

Abstract
Secreted by activated cells or passively released by damaged cells, extracellular HMGB1 is a prototypical damage-associated molecular pattern (DAMP) inflammatory mediator. During the course of developing extracorporeal approaches to treating injury and infection, we inadvertently discovered that haptoglobin, the acute phase protein that binds extracellular hemoglobin and targets cellular uptake through CD163, also binds HMGB1. Haptoglobin-HMGB1 complexes elicit the production of antiinflammatory enzymes (heme oxygenase-1) and cytokines (e.g., IL-10) in WT but not in CD163-deficient macrophages. Genetic disruption of haptoglobin or CD163 expression significantly enhances mortality rates in standardized models of intra-abdominal sepsis in mice. Administration of haptoglobin to WT and to haptoglobin gene-deficient animals confers significant protection. These findings reveal a mechanism for haptoglobin modulation of the inflammatory action of HMGB1, with significant implications for developing experimental strategies targeting HMGB1-dependent inflammatory diseases.
Haptoglobin binds and sequesters the damage-associated molecular pattern (DAMP) inflammatory mediator HMGB1 through a CD163-dependent mechanism.
Introduction
Severe sepsis, a clinical syndrome that complicates the course of infection, is a leading cause of mortality in the US (1, 2). The pathogenesis of lethal sepsis is mediated by HMGB1 and other cytokines that damage cells and impair physiological homeostasis. The inflammatory responses to infection and tissue inflammation are enhanced by exogenously derived pathogen-associated molecular pattern molecules (PAMPs), including bacterial peptidoglycan, endotoxin, and CpG-DNA, and by endogenously derived damage-associated molecular pattern molecules (DAMPs), including HMGB1 (3–9). HMGB1 is a central mediator of lethal infection and injury (5, 10). The protein harbors 3 conserved cysteine residues at position 23, 45, and 106, and the redox state of these cysteines determine whether HMGB1 functions as a chemokine or as a proinflammatory cytokine (11–14). HMGB1, actively secreted by activated immune cells or passively released from dying cells, is a mixture of redox isoforms with distinct posttranslational modifications (5, 15, 16). Administration of anti-HMGB1 antibodies confers significant protection in animal models of experimental sepsis, endotoxemia, ischemia reperfusion injury, trauma, hepatitis, and other syndromes (3–5, 17).
Another DAMP, extracellular (free) hemoglobin, is also significantly increased in trauma, burn injury, blood transfusion, cardiopulmonary bypass, infection, and other clinical syndromes (18–20). Increased extracellular hemoglobin significantly amplifies PAMP-mediated cytokine production, organ damage, and mortality (7–9, 18–20). Evolution has conferred mammals with redundant counterregulatory mechanisms to protect against the toxicity of extracellular hemoglobin, primarily by binding to haptoglobin, a 100-KDa hemoglobin binding protein produced in the liver and secreted into the circulation. Haptoglobin, comprised of two α and two β chains linked by disulfide bonds, is the product of gene polymorphisms that yield 3 common protein phenotypes, termed Hp1-1, Hp2-2, and Hp 2-1 (21). Haptoglobin binding to hemoglobin is among the strongest known protein-protein interactions, with extremely high–affinity Kd on the order of 10–15 mol/l (22). Haptoglobin-hemoglobin complexes bind CD163, a receptor expressed on macrophages and other cells that mediates endocytosis and internalization of the haptoglobin-hemoglobin complexes. This uptake mechanism also stimulates the enhanced production of macrophage hemeoxygenase-1 (HO-1) and IL-10 (23–25). Genetic deletion of haptoglobin renders animals significantly more susceptible to lethal endotoxemia (26). Supplementation of haptoglobin prevents free hemoglobin–induced hypertension and oxidative kidney damage, as well as blood transfusion–mediated vascular injury and kidney dysfunction (27, 28). Haptoglobin is approved as an adjuvant therapy for patients in Japan with trauma, burns, and transfusion-related hemolysis (29).
Accordingly, here we reasoned that removal of free hemoglobin would be protective against tissue damage and lethality in sepsis (18). To study this hypothesis, we developed an extracorporeal haptoglobin affinity chromatography method to remove extracellular hemoglobin in rodents with sepsis from cecal ligation and puncture (CLP). Surprisingly, we observed that haptoglobin-affinity chromatography extracted large amounts of HMGB1 from the blood of septic rats. Studies of underlying mechanisms indicate that haptoglobin forms a complex with HMGB1 to stimulate macrophage HO-1 and IL-10 production through a CD163-dependent mechanism that confers significant protection against the lethality of HMGB1 in sepsis.
Results
Identification of haptoglobin as an HMGB1 binding protein.
As extracellular (free) hemoglobin is elevated and worsens outcome in sepsis (18), we developed an extracorporeal hemoperfusion device by conjugating haptoglobin to sepharose beads in order to sequester the circulating free hemoglobin in septic animals induced by cecal perforation. Analysis of proteins extracted from the haptoglobin beads confirmed the presence of hemoglobin using an immunoblot assay. Surprisingly, however, SDS-PAGE gel analysis of haptoglobin binding proteins in the eluate also revealed large quantities of a 30-kDa protein, which was identified as HMGB1 by amino acid sequence analysis (data not shown) and immunoblotting (Figure 1A). To verify the HMGB1 binding properties of haptoglobin, the HMGB1-haptoglobin complexes were immune precipitated using anti-haptoglobin antibodies. The coimmunoprecipitation of HMGB1 with haptoglobin was confirmed by Western blotting using anti-HMGB1 antibodies (Figure 1B, lane 3). Using surface plasmon resonance, we further observed that the binding affinity (Kd) of HMGB1 to haptoglobin was around 64 nM regardless of whether haptoglobin or HMGB1 was immobilized on the sensor chip (Figure 1C and data not shown).
Figure 1. Haptoglobin (Hp) binds HMGB1.

(A) Rats were subjected to CLP to induce sepsis and subsequently connected to an extracorporeal pump to filter blood through human haptoglobin–conjugated (Hp) or empty beads for 1 hour. Proteins bound to Hp or empty beads were eluted and subjected to Western blot analysis using anti-HMGB1 antibodies. Data are representative from 3 repeats. (B) Human Hp and HMGB1 was mixed and applied to beads coupled with anti–human Hp antibody or control (empty beads). Proteins bound to the beads were analyzed by Western blot probed with anti-HMGB1 antibodies. Data are representative from 5 experiments. (C) Human Hp was immobilized on the sensor chip, and binding to HMGB1 was assessed. The apparent Kd is ~64 nM. Data are presented as response units over time (Sec) and representative of 3 experiments. (D) Human Hp–coupled beads were incubated with various HMGB1 isoforms. Elutes were analyzed by Western blot using anti-HMGB1 mAbs. Data are representative from 6 repeats. (E) Upper: Schematics of human HMGB1 structure. Middle/lower: nuclear magnetic resonance (NMR) analysis of disulfide (middle) or sulfonyl (lower) Δ30-HMGB1 in complex with human Hp. Chemical shift perturbation of disulfide or sulfonyl Δ30-HMGB1 in complex with Hp is plotted as a function of residue number of HMGB1. Perturbations in chemical shift above 0.1 are considered a significant interaction (46). The majority of interacting amino acids are located within HMGB1 Box A. Data are representative from 3 repeats. (F) Upper: 15N-1H-heteronuclear single quantum coherence (HSQC) spectra for disulfide Δ30-HMGB1 in the presence or absence of Hp. HMGB1 residues (10 amino acids) showing significant chemical shift perturbations due to Hp binding are labeled. Middle: Representative 15N-HSQC spectra of disulfide Δ30-HMGB1 free (black) and in complex with haptoglobin (red); residues showing chemical shift changes upon complex formation are circled. Lower: 15N-HSQC spectra of sulfonyl Δ30-HMGB1 free (black) and in complex with Hp (red). Residues showing chemical shift changes seen with disulfide Δ30-HMGB1 upon complex formation with Hp are circled for comparison. Nonoverlapping black and red resonance spectra indicate a significant chemical shift and, hence, an interaction between proteins via that amino acid. (G) Serum samples (10 μl) from healthy individuals or sepsis patients were incubated with beads conjugated to anti–human Hp mAbs or empty (20 μl drained beads). Eluates were probed with anti-HMGB1 (upper) and anti–human Hp (lower) antibodies using Western blot analysis. Data are representative from 5 samples each.
