doi: 10.1016/j.biopsych.2006.03.020. [DOI] [PubMed] [Google Scholar]
- Salahpour A, Ramsey AJ, Medvedev IO, Kile B, Sotnikova TD, Holmstrand E, Caron MG. Increased amphetamine-induced hyperactivity and reward in mice overexpressing the dopamine transporter. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(11):4405–4410. doi: 10.1073/pnas.0707646105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamone JD, Correa M. The mysterious motivational functions of mesolimbic dopamine. Neuron. 2012;76(3):470–485. doi: 10.1016/j.neuron.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeman P. Dopamine receptors and the dopamine hypothesis of schizophrenia. Synapse (New York, N.Y.) 1987;1(2):133–152. doi: 10.1002/syn.890010203. [DOI] [PubMed] [Google Scholar]
- Seiden LS, Sabol KE, Ricaurte GA. Amphetamine: effects on catecholamine systems and behavior. Annual Review of Pharmacology and Toxicology. 1993;33:639–677. doi: 10.1146/annurev.pa.33.040193.003231. [DOI] [PubMed] [Google Scholar]
- Sesack SR, Hawrylak VA, Guido MA, Levey AI. Cellular and subcellular localization of the dopamine transporter in rat cortex. Advances in Pharmacology (San Diego, Calif.) 1998;42:171–174. doi: 10.1016/s1054-3589(08)60720-6. [DOI] [PubMed] [Google Scholar]
- Shen H-W, Hagino Y, Kobayashi H, Shinohara-Tanaka K, Ikeda K, Yamamoto H, Sora I. Regional differences in extracellular dopamine and serotonin assessed by in vivo microdialysis in mice lacking dopamine and/or serotonin transporters. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology. 2004;29(10):1790–1799. doi: 10.1038/sj.npp.1300476. [DOI] [PubMed] [Google Scholar]
- Snyder SH. Amphetamine psychosis: a “model” schizophrenia mediated by catecholamines. The American Journal of Psychiatry. 1973;130(1):61–67. doi: 10.1176/ajp.130.1.61. [DOI] [PubMed] [Google Scholar]
- Sora I, Wichems C, Takahashi N, Li XF, Zeng Z, Revay R, Uhl GR. Cocaine reward models: conditioned place preference can be established in dopamine- and in serotonin-transporter knockout mice. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(13):7699–7704. doi: 10.1073/pnas.95.13.7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotnikova TD, Beaulieu J-M, Barak LS, Wetsel WC, Caron MG, Gainetdinov RR. Dopamine-independent locomotor actions of amphetamines in a novel acute mouse model of Parkinson disease. PLoS Biology. 2005;3(8):e271. doi: 10.1371/journal.pbio.0030271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Chen TK, Lau YY, Kristensen H, Rayport S, Ewing A. Amphetamine redistributes dopamine from synaptic vesicles to the cytosol and promotes reverse transport. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience. 1995;15(5 Pt 2):4102–4108. doi: 10.1523/JNEUROSCI.15-05-04102.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczypka MS, Kwok K, Brot MD, Marck BT, Matsumoto AM, Donahue BA, Palmiter RD. Dopamine production in the caudate putamen restores feeding in dopamine-deficient mice. Neuron. 2001;30(3):819–828. doi: 10.1016/s0896-6273(01)00319-1. [DOI] [PubMed] [Google Scholar]
- Takamatsu Y, Hagino Y, Sato A, Takahashi T, Nagasawa SY, Kubo Y, Ikeda K. Improvement of learning and increase in dopamine level in the frontal cortex by methylphenidate in mice lacking dopamine transporter. Current Molecular Medicine. 2015;15(3):245–252. doi: 10.2174/1566524015666150330144018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakker DR, Natt F, Hüsken D, Maier R, Müller M, van der Putten H, Cryan JF. Neurochemical and behavioral consequences of widespread gene knockdown in the adult mouse brain by using nonviral RNA interference. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(49):17270–17275. doi: 10.1073/pnas.0406214101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen M, Hall FS, Uhl GR, Caine SB. Dramatically decreased cocaine self-administration in dopamine but not serotonin transporter knock-out mice. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience. 2009a;29(4):1087–1092. doi: 10.1523/JNEUROSCI.4037-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen M, Han DD, Gu HH, Caine SB. Lack of cocaine self-administration in mice expressing a cocaine-insensitive dopamine transporter. The Journal of Pharmacology and Experimental Therapeutics. 2009b;331(1):204–211. doi: 10.1124/jpet.109.156265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilley MR, Cagniard B, Zhuang X, Han DD, Tiao N, Gu HH. Cocaine reward and locomotion stimulation in mice with reduced dopamine transporter expression. BMC Neuroscience. 2007;8:42. doi: 10.1186/1471-2202-8-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilley MR, O’Neill B, Han DD, Gu HH. Cocaine does not produce reward in absence of dopamine transporter inhibition. Neuroreport. 2009;20(1):9–12. doi: 10.1097/WNR.0b013e32831b9ce4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Enkhuizen J, Geyer MA, Halberstadt AL, Zhuang X, Young JW. Dopamine depletion attenuates some behavioral abnormalities in a hyperdopaminergic mouse model of bipolar disorder. Journal of Affective Disorders. 2014;155:247–254. doi: 10.1016/j.jad.2013.