

Abstract
Antagonists of retinol-binding protein 4 (RBP4) impede ocular uptake of serum all-trans retinol (1) and have been shown to reduce cytotoxic bisretinoid formation in the retinal pigment epithelium (RPE), which is associated with the pathogenesis of both dry age-related macular degeneration (AMD) and Stargardt disease. Thus, these agents show promise as a potential pharmacotherapy by which to stem further neurodegeneration and concomitant vision loss associated with geographic atrophy of the macula. We previously disclosed the discovery of a novel series of nonretinoid RBP4 antagonists, represented by bicyclic [3.3.0]-octahydrocyclopenta[c]pyrrolo analogue 4. We describe herein the utilization of a pyrimidine-4-carboxylic acid fragment as a suitable isostere for the anthranilic acid appendage of 4, which led to the discovery of standout antagonist 33. Analogue 33 possesses exquisite in vitro RBP4 binding affinity and favorable drug-like characteristics and was found to reduce circulating plasma RBP4 levels in vivo in a robust manner (>90%).
Graphical abstract

INTRODUCTION
Atrophic (dry) age-related macular degeneration (AMD) is a chronic and slowly progressing neurodegenerative ocular disorder that involves geographic atrophy of the central region of the retina called the macula.1 Approximately 85% to 90% of reported cases of macular degeneration are of the atrophic form,1 and this disorder is the leading cause of vision loss for individuals aged 60 years or older.2 The dry AMD patient population is estimated at 11 million individuals in the US alone and is expected to double by 2020.3 Currently, there is no FDA-approved treatment available for patients suffering from this debilitating disease.
Accumulation of lipofuscin, a granular substance comprising cytotoxic bisretinoid fluorophores, in the retinal pigment epithelium (RPE) has been implicated as one of several pathogenic factors contributing to photoreceptor cell degeneration in the macula of dry AMD patients.4 In addition, dramatic retinal accumulation of lipofuscin is also believed to be the key etiological factor in autosomal recessive Stargardt disease, an untreatable and inherited form of juvenile-onset macular degeneration caused by genetic mutations in the Abca4 gene. Abca4 is a key component of the visual retinoid cycle, and this critical gene product loss is functionally recapitulated in the Abca4−/− knockout mouse model.5 The best studied bisretinoid component of lipofuscin is N-retinide-N-retinylidene ethanolamine (A2E),4i a naturally occurring byproduct of the visual cycle derived from phosphatidylethanolamine and two molecules of all-trans retinal. A2E induces several toxic effects that may lead to the observed abnormalities in the dry AMD RPE.4h,6 Therefore, it is postulated that disruption or inhibition of de novo A2E formation in the RPE may provide a pharmacological intervention point by which to delay or retard the neurodegenerative processes associated with dry AMD and Stargardt disease.7 Currently, there are several agents in various stages of development that interfere with the visual cycle under clinical investigation for the treatment of dry AMD.3,8 We recently reported our initial efforts in this field, which are based on the hypothesis that reduction of the ocular uptake of serum all-trans retinol (retinol, vitamin A) (1) (Figure 1) via reduction in circulating levels of serum retinol binding protein 4 (RBP4) can lead to a reduced rate of bisretinoid A2E accumulation in the retina.9
Figure 1.

Retinol (1), fenretinide (2), A1120 (3), and 2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)octahydrocyclopenta[c]pyrrole-2-carboxamido)benzoic acid (4).
Retinol is transported systemically via a tertiary complex with RBP4 and transthyretin (TTR).10 Non-TTR complexed RBP4, a low molecular weight (21 kDa) lipocalin serum protein,11 is rapidly cleared from the bloodstream through glomerular filtration. RBP4-TTR complexation is retinol dependent as apo-RBP4 interacts poorly with TTR.10 It was suggested that lower serum retinol levels achieved via inhibition of RBP4 would modulate the visual cycle, which is fueled by a constant delivery of retinol to the RPE by RBP4.12 As a consequence of reduced retinol supply to fuel the visual cycle, the rate of bisretinoid formation in the RPE would be slowed or halted and the concomitant progression of geographic atrophy within the dry AMD retina would be retarded.7a Indeed, this hypothesis is supported by preclinical and clinical data obtained for the retinoid-based RBP4 antagonist fenretinide (2). In preclinical studies, fenretinide also lowered circulating plasma RBP4 levels in vivo7a,13 and significantly reduced the accumulation of lipo-fuscin bisretinoids in the Abca4−/− mouse model of enhanced retinal lipofuscinogenesis.7a In extended clinical studies, fenretinide lowered serum RBP4 levels and reduced the rate of the geographic atrophy growth in a subset of the treated AMD patients wherein an approximate 70% (or greater) reduction in circulating serum RBP4 levels was achieved.14
We have shown that chronic oral administration of the nonretinoid RBP4 antagonist A1120 (3) (previously developed by Amgen for the potential treatment of diabetes)15 reduced the accumulation of lipofuscin bisretinoids by approximately 50% in the Abca4−/− mouse model of enhanced retinal lipo-fuscinogenesis.16 This reduction was correlated with a 75% reduction in serum RBP4.16 In our more recent report,9 we described medicinal chemistry efforts conducted in the pursuit of designing novel nonretinoid RBP4 antagonists that utilized 3 as a rational design starting point. Compound 3 exhibits favorable RBP4 binding affinity and provided several key structural elements that allowed for rapid structure–ctivity relationship (SAR) exploration. However, the compound suffers from poor human liver microsomal (HLM) stability (~3% remaining after 30 min incubation), which was a major liability that needed to be addressed. Thus, our goal was to develop novel chemical matter with improved drug-like characteristics relative to 3, and our efforts led to the discovery of the novel bicyclic [3.3.0]-octahydrocyclopenta[c]pyrrolo RBP4 antagonist 4.9 Analogue 4 possesses a favorable balance of suitable in vitro RBP4 binding (scintillation proximity assay (SPA)) and functional RBP4-TTR interaction antagonist (HTRF) activity with significantly improved HLM stability (100% remaining after a 30 min incubation). Furthermore, 4 possesses good pharmacokinetic (PK) and pharmacodynamic (PD) properties, leading to robust and sustained lowering (>85%) of serum RBP4 levels in both acute and chronic rodent oral dosing studies.9
We sought to build upon our reported SAR efforts by further exploring the anthranilic acid appendage of 3 and compound 4. Specifically, we wished to investigate whether a pyrimidine-4-carboxylic acid could provide a suitable isostere for the amide of 3 while still presenting the acid group as a favorable interaction. Pyrimidine reduces the number of rotatable bonds and was expected to lead to improved RBP4 binding affinity (Figure 2). Our overarching goal was to further enhance the in vitro and in vivo RBP4 potency observed for 4 while maintaining its excellent microsomal stability and other favorable drug-like characteristics. Guided by our RBP4 computational docking models,9 we initially designed and synthesized illustrative examples containing alternative cores that link together the ortho-trifluoromethylphenyl moiety of 3 and 4 with the newly proposed 6-methylpyrimidine-4-carboxylic acid fragment. We subsequently explored the SAR of the pyrimidine-4-carboxylic acid appended to the bicyclic [3.3.0]-octahydrocyclopenta[c]-pyrrolo core featured in compound 4. Lastly, we also included within our test set a focused series of bicyclic [3.3.0]-octa-hydrocyclopenta[c]pyrrolo core analogues bearing a 6-methyl-pyrimidine-4-carboxylic acid fragment in conjunction with variable aryl head groups.
Figure 2.

Medicinal chemistry work plan for pyrimidine-4-carboxylic acid containing antagonists of RBP4.
CHEMISTRY
The synthesis of 6-methyl-2-(4-(2-(trifluoromethyl)phenyl)-piperidin-1-yl)pyrimidine-4-carboxylic acid (10) is presented in Scheme 1.9,17 Addition of the lithium salt of 1-bromo-2-(trifluoromethyl)benzene (5) to 1-benzylpiperidin-4-one gave tertiary alcohol 6, which was readily dehydrated in the presence of thionyl chloride to furnish tetrahydropyridine 7. Reduction of the olefin of 7 with concomitant N-benzyl deprotection using ammonium formate and 10% Pd/C followed by exposure to HCl provided piperidine hydrochloride salt 8. Treatment of 8 with methyl 2-chloro-6-methylpyrimidine-4-carboxylate in the presence of i-Pr2NEt in DMF at 60 °C afforded methyl ester 9, which underwent facile saponification with LiOH·H2O. Acidification afforded carboxylic acid 10 in good yield.
Scheme 1a.

aReagents and conditions: (a) (i) n-BuLi, THF, −78 °C, 40 min; (ii) 1-benzylpiperidin-4-one, THF, −78 °C; (b) SOCl2, 0 °C, 2 h; (c) (i) HCO2NH4, 10% Pd/C, CH3OH, reflux; (ii) 4.0 M HCl solution in 1,4-dioxane, CH3CN, rt, 2 h; (d) methyl 2-chloro-6-methylpyrimidine-4-carboxylate, i-Pr2NEt Et3N, DMF, 60 °C, 16 h; (e) (i) LiOH·H2O, H2O, THF, rt, 16 h; (ii) 2 N aq HCl.
The synthesis of 6-methyl-2-(4-(2-(trifluoromethyl)phenyl)-piperazin-1-yl)pyrimidine-4-carboxylic acid (14) is highlighted in Scheme 2. A palladium-catalyzed amination reaction between 1-bromo-2-(trifluoromethyl)benzene (5) and tert-butyl piperazine-1-carboxylate afforded aryl piperazine 11, which underwent smooth Boc-deprotection in the presence of HCl to provide piperazine hydrochloride 12. Treatment of 12 with methyl 2-chloro-6-methylpyrimidine-4-carboxylate gave ester 13, which underwent saponification in the presence of LiOH· H2O to give upon acidification the desired carboxylic acid 14.
Scheme 2a.

aReagents and conditions: (a) tert-butyl piperazine-1-carboxylate, Pd(OAc)2, BINAP, NaOt-Bu toluene, 110 °C, 16 h; (b) 2.0 M HCl solution in Et2O, CH2Cl2, rt, 3 h; (c) methyl 2-chloro-6-methylpyrimidine-4-carboxylate, i-Pr2NEt, DMF, 60 °C, 16 h; (d) (i) LiOH·H2O, H2O, THF, rt, 16 h; (ii) 2 N aq HCl.
The synthesis of 6-methyl-2-((3aR,6aS)-5-(2-(trifluoromethyl)-phenyl)hexahydropyrrolo[3,4-c]pyrrol-2(1H)-yl)pyrimidine-4-carboxylic acid (18) is depicted in Scheme 3. Palladium-catalyzed amination of 1-bromo-2-(trifluoromethyl)benzene (5) with (3aR,6aS)-tert-butyl hexahydropyrrolo[3,4-c]pyrrole-2(1H)-carboxylate provided intermediate 15, which underwent HCl-mediated Boc-deprotection to give amine hydrochloride 16. Treatment of 16 with methyl 2-chloro-6-methylpyrimidine-4-carboxylate gave the corresponding methyl ester 17, which was hydrolyzed with LiOH·H2O and then acidified to afford carboxylic acid 18 in good yield.
Scheme 3a.

aReagents and conditions: (a) (3aR,6aS)-tert-butyl hexahydropyrrolo[3,4-c]pyrrole-2(1H)-carboxylate, Pd(OAc)2, BINAP, NaOt-Bu, toluene, 110 °C, 16 h; (b) 2.0 M HCl solution in Et2O, CH2Cl2, rt, 3 h; (c) methyl 2-chloro-6-methylpyrimidine-4-carboxylate, i-Pr2NEt, DMF, 60 °C, 16 h; (d) (i) LiOH·H2O, H2O, THF, rt, 16 h; (ii) 2 N aq HCl.
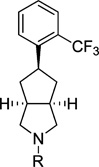
Construction of the [3.3.0]-octahydrocyclopenta[c]pyrrolo series of analogues 32–36 was achieved following our previously reported route (Scheme 4).9,18 LiAlH4 reduction of commercially available dione 19 and subsequent N-Boc protection of the resulting secondary amine gave tetrahydroisoindole 20 in good yield. NaIO4 and RuO2·H2O promoted oxidative cleavage of 20 gave ring-opened diacid 21, which underwent Dieckman condensation with concomitant decarboxylation in acetic anhydride at 120 °C to give ketone 22. Ketone 22 was converted to the corresponding racemic triflate (±)-23 in the presence of 1,1,1-trifluoro-N-phenyl-N-((trifluoromethyl)sulfonyl)meth-anesulfonamide. Intermediate (±)-23 underwent facile Suzuki Pd-catalyzed cross-coupling with (2-(trifluoromethyl)phenyl)-boronic acid conversion to give styrene (±)-24. Catalytic reduction of (±)-24 was conducted under 40 psi H2 in the presence of 10% Pd/C in CH3OH and exclusively provided endo-product 25. HCl-induced Boc-deprotection of 25 followed by reaction with a corresponding 6-substituted 2-chloropyrimidine-4-carboxylic acid methyl ester afforded analogues 27–31. Methyl esters 27–31 were subsequently hydrolyzed and acidified to the corresponding acids 32–36.
Scheme 4a.

aReagents and conditions: (a) (i) LiAlH4 (1.0 M solution in THF), THF, 70 °C, 16 h; (ii) Boc2O, CH2Cl2, rt, 16 h; (b) (i) NaIO4, RuO2·H2O, CH3CN, CCl4, H2O, rt, 24 h; (c) Ac2O, NaOAc, 120 °C, 3 h; (d) (i) LiHMDS (1.0 M solution in THF), THF, −78 °C, 30 min; (ii), PhN(SO2CF3)2, THF, −78 °C to rt, 3 h; (e) (2-(trifluoromethyl)phenyl)boronic acid, Pd(PPh3)4, 2 M Na2CO3, DME, 80 °C, 6 h; (f) H2 (40 psi), 10% Pd/C, CH3OH, rt, 16 h; (g) 2.0 M HCl in Et2O, CH2Cl2, 0 °C to rt, 24 h; (h) 2-chloro-6-substitutedpyrimidine-4-carboxylic acid methyl ester, Et3N, DMF, 60 °C, 16 h; (i) (i) 2 N aq NaOH, THF, CH3OH, rt, 16 h; (ii) 2 N HCl.
The preparation of exo-analogue 40 was achieved via our previously reported route, which is shown in Scheme 5.9,18 Reduction of des-Boc olefin (±)-37 provided a mixture of endo-and exo-isomers 38 and 39, which were isolated via reversed phase column chromatography. Treatment of exo-amine 39 with methyl 2-chloro-5-methylpyrimidine-4-carboxylate followed by saponification with 2 N NaOH and subsequent acidification provided the desired exo-acid 40.
Scheme 5a.