Haptoglobin binds the proinflammatory forms of HMGB1.
HMGB1 contains 3 cysteine residues at position 23, 45, and 106, and HMGB1 receptor interaction is determined by the cysteine redox state. Fully reduced extracellular HMGB1 is chemotactic by specific interaction and signal transduction via CXCL12/SDF1 pathways, whereas disulfide HMGB1 is proinflammatory by binding and signaling via MD2-TLR4 to enhance macrophage cytokine production. The sulfonyl (fully oxidized) molecule has no known biological activity (15). Haptoglobin effectively binds fully reduced and disulfide HMGB1 but not the fully oxidized sulfonyl HMGB1 (Figure 1D). Nuclear magnetic resonance (NMR) analysis confirmed the results observed from immunoprecipitation, revealing that haptoglobin interacts with the HMGB1 Box A (Figure 1E). NMR analyses also showed significant chemical shift perturbations for 10 amino acids residing in or near the Box A when haptoglobin binds HMGB1: (F [phenylalanine] 18, T [threonine] 22, R [arginine] 24, E [glutamic acid] 25, K [lysine] 28, H [histidine] 31, A [alanine] 54, K55, A126, and G [glycine] 130). The localization of these amino acids within the HMGB1 Box A region (amino acids 8–78) is shown in Figure 1F. Finally, we observed HMGB1-haptoglobin complexes in human serum from patients with severe sepsis by coimmunoprecipitation using haptoglobin-specific antibodies (Figure 1G). Together, these findings indicate that haptoglobin forms a protein complex with the proinflammatory, disulfide HMGB1 in sepsis.
Haptoglobin disruption impairs survival during lethal sepsis, and haptoglobin administration confers protection.
To assess the functional role of haptoglobin binding to HMGB1 in sepsis, we subjected haptoglobin KO mice to CLP-induced sepsis. Haptoglobin KO animals were significantly more susceptible to lethal sepsis as compared with WT littermates (survival = 34% in haptoglobin KO mice vs. 75% in WT mice, n = 15 per group) (Figure 2A). The disruption of haptoglobin expression led to a transient elevation of extracellular hemoglobin for 2–7 days, but HMGB1 levels were increased for at least 2 weeks (Figure 2B). Administration of haptoglobin (500 μg/mouse/day, i.p.) once daily for 3 days beginning 24 hours after CLP surgery significantly improved survival (63% survival in haptoglobin-treated vs. 33% in control group; P < 0.05) (Figure 2C, upper). Most animals treated with haptoglobin were alert and active, whereas most of the vehicle-treated control animals exhibited signs of sickness syndrome with behavioral withdrawal, piloerection, and group huddling. Administration of haptoglobin to septic haptoglobin KO animals also conferred significant protection against lethality (Figure 2D, upper). Haptoglobin administration to septic animals significantly decreased extracellular hemoglobin, HMGB1, and IL-6 levels as compared with vehicle-treated animals in both WT and haptoglobin KO mice (Figure 2, C and D, lower). Administration of neutralizing anti-HMGB1 mAbs also conferred significant protection in haptoglobin KO mice (12% survival rate in PBS-treated CLP sepsis mice vs. 56% in animals that received anti-HMGB1 antibody; P < 0.05, n = 16 mice per group) (Figure 2E). Together, these findings indicate that haptoglobin confers protection against sepsis by attenuating HMGB-mediated lethality.
Figure 2. Protective role of haptoglobin in lethal sepsis.

(A) Disruption of haptoglobin expression renders mice more susceptible to lethal sepsis. Male WT (C57BL/6) and haptoglobin (Hp) KO mice (8–12 weeks old) were subjected to cecal ligation and puncture (CLP) surgery, and survival was monitored for 2 weeks (n = 15 mice per group). *P < 0.05 vs. WT group (Fisher’s exact test). (B) KO of Hp prolongs CLP-induced systemic accumulation of HMGB1 but not free hemoglobin. Male WT (C57BL/6) and Hp KO mice were subjected to CLP and euthanized at the indicated time points; serum-free hemoglobin and HMGB1 levels were measured. Data are from 3 (for day 21), 6 (for days 0, 2, and 14), and 7 (for day 7) mice at each time point. *P < 0.05, ***P < 0.001 vs. WT group (t test). (C and D) Administration of haptoglobin confers protection against sepsis lethality and reduces inflammation in WT and Hp KO mice. Upper: Male BALB/c or Hp KO mice were subjected to CLP surgery, and Hp was administered at 0.5 mg/mouse (for WT mice) or 0.2 mg/mouse (for Hp KO mice) in 200 μl volume, injected i.p. once a day for 3 days starting at 24 hours after CLP surgery. Mice in control group received vehicle (PBS) only. Survival was monitored for 2 weeks (n = 22 mice per group). *P < 0.05 vs. vehicle-treated group (Fisher’s exact test). Lower: Male WT (C57BL/6) or Hp KO mice were subjected to CLP surgery and received Hp (0.5 mg/mouse) or vehicle (PBS) i.p. at 24 and 36 hours after CLP surgery, and mice were euthanized at 48 hours after CLP. Sera were isolated for analyses. n = 7 (for WT group) and 8 (for Hp KO group) mice per group. *P < 0.05 vs. CLP-vehicle group (t test). (E) HMGB1-neutralizing antibodies confer protection against lethal sepsis in Hp KO mice. Hp KO mice (male, 8–12 weeks old) were subjected to CLP surgery, and neutralizing anti-HMGB1 mAbs or nonimmune mouse IgG were administered at 50 μg/mouse in 200 μl PBS, injected i.p. once a day for 3 days starting at 24 hours after CLP surgery. Survival was monitored for 2 weeks (n = 16 per group). *P < 0.05 vs. IgG control group (Fisher’s exact test).
Haptoglobin inhibits HMGB1-induced cytokine release from macrophages via CD163.
To elucidate the mechanism of haptoglobin-HMGB1 complex in sepsis, we studied haptoglobin-HMGB1 complexes in murine macrophage-like RAW 264.7 cells and primary human macrophages. Although HMGB1 alone did not induce apoptosis and haptoglobin alone did not affect TNF release, haptoglobin dose-dependently inhibited HMGB1-induced TNF mRNA expression and TNF and IL-8 protein secretion (Figure 3, A and B). The haptoglobin concentration that inhibited 50% of TNF and IL-8 secretion (EC50) was approximately 20 μg/ml when macrophages were stimulated by HMGB1 at 2 μg/ml (Figure 3B). Haptoglobin also significantly reduced HMGB1-induced cytokines or chemokines such as IL-6, MIG, and MCP-2, but did not significantly alter others (e.g., growth-related oncogene, GRO) (data not shown). Reasoning that CD163 is an evolutionarily conserved scavenger receptor for haptoglobin-mediated removal of hemoglobin, we investigated whether the genetic deletion of CD163 would affect haptoglobin-HMGB1 in sepsis (25). Disulfide HMGB1 alone induced IL-10 release from WT and CD163 KO macrophages, indicating that CD163 deficiency does not impair TLR4/MD2 signaling (30) (Figure 4A). However, genetic deletion of CD163 inhibited haptoglobin-HMGB1 complex–induced IL-10 secretion and almost completely abolished haptoglobin- and HMGB1-induced HO-1 expression (Figure 4A). Taken together, these experimental data indicate that CD163 is required for haptoglobin-HMGB1 complex–induced IL-10 and HO-1 production. CD163 KO mice were significantly more susceptible to sepsis lethality as compared with age-, sex-, and genetic background–matched WT mice (survival = 80% in WT animals vs. 33% in CD163 KO mice; P < 0.05, n = 18 or 20 per group, Figure 4B). The CD163 KO mice died between days 4–14 after the initiation of sepsis, a time frame that corresponds closely with the period of increased HMGB1 levels in these CD163 KO mice (Figure 4C). Serum IL-6 levels were comparable between CD163 KO and CD163-competent mice (Figure 4C), whereas serum levels of IL-10, IL-1β, and TNF were not detectable in these animals. Together with the other results, these findings indicate that CD163 is required for the haptoglobin-dependent protection against HMGB1-mediated lethality in sepsis.