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Enkhuizen J, Geyer MA, Kooistra K, Young JW. Chronic valproate attenuates some, but not all, facets of mania-like behaviour in mice. The International Journal of Neuropsychopharmacology / Official Scientific Journal of the Collegium Internationale Neuropsychopharmacologicum (CINP) 2013;16(5):1021–1031. doi: 10.1017/S1461145712001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent SG, Waddell AE, Caron MG, Walker JKL, Fisher JT. A murine model of hyperdopaminergic state displays altered respiratory control. FASEB Journal : Official Publication of the Federation of American Societies for Experimental Biology. 2007;21(7):1463–1471. doi: 10.1096/fj.06-7248com. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wang G-J, Swanson JM. Dopamine in drug abuse and addiction: results from imaging studies and treatment implications. Molecular Psychiatry. 2004;9(6):557–569. doi: 10.1038/sj.mp.4001507. [DOI] [PubMed] [Google Scholar]
- Wilson JM, Levey AI, Bergeron C, Kalasinsky K, Ang L, Peretti F, Kish SJ. Striatal dopamine, dopamine transporter, and vesicular monoamine transporter in chronic cocaine users. Annals of Neurology. 1996;40(3):428–439. doi: 10.1002/ana.410400312. [DOI] [PubMed] [Google Scholar]
- Wong P, Chang CCR, Marx CE, Caron MG, Wetsel WC, Zhang X. Pregnenolone rescues schizophrenia-like behavior in dopamine transporter knockout mice. PloS One. 2012;7(12):e51455. doi: 10.1371/journal.pone.0051455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong P, Sze Y, Chang CCR, Lee J, Zhang X. Pregnenolone sulfate normalizes schizophrenia-like behaviors in dopamine transporter knockout mice through the AKT/GSK3β pathway. Translational Psychiatry. 2015;5:e528. doi: 10.1038/tp.2015.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T-X, Ma Q, Spealman RD, Yao W-D. Amphetamine modulation of long-term potentiation in the prefrontal cortex: dose dependency, monoaminergic contributions, and paradoxical rescue in hyperdopaminergic mutant. Journal of Neurochemistry. 2010;115(6):1643–1654. doi: 10.1111/j.1471-4159.2010.07073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T-X, Sotnikova TD, Liang C, Zhang J, Jung JU, Spealman RD, Yao W-D. Hyperdopaminergic tone erodes prefrontal long-term potential via a D2 receptor-operated protein phosphatase gate. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience. 2009;29(45):14086–14099. doi: 10.1523/JNEUROSCI.0974-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, Sakakibara Y, Hall FS, Numachi Y, Yoshida S, Kobayashi H, Sora I. Impaired cliff avoidance reaction in dopamine transporter knockout mice. Psychopharmacology. 2013;227(4):741–749. doi: 10.1007/s00213-013-3009-9. [DOI] [PubMed] [Google Scholar]
- Yao W-D, Gainetdinov RR, Arbuckle MI, Sotnikova TD, Cyr M, Beaulieu J-M, Caron MG. Identification of PSD-95 as a regulator of dopamine-mediated synaptic and behavioral plasticity. Neuron. 2004;41(4):625–638. doi: 10.1016/s0896-6273(04)00048-0. [DOI] [PubMed] [Google Scholar]
- Yin HH, Zhuang X, Balleine BW. Instrumental learning in hyperdopaminergic mice. Neurobiology of Learning and Memory. 2006;85(3):283–288. doi: 10.1016/j.nlm.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Young JW, Goey AKL, Minassian A, Perry W, Paulus MP, Geyer MA. The mania-like exploratory profile in genetic dopamine transporter mouse models is diminished in a familiar environment and reinstated by subthreshold psychostimulant administration. Pharmacology, Biochemistry, and Behavior. 2010;96(1):7–15. doi: 10.1016/j.pbb.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JW, van Enkhuizen J, Winstanley CA, Geyer MA. Increased risk-taking behavior in dopamine transporter knockdown mice: further support for a mouse model of mania. Journal of Psychopharmacology (Oxford, England) 2011;25(7):934–943. doi: 10.1177/0269881111400646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Saur T, Duke AN, Grant SGN, Platt DM, Rowlett JK, Yao W-D. Motor impairments, striatal degeneration, and altered dopamine-glutamate interplay in mice lacking PSD-95. Journal of Neurogenetics. 2014;28(1–2):98–111. doi: 10.3109/01677063.2014.892486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Vinuela A, Neely MH, Hallett PJ, Grant SGN, Miller GM, Yao W-D. Inhibition of the dopamine D1 receptor signaling by PSD-95. The Journal of Biological Chemistry. 2007;282(21):15778–15789. doi: 10.1074/jbc.M611485200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Xu T-X, Hallett PJ, Watanabe M, Grant SGN, Isacson O, Yao W-D. PSD-95 uncouples dopamine-glutamate interaction in the D1/PSD-95/NMDA receptor complex. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience. 2009;29(9):2948–2960. doi: 10.1523/JNEUROSCI.4424-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Bearer EL, Boulat B, Hall FS, Uhl GR, Jacobs RE. Altered neurocircuitry in the dopamine transporter knockout mouse brain. PloS One. 2010;5(7):e11506. doi: 10.1371/journal.pone.0011506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang X, Oosting RS, Jones SR, Gainetdinov RR, Miller GW, Caron MG, Hen R. Hyperactivity and impaired response habituation in hyperdopaminergic mice. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(4):1982–1987. doi: 10.1073/pnas.98.4.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]