aReagents and conditions: (a) TFA, CH2Cl2, rt, 3 h; (b) H2 (50 psi), 10% Pd/C, CH3OH, rt, 6 h; (c) (i) 2-chloro-6-methylpyrimidine-4-carboxylic acid, Et3N, DMF, 60 °C, 16 h; (ii) 2 N aq NaOH, THF, CH3OH, rt, 16 h; (iii) 2 N aq HCl.
Pyrimidine analogues 41–44 were accessed via intermediate 26 (Scheme 6). Compound 41 was furnished via a palladium-catalyzed amination of 2,4-dichloro-5,6-dimethylpyrimidine with 26, followed by carbonylation with Mo(CO)6 and subsequent saponification of the resulting methyl ester. Treatment of 26 with methyl 2-chloro-5-methylpyrimidine-4-carboxylate followed by hydrolysis and acidification gave acid 42. Analogues 43 and 44 were prepared via treatment of 26 with either methyl 2-chloro-4-methylpyrimidine-5-carboxylate or methyl 2-chlor-opyrimidine-5-carboxylate followed by saponification and acidification of the corresponding methyl esters.
Scheme 6a.

aReagents and conditions: (a) (i) 2,4-dichloro-5,6-dimethylpyrimidine, Pd(OAc)2, Cs2CO3, JohnPhos, toluene, reflux, 16 h; (ii) Mo(CO)6, Pd(OAc)2, BINAP, CH3OH, CH3CN, 80 °C, 16 h; (iii) LiOH·H2O, THF, CH3OH, H2O, rt, 16 h; (b) (i) methyl 2-chloro-5-methylpyrimidine-4-carboxylate, Et3N, DMF, 60 °C, 16 h; (ii) 2 N aq NaOH, THF, CH3OH, rt, 16 h; (c) (i) methyl 2-chloro-4-methylpyrimidine-5-carboxylate or methyl 2-chloropyrimidine-5-carboxylate, i-Pr2NEt, 70 °C (microwave irradiation), 1.5 h or Et3N, DMF, 70 °C, 16 h; (ii) LiOH·H2O, THF, H2O, rt, 16 h or 2 N aq NaOH, THF, CH3OH, rt, 16 h; (iii) 2 N aq HCl.
The preparation of the carboxylic acid isosteres carboxamide 46 and tetrazole 47 are depicted in Scheme 7. Treatment of 26 with 2-chloro-6-methylpyrimidine-4-carbonitrile provided nitrile intermediate 45, which was hydrolyzed with LiOH·H2O in THF and H2O at 70 °C to provide carboxamide 46. Nitrile 45 also underwent cycloaddition with NaN3 to furnish tetrazole 47 in good yield.
Scheme 7a.

aReagents and conditions: (a) (i) 2-chloro-6-methylpyrimidine-4-carbonitrile, i-Pr2NEt, DMF, 60 °C, 16 h; (b) LiOH·H2O, THF, H2O, 70 °C, 18 h; (c) NaN3, NH4Cl, DMF, 130 °C (microwave irradiation), 1 h.
A series of diverse analogues (48–54) designed to further explore pyrimidine carboxylic acid SAR is shown in Scheme 8. The corresponding substituted chloro or bromo-substituted picolinic, nicotinic, isonicotinic, or benzoic acid methyl esters were aminated with amine 26 using a combination of either Pd(OAc)2 or Pd2(dba)3 and ligands XantPhos or JohnPhos in the presence of Cs2CO3 in refluxing toluene or 1,4-dioxane. The intermediary methyl esters were subsequently hydrolyzed and acidified to give the corresponding acid analogues 48–54.
Scheme 8a.

aReagents and conditions: (a) (i) methyl 6-chloro-4-methylpicolinate, Pd(OAc)2, XantPhos, Cs2CO3, toluene, 110 °C, 16 h; (ii) 2 N aq NaOH, THF, CH3OH, rt, 16 h; (iii) 2 N aq HCl; (b) (i) methyl 2-chloro-6-methylisonicotinate, Pd(OAc)2, XantPhos, Cs2CO3, toluene, 110 °C, 16 h; (ii) 2 N aq NaOH, THF, CH3OH, rt, 16 h; (iii) 2 N aq HCl; (c) (i) methyl 6-chloro-3-methylpicolinate, Pd(OAc)2, JohnPhos, Cs2CO3, toluene, reflux, 14 h; (ii) LiOH·H2O, THF, CH3OH, H2O, rt, 72 h; (iii) 2 N aq HCl; (d) (i) methyl 5-bromonicotinate, Pd2(dba)3, XantPhos, Cs2CO3, 1,4-dioxane, reflux, 16 h; (ii) LiOH·H2O, THF, CH3OH, H2O, rt, 4 h; (iii) 2 N aq HCl; (e) methyl 3-bromobenzoate, Pd(OAc)2, BINAP, NaOt-Bu, toluene, 110 °C, 16 h; (ii) LiOH·H2O, THF, CH3OH, H2O, rt, 16 h; (iii) 2 N aq HCl; (f) (i) methyl 2-chloro-6-methylnicotinate, Pd2(dba)3, XantPhos, Cs2CO3, toluene, 100 °C, 16 h; (ii) LiOH·H2O, THF, CH3OH, H2O, 50 °C, 30 min; (iii) 2 N aq HCl; (g) (i) methyl 2-chloronicotinate, Pd(OAc)2, XantPhos, Cs2CO3, 1,4-dioxane, 110 °C, 16 h; (ii) LiOH·H2O, THF, CH3OH, H2O, rt, 16 h; (iii) 2 N aq HCl.
The syntheses of alternative heteroaromatic analogues 55–57 are highlighted in Scheme 9. Methyl 6-chloropyrazine-2-carboxylate, methyl 6-chloropyridazine-4-carboxylate, and methyl 2-chloro-5-methylthiazole-4-carboxylate were independently reacted with amine 26 in the presence of either Et3N or i-Pr2NEt in DMF at 60 °C followed by saponification of the resulting intermediary methyl esters with LiOH·H2O and acidification to provide analogues 55, 56, and 57, respectively.
Scheme 9a.

aReagents and conditions: (a) (i) methyl 2-chloropyrazine-6-carboxylate or methyl 3-chloropyridazine-5-carboxylate or methyl 2-chloro-5-methylthiazole-4-carboxylate, Et3N or i-Pr2NEt, DMF, 60 °C, 16 h or i-Pr2NEt, DMSO, 110 °C, 24 h; (ii) LiOH·H2O, THF, CH3OH, H2O, rt, 30 min-16 h; (iii) 2 N aq HCl.
The synthesis of a series of analogues possessing varied aryl head groups (61–66) is depicted in Scheme 10. Palladium-catalyzed Suzuki cross-coupling of triflate (±)-23 with the appropriately substituted aryl boronic acids provided the corresponding styrene intermediates represented by (±)-58a–f in good yield. Hydrogenation of the olefins followed by HCl-promoted Boc-deprotection afforded the endo-amine hydrochloride intermediates 60a–f. Treatment of these secondary amines with 2-methyl 2-chloro-6-methylpyrimidine-4-carboxylate followed by hydrolysis and acidification of the corresponding methyl esters provided the desired acids 61–66.
Scheme 10a.

aReagents and conditions: (a) (i) substituted phenyl or pyridyl boronic acid, Pd(PPh3)4, 2.0 M Na2CO3, DME, 80 °C, 6 h; (b) H2 (40 psi), 10% Pd/C, CH3OH, rt, 16 h; (c) 2.0 M HCl in Et2O, CH2Cl2, 0 °C to rt, 24 h; (d) (i) methyl 2-chloro-6-methylpyrimidine-4-carboxylate, Et3N, DMF, 60 °C, 16 h; (ii) 2 N aq NaOH, THF, CH3OH, rt, 16 h; (iii) 2 N aq HCl.
RESULTS AND DISCUSSION
Computational docking of an initial proposed set of 6-methyl-pyrimidine-4-carboxylic acid ligands was conducted within our model derived from the cocrystal structure of 3 bound to RBP4 (PDB code: 3fmz)15 using standard precision (SP) mode. The docked poses were then refined using the extra precision (XP) mode of Glide (version 5.8, Schrödinger, LLC.). These docked poses of the proposed ligands were minimized within the binding site of the 3fmz model. All molecular mechanics (MM) minimization used Impact (version 5.8, Schrodinger, LLC).
We modified the binding site by incorporating a structural water molecule within the anthranilic acid binding region of 3fmz. Several reported RBP4 crystal structures show two water molecules (W1 and W2) housed within the anthranilic acid region of the RBP4 binding site, where they are engaged in H-bond interactions with Arg121, Gln98, and Tyr90. However, 3fmz is devoid of these water molecules likely because the region is surface exposed, and the solvent is likely quite mobile. However, our docking and minimization procedures would be less accurate if a solvent hole were left in this fairly polar portion of the site. Therefore, to account for partially structured water molecules, we added structural waters from other RBP4 crystal structures. When the structural water W2 of 1fel (RBP4 cocrystallized with fenretinide) was minimized within 3fmz, strong H-bond interactions among it, compound 3, and Arg121 were observed (data not shown). This observation suggests that structural water W2 could be playing a critical role in the binding of 3 to RBP4. Therefore, we have incorporated this structural water within our 3fmz docking model so as to ensure that any key differences with regard to critical H-bond interactions are observed for all newly proposed analogues and so that ligand poses do not fill a nonexistent hole.
As previously reported, the functional antagonist activity observed for 3 is attributed to conformational changes it induces in RBP4 loops β3-β4 and β5-β6.9,15 These changes block complex formation with TTR, leading to reduced RBP4 plasma levels as a result of increased glomerular excretion of the uncomplexed protein. The anthranilic carboxylic acid fragment of 3 is positioned at the opening of the RBP4 binding cavity where it is engaged in several key binding interactions; the carboxamido carbonyl oxygen atom of 3 accepts an H-bond from the backbone of Leu37, and the carboxylic acid forms a salt bridge with Arg121 and two H-bonds with Tyr90 and Gln98.9,15 These key interactions are potentially playing critical roles in inducing and stabilizing the conformational changes observed within the aforementioned RBP4 loops. To see if the 6-methylpyrimidine-4-carboxylic acid moiety could provide a suitable isosteric replacement for the anthranilic acid, we initially conducted a 3fmz docking overlay of 3 with proposed comparator 10. Indeed, analogue 10 presented a similar docking geometry relative to that of 3 (Figure 3); both compounds are shown extending through the β-barrel core of the RBP4 binding cavity with their respective ortho-trifluoromethylphenyl fragments projecting into the internal lipophilic β-ionone pocket. The overlays also show the carboxylic acid moieties of both compounds positioned within close proximity to one another and engaged in the same key H-bond interactions with Arg121, Gln98, and Tyr90. It should be noted that the geometry of the pyrimidine moiety suggests the unexpected replacement of the H-bond donor amide with the H-bond acceptor pyrimidine. The geometry shown in Figure 3B likely induces some very interesting protonation states among the acid group, Tyr90, Arg121, the modeled water molecule, and the pyrimidine, likely stabilized by Gln98. The movement of Gln98 to this region appears to be a driving force for the blocking motion of loops β3-β4 and β5-β6. These conformations are significantly different (2.7 Å) from those in the fenretinide antagonist structure 1fel. Buoyed by the favorable geometric alignment observed in this docking experiment, we sought to synthesize compound 10 and other similar analogues that contain alternative cores linking together the ortho-trifluoro-methylphenyl and 6-methylpyrimidine-4-carboxylic acid fragments. Acknowledging our previous success with the bicyclic [3.3.0]-octahydrocyclopenta[c]pyrrole core used to generate the aforementioned RBP4 antagonist 4,9 we ensured that this core scaffold was included within our initial sample set of analogues.
Figure 3.