Figure 3. Haptoglobin attenuates HMGB1-induced TNF expression and secretion in macrophages.

(A) Upper: RAW 264.7 cells in 96-well culture plates were stimulated with HMGB1 (2 μg/ml) plus various amounts of haptoglobin as indicated for 16 hours. TNF release was measured (n = 5 experiments). Lower: For TNF mRNA measurement, RAW 264.7 cells were incubated with HMGB1 and various amounts of haptoglobin for 12 hours. TNF mRNA expression in the cells was measured by real-time quantitative PCR. Housekeeping gene HPRT was used as a control for equal loading and integrity of RNA (n = 8 experiments). *P < 0.05, ***P < 0.001 vs. HMGB1 alone (t test). (B) Haptoglobin inhibits HMGB1-induced secretion of IL-8 and TNF in human macrophages. Primary human macrophages were incubated with HMGB1 (2 μg/ml) in the presence of haptoglobin at various concentrations as indicated for 16 hours. TNF and IL-8 released were measured. Data are from 8 (for TNF) or 5 (for IL-8) experiments. **P < 0.01, ***P < 0.001 vs. HMGB1 alone (t test).
Figure 4. Disruption of CD163 expression impairs haptoglobin-HMGB1 complex–induced IL-10 release and HO-1 expression and worsens CLP-induced sepsis.

(A) KO of CD163 impairs haptoglobin-HMGB1–induced HO-1 expression and IL-10 release. Residential peritoneal macrophages from CD163 KO and WT (male C57BL/6) mice were stimulated with HMGB1 (1 μg/ml) alone or plus haptoglobin (3 μg/ml) for 16 hours. Extracellular levels of IL-10 and intracellular HO-1 expression levels were measured by ELISAs (n = 3 experiments). *P < 0.05, ***P < 0.001 vs. WT (t test). (B) Disruption of CD163 expression worsens lethal sepsis. WT (C57BL/6) or CD163 KO (male, 8–12 weeks of age) were subjected to CLP surgery, and survival was monitored for 2 weeks (n = 18 [WT] or 20 [CD163 KO] per group). *P < 0.05 vs. WT (Fisher’s exact test). (C) Deletion of CD163 sustains sepsis-induced elevation of circulating HMGB1 levels. WT (C57BL/6) and CD163 KO mice (male, 8–12 weeks old) were subjected to CLP and euthanized at indicated time points; serum HMGB1 and IL-6 levels were measured. n = 6 (for day 3), 7 (for days 0, 7, and 14), and 8 (for days 1 and 20) mice at each time point. *P < 0.05, **P < 0.01, ***P < 0.001 vs. WT group (t test).
Haptoglobin-HMGB1 complexes bind CD163 to induce HO-1 and IL-10 expression.
Surface plasmon resonance analysis revealed that HMGB1 and haptoglobin complexes (1:1 molar ratio) bind to CD163 in a concentration-dependent manner with an apparent Kd of 130 nM. In contrast, and as previously reported, haptoglobin alone did not bind to CD163 (25). In the absence of haptoglobin, HMGB1 bound to CD163 with a much lower affinity (Kd >1 μM) (Figure 5A and data not shown). To further understand the biological consequence of haptoglobin and HMGB1 interaction, we examined the effect of these complexes on HO-1 and IL-10 expression in human monocyte THP-1 cells. Similar to the hemoglobin and haptoglobin complexes, the HMGB1 and haptoglobin complexes also induced a significant increase of IL-10 secretion and HO-1 expression, as compared with HMGB1 stimulation alone (Figure 5B). We also studied human macrophages transduced with CD163-specific shRNA and observed that the levels of CD163 expression were reduced by >90% at 72 hours after transduction; subsequent HMGB1- and haptoglobin-induced HO-1 expression and IL-10 secretion were almost completely abolished (Figure 5C). Flow cytometry gated analysis revealed that human primary macrophages exposed to FITC-labeled HMGB1 demonstrated intracellular internalization of the labeled HMGB1 in 2% of the macrophages, while 17% of the cells contained fluorescent HMGB1 after exposure to haptoglobin and HMGB1 complexes (Figure 6A). In contrast, knockdown of CD163 expression almost completely inhibited the endocytosis of haptoglobin and HMGB1 complexes (Figure 6A).
Figure 5. HMGB1 and haptoglobin complexes signal through CD163 to induce HO-1 and IL-10 expression in macrophages.

(A) Surface plasmon resonance analysis of HMGB1 and haptoglobin (Hp) binding to CD163. Recombinant human CD163 was coated on the sensor chip; the mixture of HMGB1 and Hp (1:1 molar ratio) at concentrations of 32, 65, 125, or 250 nM flowed over the chip to measure the binding affinity to CD163 (Kd is approximately 130 nM). Data are representative of 3 experiments. (B) Hp enhances HMGB1-induced HO-1 expression and IL-10 release. Human monocytic THP-1 cells were treated with dexamethasone (2.5 × 10–7 M) for 2 days to induce CD163 expression and then stimulated with HMGB1 alone (5 μg/ml) or plus haptoglobin (15 μg/ml) at 37°C for 16 hours. After incubation, extracellular levels of IL-10 were measured by ELISA. Intracellular levels of HO-1 and β-actin were measured by Western blot. Data are expressed as folds of unstimulated group after normalization to β-actin (n =3 experiments). **P < 0.01, ***P < 0.001 vs. HMGB1 alone (t test). (C) Knockdown of CD163 impairs Hp and HMGB1-induced IL-10 and HO-1 expression. Primary human macrophages were transduced with specific shRNA lentiviral particles targeting CD163 or vector alone (control). At 72 hours after transduction, cells were stimulated with HMGB1 (1 μg/ml) with or without Hp (3 μg/ml) for 16 hours. The expression of CD163, HO-1, and β-actin in cell lysate was measured by Western blot. Left: representative blots. Right: Data for HO-1 expression are presented as folds of unstimulated group after normalization to β-actin. Supernatants were collected to measure IL-10 release (n = 3 experiments). ***P < 0.001 vs. control (t test).
Figure 6. Modulation of CD163 expression divergently affects haptoglobin-mediated HMGB1 endocytosis.

(A) Knockdown of CD163 impairs endocytosis of haptoglobin and HMGB1 complexes. The endocytosis of HMGB1 and haptoglobin complexes was measured by flow cytometric analysis. Primary human macrophages were transduced with CD163 shRNA lentiviral or vector alone (control) and incubated with FITC-labeled HMGB1 (1 μg/ml) plus haptoglobin (3 μg/ml) for 30 minutes. Endocytosis of FITC-HMGB1 was evaluated by measuring the fluorescence intensity of FITC-labeled protein as a function of CD163-PE signals. Data are representative of 5 experiments. (B) Overexpression of CD163 enhances endocytosis of haptoglobin and HMGB1 complexes. HEK 293 cells were transduced with human CD163 plasmid and incubated with FITC-HMGB1 alone (1 μg/ml) or a mixture of FITC-HMGB1 (1 μg/ml) and haptoglobin (3 μg/ml) for 30 minutes at 37°C. Endocytosis of FITC-HMGB1 was evaluated as described above, and data are representative of 4 repeats. (C) Dynasore (DYN) inhibits endocytosis of HMGB1 and haptoglobin complexes. Human macrophages on cover slips were incubated with Alexa Fluor 555–labeled HMGB1 alone (1 μg/ml) or in complex with haptoglobin (3 μg/ml) for 2 hours at 37°C. Dynasore (8 μM) was preincubated with cells for 30 minutes before the addition of HMGB1 and/or haptoglobin. Upper: Endocytic uptake of HMGB1 was visualized via Carl Zeiss fluorescence microscope (red). Nuclei were counterstained with DAPI (blue). Lower: Corresponding phase contrast image of cells. Scale bars: 10 µm. Data are representative from 8 independent experiments.
Finally, we studied human embryonic kidney cells 293 (HEK 293 cells) transduced with a CD163 plasmid to study CD163-dependent endocytosis of HMGB1 and haptoglobin complexes. Flow cytometry studies demonstrated that nontransduced HEK 293 cells, without CD163, failed to endocytose FITC-HMGB1 or FITC-HMGB1 and haptoglobin complexes. CD163-transduced cells endocytosed FITC-HMGB1 and haptoglobin complexes (17% of the cells) vs. FITC-HMGB1 alone (2%) (Figure 6B). In agreement with these findings, fluorescent-conjugated (Alexa Fluor 555–conjugated) HMGB1 was internalized by primary human macrophages when incubated for 2 hours at 37°C compared with untreated cells (Figure 6C). This cellular uptake of Alexa Fluor 555–HMGB1 was further enhanced by haptoglobin but attenuated by pretreatment with dynasore, an endocytosis inhibitor. Together, these results indicate that CD163 mediates endocytic uptake of HMGB1-haptoglobin complexes as a protective mechanism against HMGB1 lethality in sepsis.
Discussion
“Haptoglobin” is derived from the Greek word haptein, meaning to fasten or bind, reflecting the exceptional capacity to capture free hemoglobin. Extracellular haptoglobin serves as the major plasma protein in capturing hemoglobin, and the haptoglobin-hemoglobin complexes are subsequently removed from the circulation by CD163-positive macrophages in the liver, spleen, and BM. Once ingested, hemoglobin is transferred to early endosomes and degraded into amino acids and heme, which is further metabolized by the HO-1 enzyme to produce antiinflammatory metabolites (carbon monoxide [CO], ferrous iron [Fe2+], and biliverdin) (31). As reported here, we discovered that haptoglobin also binds HMGB1 and confers protection against HMGB1-mediated lethality in sepsis through a CD163-dependent mechanism (Figure 7).
Figure 7. Haptoglobin as a negative regulator of HMGB1.
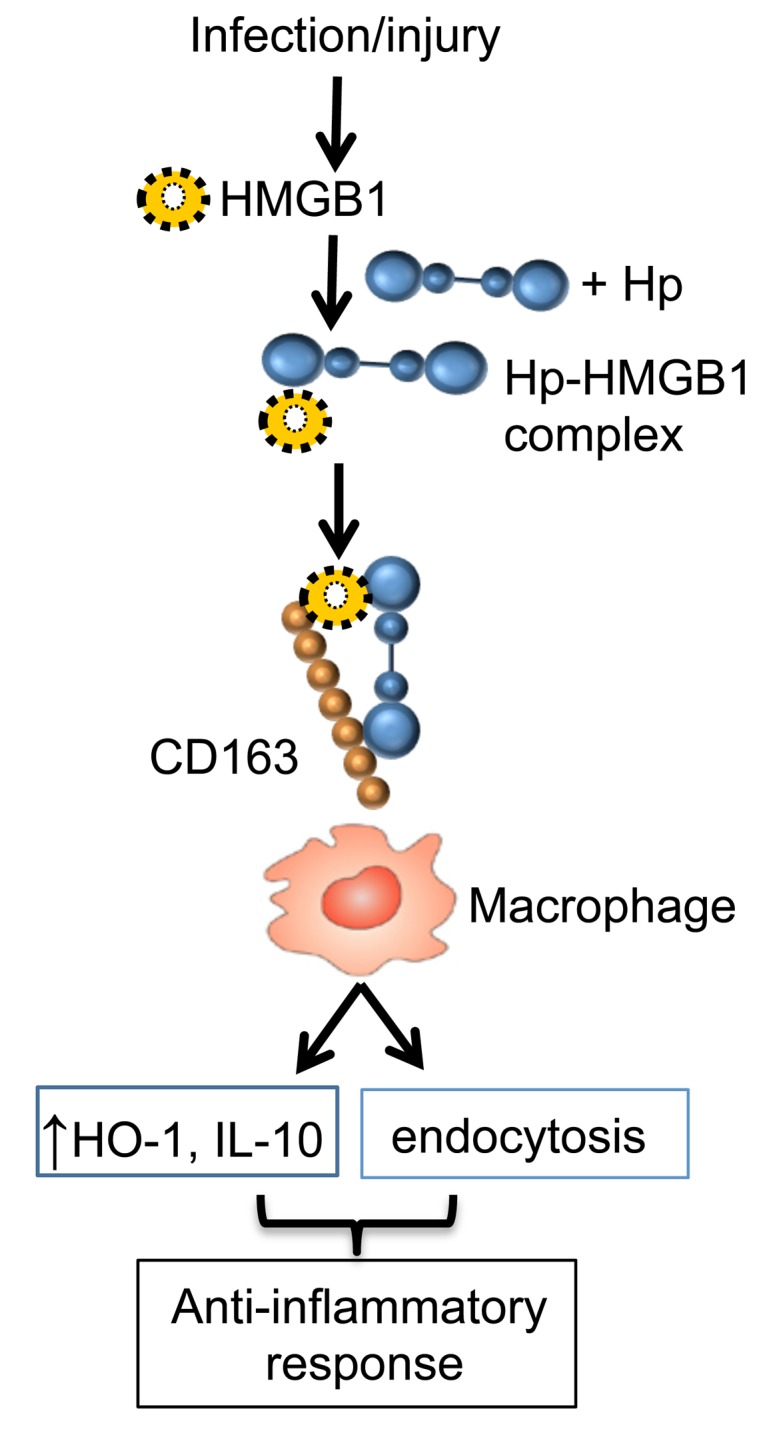
During infection or injury, HMGB1 is actively secreted or passively released outside of the cells. In the presence of haptoglobin, HMGB1 is captured; the haptoglobin and HMGB1 complexes act via CD163 to elicit an anti-inflammatory response and parallel endocytosis of HMGB1-haptoglobin complexes. Thus, haptoglobin, a liver-derived acute phase protein, acts as an endogenous HMGB1 inhibitor via a CD163-dependent antiinflammatory pathway.
The synthesis of haptoglobin and other acute phase protein in the liver during infection and injury is upregulated by IL-1, IL-6, and other cytokines. We found that disruption of haptoglobin expression significantly increased the mortality of sepsis and prolonged the exposure time to elevated HMGB1 levels as compared with WT controls. Administration of haptoglobin conferred significant protection against lethal sepsis in both WT and haptoglobin KO mice. Although it remains elusive whether there is a preference for any haptoglobin phenotype (Hp 1-1, 2-1, and 2-2) to bind HMGB1. A large body of prior work established the pathogenic role of extracellular HMGB1 in sepsis and other animal models of infectious and sterile inflammatory diseases (5, 6). HMGB1 antagonists including neutralizing antibodies and peptide antagonists (e.g., the recombinant Box A domain), as well as small chemical inhibitors of HMGB1 release or actions, confer protection and have established the damaging role of HMGB1 in pathogenesis (3, 30). Here, administration of HMGB1-neutralizing antibodies to haptoglobin KO mice conferred significant protection by targeting the persistently elevated HMGB1 levels.