(A) Overlay of minimized bound conformations of RBP4 antagonist 10 (orange) and 3 (green) within 3fmz. (B) Expanded view of the anthranilic acid and 6-methylpyrimidine-4-carboxylic acid fragments of 3 and analogue 10. Both moieties are shown engaging in H-bond interactions with Arg121, Gln98, and Tyr90. H-bond interactions to 10 are indicated with blue lines. Residues making contact with ligands are labeled and illustrated in stick format. Molecular surface is colored by hydrophobicity (green) and hydrophilicity (purple) using contact preferences (MOE, Chemical Computing Group, Inc., Montreal, CA).
In Vitro Binding of Compounds to RBP4
Analogue binding affinities for RBP4 were measured using our previously described scintillation proximity assay (SPA).16 Nonlinear regression analysis conducted following saturation binding of retinol to RBP4 revealed a Kd of 62.5 nM in our assay conditions, which is in line with the previously reported 70–190 nM range of values.10d,19 For compound characterization, we measured the competitive displacement of radiolabeled retinol added to the reaction mix at 10 nM which roughly corresponds to the generally recommended 1/10 of the Kd concentration of radioligand. The IC50 values were calculated from the 12-point compound titrations using a four-parameter logistic equation. Compounds with appreciable RBP4 binding potency (generally, with IC50 <150 nM) were further assessed for the ability to antagonize the retinol-dependent RBP4-TTR interaction using the HTRF assay.16
Assessment of Antagonistic Activity in the HTRF RBP4-TTR Interaction Assay
The ability of an analogue to disrupt the retinol-induced interaction of RBP4 with TTR was examined using the previously described HTRF assay16 that probes retinol-dependent RBP4-TTR interaction. We used bacterially expressed maltose binding protein (MBP)-tagged RBP4 and commercially available TTR labeled directly with Eu3+ cryptate along with a d2-conjugated anti-MBP monoclonal antibody in order to implement this assay. Retinol added to the reaction mix at 1 µM concentration stimulates RBP4-TTR interaction which brings europium in close proximity to the d2 dye with the following fluorescence energy transfer (FRET) to d2. The FRET signal, measured as a 668 nm emission, was normalized using the 620 nm europium emission to compensate for the pipetting and dispensing errors. Following a 12-point dose titration (20 µM-0.1 nM), the IC50 values were calculated using a standard four parameter logistic nonlinear regression model equation. In order to monitor a correlation between the binding affinity of compounds for RBP4 and their potency as antagonists of the retinol-dependent RBP4-TTR interaction, we tracked a ratio of IC50 values in the HTRF and SPA assays. Despite the difference in binding potency, for the majority of analogues, this ratio tended to be within the 5–20 range.
Structure–Activity Relationships
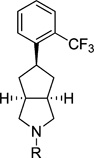
An initial series of analogues containing both the 6-methylpyrimidine-4-carboxylic acid and corresponding methyl ester moieties were prepared with the following core structures; piperidine (9 and 10), piperazine (13 and 14), bicyclic [3.3.0]-octahydropyrrolo[3,4-c]-pyrrole (17 and 18), and bicyclic [3.3.0]-octahydrocyclopenta-[c]pyrrole (28 and 33). The RBP4 SPA binding affinity, HTRF functional antagonist activity, and microsomal stability data for these new analogues is shown in Table 1. Notably, the most direct comparator of 3, analogue 10, exhibited similar in vitro RBP4 binding affinity and functional antagonist activity (RBP4 SPA IC50 = 35.3 nM; RBP4 HTRF IC50 = 188 nM), verifying that the 6-methylpyrimidine-4-carboxylic acid fragment was indeed a suitable isostere for anthranilic acid. Interestingly, the pyrimidine-4-carboxylic acid of 10 also conferred the additional benefit of significantly improving human liver microsomal stability (68% remaining after 30 min incubation) relative to that of 3. The corresponding methyl ester of 10 (9) was found to be ~3-fold more potent than 3, and the remaining methyl esters 13, 17, and 28 also exhibited appreciable in vitro RBP4 potency in both assays. However, methyl esters 13 and 28 suffered from extensive degradation in the presence of human (HLM) and rat (RLM) liver microsomes. Piperazine acid analogue 14 was less potent than 3 (RBP4 SPA IC50 = 89.5 nM; RBP4 HTRF IC50 = 511 nM), and it presented only a modest improvement in HLM stability, whereas bicyclic [3.3.0]-octahydropyrrolo[3,4-c]pyrrole 18 was slightly more potent and exhibited better HLM stability. Incorporation of the bicyclic [3.3.0]-octahydrocyclopenta[c]pyrrole core provided standout analogue 33, which exhibited significantly improved in vitro RBP4 potency (RBP4 SPA IC50 = 12.8 ± 0.4 nM; RBP4 HTRF IC50 = 43.6 ± 10.5 nM) and excellent microsomal stability. Subsequent docking analysis of 33 revealed favorable alignment within the RBP4 binding cavity (Figure 4). The 6-methylpyrimidine-4-carboxylic acid appendage of 33 is engaged in key H-bond interactions as the bicyclic [3.3.0]-octahydrocyclopenta[c]pyrrolo core extends through the β-barrel channel and projects the ortho-trifluoromethylphenyl ring slightly farther into the lipophilic hydrophobic β-ionone cavity than 3. We postulate that the excellent RBP4 potency observed for 33 potentially results from two possible factors (or a combination thereof): (1) more favorable van der Waals interactions resulting from the deeper extension and optimal angle of projection of the ortho-trifluoromethylphenyl ring into the lipophilic β-ionone cavity and/or (2) more fortified key H-bond interactions due to increased rotational restriction of the 6-methylpyrimidine-4-carboxylic acid relative to the anthranilic acid of 3 and 4 (two fewer rotatable bonds). In addition, the exo-isomer 40 was also prepared and found to be devoid of RBP4 activity (SPA IC50 > 3 µM, data not shown). The RBP4 activity differences observed between potent endo-isomer 33 and inactive exo-isomer 40 were consistent with our previously reported findings for these geometric isomers of this scaffold class.9 The results obtained from this initial sample set of probe compounds prompted us to further explore the SAR of analogue 33.
Table 1.
RBP4 SPA Binding Affinity, RBP4 HTRF, and Liver Microsomal Stability Data for 6-Methylpyrimidine-4-carboxylic Acid and Methyl Ester Analogues
 | ||||
|---|---|---|---|---|
| microsomal stability (% remaining) |
||||
| compd | RBP4 SPAa IC50 (nM)c | RBP4 HTRFb IC50 (nM)c | HLMd | RLMe |
| 3 | 15.0 ± 0.005 | 122 ± 0.035 | 3 | 85 |
| 4 | 72.7 | 294 | 100 | 75 |
| 9 | 4.1 | 132 | ND | ND |
| 10 | 35.3 | 188 | 68 | 96 |
| 13 | 9.7 | 1402 | 0.0 | 0.0 |
| 14 | 89.5 | 511 | 18 | 63 |
| 17 | 3.9 | 74.0 | ND | ND |
| 18 | 57.4 | 140 | 80 | 76 |
| 28 | 6.3 | 100 | 0.2 | 9.8 |
| 33 | 12.8 ± 0.4 | 43.6 ± 10.5 | 94 | 93 |
IC50 values for the SPA assay obtained in the presence of a fixed, 10 nM concentration of 3H-retinol.
IC50 values for the HTRF assay obtained in the presence of 1 µM concentration of retinol.
For compounds tested multiple times (more than twice), the IC50 data are represented as the mean ± standard deviation. For those compounds that were only tested twice, the IC50 data are shown as the mean of two independent experiments and not as the mean ± standard deviation.
HLM = human liver microsomes.
RLM = rat liver microsomes. Compound concentration was 10 µM, and incubation time with either human or rat microsomes was 30 min. ND = not determined.
Figure 4.

(A) Overlay of minimized bound conformations of RBP4 antagonist 33 (magenta) and 3 (orange) within 3fmz. (B) Expanded view of the anthranilic acid and 6-methylpyrimidine-4-carboxylic acid fragments of 3 (green) and analogue 33 (magenta). Both moieties are shown engaging in H-bond interactions with Arg121, Gln98, and Tyr90. H-bond interactions to 33 are indicated with blue lines. Residues making contact with ligands are labeled and illustrated in stick format. The molecular surface is colored by hydrophobicity (green) and hydrophilicity (purple) using contact preferences (MOE, Chemical Computing Group, Inc., Montreal, CA).
The SAR of the 6-methylpyrimidine-4-carboxylic acid region of analogue 33 is summarized in Table 2. Initial efforts within Table 2 focused on examining the impact on potency of incorporating various chemotypes at the 6-position of the pyrimidine-4-carboxylic acid ring. Limited exploration of this region revealed that the size of the substituent was important and that in vitro binding affinity and functional antagonist activity trends do not generally correlate. For example, removal of the 6-methyl group of analogue 33 to furnish 32 led to a ~2-fold diminishment in potency in both the SPA (RBP4 SPA IC50 = 20.8 ± 0.5 nM) and HTRF (RBP4 HTRF IC50 = 79.7 nM) assays. However, when a trifluoromethyl group was introduced to the 6-position (34), the SPA binding affinity was recovered (RBP4 SPA IC50 = 9.6 nM), yet HTRF potency remained roughly 2-fold lower (RBP4 HTRF IC50 = 88.0 nM). Introduction of the larger isopropyl group (36) led to a significant diminishment in potency relative to that of 33, which may be indicative of a size/steric tolerability limit in that region of the binding space. In addition, there was no differentiation between substituents possessing electron-donating or electron-withdrawing character, as trifluoromethyl analogue 34 and methoxy analogue 35 exhibited similar binding affinity. The incorporation of an additional methyl group at the 5-position to give 5,6-dimethyl analogue 41 resulted in a 5-fold decrease in potency, and monosubstitution of a methyl group at the 5-position (42) led to a further diminishment in potency. These data suggest that the most potent analogues of this series require one substituent of limited size to be positioned meta to the pyrimidine-4-carboxylic acid and that substitution ortho to the carboxylic acid is disfavored. We also examined the impact of repositioning the carboxylic acid of 33 from the 4- to the 5-position on the pyrimidine ring. Interestingly, analogues 43 and 44 were tolerated and exhibited appreciable binding affinity (RBP4 SPA IC50 = 40.8 nM and 171 nM, respectively); however, both analogues were significantly less potent than 33 in the HTRF assay (RBP4 HTRF IC50 = 523 nM and 1864 nM, respectively). Lastly, primary carboxamide 46 and tetrazole 47 served as suitable carboxylic acid isosteres within this series as both compounds displayed RBP4 binding affinities comparable to that of 33. However, 46 and 47 were found to be significantly less potent in the HTRF assay (RBP4 HTRF IC50 = 248 nM and 175 nM, respectively).
Table 2.
RBP4 SPA Binding Affinity and RBP4 HTRF for Pyrimidine Carboxylic Acid Analogues
 | |||
|---|---|---|---|
| Compound | R | RBP4 SPAa IC50 (nM)c |
RBP4 HTRFb IC50 (nM)c |
| 33 | 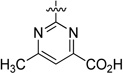 |
12.8 ± 0.4 | 43.6 ± 10.5 |
| 32 | 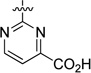 |
20.8 ± 0.5 | 79.7 |
| 34 | 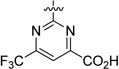 |
9.6 | 88.0 |
| 35 | 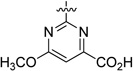 |
12.8 ± 1.6 | 81.2 ± 7.6 |
| 36 | 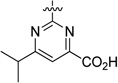 |
153 ± 0.6 | 202 |
| 41 | 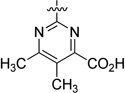 |
48.8 | 123 |
| 42 | 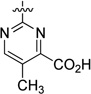 |
90.1 | 595 |
| 43 | 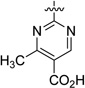 |
40.8 | 523 |
| 44 | 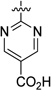 |
171 | 1864 |
| 46 | 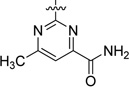 |
3.3 | 248 |
| 47 | 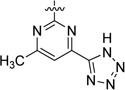 |
7.5 | 175 |
IC50 values for the SPA assay obtained in the presence of a fixed, 10 nM concentration of 3H-retinol.
IC50 values for the HTRF assay obtained in the presence of 1 µM concentration of retinol.
For compounds tested multiple times (more than twice), the IC50 data are represented as the mean ± standard deviation. For those compounds that were only tested twice, the IC50 data are shown as the mean of two independent experiments and not as the mean ± standard deviation.
In parallel, we evaluated replacing the pyrimidine-4-carboxylic acid of 33 with picolinic, isonicotinic, nicotinic, and benzoic acid moieties (Table 3). 4-Methyl picolinic acid 48 and 2-methyl isonicotinic acid 49 exhibited binding affinities within a 5-fold range of 33; however, they were both nearly 10-fold less potent in the HTRF assay (RBP4 HTRF IC50 = 295 nM and 228 nM, respectively). In contrast, 3-methyl picolinic acid 50 was significantly less potent in both assays and the unsubstituted nicotinic acid 51 was devoid of RBP4 activity. Removal of all nitrogen atoms to furnish benzoic acid 52 provided an analogue with appreciable binding affinity yet was very weakly potent in the HTRF assay (RBP4 SPA IC50 = 83.2 nM; RBP4 HTRF IC50 = 3956 nM). These data suggest that while the methyl-substituted pyridine congeners of 33 (48 and 49) exhibit good binding affinity, both nitrogen atoms of pyrimidine 33 appear to be required for optimal functional antagonist activity in the HRTF assay. In addition, the positioning of a methyl group ortho to the carboxylic acid within this pyridine series (50) had the same deleterious effect on SPA binding and HTRF potency that was observed for 5-methyl substituted pyrimidine analogue 42. We next investigated the effect of repositioning the carboxylic acid adjacent to the bicyclic [3.3.0]-octahydrocyclopenta[c]pyrrolo core, as docking of nicotinic acid analogue 54 indicated favorable geometric alignment within the RBP4 binding cavity (Figure 5). Indeed, analogues 53 and 54 were tolerated and exhibited comparable SPA binding affinities relative to one another (RBP4 SPA IC50 = 68.4 nM and 43.8 nM, respectively); however, 54 was found to be 5-fold more potent than 53 in the HTRF assay. Interestingly, the HTRF trend observed between nicotinic acids 53 and 54 suggests that substitution on this heteroaromatic system in a manner analogous to the 6-methyl group of pyrimidine 33 is not favorable with regard to functional antagonist activity. Lastly, we also pursued alternative heteroaromatic analogues pyrazine 55, pyridazine 56, and thiazole 57. Of this set, pyridazine 56 and thiazole 57 were found to possess good binding affinity (RBP4 SPA IC50 = 23.0 nM and 29.3 nM, respectively); however, both analogues were significantly less potent than benchmark 33 in the HTRF assay (RBP4 HTRF IC50 = 437 nM and 495 nM, respectively).
Table 3.
RBP4 SPA Binding Affinity and RBP4 HTRF for Pyridine and Other Heteroaromatic Carboxylic Acid Analogues
 | |||
|---|---|---|---|
| Compound | R | RBP4 SPAa IC50 (nM)c |
RBP4 HTRFb IC50 (nM)c |
| 33 | 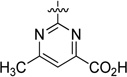 |
12.8 ± 0.4 | 43.6 ± 10.5 |
| 48 | 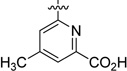 |
14.1 | 295 |
| 49 |  |
53.3 | 228 |
| 50 | 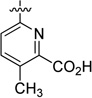 |
584 | 4908 |
| 51 | 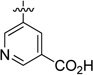 |
>3000 | ND |
| 52 | 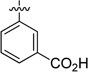 |
83.2 | 3956 |
| 53 | 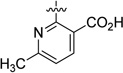 |
68.4 | 2005 |
| 54 | 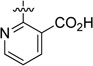 |
43.8 | 381 |
| 55 | 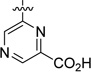 |
166 | ND |
| 56 | 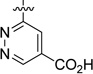 |
23.0 | 437 |
| 57 | 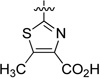 |
29.3 | 495 |
IC50 values for the SPA assay obtained in the presence of a fixed, 10 nM concentration of 3H-retinol.
IC50 values for the HTRF assay obtained in the presence of 1 µM concentration of retinol.
For compounds tested multiple times (more than twice), the IC50 data are represented as the mean ± standard deviation. For those compounds that were only tested twice, the IC50 data are shown as the mean of two independent experiments and not as the mean ± standard deviation. ND = not determined.
Figure 5.

(A) Overlay of minimized bound conformations of RBP4 nicotinic acid antagonist 54 (yellow) and 3 (A1120, green) within 3fmz. (B) Expanded view of the anthranilic acid and nicotinic acid fragments of 3 (green) and analogue 54 (yellow). Both moieties are shown engaging in H-bond interactions with Arg121, Gln98, and Tyr90. H-bond interactions to 54 are indicated with blue lines. Residues making contact with ligands are labeled and illustrated in stick format. The molecular surface is colored by hydrophobicity (green) and hydrophilicity (purple) using contact preferences (MOE, Chemical Computing Group, Inc., Montreal, CA).
We also probed the SAR associated with the aryl headgroup region of compound 33 with a focused set of substituted aromatic and heteroaromatic analogues that are shown in Table 4. 5-Fluoro-2-(trifluoromethyl)phenyl analogue 61 exhibited potency comparable to that of parent 33 (RBP4 SPA IC50 = 46.3 ± 1.1 nM; RBP4 HTRF IC50 = 33.4 ± 1.8 nM). In contrast, incorporation of a fluorine ortho to the trifluoromethyl group to give 3-fluoro-2-(trifluoromethyl)phenyl analogue 62 was not well tolerated and led to a 20-fold loss in SPA binding potency (RBP4 SPA IC50 = 196 nM). Replacing the ortho-trifluoromethyl group of 61 and 62 with chlorine to give analogues 63 and 64 led to a precipitous drop in potency (SPA IC50 = 532 ± 198 nM and 137 nM; HTRF IC50 = 347 nM and 504 nM, respectively). Lastly, introduction of polarity by means of pyridine analogues 65 and 66 yielded appreciable SPA binding affinity; however, the compounds were weakly potent in the HRTF assay.
Table 4.
RBP4 SPA Binding Affinity and RBP4 HTRF for Pyridine and Other Heteroaromatic Carboxylic Acid Analogues
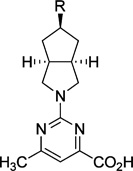 | |||
|---|---|---|---|
| Compound | R | RBP4 SPAa IC50 (nM)c |
RBP4 HTRFb IC50 (nM)c |
| 33 | 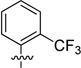 |
12.8 ± 0.4 | 43.6 ± 10.5 |
| 61 | 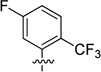 |
46.3 ± 1.1 | 33.4 ± 1.8 |
| 62 | 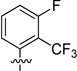 |
196 | ND |
| 63 | 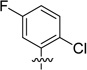 |
532 ± 198 | 347 |
| 64 | 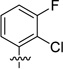 |
137 | 504 |
| 65 | 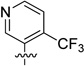 |
51.8 ± 1.5 | 821 ± 142 |
| 66 | 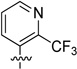 |
31.1 ± 1.9 | 719 ± 22.4 |
IC50 values for the SPA assay obtained in the presence of a fixed, 10 nM concentration of 3H-retinol.
IC50 values for the HTRF assay obtained in the presence of 1 µM concentration of retinol.
For compounds tested multiple times (more than twice), the IC50 data are represented as the mean ± standard deviation. For those compounds that were only tested twice, the IC50 data are shown as the mean of two independent experiments and not as the mean ± standard deviation. ND = not determined.
In light of the excellent RBP4 potency and robust microsomal stability observed for analogue 33, the compound was selected for further in vitro and in vivo evaluation. The in vitro pharmacological profile is presented in Table 5. Compound 33 exhibited excellent kinetic solubility in phosphate buffered saline (PBS) (pH 7.4) and was classified as highly permeable in the MDR1-MDCK permeability assay. The observed CLint values suggest very low predicted hepatic clearance, and the % plasma protein binding (PPB) data indicate low unbound fractions. Interestingly, unlike previously reported anthranilic acid analogue 4, which exhibited significant CYP2C9 inhibitory activity (IC50 = 340 nM),9 33 was found to be clean in a standard CYP panel (all CYP IC50 values >34 µM). Additional selectivity profiling revealed no significant off-target activity at the hERG channel or within a standard screening panel of fifty-five GPCRs, enzymes, ion channels, and transporters (data not shown). Lastly, 33 did not induce human PXR activation (data not shown).
Table 5.
In Vitro ADMET Profile for 33
| hepatic CLint (µL/mL/min)b | % PPBd | MDCK-MDR1 permeability Papp (×10−6 cm/s) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| solubilitya | H | R | M | cyno | CYP inhibition (IC50) 2C9, 2C19, 2D6, 3A4 | hERGc (IC50) |
H | R | A-B | B-A | class |
| >100 µM | <0.7 | <0.7 | <0.7 | 0.7 | All >34 µM | >30 µM | 99.9 | 99.4 | 74.7 | 82.2 | high permeability |
Kinetic solubility measured in PBS (pH 7.4).
Hepatic intrinsic clearance; H = human; R = rat; M = mouse; cyno = cynomolgus monkey.
Patch-Xpress’ patch-clamp assay; compounds were tested (n = 3) in a five-point concentration–response on HE293 cells stably expressing the hERG channel.
% PPB = plasma protein binding.
In Vivo Activity: PK Characteristics of 33 in Rats
Single dose PK studies conducted with Sprague–Dawley rats at 5.1 µmol/kg (2 mg/kg) IV and 18.2 µmol/kg (5 mg/kg) PO doses showed very low plasma clearance (5.06 mL/min/kg) and a very long half-life of 22 h (Table 6). The compound was well absorbed and slowly eliminated from plasma after oral administration with an observed Cmax of 62.2 µM and corresponding Tmax at 2.67 h. Very high exposures were observed (AUCINF was 3636 µM·h) and the estimated % F exceeded 100%. The greater than 100% oral bioavailability may be related to the very low plasma clearance of 33 and some nonlinear pharmacokinetics over the 5.1 to 12.7 µmol/kg dose range.
Table 6.
In Vivo PK Data for Analogue 33 Following IV and PO Administration in Ratsa
| route | dose (µmol/kg) | C0b (µM) | CLc (mL/h/kg) | t1/2d (h) | Vsse (mL/kg) | AUClastf (µM·h) | AUCINFg (µM·h) | %Fi |
|---|---|---|---|---|---|---|---|---|
| IV | 5.1 | 58.6 (1.8) | 5.06 (0.49) | 22.0 (3.0) | 156 (9.0) | 795 (48) | 1017 (93) | NA |
| route | dose (µmol/kg) | Cmaxh (µM) | Tmaxh (h) | t1/2d (h) | Vsse (mL/kg) | AUClastf (µM·h) | AUCINFg (µM·h) | %Fi |
| PO | 12.7 | 62.2 (4.3) | 2.67 (1.15) | 38.8 (17.3) | NA | 2011 (326) | 3636 (1588) | 143 (62.4) |
Data are represented as the mean with standard deviation in parentheses (mean (SD)). Dosing groups consisted of three drug naïve adult male Sprague–Dawley rats. IV administration: the test article was administered at the 5.1 µmol/kg (2 mg/kg) dose; test article vehicle = 3% DMA/45% PEG300/12% EtOH/40% sterile H2O; PO administration, test article was administered at the 12.7 µmol/kg (5 mg/kg) dose; vehicle = 2% Tween 80 in 0.9% saline. Bioavailability; F = (AUCINFpo × doseiv) ÷ AUCINFiv × dosepo).
Observed initial concentration of compound in plasma at time zero.
Total body clearance.
Apparent half-life of the terminal phase of elimination of the compound from plasma.
Volume of distribution at steady state.
Area under the plasma concentration versus time curve from 0 to the last time point that the compound was quantifiable in plasma.
Area under the plasma concentration versus time curve from 0 to infinity.
Maximum observed concentration of the compound in plasma.
Time of maximum observed concentration of the compound in plasma.
We also conducted a subsequent 7-day, subchronic rodent oral dosing PK–PD study with 33 (Figure 6). Sprague–Dawley rats received either a single 18.2 µmol/kg (5 mg/kg) oral dose or a once daily (QD) 18.2 µmol/kg oral dose of 33 over a period of 7-days. Blood was collected from the single dose-treated rats (Group 1) at predose, 15 and 30 min, and 1, 2, 4, 8, 12, 24, 36, and 48 h post dose on Day 1. Blood was collected from the 7-day QD dose-treated rats (Group 3) at predose (Days 1–6; for RBP4 biomarker analysis only) and predose, 15 and 30 min, and 1, 2, 4, 8, 12, 24, 36, and 48 h post-last dose on Day 7. A vehicle dose group (Group 2) was also included in the study and blood was collected on Days 1 and 7 as a control group for RBP4 biomarker analysis. The plasma exposure levels of 33 were high in all samples of Groups 1 and 3. On Day 1, compound 33 reached the peak concentration rapidly after dose administration and declined slowly thereafter (Figure 6, A). Mean plasma levels of 33 were approximately 20 µM at the 48 h time-point. As a result of the slow clearance of 33 from plasma, the mean predose plasma concentration on Day 7 was 148.1 ± 58.2 µM (Figure 6B). After the administration of the seventh daily dose, the mean plasma level increased to 178.9 ± 59.4 µM at the 4 h time-point (Figure 6A). At 48 h after this dose, the mean concentration of compound 33 remained high at 97.1 ± 48.6 µM. The PK parameters for the single and 7-day doses were also determined. The observed Cmax values were 66.5 ± 23.6 µM with a Tmax of 2.75 ± 2.17 h and 183.1 ± 55.2 µM with a Tmax of 2.17 ± 1.76 h on Days 1 and 7, respectively. Exposure levels based on AUClast were 1697 ± 279 µM·h on Day 1 and 6385 ± 2501 µM·h on Day 7. The elimination phase t1/2 values were calculated to be 31.9 ± 17.7 h and 49.4 ± 17.7 h for Day 1 and Day 7, respectively. Notably, plasma collected before the administration of the final dose on Day 7 contained approximately 150 µM of 33, which is consistent with the observed long t1/2 and suggests that steady state levels were achieved after 6 to 7 days of QD oral administration (Figure 6, B).
Figure 6.

(A) Plasma exposure levels of analogue 33 in Sprague–Dawley rats following a single (Day 1) and 7-day (Day 7) QD oral dose administration at 18.2 µmol/kg (5 mg/kg). Data represented as the mean (SD). Each dosing group consisted of three drug naïve adult male Sprague–Dawley rats; the test article vehicle was 2% Tween 80 in 0.9% saline. The difference in plasma compound concentration between Day 1 and Day 7 samples was statistically significant at all time points studied (P < 0.05; two-tailed unpaired Student’s t test). (B) Plasma concentration of analogue 33 at predose time points during the 7-day QD oral dose administration in three rats from Group 3. Data represented as the mean (SD). The predose compound concentration was statistically significantly increased at Day 7 in comparison to the Day 2 predose level (P < 0.05; two-tailed unpaired Student’s t test). (C) Pharmacokinetic parameters for 33 in Sprague–Dawley rats after a single dose (Day 1) and seven QD oral doses (Day 7) at 18.2 µmol/kg. Data are represented as the mean with standard deviation shown in parentheses.
In Vivo Activity: PK–PD Correlations of Analogue 33
In order to establish the in vivo target engagement, provide proof of in vivo activity, and document PK–PD correlations, we studied the effect of single and 7-day dosing of 33 on the level of plasma RBP4 in rats. Aliquots of plasma samples collected during the PK experiments were used to analyze plasma RBP4 concentrations. After a single 18.2 µmol/kg (5 mg/kg) oral dose of 33, a maximum of 85% decrease in plasma RBP4 was observed (Figure 7, A), while the 7-day QD oral administration in rats at 18.2 µmol/kg induced an approximately 95% reduction in plasma RBP4 (Figure 7, A). Comparison of the dynamics of RBP4 lowering in the compound 33-treated animals with that of the vehicle-treated rats over 7 days of QD dosing confirmed the magnitude of the compound effect on the level of plasma RBP4 (Figure 7, B). The extent of the RBP4 lowering (Figure 7) shows a clear correlation with plasma compound levels (Figure 6). With regard to this effect, even though the plasma protein binding of 33 is significant (rat 99.4%, Table 5), the high circulating plasma levels, long exposure, and very low clearance mean that the projected plasma free drug concentration of 33 significantly exceeds that required to antagonize RBP4/TTR interaction measured by either the SPA or HTRF binding assays (Table 4). Antagonists of the RBP4-TTR interaction from different structural classes had previously established the correlation among RBP4 lowering, serum retinol lowering, and bisretinoid reduction in the Abca4−/− mouse model of Stargardt disease.7a,15 While we did not measure retinol lowering by 33 in our in vivo studies, it seemed reasonable to expect that 33 would reduce serum retinol and show the desired preclinical chronic efficacy in the Abca4−/− mice given its pronounced RBP4-lowering activity (Figure 7) and the established correlations between levels of serum RBP4 and serum retinol.7a,15,20 This and additional data describing the nonrodent pharmacology of compound 33 will be published elsewhere.
Figure 7.