CD163 is a scavenger receptor expressed on monocytes/macrophages and in relatively low abundance in quiescent circulating monocytes. CD163 expression is enhanced by glucocorticoids, antiinflammatory cytokines (e.g., IL-10), or growth factors that can promote monocyte-to-macrophage differentiation (e.g., macrophage CSF, M-CSF) (32, 33). The expression of CD163 is downregulated by proinflammatory cytokines (e.g., IFN-γ) and growth factors that induce monocyte to DC differentiation (e.g., granulocyte M-CSF, GM-CSF) (34). CD163 is abundant in mature tissue macrophages including Kupffer cells in the liver, red pulp macrophages in the spleen, resident BM macrophages, and alveolar macrophages in the lungs (34, 35). CD163 is also a receptor for cytokine TNF-like weak inducer of apoptosis (TWEAK, a secreted cytokine belonging to the TNF super-family) (36, 37). CD163 can be shed as a soluble protein, but little is known about the biological functions of the soluble CD163 (reviewed by ref. 37). At present, our in vivo studies do not exclude the possibility that free hemoglobin and HMGB1 synergize to activate proinflammatory responses, which could be similarly downmodulated by the common haptoglobin-CD163 axis. Other plasma proteins that can bind HMGB1 and confer protection include thrombomodulin (38), soluble receptor for advanced glycation end products (s-RAGE) (39), and Siglec-10/CD24 (40). Our investigation does reveal a mechanism by which haptoglobin negatively modulates the proinflammatory properties of HMGB1, a critically important mediator of infection- and injury-elicited inflammatory diseases (10).
Thus, haptoglobin is an endogenous HMGB1 binding protein that directs HMGB1 to a CD163-dependent receptor pathway that activates antiinflammatory signaling in the monocyte-macrophage lineage. Since the administration of haptoglobin conferred significant protection against CLP-induced sepsis lethality in both WT and haptoglobin KO mice, it will be interesting to assess the efficacy of haptoglobin-based therapies in other HMGB1-dependent inflammatory diseases.
Methods
Reagents.
Human haptoglobin (from pooled human plasma, a mixture of 1-1, 2-1, and 2-2 phenotype, catalog 3536), REDExtract-N-Amp Tissue PCR kit (catalog XNATS), Triton X-114, imidazole, monoclonal anti-human haptoglobin antibodies (catalog H6395), nonimmune mouse IgG, Drabkin’s reagents (catalog D5941), human M-CSF, DAPI, and dexamethasone (catalog D2915) were purchased from Sigma-Aldrich. TACS MTT Cell proliferation assay kit was from Trevigen. Trizol reagent was from Invitrogen. The RevertAid First Strand cDNA Synthesis Kit was from Fermentas. SYBR Premix Ex Taq II was from Takara Bio Inc. Thioglycollate medium was purchased from BD Biosciences. Primers for quantitative PCR (qPCR), trypsin-EDTA, and carbenicillin were from Invitrogen. Protein A/G agarose and isopropyl-D-thiogalactopyranoside (IPTG) were purchased from Pierce. NHS-activated Sepharose 4 fast flow beads were obtained from GE Healthcare (catalog 17-0906-01). E. coli strain DH5α was from Novagen. Recombinant human CD163 protein, CD163 expression plasmid (SC117495, NM_004244.3), and MegaTran 1.0 transfection reagent were purchased from Origene. Antibodies for human HO-1 (catalog ab52946) and CD163 (catalog MCA1853) were from Abcam and Serotec, respectively. Anti–human CD163-PE antibody was from BioLegend. FITC antibody labeling kit was from Thermo Scientific. Dynasore was from Tocris Bioscience.
Preparations of recombinant HMGB1, redox-modified HMGB1, and HMGB1 mAbs.
Recombinant HMGB1 was expressed in E. coli and purified to homogeneity as previously described (41, 42). HMGB1-FITC was prepared by using a Pierce FITC Antibody Labeling Kit (Thermo Scientific). Anti-HMGB1 mAbs were generated as described previously (17). Nonimmune mouse IgG was used as isotype control in experiments when anti-HMGB1 mAb was used. HMGB1 with redox modifications was created as described previously (14).
Cell culture.
Murine macrophage–like RAW 264.7, human acute monocytic leukemia cell line THP-1, and HEK 293 cells were obtained from ATCC. Cells were used at 90% confluence, and treatment was carried out in serum-free Opti-MEM I medium (Invitrogen). Human primary monocytes were purified by density gradient centrifugation through Ficoll from blood donated by normal individuals to the Long Island Blood Bank (New York Blood Center, Melville, New York, USA). Cells were allowed to differentiate into macrophages for 7 days in complete DMEM medium and 1 ng/ml M-CSF as described previously (43). Primary mouse residential macrophages were obtained by washing the peritoneal cavity with 5 ml sterile PBS. Thioglycollate-elicited mouse macrophages were obtained as previously described (43). Cells were plated in culture dishes and used after seeding overnight. Experiments were carried out in Opti-MEM I medium. Cell apoptosis was measured using TACS MTT cell proliferation assay.
Measurements of CD163 and HO-1 expression, levels of cytokines, serum hemoglobin, and haptoglobin.
THP-1 cells were incubated with dexamethasone for 2 days to induce CD163 expression. Cells were exposed to HMGB1 in the presence or absence of haptoglobin for 16 hours, and the expression of HO-1 and CD163 in cell lysate was measured by Western blot analysis as previously described (24). In some experiments, HO-1 expression in cell lysate was measured using ELISA kits (Enzo Life Science). HMGB1, TNF, IL-10, and IL-8 released in the cell culture supernatants were measured by commercially obtained ELISA kits per manufacturer’s instructions (IBL International and R&D Systems Inc.). In some experiments, HMGB1 levels were measured by Western immunoblotting analysis as previously described (3, 41). The levels of HMGB1 were calculated with reference to standard curves generated with purified HMGB1. Cytokine expression profile from primary human macrophages was determined by human cytokine array C1 (Raybiotech) according to the manufacturer’s instructions. Twenty-two cytokines or chemokines were determined simultaneously. Serum hemoglobin levels were determined using Drabkin’s reagents (44) following the manufacturer’s instructions (Sigma-Aldrich). The amounts of hemoglobin in the samples were calculated with reference to standard curves generated with purified mouse hemoglobin. Haptoglobin levels were measured by quantitative ELISA kits following the manufacturer’s instructions (Genway Biotech Inc.) or by Western blot.
Immunoprecipitation assay.
Recombinant disulfide HMGB1 (10 μg) was precleared with protein A/G agarose (20 μl beads, 20 minutes at 4°C) and incubated with human haptoglobin (5 μg) at 4°C for 4 hours with gentle shaking. The mixture was added to beads (30 μl drained beads) containing either A/G agarose alone (control) or the same agarose coupled with anti-haptoglobin mAb overnight at 4°C. After washing extensively with PBS containing 0.1% Triton X-100, beads were boiled for 5 minutes and proteins bound to the beads were analyzed by Western blot probed with anti-HMGB1 mAbs.
To test binding of haptoglobin to isoforms of HMGB1, human haptoglobin was cross-linked to NHS-activated Sepharose 4 Fast Flow beads according to the manufacturer’s instructions (GE Healthcare). Approximately 16 mg haptoglobin is bound to 1 ml of drained beads. Control (empty) beads were prepared the same way without adding the haptoglobin. Various HMGB1 isoforms (2 μg) were added to haptoglobin-containing beads (10 μl per reaction) or empty beads for 2 hours at room temperature with gentle rotation. Samples were centrifuged, and beads were washed extensively with PBS containing 0.1% Triton X-100. After washing, elutes from the beads were analyzed by Western blot probed with anti-HMGB1 mAbs.