(A) Plasma RBP4 levels (µg/mL) in male Sprague–Dawley rats following single (Day 1, Group 1) and 7-day (Day 7, Group 3) 18.2 µmol/kg (5 mg/kg) QD dose administration of analogue 33. Data represented as the mean (SD). (B) Plasma RBP4 levels (µg/mL) at predose time-points during the 7-day 5 mg/kg QD oral dose administration of analogue 33 (Group 3) or test article vehicle (Group 2) in Sprague–Dawley rats. Data represented as the mean (SD).
CONCLUSIONS
We sought to investigate whether a 6-methylpyrimidine-4-car-boxylic acid fragment could serve as a suitable isostere for the anthranilic acid moiety of 3 and the previously reported bicyclic [3.3.0]-octahydrocyclopenta[c]pyrrolo RBP4 antagonist 4 with the goal of further improving RBP4 potency and druglike characteristics. We initially investigated probe analogues (10, 14, 18, and 33) that featured diverse cores in which the ortho-trifluoromethylphenyl headgroup of 3 and 4 was retained. Of this focused sample set of compounds, bicyclic [3.3.0]-octahydrocyclopenta[c]pyrrolo 33 emerged as a standout analogue exhibiting exquisite in vitro RBP4 potency (SPA IC50 = 12.8 ± 0.4 nM and HTRF IC50 = 43.6 ± 10.5 nM) and excellent in vitro microsomal stability. Follow-up analogues of 33 that explored the 6-methylpyrimidine-4-carboxylic acid region of the scaffold were subsequently pursued. Although these analogues displayed intriguing SAR trends, none of them proved to be better than 33 in terms of in vitro RBP4 potency. Concurrent with this campaign, SAR exploration of the aryl headgroup of 33 was also conducted. Similar to the aforementioned 6-methylpyrimidine-4-carboxylic acid SAR effort, the aryl headgroup campaign did not yield a more potent compound than 33.
Analogue 33 was further evaluated in vitro and in vivo and was found to possess a favorable ADME profile, no limiting off-target in vitro pharmacology, and excellent PK characteristics. In addition, the compound induced a robust and sustained lowering (>90%) of serum RBP4 levels upon 7-day QD oral dosing studies conducted in rats. Taken collectively, these data suggest that compound 33 shows particular promise as a potential oral treatment for dry AMD and Stargardt disease.
EXPERIMENTAL SECTION
General Chemistry
All reactions were performed under a dry atmosphere of nitrogen unless otherwise specified. Indicated reaction temperatures refer to the reaction bath, while room temperature (rt) is noted as 25 °C. Commercial grade reagents and anhydrous solvents were used as received from vendors, and no attempts were made to purify or dry these components further. Removal of solvents under reduced pressure was accomplished with a Buchi rotary evaporator at approximately 28 mmHg pressure using a Teflon-linked KNF vacuum pump. Thin layer chromatography was performed using 1″ × 3″ AnalTech No. 02521 silica gel plates with fluorescent indicator. Visualization of TLC plates was made by observation with short wave UV light (254 nm lamp), 10% phosphomolybdic acid in ethanol, or in iodine vapors. Preparative thin layer chromatography was performed using Analtech, 20 × 20 cm, 1000 µm preparative TLC plates. Flash column chromatography was carried out using a Teledyne Isco CombiFlash Companion Unit with RediSep Rf silica gel columns. If needed, products were purified by reverse phase chromatography, using a Teledyne Isco CombiFlash Companion Unit with RediSep Gold C18 reverse phase column. Proton NMR spectra were obtained either on a 300 MHz Bruker Nuclear Magnetic Resonance Spectrometer or on 500 MHz Bruker Nuclear Magnetic Resonance Spectrometer, and chemical shifts (δ) are reported in parts per million (ppm). Coupling constant (J) values are given in Hz, with the following spectral pattern designations: s, singlet; d, doublet; t, triplet, q, quartet; dd, doublet of doublets; m, multiplet; and br, broad. Tetramethylsilane was used as an internal reference. Melting points are uncorrected and were obtained using a MEL-TEMP electrothermal melting point apparatus. Mass spectroscopic analyses were performed using positive mode electron spray ionization (ESI) on a Varian ProStar LC-MS with a 1200L quadrapole mass spectrometer. High pressure liquid chromatography (HPLC) purity analysis was performed using a Varian Pro Star HPLC system with a binary solvent system A and B using gradient elusion [A, H2O with 0.05% trifluoroacetic acid (TFA); B, CH3CN with 0.05% TFA] and a flow rate = 1 mL/min, with UV detection at 223 nm. All final compounds were purified to ≥95% purity, and these purity levels were measured by a Varian Pro Star HPLC system. The following Varian Pro Star HPLC methods were used to establish compound purity: (A) Phenomenex C18(2) column (3.0 × 250 mm); mobile phase, A = H2O with 0.05% TFA and B = CH3CN with 0.05% TFA; gradient, 0–90% B (0.0–20.0 min); UV detection at 254 nm. (B) Phenomenex C18(2) column (3.0 × 250 mm); mobile phase, A = H2O with 0.05% TFA and B = CH3CN with 0.05% TFA; gradient, 0–100% B (0.0–20 min); UV detection at 254 nm.
6-Methyl-2-(4-(2-(trifluoromethyl)phenyl)piperidin-1-yl)-pyrimidine-4-carboxylic Acid (10)
Step A
To a solution of 1-bromo-2-(trifluoromethyl)benzene (5, 35.0 g, 156 mmol) in THF (350 mL) cooled to −78 °C was slowly added a solution of n-BuLi (70.4 mL, 2.5 M in THF, 176 mmol) over a period of 15 min. The mixture stirred at −78 °C for 40 min was allowed to warm to 0 °C and then cooled back down to −78 °C. To this was added a solution of 1-benzylpiperidin-4-one (22.1 g, 117 mmol) in THF (80 mL) over a period of 10 min. The resulting mixture continued to stir at −78 °C for 2 h. The reaction was carefully quenched with aqueous, saturated aq NH4Cl solution (500 mL), and the mixture was extracted with EtOAc (300 mL). The organic extract was washed with H2O, brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (Isco CombiFlash Companion unit, 330 g RediSep column, 0–30% EtOAc in hexanes) to give 1-benzyl-4-(2-(trifluoromethyl)phenyl)piperidin-4-ol (6) as a light-yellow oil (29.2 g, 74%): 1H NMR (500 MHz, CDCl3) δ 7.78 (d, J = 1.6 Hz, 1H), 7.59 (m, 1H), 7.47 (m, 1H), 7.36 (m, 5H), 7.31 (m, 2H), 3.58 (s, 2H), 2.80 (m, 2H), 2.55 (m, 2H), 2.27 (m, 2H), 1.88 (m, 2H); MS (ESI+) m/z 336 [M + H]+.
Step B
A 0 °C cooled solution of 1-benzyl-4-(2-(trifluoromethyl)-phenyl)piperidin-4-ol (6, 29.2 g, 87.1 mmol) in SOCl2 (60 mL) was stirred for 2 h and was then diluted with CH2Cl2 (250 mL). The mixture was carefully poured into a saturated aq NaHCO3 solution (200 mL). The biphasic mixture was separated, and the aqueous layer was further extracted with CH2Cl2 (400 mL). The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated. The resulting residue was chromatographed over silica gel (Isco CombiFlash Companion unit, 330 g RediSep column, 0–30% EtOAc in hexanes) to give 1-benzyl-4-(2-(trifluoromethyl)phenyl)-1,2,3,6-tetrahydropyridine (7) as a light-yellow oil (13.5 g, 49%): 1H NMR (500 MHz, CDCl3) δ 7.63 (d, J = 1.6 Hz, 1H), 7.48 (m, 1H), 7.39 (m, 5H), 7.28 (m, 2H), 5.56 (s, 1H), 0.68 (s, 2H), 3.14 (m, 2H), 2.70 (m, 2H), 2.39 (m, 2H); MS (ESI+) m/z 318 [M + H]+.
Step C
A mixture of 1-benzyl-4-(2-(trifluoromethyl)phenyl)-1,2,3,6-tetrahydropyridine (7, 13.6 g, 42.5 mmol), 10% Pd/C (3.0 g), and ammonium formate (26.8 g, 425 mmol) in CH3OH (800 mL) was heated at reflux for 2 h. The mixture was cooled to rt and was filtered over Celite. The filtrate was concentrated, and the resulting residue was chromatographed over silica gel (Isco CombiFlash Companion unit, 330 g RediSep column, 0–10% CH3OH with 1% NH4OH in CH2Cl2) to give 4-(2-(trifluoromethyl)phenyl)piperidine as a colorless oil (2.0 g, 21%): 1H NMR (500 MHz, CDCl3) δ 7.61 (d, J = 1.7 Hz, 1H), 7.52 (m, 2H), 7.29 (m, 1H), 3.21 (m, 2H), 3.07 (m, 1H), 2.80 (m, 2H), 2.33 (bs, 1H), 1.77 (m, 4H); MS (ESI+) m/z 230 [M + H]+. To a solution of 4-(2-(trifluoromethyl)phenyl)piperidine (5.6 g, 24.5 mmol) in CH3CN (30 mL) was added a 4 M solution of HCl in 1,4-dioxane (6.1 mL, 24.5 mmol) at rt. The mixture was stirred for 2 h and was then concentrated under reduced pressure to give 4-(2-(trifluoromethyl)-phenyl)piperidine hydrochloride (8) as a white solid (6.4 g, >99%): MS (ESI+) m/z 230 [M + H]+.
Step D
A mixture of 4-(2-(trifluoromethyl)phenyl)piperidine hydrochloride (8, 0.050 g, 0.19 mmol), methyl 2-chloro-6-methylpyr-imidine-4-carboxylate (0.046 g, 0.21 mmol), and i-Pr2NEt (0.04 mL, 0.23 mmol) in DMF (2 mL) was heated at 60 °C for 16 h. The reaction was concentrated under reduced pressure, and the resulting residue was chromatographed over silica gel (0% to 100% EtOAc in hexanes) to give methyl 6-methyl-2-(4-(2-(trifluoromethyl)phenyl)-piperidin-1-yl)pyrimidine-4-carboxylate (9) as an off-white solid (0.049 g, 68%): mp 109–112 °C; 1H NMR (500 MHz, CDCl3) δ 7.70–7.61 (m, 3H), 7.43 (m, 1H), 7.02 (s, 1H), 4.94 (m, 2H), 3.85 (s, 3H), 3.16 (m, 1H), 2.99 (m, 2H), 2.38 (s, 3H), 1.79–1.68 (m, 4H); MS (ESI+) m/z 380 [M + H]+; HPLC >99% (AUC), (Method A), tR=16.8 min.
Step E
A solution of methyl 6-methyl-2-(4-(2-(trifluoromethyl)-phenyl)piperidin-1-yl)pyrimidine-4-carboxylate (9, 0.049 g, 0.13 mmol) and LiOH·H2O (0.030 g, 0.72 mmol) in THF (2 mL) and H2O (2 mL) was stirred at rt for 16 h. The mixture was acidified to pH 5 with 2 N aq HCl and extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure to give 6-methyl-2-(4-(2-(trifluoromethyl)phenyl)piperidin-1-yl)pyrimidine-4-carboxylic acid (10) as an off-white solid (0.043 g, 91%): mp 166–169 °C; H NMR (300 MHz, DMSO-d6) δ 13.27 (bs, 1H), 7.70–7.61 (m, 3H), 7.43 (m, 1H), 6.99 (s, 1H), 4.98 (m, 2H), 3.13 (m, 1H), 2.98 (m, 2H), 2.37 (s, 3H), 1.75–1.72 (m, 4H); MS (ESI+) m/z 366 [M + H]+; HPLC >99% (AUC), (Method A), tR = 15.1 min.
6-Methyl-2-(4-(2-(trifluoromethyl)phenyl)piperazin-1-yl)-pyrimidine-4-carboxylic Acid (14)
Step A
To a mixture of tert-butyl piperazine-1-carboxylate (1.0 g, 5.37 mmol), 1-bromo-2-(trifluoromethyl)-benzene (5, 1.0 mL, 7.34 mmol), BINAP (0.135 g, 0.22 mmol), and NaOt-Bu (0.670 g, 6.57 mmol) in toluene (20 mL) was added Pd(OAc)2 (0.024 g, 0.11 mmol). The mixture was heated at 110 °C for 16 h then allowed to cool to rt. The mixture was filtered over Celite, and the filter cake was washed with EtOAc. The filtrate was concentrated under reduced pressure, and the resulting residue was purified by flash column chromatography (Isco CombiFlash Rf unit, 40 g Gold RediSep column, 0%-100% EtOAc in hexanes) to give tert-butyl 4-(2-(trifluoromethyl)-phenyl)piperazine-1-carboxylate (11) as an off-white solid (1.63 g, 92%): 1H NMR (300 MHz, CDCl3) δ 7.64 (m, 1H), 7.55 (m, 1H), 7.33 (m, 1H), 7.21 (m, 1H), 3.57 (m, 4H), 2.88 (m, 4H), 1.48 (s, 9H); MS (ESI+) m/z 331 [M + H]+.
Step B
A solution of tert-butyl 4-(2-(trifluoromethyl)phenyl)-piperazine-1-carboxylate (11, 1.63 g, 4.93 mmol) and 2.0 M HCl in Et2O (10 mL, 20 mmol) in CH2Cl2 (10 mL) was stirred at rt for 3 h. The mixture was concentrated under reduced pressure to provide 1-(2-(trifluoromethyl)phenyl)piperazine hydrochloride (12) as an off-white solid (1.23 g, 94%): 1H NMR (300 MHz, DMSO-d6) δ 9.18 (bs, 1H), 7.74 (m, 2H), 7.55 (m, 1H), 7.38 (m, 1H), 3.36 (m, 4H), 3.07 (m, 4H); MS (ESI+) m/z 231 [M + H]+.
Step C
A mixture of 1-(2-(trifluoromethyl)phenyl)piperazine hydrochloride (12, 0.100 g, 0.37 mmol), methyl 2-chloro-6-methyl-pyrimidine-4-carboxylate (0.076 g, 0.41 mmol), and i-Pr2NEt (0.15 mL, 0.86 mmol) in DMF (3 mL) was heated at 60 °C for 16 h. The reaction was concentrated under reduced pressure, and the resulting residue was chromatographed over silica gel (0% to 100% EtOAc in hexanes) to give methyl 6-methyl-2-(4-(2-(trifluoromethyl)phenyl)piperazin-1-yl)-pyrimidine-4-carboxylate (13) as an off-white solid (0.089 g, 63%): mp 103–106 °C; 1H NMR (300 MHz, DMSO-d6) δ 7.71–7.57 (m, 3H), 7.39 (m, 1H), 7.06 (s, 1H), 3.90 (m, 4H), 3.85 (s, 3H), 2.94 (m, 4H), 2.39 (s, 3H); MS (ESI+) m/z 381 [M + H]+; HPLC 98.9% (AUC), (Method A), tR = 16.5 min.