To measure HMGB1 and haptoglobin in septic human serum using immunoprecipitation assay, serum samples were obtained from patients with clinical signs and symptoms of sepsis after admission to the Department of Emergency Medicine, Northwell Health. As controls, blood samples were collected from healthy volunteers. The study was conducted in accordance with the declaration of Helsinki and was approved by the IRB at the Feinstein Institute for Medical Research. Samples (10 μl, precleared with protein A/G agarose) were incubated with A/G agarose alone (control) or the same agarose coupled with anti–human haptoglobin mAbs overnight at 4°C. After washing extensively with PBS with 0.1% Triton X-100, beads were boiled for 5 minutes and proteins bound to the beads were analyzed by Western blot probed with monoclonal anti-HMGB1 or anti–human haptoglobin antibodies.
Endocytosis of HMGB1 and haptoglobin complexes by CD163-expressing HEK 293 cells or primary human macrophages.
Transduction of CD163 plasmid to HEK 293 cells was performed according to the manufacturer’s instructions. Briefly, HEK 293 cells in a 6-well plate were transduced with CD163 plasmid DNA (in MegaTran 1.0 reagent) for 3 hours at 37°C. The transduction reaction was terminated by washing the cells 3 times with PBS. At 72 hours after transduction, cells were incubated with FITC-HMGB1 alone (1 μg/ml) or plus haptoglobin (3 μg/ml) for 30 minutes at room temperature. After washing to remove any nonspecific binding of FITC-HMGB1, cells were stained with anti–CD163-PE antibody at 4°C for 45 minutes. Cells were washed with PBS and then analyzed by a BD LSRFortessa cytometer (BD Biosciences).
Fluorescence microscopy.
Alexa Fluor 555–labeled HMGB1 was performed according to the manufacturer’s instructions (Molecular Probes). To identify the haptoglobin-induced HMGB1 endocytosis process, primary human macrophages on coverslips were preincubated with endocytosis inhibitor Dynasore (8 μM) for 30 minutes (45) and then incubated with Alexa Fluor 555–labeled HMGB1 (1 μg/ml) in the presence and absence of haptoglobin (3 μg/ml) for 2 hours at 37°C in a 5% CO2 incubator. The cells were rinsed with PBS and fixed using 4% PFA (paraformaldehyde) for 30 minutes at room temperature, stained with DAPI for nuclear staining, and mounted on the slide using permanent mounting medium (Vecta mount). Images were taken using a Carl Zeiss fluorescence microscope with a ×40 objective.
Human primary macrophages were incubated with FITC-HMGB1 (1 μg/ml) alone or plus haptoglobin (3 μg/ml) for 30 minutes at 37°C. After washing with PBS to remove nonspecific binding, macrophages were stained with anti–CD163-PE antibody at 4°C for 45 minutes and then analyzed by a FACS Calibur flow cytometer. The optimal ratio of HMGB1 to haptoglobin was determined by measuring the endocytosis of FITC-HMGB1 (1 μg/ml) plus increasing amounts of haptoglobin (0–10 μg/ml), and we observed the optimal binding is 1:1 molar ratio for HMGB1 to haptoglobin.
Surface plasmon resonance analysis.
Surface plasmon resonance analysis of binding of HMGB1 to haptoglobin was conducted using the BIAcore 3000 or T200 instrument as previously described (14, 30). For HMGB1-haptoglobin binding analyses, purified human haptoglobin (1 μM, in 10 mM sodium acetate, pH 4.5) was immobilized onto the sensor chip. To evaluate binding, a series of concentrations of analytes (HMGB1 0–500 nM) were then passed over the sensor chip. The association of analyte and ligand was recorded respectively by surface plasmon resonance. Results were analyzed using the software BIAeval 3.2 (BIAcore Inc.). For the binding studies of CD163 to the complexes of haptoglobin and HMGB1, human CD163 protein was coated on the sensor chip. Increasing concentrations of the complexes of HMGB1 and haptoglobin formed at 1:1 molar ratio were flowed over the sensor chip; analyses were performed in a similar manner as described above.
NMR analyses to determine the interacting amino acid residues between redox isoforms of HMGB1 and haptoglobin.
Recombinant Δ30-HMGB1 (HMGB1 expressed minus 30 C-terminal amino acids) of various redox states was produced in complex with haptoglobin, and the nature of the interaction was examined by NMR. Standard heteronuclear single quantum correlation–based (HSQC-based) 3D triple resonance (1H, 15N, and 13C) was used to assign the backbone residues as described previously (13, 46). 3D HNCO (provides the connectivity between the amide of a residue with the carbonyl carbon of the preceding residues) and HNCACO (correlates the amide resonance of a residue with the carbonyl carbon of the same residue, as well as that of the preceding residues) experiments were used to sequentially assign the backbone NH via the carboxyl group. CBCACONH and HNCACB experiments were used to assign the backbone NH via the backbone Cα and side-chain Cβ groups. 15N- and/or 13C-labeled recombinant proteins were expressed in minimal media and purified by ion-exchange and size-exclusion chromatography. NMR samples were prepared at 10–100 μM concentrations in PBS with a final concentration of 10% D2O (deuterium oxide, 2H2O). Samples were placed in standard 5-mm NMR tubes. Chemical shift perturbation studies were carried out using either a Bruker Avance III 600 or 800 MHz NMR system equipped with triple (TCI) resonance cryoprobes at 25oC (298K). All data were processed using Topspin 3.1 (Bruker) and analyzed using CCPN (collaborative computing project for NMR) analysis. The redox status of cysteines on HMGB1 was confirmed by liquid chromatography tandem mass spectrometry as previously described (14).
CD163 shRNA lentiviral transduction of human macrophages.
Ficoll gradient–separated human monocytes were differentiated for 7 days in complete DMEM medium containing 10% heat-inactivated human serum, 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 ng/ml M-CSF in 24-well Primaria tissue culture plates at 5 × 105 cells/well. After screening of 5 human CD163 (NM_004244) Mission shRNA lentiviral clones using FACS analysis, the clone that showed the best knockdown efficiency (TRCN0000421748) was selected. The selected clone was transduced into human macrophages by spinoculation according to the manufacturer’s instructions (Sigma-Aldrich).
Animal experiments.
Male Sprague-Dawley rats (8–12 weeks old) and male BALB/c, or C57BL/6 mice (8–12 weeks old) were obtained from Taconic Laboratories and allowed to acclimate for 7 days before use in the experiment.
KO (haptoglobin or CD163) and WT mice.
Haptoglobin KO C57BL/6 mice were generated and provided by Dominique P.V. de Kleijn (Utrecht, the Netherlands). CD163 KO C57BL/6 mice were purchased from KOMP repository (University of California-Davis, Davis, California, USA). Since the KO mice are all derived from C57BL/6 mice, small colonies of WT C57BL/6 were maintained under the same conditions as the KO mice. In some experiments, BALB/c mice were used. To ascertain that newly bred animals had the expected gene deficiencies, we carried out genotyping on genomic DNA obtained from tail snips (26). Animals were anesthetized using EMLA cream (Lidocaine and prilocaine mixture, AstraZeneca LP) applied to the intact skin, and 1- to 2-mm tail snips were obtained. Genomic DNA was extracted using a tissue PCR kit. The PCR primers used for haptoglobin KO were: Neo (544 bp) forward: 5′-AGGCTATTCGGCTATGACTGG-3′ and reverse: 5′-CCACCATGATATTCGGCAAGC-3′. Haptoglobin (319 bp) forward: 5′-CTGTGGAGTTGGGCAATGATG-3′ and reverse: 5′-AACCAAGTGCTCCACATAGCC-3′. The PCR amplification conditions include 95°C for 4 minutes, followed by 35 cycles of 94°C for 1 minute, 60°C for 1 minute, 72°C for 40 seconds, and finally 10 minutes at 72°C (Applied Biosystems). The PCR primers for CD163 KO mice were: Neo (632 bp) forward: 5′-GCAGCCTCTGTTCCACATACACTTCA-3′ and reverse: 5′-ATTCTAGGCTTGCCTGCTCTCTATCG-3′. CD163 (385 bp) forward: 5′-TCACTCCTGGGCTGCACGTAAAC-3′ and reverse: 5′-GATGTTATTTGCCATACAGGAGAATTG-3′. The PCR amplification conditions were: 94°C for 3 minutes, followed by 35 cycles of 94°C for 30 seconds, 58°C for 30 seconds, 72°C for 40 seconds, and final extension at 72°C for 5 minutes.