Step D
A mixture of methyl 6-methyl-2-(4-(2-(trifluoromethyl)-phenyl)piperazin-1-yl)pyrimidine-4-carboxylate (13, 0.080 g 0.21 mmol) and LiOH·H2O (0.050 g, 1.19 mmol) in THF (2 mL) and H2O (2 mL) was stirred at rt for 16 h. The mixture was acidified to pH 5 with 2 N aq HCl and extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure to give 6-methyl-2-(4-(2-(trifluoromethyl)-phenyl)piperazin-1-yl)pyrimidine-4-carboxylic acid (14) as an off-white solid (0.043 g, 91%): 1H NMR (300 MHz, DMSO-d6) δ 13.53 (bs, 1H), 7.91–7.80 (m, 3H), 7.58 (m, 1H), 7.01 (s, 1H), 4.12 (m, 4H), 2.70 (m, 4H), 2.37 (s, 3H); MS (ESI+) m/z 367 [M + H]+; HPLC >99% (AUC), (Method A), tR = 14.8 min.
6-Methyl-2-((3aR,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydropyrrolo[3,4-c]pyrrol-2(1H)-yl)pyrimidine-4-carboxylic Acid (18)
Step A
To a mixture of (3aR,6aS)-tert-butyl hexahydropyrrolo[3,4-c]-pyrrole-2(1H)-carboxylate (1.0 g, 4.70 mmol), 1-bromo-2-(trifluoro-methyl)benzene (5, 1.0 mL, 7.34 mmol), BINAP (0.117 g, 0.19 mmol), and NaOt-Bu (0.587 g, 6.11 mmol) in toluene (10 mL) was added Pd(OAc)2 (0.021 g, 0.10 mmol). The mixture was heated at 110 °C for 16 h then allowed to cool to rt. The mixture was filtered over Celite, and the filter cake was washed with EtOAc. The filtrate was concentrated under reduced pressure, and the resulting residue was purified by flash column chromatography (Isco CombiFlash Rf unit, 40 g Gold RediSep column, 0%-100% EtOAc in hexanes) to give (3aR,6aS)-tert-butyl 5-(2-(trifluoromethyl)phenyl)hexahydropyrrolo[3,4-c]pyrrole-2(1H)-carbox-ylate (15) as an off-white solid (1.35 g, 81%): 1H NMR (300 MHz, CDCl3) δ 7.61 (m, 1H), 7.46 (m, 1H), 7.17 (m, 1H), 7.02 (m, 1H), 3.70 (m, 2H), 3.38–3.28 (m, 4H), 3.14 (m, 2H), 2.93 (m, 2H); MS (ESI+) m/z 357 [M + H]+.
Step B
A solution of (3aR,6aS)-tert-butyl 5-(2-(trifluoromethyl)-phenyl)hexahydropyrrolo[3,4-c]pyrrole-2(1H)-carboxylate (15, 1.37 g, 3.84 mmol) and 2.0 M HCl in Et2O (10 mL, 20 mmol) in CH2Cl2 (10 mL) was stirred at rt for 3 h. The mixture was concentrated under reduced pressure to provide (3aR,6aS)-2-(2-(trifluoromethyl)phenyl)-octahydropyrrolo[3,4-c]pyrrole hydrochloride (16) as an off-white solid (0.968 g, 86%): 1H NMR (300 MHz, CDCl3) δ 9.18 (bs, 1H), 10.08 (bs, 1H), 9.56 (bs, 1H), 7.64 (m, 1H), 7.53 (m, 1H), 7.43 (m, 1H), 7.21 (m, 1H), 3.72 (m, 2H), 3.12 (m, 8H); MS (ESI+) m/z 257 [M + H]+.
Step C
A mixture of (3aR,6aS)-2-(2-(trifluoromethyl)phenyl)-octahydropyrrolo[3,4-c]pyrrole hydrochloride (16, 0.100 g, 0.34 mmol), methyl 2-chloro-6-methylpyrimidine-4-carboxylate (0.070 g, 0.38 mmol), and i-Pr2NEt (0.1 mL, 0.46 mmol) in DMF (4 mL) was heated at 60 °C for 16 h. The reaction was concentrated under reduced pressure, and the resulting residue was chromatographed over silica gel (0% to 10% CH3OH in CH2Cl2) to give methyl 6-methyl-2-((3aR,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydropyrrolo[3,4-c]pyrrol-2(1H)-yl)-pyrimidine-4-carboxylate (17) as an off-white solid (0.039 g, 28%): 1H NMR (300 MHz, DMSO-d6) δ 7.60–7.51 (m, 3H), 7.34 (m, 1H), 7.08 (s, 1H), 3.87 (m, 5H), 3.47–3.38 (m, 4H), 3.14–3.07 (m, 4H), 2.37 (s, 3H); MS (ESI+) m/z 407 [M + H]+; HPLC >99% (AUC), (Method A), tR = 19.1 min.
Step D
A mixture of 6-methyl-2-((3aR,6aS)-5-(2-(trifluoromethyl)-phenyl)hexahydropyrrolo[3,4-c]pyrrol-2(1H)-yl)pyrimidine-4-carbox-ylate (17, 0.031 g, 0.076 mmol) and LiOH · H2O (0.031 g, 0.74 mmol) in THF (2 mL) and H2O (1 mL) was stirred at rt for 16 h. The mixture was acidified to pH 5 with 2 N aq HCl and extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure to give 6-methyl-2-(4-(2-(trifluoromethyl)phenyl)-piperazin-1-yl)pyrimidine-4-carboxylic acid (18) as an off-white solid (0.043 g, 91%): 1H NMR (300 MHz, DMSO-d6) δ 13.53 (bs, 1H), 7.61–7.58 (m, 2H), 7.34 (m, 1H), 7.10 (m, 1H), 6.99 (s, 1H), 3.86 (m, 2H), 3.47–3.35 (m, 4H), 3.14 (m, 4H), 2.36 (s, 3H); MS (ESI+) m/z 393 [M + H]+; HPLC 97.6% (AUC), (Method A), tR = 16.8 min.
(3aR,5r,6aS)-5-(2-(Trifluoromethyl)phenyl)octahydrocyclopenta-[c]pyrrole Hydrochloride (26)
Step A (i)
To a 0 °C cooled solution of LiAlH4 in THF (1.0 M, 800 mL, 800 mmol) in THF (800 mL) was carefully added (3aR,7aS)-3a,4,7,7a–tetrahydro-1H–isoindole-1,3(2H)-dione (19, 53.7 g, 0.35 mol) portion-wise. An exotherm of ~5 °C occurred upon each addition of 19. Upon complete addition, the mixture was allowed to warm to rt followed by heating at 70 °C for 16 h. The mixture was allowed to cool back to rt and then further cooled to 0 °C. The reaction was carefully quenched by slow addition of H2O (30 mL), 15% aq NaOH solution (30 mL), followed by another bolus of H2O (90 mL). The rate of quenching was done carefully so as to maintain an internal temperature below 25 °C. The mixture was stirred for 1 h and was filtered through Celite. The aqueous filtrate was extracted with Et2O (2 × 100 mL), and the organic extracts were combined and concentrated under reduced pressure. The resulting residue was purified using a Kugelrohr distillation apparatus to give (3aR,7aS)-2,3,3a,4,7,7a–hexahydro-1H–isoindole as a clear, colorless oil (19.45 g, 44%): 1H NMR (500 MHz, CDCl3) δ 5.29 (s, 2H), 3.88 (bs, 1H), 3.26 (m, 2H), 2.82 (m, 2H), 2.41–2.19 (m, 4H), 1.96 (m, 2H).
Step B
To a 0 °C cooled solution of (3aR,7aS)-2,3,3a,4,7,7a–hexahydro-1H–isoindole (11.5 g, 93.5 mmol) in CH2Cl2 (200 mL) was added Boc2O (24.5 g, 112 mmol) and the mixture stirred at rt for 16 h. The mixture was washed with H2O (100 mL), brine (100 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography (Isco CombiFlash Rf unit, 330 g RediSep column, 0% to 30% EtOAc in hexanes) to give (3aR,7aS)-tert-butyl 3a,4,7,7a–tetrahydro-1H–isoindole-2(3H)-carboxylate (20) as an oil (20.10 g, 49%): 1H NMR (500 MHz, CDCl3) δ 5.64 (s, 2H), 3.39 (m, 2H), 3.20 (m, 2H), 3.15 (m, 2H), 2.23–2.19 (m, 4H), 1.97 (m, 2H), 1.57 (s, 9H).
Step C
To a 0 °C cooled mixture of (3aR,7aS)-tert-butyl 3a,4,7,7a–tetrahydro-1H–isoindole-2(3H)-carboxylate (20, 66.78 g, 224 mmol) in CH3CN (600 mL), CCl4 (400 mL), and H2O (800 mL) was added NaIO4 (192.3 g, 899 mmol) followed by RuO2·H2O (1.19 g, 8.94 mmol). The mixture was stirred at rt for 24 h with mechanical stirring and then filtered through Celite. The filter cake was washed with 10% CH3OH in CH2Cl2 (200 mL), and the biphasic mother liquor was separated. The aqueous phase was further extracted with CH2Cl2 (3 × 150 mL), and the combined organic extracts were washed with H2O (100 mL), brine (100 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was filtered through a plug of silica gel using a CH3OH/CH2Cl2 eluent system (2%-10% CH3OH in CH2Cl2). The filtrate was concentrated under reduced pressure to give 2,2′-((3S,4R)-1-(tert-butoxycarbonyl)pyrrolidine-3,4-diyl)diacetic acid (21) as a solid (46.75 g, 72%): 1H NMR (500 MHz, DMSO-d6) δ 12.2 (s, 2H), 3.38 (m, 2H), 3.02 (m, 2H), 2.49 (m, 2H), 2.32 (m, 2H), 2.29 (m, 2H), 1.42 (s, 9H).
Step D
To a suspension of 2,2′-((3S,4R)-1-(tert-butoxycarbonyl)-pyrrolidine-3,4-diyl)diacetic acid (21, 6.97 g, 24.31 mmol) in Ac2O (50 mL) was added NaOAc (1.99 g, 24.31 mmol), and the mixture was heated at 120 °C for 3 h. The mixture was allowed to cool to rt and filtered through Celite. The filter cake was washed with Et2O (5 × 50 mL), and the mother liquor was concentrated under reduced pressure. The resulting residue was purified by flash column chromatography (Isco CombiFlash Rf unit, 120 g RediSep column, 0%-30% EtOAc in hexanes) to give (3aR,6aS)-tert-butyl 5-oxohexahydro-cyclopenta[c]pyrrole-2(1H)-carboxylate (22) as a white foam (2.17 g, 40%): 1H NMR (500 MHz, CDCl3) δ 3.69 (m, 2H), 3.22 (m, 2H), 2.91 (m, 2H), 2.50 (m, 2H), 2.17 (m, 2H), 1.46 (s, 9H).
Step E
To a −78 °C cooled solution of (3aR,6aS)-tert-butyl 5-oxohexahydrocyclopenta[c]pyrrole-2(1H)-carboxylate (22, 22.35 g, 99.2 mmol) in THF (500 mL) was slowly added a solution of LiHMDS in THF (1.0 M, 129 mL). The mixture continued to stir at −78 °C for 30 min, then a solution of 1,1,1-trifluoro-N-phenyl-N-((trifluoromethyl)sulfonyl)methanesulfonamide (49.65 g, 139 mmol) in THF (150 mL) was slowly added. The mixture was stirred for an additional 1 h at −78 °C and was then allowed to stir at rt for 2 h. The mixture was concentrated under reduced pressure, and the residue was purified by flash column chromatography (Isco CombiFlash Rf unit, 330 g RediSep column, 0%-50% EtOAc in hexanes) to give (±)-(3aS,6aS)-tert-butyl 5-(((trifluoromethyl)sulfonyl)oxy)-3,3a,6,6a–tetrahydrocyclopenta[c]pyrrole-2(1H)-carboxylate ((±)-23) as a clear, viscous oil (1.56 g, quantitative): 1H NMR (500 MHz, CDCl3) δ 5.58 (s, 1H), 3.62 (m, 1H), 3.53 (m, 1H), 3.46 (m, 2H), 3.19 (m, 1H), 2.95 (m, 2H), 2.46 (m, 1H), 1.47 (s, 9H).
Step F
To an N2 degassed mixture of (±)-(3aS,6aS)-tert-butyl 5-(((trifluoromethyl)sulfonyl)oxy)-3,3a,6,6a–tetrahydrocyclopenta[c]-pyrrole-2(1H)-carboxylate ((±)-23, 14.79 g, 41.4 mmol), 2-trifluor-omethylphenylboronic acid (19.70 g, 104 mmol), and a 2 M aqueous solution of Na2CO3 (250 mL) in DME (500 mL) was added Pd(PPh3)4 (4.80 g, 4.16 mmol). The mixture was heated at 80 °C for 6 h, then cooled to rt, and diluted with H2O (500 mL). The aqueous mixture was extracted with EtOAc (2 × 200 mL), and the combined organic extracts were washed with H2O (200 mL), brine (200 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography (Isco CombiFlash Rf unit, 330 g RediSep column, 0%-10% EtOAc in hexanes) to give (±)-(3aR,6aS)-tert-butyl 5-(2-(trifluoromethyl)-phenyl)-3,3a,6,6a–tetrahydrocyclopenta[c]pyrrole-2(1H)-carboxylate ((±)-24 as a clear, viscous oil (13.70 g, 94%): 1H NMR (500 MHz, CDCl3) δ 7.65 (m, 1H), 7.47 (m, 2H), 7.25 (m, 1H), 5.58 (s, 1H), 3.85–3.42 (m, 4H), 3.23 (m, 1H), 2.98 (m, 2H), 2.49 (m, 1H), 1.47 (s, 9H).
Step G
A mixture of (±)-(3aR,6aS)-tert-butyl 5-(2-(trifluoromethyl)-phenyl)-3,3a,6,6a–tetrahydrocyclopenta[c]pyrrole-2(1H)-carboxylate ((±)-24, 8.63 g, 24.41 mmol) and 10% Pd/C (1.57 g, wet, 10% w/w) in CH3OH (50 mL) was subjected to an atmosphere of H2 gas (40 psi) using a Parr Shaker apparatus at rt for 16 h. The mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure. The resulting residue was purified by flash column chromatography (Isco CombiFlash Rf unit, 40 g RediSep column, 0%-30% EtOAc in hexanes) to give (3aR,5r,6aS)-tert-butyl 5-(2-(trifluoromethyl)-phenyl)hexahydrocyclopenta[c]pyrrole-2(1H)-carboxylate (25) as a clear, viscous oil (0.910 g, 85%): 1H NMR (500 MHz, CDCl3) δ 7.69 (m, 1H), 7.51 (m, 2H), 7.25 (m, 1H), 3.49 (m, 5H), 2.75 (m, 2H), 2.92 (m, 2H), 1.52 (m, 2H), 1.48 (s, 9H).
Step H
To a 0 °C cooled solution of (3aR,5r,6aS)-tert-butyl 5-(2-(trifluoromethyl)phenyl)hexahydrocyclopenta[c]pyrrole-2(1H)-car-boxylate (25, 7.94 g, 22.32 mmol) in CH2Cl2 (60 mL) was added a 2.0 M HCl solution in Et2O (60 mL), and the mixture was allowed to stir at rt for 24 h. The mixture was diluted with Et2O (200 mL), and the precipitated product was filtered to give (3aR,5r,6aS)-5-(2-(trifluo-romethyl)phenyl)octahydrocyclopenta[c]pyrrole hydrochloride (26) as a white solid (5.90 g, 91%): 1H NMR (500 MHz, CDCl3) δ 10.17 (bs, 1H), 8.06 (m, 1H), 7.59 (m, 1H), 7.53 (m, 1H), 7.27 (m, 1H), 3.42 (m, 2H), 3.38 (m, 3H), 3.01 (m, 2H), 2.36 (m, 2H), 1.96 (m, 2H); MS (ESI+) m/z 256 [M + H]+.
Methyl 2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)pyrimidine-4-carboxylate (27)
Step A
To a solution of (3aR,5R,6aS)-5-(2-(trifluoromethyl)phenyl)octa-hydrocyclopenta[c]pyrrole hydrochloride (26, 0.050 g, 0.17 mmol) and Et3N (0.05 mL, 0.51 mmol) in DMF (10 mL) was added methyl 2-chloropyrimidine-4-carboxylate (0.029 g, 0.17 mmol), and the resulting solution was stirred at 60 °C for 16 h. The reaction was diluted with H2O (200 mL) and extracted with EtOAc (3 × 100 mL). The combined organic extracts were washed with H2O (3 × 100 mL), brine (100 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0% to 30% EtOAc in hexanes) to give methyl 2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydrocyclopenta[c]-pyrrol-2(1H)-yl)pyrimidine-4-carboxylate (27) as an off-white solid (0.055 g, 77%): MS (ESI+) m/z 392 [M + H]+.