CLP.
WT (male, C57BL/6 or BALB/c), haptoglobin, or CD163 KO mice (male, 8–12 weeks of age) were subjected to CLP procedure (3). In some experiments, survival was monitored for 2 weeks. In separate experiments, mice were euthanized at 1–21 days after CLP surgery through overexposure to CO2. Sera were isolated and stored at –20ºC until analysis. In some experiments, male WT (C57BL/6) or haptoglobin KO mice were subjected to CLP surgery and received haptoglobin treatment (500 μg/mouse i.p. at 24 and 36 hours after CLP surgery), and mice were euthanized at 48 hours after CLP. Sera were isolated for analyses. For other treatment studies, administration of haptoglobin or monoclonal anti-HMGB1 antibody was given i.p., and survival was monitored for 2 weeks.
Perfusion studies using haptoglobin in an extracorporeal device in septic rats.
CLP was performed in rats as previously described (3). At 48 hours after CLP, the septic rats were anesthetized with isofluran; both femoral veins were cannulated using a 24-gauge angiocatheter and connected to a sterilized miniaturized circuit that had been primed with heparinized saline (10 units/ml). The blood was pumped through the filter linked with haptoglobin or empty beads (1.5 ml beads) for 1 hour at a flow rate of 1 ml/min. The filter beads were then washed extensively with PBS (5 times); 2× Laemmli buffer was added to the beads to elute bound proteins. Western blot was performed on bead eluate using anti-HMGB1 antibodies. The blood was pumped through the filter linked with haptoglobin or empty beads (1.5 ml beads) for 1 hour at a flow rate of 1 ml/min. The filter beads were then washed extensively with saline. After washing with PBS 5 times, 2× Laemmli buffer was added to the beads to elute bound proteins. Western blot was performed on bead eluate using anti-HMGB1 antibodies.
Statistics.
Data are presented as means ± SEM unless otherwise stated. Differences between treatment groups were determined by Student’s t test. Differences between treatment groups in animal survival studies were determined using 2-tailed Fisher’s exact test. P values less than 0.05 were considered statistically significant.
Study Approval.
All animal procedures were approved by the Institutional Animal Care and Use Committee. Mice were housed in the animal facility of the Feinstein Institute for Medical Research under standard temperature and light and dark cycles.
Author contributions
HY, SZ, YAL, MKG, YW, and MA performed the experiments. YAA and MH conducted SPR (BIAcore) analysis. JL provided recombinant HMGB1 and anti-HMGB1 mAbs. DPVDK provided the haptoglobin KO mice. HY, HW, WL, YW, YAL, SSC, UA, KJT, MA, PSO, and HSW analyzed data. DJA coordinated the NMR analysis and interpretation. HY, HW, JR, UA, and KJT wrote the paper.
Acknowledgments
This work was supported by grants from the NIH, R01GM62508 (to K.J. Tracey), R01AT005076 and R01GM063075 (to H. Wang), and R01GM098446 (to H. Yang). D.J. Antoine would like to acknowledge support from the Wellcome Trust and the University of Liverpool Technology Directorate voucher scheme. We would like to thank Zhongliang Ju for his technical assistance.
Footnotes
Yaakov A. Levine’s present address is SetPoint Medical Inc., Valencia, California, USA.
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:JCI Insight. 2016;1(7):e85375. doi:10.1172/jci.insight.85375.
Contributor Information
Huan Yang, Email: HYang@NSHS.edu.
Haichao Wang, Email: hwang@nshs.edu.
Yongjun Wang, Email: ywang12@nshs.edu.
Meghan Addorisio, Email: maddorisio@nshs.edu.
Shu Zhu, Email: szhu@nshs.edu.
Wei Li, Email: wli2@nshs.edu.
Jianhua Li, Email: jli@nshs.edu.
H. Shaw Warren, Email: warren@helix.mgh.harvard.edu.
Mingzhu He, Email: mhe@nshs.edu.
Yousef Al-Abed, Email: yalabed@nshs.edu.
Jesse Roth, Email: jesserothmd@hotmail.com.
Sangeeta S. Chavan, Email: schavan@nshs.edu.
Ulf Andersson, Email: Ulf.Andersson@ki.se.
Kevin J. Tracey, Email: kjtracey@nshs.edu.
References
- 1.Angus DC, Wax RS. Epidemiology of sepsis: an update. Crit Care Med. 2001;29(7 Suppl):S109–S116. doi: 10.1097/00003246-200107001-00035. [DOI] [PubMed] [Google Scholar]
- 2.Lagu T, Rothberg MB, Shieh MS, Pekow PS, Steingrub JS, Lindenauer PK. Hospitalizations, costs, and outcomes of severe sepsis in the United States 2003 to 2007. Crit Care Med. 2012;40(3):754–761. doi: 10.1097/CCM.0b013e318232db65. [DOI] [PubMed] [Google Scholar]
- 3.Yang H, et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci U S A. 2004;101(1):296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang H, Tracey KJ. Targeting HMGB1 in inflammation. Biochim Biophys Acta. 2010;1799(1–2):149–156. doi: 10.1016/j.bbagrm.2009.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Ann Rev Immunol. 2011;29:139–162. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsung A, Tohme S, Billiar TR. High-mobility group box-1 in sterile inflammation. J Intern Med. 2014;276(5):425–443. doi: 10.1111/joim.12276. [DOI] [PubMed] [Google Scholar]
- 7.Bloom O, Wang H, Ivanova S, Vishnubhakat JM, Ombrellino M, Tracey KJ. Hypophysectomy, high tumor necrosis factor levels, and hemoglobinemia in lethal endotoxemic shock. Shock. 1998;10(6):395–400. doi: 10.1097/00024382-199812000-00003. [DOI] [PubMed] [Google Scholar]
- 8.Yang H, et al. Globin attenuates the innate immune response to endotoxin. Shock. 2002;17(6):485–490. doi: 10.1097/00024382-200206000-00008. [DOI] [PubMed] [Google Scholar]
- 9.Kaca W, Roth RI, Levin J. Hemoglobin, a newly recognized lipopolysaccharide (LPS)-binding protein that enhances LPS biological activity. J Biol Chem. 1994;269(40):25078–25084. [PubMed] [Google Scholar]
- 10.Yang H, Wang H, Chavan SS, Andersson U. High mobility group box protein 1 (HMGB1): The prototypical endogenous danger molecule. Mol Med. 2015;21(Suppl 1):S6–S12. doi: 10.2119/molmed.2015.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Antoine DJ, Harris HE, Andersson U, Tracey KJ, Bianchi ME. A systematic nomenclature for the redox states of high mobility group box (HMGB) proteins. Mol Med. 2014;20:135–137. doi: 10.2119/molmed.2014.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang D, Billiar TR, Lotze MT. A Janus tale of two active high mobility group box 1 (HMGB1) redox states. Mol Med. 2012;18:1360–1362. doi: 10.2119/molmed.2012.00314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Venereau E, et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med. 2012;209(9):1519–1528. doi: 10.1084/jem.20120189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang H, et al. Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1) Mol Med. 2012;18:250–259. doi: 10.2119/molmed.2011.00389. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Yang H, Antoine DJ, Andersson U, Tracey KJ. The many faces of HMGB1: molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J Leukoc Biol. 2013;93(6):865–873. doi: 10.1189/jlb.1212662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang Y, et al. Regulation of posttranslational modifications of HMGB1 during immune responses. Antioxid Redox Signal. 2016;24(12):620–624. doi: 10.1089/ars.2015.6409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qin S, et al. Role of HMGB1 in apoptosis-mediated sepsis lethality. J Exp Med. 2006;203(7):1637–1642. doi: 10.1084/jem.20052203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Larsen R, et al. A central role for free heme in the pathogenesis of severe sepsis. Sci Transl Med. 2010;2(51):e85375. doi: 10.1126/scitranslmed.3001118. [DOI] [PubMed] [Google Scholar]