Methyl 6-Methyl-2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)-hexahydrocyclopenta[c]pyrrol-2(1H)-yl)pyrimidine-4-carboxylate (28)
Compound 28 was prepared according to a similar procedure described for the synthesis of 27. 1H NMR (300 MHz, CDCl3) δ 7.61 (m, 1H), 7.58 (m, 2H), 7.23 (m, 1H), 7.05 (s, 1H), 3.95 (s, 3H), 3.82 (m, 4H), 3.59 (m, 1H), 2.92 (m, 2H), 2.44 (s, 3H), 2.40 (m, 2H), 1.69 (m, 2H); MS (ESI+) m/z 406 [M + H]+; HPLC >99% purity (Method C), tR = 16.3 min.
Methyl 6-(trifluoromethyl)-2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)-phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)pyrimidine-4-carboxylate (29)
Compound 29 was prepared according to a similar procedure described for the synthesis of 27. MS (ESI+) m/z 460 [M + H]+.
Methyl 6-methoxy-2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)-hexahydrocyclopenta[c]pyrrol-2(1H)-yl)pyrimidine-4-carboxylate (30)
Compound 30 was prepared according to a similar procedure described for the synthesis of 27. MS (ESI+) m/z 422 [M + H]+.
Methyl 6-isopropyl-2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)-hexahydrocyclopenta[c]pyrrol-2(1H)-yl)pyrimidine-4-carboxylate (31)
Compound 31 was prepared according to a similar procedure described for the synthesis of 27. MS (ESI+) m/z 434 [M + H]+.
2-((3aR,5r,6aS)-5-(2-(Trifluoromethyl)phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)pyrimidine-4-carboxylic Acid (32)
Step A
A solution of methyl 2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)-phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)pyrimidine-4-carboxylate (27, 0.050 g, 0.12 mmol) and 2 N aq NaOH (5 mL) in a 1:1 mixture of CH3OH/THF (10 mL) stirred at rt for 16 h. The mixture was neutralized at 0 °C with 2 N aq HCl and extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0% to 10% CH3OH in CH2Cl2) to give 2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)-phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)pyrimidine-4-carboxylic acid (32) as an off-white solid (0.044 g, 92%): mp 183–187 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.56 (s, 1H), 7.71 (m, 1H), 7.65 (m, 2H), 7.39 (m, 1H), 7.06 (s, 1H), 3.73 (m, 2H), 3.64 (m, 2H), 3.43 (m, 1H), 2.86 (m, 2H), 2.30 (m, 2H), 1.64 (m, 2H); MS (ESI+) m/z 378 [M + H]+; HPLC >99% purity (Method A), tR = 10.5 min.
6-Methyl-2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)-hexahydrocyclopenta[c]pyrrol-2(1H)-yl)pyrimidine-4-carboxylic Acid (33)
Compound 33 was prepared according to a similar procedure described for the synthesis of 32. Mp 129–131 °C; 1H NMR (300 MHz, DMSO-d6) δ 7.73 (m, 1H), 7.64 (m, 2H), 7.36, (m, 1H), 6.51, (s, 1H), 3.90 (s, 3H), 3.76 (m, 2H), 3.72, (m, 2H), 2.88 (m, 2H), 2.22 (m, 2H), 1.67 (m, 2H); 13C NMR (300 MHz, DMSO-d6) δ 169.02, 166.47, 160.56, 157.04, 142.84, 132.72, 128.29, 127.09, 126.71, 126.34, 125.28, 125.19, 122.80, 107.82, 52.08, 42.65, 42.28, 41.19, 23.94; MS (ESI+) m/z 408 [M + H]+; HRMS (ESI+) for C20H21F3N3O2 [M + H]+ calcd 392.1586, found 392.1570; HPLC >99% purity (Method A), tR = 14.3 min.
6-(Trifluoromethyl)-2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)-hexahydrocyclopenta[c]pyrrol-2(1H)-yl)pyrimidine-4-carboxylic Acid (34)
Compound 34 was prepared according to a similar procedure described for the synthesis of 32. Mp 155–156 °C; 1H NMR (500 MHz, DMSO-d6) δ 13.92 (bs, 1H), 7.57 (m, 1H), 7.65 (m, 1H), 7.59 (m, 1H), 7.40 (m, 1H), 7.33 (s, 1H), 3.78 (m, 2H), 3.68 (m, 2H), 3.30 (m, 1H), 2.93 (m, 2H), 2.29 (m, 2H), 1.64 (m, 2H); MS (ESI+) m/z 446 [M + H]+; HPLC >99% purity (Method A), tR = 12.9 min.
6-Methoxy-2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)-hexahydrocyclopenta[c]pyrrol-2(1H)-yl)pyrimidine-4-carboxylic Acid (35)
Compound 35 was prepared according to a similar procedure described for the synthesis of 32. Mp 171–174 °C; 1H NMR (300 MHz, CDCl3) δ 7.73 (m, 1H), 7.64 (m, 2H), 7.36, (m, 1H), 6.51, (s, 1H), 3.90 (s, 3H), 3.76 (m, 2H), 3.72, (m, 2H), 2.88 (m, 2H), 2.22 (m, 2H), 1.67 (m, 2H); MS (ESI+) m/z 408 [M + H]+; HPLC >99% purity (Method A), tR = 16.2 min.
6-Isopropyl-2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)-hexahydrocyclopenta[c]pyrrol-2(1H)-yl)pyrimidine-4-carboxylic Acid (36)
Compound 36 was prepared according to a similar procedure described for the synthesis of 32. 1H NMR (300 MHz, DMSO-d6) δ 13.21 (bs, 1H), 7.72 (m, 1H), 7.65, (m, 2H), 7.39 (m, 2H), 7.01, (s, 1H), 3.75 (m, 2H), 3.71, (m, 2H), 3.42 (m, 1H), 2.90, (m, 3H), 2.49 (m, 2H), 1.66 (m, 2H), 1.23 (s, 6H); MS (ESI+) m/z 420 [M + H]+; HPLC >99% purity (Method A), tR = 13.9 min.
6-Methyl-2-((3aR,5s,6aS)-5-(2-(trifluoromethyl)phenyl)-hexahydrocyclopenta[c]pyrrol-2(1H)-yl)pyrimidine-4-carboxylic Acid (40)
Step A
To a solution of (±)-(3aR,6aS)-tert-butyl 5-(2-(trifluoromethyl)phenyl)-3,3a,6,6a–tetrahydrocyclopenta[c]pyrrole-2(1H)-carboxylate ((±)-24, 120 mg, 0.34 mmol) in CH2Cl2 (3 mL) was added TFA (3 mL) and the resulting solution stirred at rt for 3 h. The residue was dissolved in CH2Cl2 (25 mL) and washed with saturated aq NaHCO3 solution (25 mL), brine (25 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to provide (±)-(3aS,6aR)-5-(2-(trifluoromethyl)phenyl)-1,2,3,3a,4,6a–hexahydro-cyclopenta[c]pyrrole ((±)-37) as an off-white solid (0.146 mg, > 99%): 1H NMR (300 MHz, CDCl3) δ 9.21 (br s, 1H), 8.18 (br s, 1H), 7.67 (d, J = 7.8 Hz, 1H), 7.51–7.34 (m, 3H), 5.57 (s, 1H), 3.82 (br s, 1H), 3.62–3.54 (m, 2H), 3.39–3.09 (m, 4H), 2.62–2.56 (m, 1H).
Step B
To a solution of (±)-(3aS,6aR)-5-(2-(trifluoromethyl)-phenyl)-1,2,3,3a,4,6a–hexahydrocyclopenta[c]pyrrole ((±)-37, 0.680 g, 1.92 mmol) in CH3OH (25 mL) was added Pd/C (10% w/w, Degussa type E101 NE/W, 0.140 g). The mixture was subjected to an atmosphere of H2 (50 psi) at rt for 6 h and was filtered through Celite. The filtrated was concentrated under reduced pressure, and the resulting residue was purified by reversed phase column chromatography (Isco C18 Reversed Phase Gold column, 10%-30% CH3CN in H2O with 0.05% TFA). The resulting material was dissolved in CH2Cl2 and washed with saturated aqueous NaHCO3, dried over Na2SO4, filtered, and concentrated under reduced pressure to give (3aR,5s,6aS)-5-(2-(trifluoromethyl)phenyl)octahydrocyclopenta[c]pyrrole (39) as a white solid (0.070 g, 14%): 1H NMR (300 MHz, CDCl3) δ 7.61 (d, J = 7.8 1H), 7.50 (m, 2H), 7.30–7.24 (m, 1H), 3.54–3.42 (m, 1H), 3.32–3.26 (m, 2H), 2.81–2.68 (m, 2H), 2.51–2.46 (m, 2H), 1.84–1.76 (m, 4H); MS (ESI+) m/z 256 [M + H]+.
Step C
To a solution of (3aR,5s,6aS)-5-(2-(trifluoromethyl)-phenyl)octahydrocyclopenta[c]pyrrole (39, 0.100 g, 0.39 mmol) and Et3N (0.16 mL, 1.17 mmol) in DMF (10 mL) was added methyl 2-chloropyrimidine-4-carboxylate (0.067 g, 0.39 mmol), and the resulting solution was stirred at 60 °C for 16 h. The reaction was diluted with H2O (200 mL) and extracted with EtOAc (3 × 100 mL). The combined organic extracts were washed with H2O (3 × 100 mL), brine (100 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0% to 30% EtOAc in hexanes) to give methyl 6-methyl-2-((3aR,5s,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydrocyclopenta[c]-pyrrol-2(1H)-yl)pyrimidine-4-carboxylate as an off-white solid (0.123 g, 78%): MS (ESI+) m/z 406 [M + H]+.
Step D
A solution of methyl 6-methyl-2-((3aR,5s,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)-pyrimidine-4-carboxylate (0.100 g, 0.24 mmol) and 2 N aq NaOH (5 mL) in a 1:1 mixture of CH3OH/THF (10 mL) stirred at room temperature for 16 h. The mixture was carefully at 0 °C with 2 N aq HCl and extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0% to 10% CH3OH in CH2Cl2) to give 6-methyl-2-((3aR,5s,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydro-cyclopenta[c]pyrrol-2(1H)-yl)pyrimidine-4-carboxylic acid (40) as an off-white solid (0.089 g, 93%): mp 175–178 °C; 1H NMR (300 MHz, CDCl3) δ 7.68–7.45 (m, 3H), 7.29 (m, 1H), 7.15 (s, 1H), 4.00 (m, 2H), 3.75 (m, 1H), 3.46 (m, 2H), 3.17 (m, 2H), 2.48 (s, 3H), 2.17–1.90 (m, 4H); MS (ESI+) m/z 392 [M + H]+; HPLC >99% purity (Method A), tR = 14.2 min.
5,6-Dimethyl-2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)-hexahydrocyclopenta[c]pyrrol-2(1H)-yl)pyrimidine-4-carboxylic Acid (41)
Step A
To a mixture of (3aR,5r,6aS)-5-(2-(trifluoromethyl)-phenyl)octahydrocyclopenta[c]pyrrole hydrochloride (26, 0.400 g, 1.37 mmol), 2,4-dichloro-5,6-dimethylpyrimidine (0.242 g 1.37 mmol), JohnPhos (0.048 g, 0.137 mmol), and Cs2CO3 (1.34 g, 4.11 mmol) in toluene (10 mL) was added Pd(OAc)2 (0.015 g, 0.068 mmol). The mixture was heated at reflux for 16 h then allowed to cool to rt. The mixture was filtered over Celite, and the filter cake was washed with EtOAc. The filtrate was concentrated under reduced pressure, and the resulting residue was purified by flash column chromatography (Isco CombiFlash Rf unit, 40 g Gold RediSep column, 0%-15% EtOAc in hexanes) to give (3aR,5r,6aS)-2-(4-chloro-5,6-dimethylpyrimidin-2-yl)-5-(2-(trifluoromethyl)phenyl)octahydrocyclopenta[c]pyrrole as a white solid (0.271 g, 50%): MS (ESI+) m/z 396 [M + H]+.
Step B
To a mixture of (3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)-octahydrocyclopenta[c]pyrrole hydrochloride (26, 0.200 g, 0.50 mmol), Mo(CO)6 (0.160 g, 0.60 mmol), and BINAP (0.009 g, 0.05 mmol) in CH3OH (10 mL), and CH3CN (10 mL) was added Pd(OAc)2 (0.005 g, 0.024 mmol). The mixture was heated at 80 °C for 16 h then allowed to cool to rt. The mixture was filtered over Celite, and the filter cake was washed with EtOAc. The filtrate was concentrated under reduced pressure, and the resulting residue was purified by flash column chromatography (Isco CombiFlash Rf unit, 40 g Gold RediSep column, 0%-15% EtOAc in hexanes) to give methyl 5,6-dimethyl-2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydrocyclopenta[c]-pyrrol-2(1H)-yl)pyrimidine-4-carboxylate as a white solid (0.132 g, 63%): MS (ESI+) m/z 420 [M + H]+.
Step C
A mixture of 5,6-dimethyl-2-((3aR,5r,6aS)-5-(2-(trifluoro-methyl)phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)pyrimidine-4-carboxylate (0.130 g 0.309 mmol) and LiOH·H2O (0.038 g, 0.929 mmol) in THF (4 mL), CH3OH (4 mL), and H2O (4 mL) stirred at rt for 16 h. The mixture was acidified to pH 6 via the addition of 2 N aq HCl and then extracted with CH2Cl2 (3 × 50 mL). The combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by flash column chromatography (Isco CombiFlash Rf unit, 40 g RediSep column, 0%-8% CH3OH in CH2Cl2) to give 5,6-dimethyl-2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)-pyrimidine-4-carboxylic acid (41) as a white solid (0.115 g, 92%): mp 140–142 °C; 1H NMR (300 MHz, DMSO-d6) δ 7.72 (m, 3H), 7.32 (m, 1H), 3.62 (m, 4H), 2.89 (m, 2H), 2.43 (s, 3H), 2.23 (m, 2H), 2.09 (s, 3H), 1.66 (m, 2H); MS (ESI+) m/z 406 [M + H]+; HPLC >99% (AUC), (Method A), tR = 13.3 min.
5-Methyl-2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)-hexahydrocyclopenta[c]pyrrol-2(1H)-yl)pyrimidine-4-carboxylic Acid (42)
Step A
To a solution of (3aR,5R,6aS)-5-(2-(trifluoromethyl)-phenyl)octahydrocyclopenta[c]pyrrole hydrochloride (26, 0.050 g, 0.17 mmol) and Et3N (0.05 mL, 0.51 mmol) in DMF (10 mL) was added methyl 2-chloro-5-methylpyrimidine-4-carboxylate (0.031 g, 0.17 mmol), and the resulting solution was stirred at 60 °C for 16 h. The reaction was allowed to cool to rt and then diluted with H2O (200 mL). The aqueous mixture was extracted with EtOAc (3 × 100 mL), and the combined organic extracts were washed with H2O (3 × 100 mL), brine (100 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0% to 30% EtOAc in hexanes) to give methyl 5-methyl-2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydrocyclopenta[c]-pyrrol-2(1H)-yl)pyrimidine-4-carboxylate as an off-white solid (0.015 g, 22%): MS (ESI+) m/z 406 [M + H]+.