- 19.Bornside GH, Bouis PJ, Jr, Cohn I., Jr Enhancement of Escherichia coli infection and endotoxic activity by hemoglobin and ferric ammonium citrate. Surgery. 1970;68(2):350–355. [PubMed] [Google Scholar]
- 20.Bornside GH, Cohn I., Jr Hemoglobin as a bacterial virulence-enhancing factor in fluids produced in strangulation intestinal obstruction. Am Surg. 1968;34(1):63–67. [PubMed] [Google Scholar]
- 21.Yueh SC, Lai YA, Chen WL, Hsu HH, Mao SJ. An improved method for haptoglobin 1-1, 2-1, and 2-2 purification using monoclonal antibody affinity chromatography in the presence of sodium dodecyl sulfate. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;845(2):210–217. doi: 10.1016/j.jchromb.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 22.Lim SK, et al. Increased susceptibility in Hp knockout mice during acute hemolysis. Blood. 1998;92(6):1870–1877. [PubMed] [Google Scholar]
- 23.Schaer CA, Schoedon G, Imhof A, Kurrer MO, Schaer DJ. Constitutive endocytosis of CD163 mediates hemoglobin-heme uptake and determines the noninflammatory and protective transcriptional response of macrophages to hemoglobin. Circ Res. 2006;99(9):943–950. doi: 10.1161/01.RES.0000247067.34173.1b. [DOI] [PubMed] [Google Scholar]
- 24.Moreno JA, et al. The CD163-expressing macrophages recognize and internalize TWEAK: potential consequences in atherosclerosis. Atherosclerosis. 2009;207(1):103–110. doi: 10.1016/j.atherosclerosis.2009.04.033. [DOI] [PubMed] [Google Scholar]
- 25.Kristiansen M, et al. Identification of the haemoglobin scavenger receptor. Nature. 2001;409(6817):198–201. doi: 10.1038/35051594. [DOI] [PubMed] [Google Scholar]
- 26.Arredouani MS, Kasran A, Vanoirbeek JA, Berger FG, Baumann H, Ceuppens JL. Haptoglobin dampens endotoxin-induced inflammatory effects both in vitro and in vivo. Immunology. 2005;114(2):263–271. doi: 10.1111/j.1365-2567.2004.02071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boretti FS, et al. Sequestration of extracellular hemoglobin within a haptoglobin complex decreases its hypertensive and oxidative effects in dogs and guinea pigs. J Clin Invest. 2009;119(8):2271–80. doi: 10.1172/JCI39115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baek JH, et al. Hemoglobin-driven pathophysiology is an in vivo consequence of the red blood cell storage lesion that can be attenuated in guinea pigs by haptoglobin therapy. J Clin Invest. 2012;122(4):1444–1458. doi: 10.1172/JCI59770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Irwin DC, et al. Hemoglobin-induced lung vascular oxidation, inflammation, and remodeling contribute to the progression of hypoxic pulmonary hypertension and is attenuated in rats with repeated-dose haptoglobin administration. Free Radic Biol Med. 2015;82:50–62. doi: 10.1016/j.freeradbiomed.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang H, et al. MD-2 is required for disulfide HMGB1-dependent TLR4 signaling. J Exp Med. 2015;212(1):5–14. doi: 10.1084/jem.20141318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moestrup SK, Moller HJ. CD163: a regulated hemoglobin scavenger receptor with a role in the anti-inflammatory response. Ann Med. 2004;36(5):347–354. doi: 10.1080/07853890410033171. [DOI] [PubMed] [Google Scholar]
- 32.Komori H, et al. alpha(1)-Acid glycoprotein up-regulates CD163 via TLR4/CD14 protein pathway: possible protection against hemolysis-induced oxidative stress. J Biol Chem. 2012;287(36):30688–30700. doi: 10.1074/jbc.M112.353771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams L, Jarai G, Smith A, Finan P. IL-10 expression profiling in human monocytes. J Leukoc Biol. 2002;72(4):800–809. [PubMed] [Google Scholar]
- 34.Buechler C, Ritter M, Orso E, Langmann T, Klucken J, Schmitz G. Regulation of scavenger receptor CD163 expression in human monocytes and macrophages by pro- and antiinflammatory stimuli. J Leukoc Biol. 2000;67(1):97–103. [PubMed] [Google Scholar]
- 35.Bachli EB, Schaer DJ, Walter RB, Fehr J, Schoedon G. Functional expression of the CD163 scavenger receptor on acute myeloid leukemia cells of monocytic lineage. J Leukoc Biol. 2006;79(2):312–318. doi: 10.1189/jlb.0605309. [DOI] [PubMed] [Google Scholar]
- 36.Bover LC, et al. A previously unrecognized protein-protein interaction between TWEAK and CD163: potential biological implications. J Immunol. 2007;178(12):8183–8194. doi: 10.4049/jimmunol.178.12.8183. [DOI] [PubMed] [Google Scholar]
- 37.Van Gorp H, Delputte PL, Nauwynck HJ. Scavenger receptor CD163, a Jack-of-all-trades and potential target for cell-directed therapy. Mol Immunol. 2010;47(7–8):1650–1660. doi: 10.1016/j.molimm.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 38.Abeyama K, et al. The N-terminal domain of thrombomodulin sequesters high-mobility group-B1 protein, a novel antiinflammatory mechanism. J Clin Invest. 2005;115(5):1267–1274. doi: 10.1172/JCI22782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song F, et al. RAGE regulates the metabolic and inflammatory response to high-fat feeding in mice. Diabetes. 2014;63(6):1948–1965. doi: 10.2337/db13-1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen GY, Tang J, Zheng P, Liu Y. CD24 and Siglec-10 selectively repress tissue damage-induced immune responses. Science. 2009;323(5922):1722–1725. doi: 10.1126/science.1168988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang H, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285(5425):248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 42.Li J, et al. Recombinant HMGB1 with cytokine-stimulating activity. J Immunol Methods. 2004;289(1–2):211–223. doi: 10.1016/j.jim.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 43.Yang H, et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci U S A. 2010;107(26):11942–11947. doi: 10.1073/pnas.1003893107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rice EW. Rapid determination of total hemoglobin as hemiglobin cyanide in blood containing carboxyhemoglobin. Clin Chim Acta. 1967;18(1):89–91. doi: 10.1016/0009-8981(67)90254-9. [DOI] [PubMed] [Google Scholar]
- 45.Saenz R, et al. TLR4-dependent activation of dendritic cells by an HMGB1-derived peptide adjuvant. J Transl Med. 2014;12:e85375. doi: 10.1186/1479-5876-12-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi HW, et al. Aspirin’s active metabolite salicylic acid targets high mobility group box 1 to modulate inflammatory responses. Mol Med. 2015;21:526–535. doi: 10.2119/molmed.2015.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]