Step B
A solution of methyl 5-methyl-2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)-pyrimidine-4-carboxylate (0.015 g, 0.04 mmol) and 2 N aq NaOH (5 mL) in a 1:1 mixture of CH3OH/THF (10 mL) stirred at rt for 16 h. The mixture was carefully neutralized at 0 °C with 2 N aq HCl and extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0% to 10% CH3OH in CH2Cl2) to 5-methyl-2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydro-cyclopenta[c]pyrrol-2(1H)-yl)pyrimidine-4-carboxylic acid (42) as an off-white solid (0.013 g, 92%): 1H NMR (300 MHz, CD3OD) δ 8.12 (s, 1H), 7.67–7.78 (m, 3H), 7.31 (m, 1H), 3.41–3.61 (m, 5H), 2.91 (m, 2H), 2.34 (m, 4H), 2.15 (s, 3H), 1.67 (m, 2H); MS (ESI+) m/z 392 [M + H]+; HPLC >99% (AUC), (Method A), tR = 14.2 min.
4-Methyl-2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)-hexahydrocyclopenta[c]pyrrol-2(1H)-yl)pyrimidine-5-carboxylic Acid (43)
Step A
A solution of (3aR,6aS)-5-(2-(trifluoromethyl)-phenyl)octahydrocyclopenta[c]pyrrole hydrochloride (26, 0.100 g, 0.340 mmol), ethyl 2-chloro-6-methylpyrimidine-5-carboxylate (76 mg, 0.38 mmol), and i-Pr2NEt (0.15 mL, 0.86 mmol) in DMF (5 mL) was heated at 70 °C for 90 min via microwave irradiation. The mixture was allowed to cool to rt then concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (Isco CombiFlash Companion unit, 12 g Redisep column, 0–100% EtOAc in hexanes) to provide methyl 4-methyl-2-((3aR,6aS)-5-(2-(trifluoro-methyl)phenyl)hexahydrocyclopenta[c]pyrrole-2(1H)-yl)pyrimidine-5-carboxylate as a colorless semisolid (99 mg, 69%): 1H NMR (300 MHz, CDCl3) δ 8.87 (s, 1H), 7.61 (dd, J = 7.8 Hz, 1H), 7.50–7.48 (m, 2H), 4.36–4.29 (m, 2H) 3.88–3.77 (m, 4H), 3.59–3.56 (m, 1H), 3.06–2.94 (m, 2H), 2.71 (s, 3H), 2.44–2.35 (m, 2H), 1.69–1.56 (m, 3H), 1.39–1.35 (m, 3H); MS (ESI+) m/z 420 [M + H]+.
Step B
A mixture of methyl 4-methyl-2-((3aR,6aS)-5-(2-(trifluo-romethyl)phenyl)hexahydro cyclopenta[c]pyrrole-2(1H)-yl)-pyrimidine-5-carboxylate (0.099 g, 0.241 mmol) and LiOHH2O (0.100 g, 2.38 mmol) in THF (4 mL) and H2O (2 mL) stirred at rt for 18 h and was then heated at 40 °C for 7 h. The mixture was allowed to cool to rt and was diluted with H2O (4 mL) and acidified with 2 N aq HCl to pH 6. The mixture was extracted with EtOAc (3 × 10 mL), and the combined extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure to provide 4-methyl-2-((3aR,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)-pyrimidine-5-carboxylic acid (43) as a white solid (0.020 g, 21%): mp 82–85 °C; 1H NMR (300 MHz, DMSO-d6) δ 12.57 (br s, 1H), 8.73 (s, 1H), 7.73–7.61 (m, 3H), 7.41–7.38 (m, 1H), 3.82–3.62 (m, 4H), 2.86 (br s, 2H), 2.58 (s, 3H), 2.26 (br s, 2H), 1.63 (br s, 2H); MS (ESI+) m/z 392 [M + H]+; HPLC 98.5% purity (Method H), tR = 17.4 min.
2-((3aR,5r,6aS)-5-(2-(Trifluoromethyl)phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)pyrimidine-5-carboxylic Acid (44)
Step A
A mixture of (3aR,5R,6aS)-5-(2-(trifluoromethyl)phenyl)-octahydrocyclopenta[c]pyrrole hydrochloride (26, 0.050 g, 0.172 mmol) methyl 2-chloropyrimidine-5-carboxylate (0.029 g, 0.17 mmol), and Et3N (0.05 mL, 0.51 mmol) in DMF (5 mL) was heated at 70 °C for 16 h. The mixture was allowed to cool to rt and was diluted with H2O (20 mL). The aqueous mixture was extracted with EtOAc (3 × 20 mL), and the combined organic extracts were washed with H2O (3 × 100 mL), brine (100 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0% to 30% EtOAc in hexanes) to give methyl 2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydro-cyclopenta[c]pyrrol-2(1H)-yl)pyrimidine-5-carboxylate as an off-white solid (0.035 g, 52%): MS (ESI+) m/z 392 [M + H]+.
Step B
A solution of methyl 2-((3aR,5r,6aS)-5-(2-(trifluoro-methyl)phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)pyrimidine-5-carboxylate (0.030 g, 0.071 mmol) and 2 N aq NaOH (5 mL) in a 1:1 mixture of CH3OH/THF (10 mL) stirred at room temperature for 16 h. The mixture was carefully neutralized at 0 °C with 2 N HCl and extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0% to 10% CH3OH in CH2Cl2) to 2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)-pyrimidine-5-carboxylic acid (44) as an off-white solid (0.026 g, 91%): mp 218–222 °C; 1H NMR (500 MHz, CDCl3) δ 8.90 (bs, 1H), 7.32–7.51 (m, 3H), 7.21 (m, 3H), 3.41–3.91 (m, 5H), 2.89 (m, 2H), 2.32 (m, 2H), 1.64 (m, 2H); MS (ESI+) m/z 378 [M + H]+; HPLC >99% (AUC), (Method A), tR = 17.6 min.
6-Methyl-2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)-hexahydrocyclopenta[c]pyrrol-2(1H)-yl)pyrimidine-4-carboxamide (46)
Step A
A mixture of 5-(2-(trifluoromethyl)phenyl)octahy-drocyclopenta[c]pyrrole hydrochloride (26, 0.068 g, 0.355 mmol), 2-chloro-6-methylpyrimidine-4-carbonitrile (0.054 g, 0.352 mmol), and i-Pr2NEt (0.14 mL, 1.05 mmol) in DMF (2 mL) was heated at 60 °C for 16 h. The mixture was allowed to cool to rt and then poured into brine (50 mL). The aqueous mixture was extracted with EtOAc (3 × 50 mL), and the combined organic extracts were washed with saturated NaHCO3 (3 × 50 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0–50% EtOAc in hexanes) to afford 6-methyl-2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydro-cyclopenta[c]pyrrol-2(1H)-yl)pyrimidine-4-carbonitrile (45) as an off-white solid (0.072 g, 55%): ESI MS m/z 373 [M + H]+.
Step B
A mixture of 6-methyl-2-((3aR,5r,6aS)-5-(2-(trifluoro-methyl)phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)pyrimidine-4-carbonitrile (45, 0.049 g, 0.133 mmol) LiOH·H2O (0.030 g, 0.665 mmol) in THF (4 mL) and H2O (2 mL) was heated at 70 °C for 18 h. The mixture was allowed to cool to rt and was then acidified to pH 6 with 2 N aq HCl. The aqueous mixture was extracted with CH2Cl2 (3 × 50 mL), and the combined organics were dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was chromatographed over silica gel (0–5% methanol in CH2Cl2) to yield 6-methyl-2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)-hexahydrocyclopenta[c]pyrrol-2(1H)-yl)pyrimidine-4-carboxamide (46) as an off-white solid (0.041 g, 78%): mp 183–187 °C; 1H NMR (300 MHz, CDCl3) δ 7.77 (s, 1H), 7.66–7.58 (m, 1H), 7.58–7.41 (m, 2H), 7.20 (s, 1H), 5.57 (s, 1H), 3.85–3.64 (m, 4H) 3.64–3.45 (m, 1H), 3.01–3.85 (m, 2H), 2.46 (s, 3H), 2.45–2.32 (m, 2H), 1.72–1.56 (m, 2H), aromatic CH obscured by residual peak; ESI MS m/z: 254.1 [M + H]+; HPLC >99% (AUC), (Method B), tR = 17.5 min.
(3aR,5r,6aS)-2-(4-Methyl-6-(1H-tetrazol-5-yl)pyrimidin-2-yl)-5-(2-(trifluoromethyl)phenyl)octahydrocyclopenta[c]pyrrole (47)
Step A
A mixture of 6-methyl-2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)-phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)pyrimidine-4-carbonitrile (45, 0.093 g, 0.250 mmol), NaN3 (0.195 g, 3.0 mmol), and NH4Cl (0.160 g, 3.0 mmol) in DMF (5 mL) was heated at 130 °C in a sealed tube via microwave irradiation for 1 h. The mixture was allowed to cool to rt and then diluted with H2O (50 mL). The aqueous mixture was extracted with EtOAc (3 × 50 mL), and the combined organic extracts were washed with H2O (3 × 50 mL) and brine. The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by reversed phase column chromatography (Isco RediSep 12 g Gold C18 reverse phase column, 0%-100% CH3CN in H2O) to provide (3aR,5r,6aS)-2-(4-methyl-6-(1H–tetrazol-5-yl)pyrimidin-2-yl)-5-(2-(trifluoromethyl)-phenyl)octahydrocyclopenta[c]pyrrole (47) as a white solid (0.074 g, 71%): mp 150–158 °C; 1H NMR (500 MHz, DMSO-d6) δ 7.71–7.59 (m, 3H), 7.41 (m, 1H), 7.21 (s, 1H), 3.85–3.65 (m, 4H), 3.49 (m, 1H), 2.98 (m, 2H), 2.42 (s, 3H), 2.32 (m, 2H), 1.62 (m, 2H); MS (ESI+) m/z 416 [M + H]+; HPLC 98.3% (AUC), (Method A), tR = 18.0 min.
4-Methyl-6-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)-hexahydrocyclopenta[c]pyrrol-2(1H)-yl)picolinic Acid (48)
Step A
A mixture of (3aR,5R,6aS)-5-(2-(trifluoromethyl)phenyl)octahydro-cyclopenta[c]pyrrole hydrochloride (26, 0.158 g, 0.54 mmol), methyl 2-chloro-4-methylnicotinate (0.100 g, 0.54 mmol), Pd(OAc)2 (0.011 g, 0.05 mmol), Xantphos (0.011 g), and Cs2CO3 (0.050 g, 0.15 mmol) in toluene (10 mL) was heated at 110 °C for 16 h. The reaction was allowed to cool to rt then diluted with H2O (200 mL) and extracted with EtOAc (3 × 100 mL). The combined organic extracts were washed with H2O (3 × 100 mL), brine (100 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0% to 30% EtOAc in hexanes) to give methyl 4-methyl-6-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)-hexahydrocyclopenta[c]pyrrol-2(1H)-yl)picolinate as an off-white solid (0.129 g, 59%).
Step B
A solution of methyl 4-methyl-6-((3aR,5r,6aS)-5-(2-(trifluo-romethyl)phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)picolinate (0.129 g, 0.07 mmol) and 2 N aq NaOH (10 mL) in a 1:1 mixture of CH3OH/THF (20 mL) stirred at room temperature for 16 h. The mixture was carefully neutralized at 0 °C with 2 N aq HCl and extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0% to 10% CH3OH in CH2Cl2) to give 4-methyl-6-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydrocyclopenta[c]-pyrrol-2(1H)-yl)picolinic acid (48) as an off-white solid (0.106 g, 88%): mp 152–155 °C; 1H NMR (500 MHz, DMSO-d6) δ 12.51 (bs, 1H), 7.71 (m, 3H), 7.61 (m, 1H), 7.14 (s, 1H), 6.62 (s, 1H), 3.55 (m, 4H), 3.34 (m, 4H), 2.88 (m, 2H), 2.29 (m, 5H), 1.62 (m, 2H); MS (ESI+) m/z 391 [M + H]+; HPLC >99% (AUC), (Method A), tR = 13.4 min.
2-Methyl-6-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)-hexahydrocyclopenta[c]pyrrol-2(1H)-yl)isonicotinic Acid (49)
Step A
A mixture of (3aR,5R,6aS)-5-(2-(trifluoromethyl)phenyl)octahydro-cyclopenta[c]pyrrole hydrochloride (26, 0.160 g, 0.55 mmol), methyl 2-chloro-6-methylisonicotinate (0.101 g, 0.55 mmol), Pd(OAc)2 (0.011 g, 0.05 mmol), Xantphos (0.011 g), and Cs2CO3 (0.050 g, 0.15 mmol) in toluene (10 mL) was stirred at 110 °C for 16 h. The reaction was allowed to cool to rt and diluted with H2O (200 mL) and extracted with EtOAc (3 × 100 mL). The combined organic extracts were washed with H2O (3 × 100 mL), brine (100 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0% to 30% EtOAc in hexanes) to give methyl 2-methyl-6-((3aR,5r,6aS)-5-(2-(trifluoro-methyl)phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)isonicotinate as an off-white solid (0.140 g, 63%).
Step B
A solution of methyl 2-methyl-6-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)-isonicotinate (0.140 g, 0.08 mmol) and 2 N aq NaOH (10 mL) in a 1:1 mixture of CH3OH/THF (20 mL) was stirred at room temperature for 16 h. The mixture was carefully neutralized at 0 °C with 2 N aq HCl and extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0% to 10% CH3OH in CH2Cl2) to give 2-methyl-6-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)-hexahydrocyclopenta[c]pyrrol-2(1H)-yl)isonicotinic acid (49) as an off-white solid (0.124 g, 86%): mp 121–124 °C; 1H NMR (500 MHz, DMSO-d6) δ 12.81 (bs, 1H), 7.77 (s, 1H), 7.66 (m, 3H), 7.40 (m, 1H), 6.60 (s, 1H), 3.52 (m, 4H), 3.45 (m, 1H), 2.80 (m, 2H), 2.36 (s, 3H), 2.24 (m, 2H), 1.60 (m, 2H); MS (ESI+) m/z 391 [M + H]+; HPLC >99% (AUC), (Method A), tR = 10.1 min.
3-Methyl-6-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)-hexahydrocyclopenta[c]pyrrol-2(1H)-yl)picolinic Acid (50)
Step A
To a mixture of (3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)octahydro-cyclopenta[c]pyrrole hydrochloride (26, 0.400 g, 1.37 mmol), 6-chloro-3-methylpicolinic acid (0.254 g, 1.37 mmol), JohnPhos (0.048 g, 0.137 mmol), and Cs2CO3 (1.34 g, 4.11 mmol) in toluene (7 mL) was added Pd(OAc)2 (0.015 g, 0.068 mmol). The mixture was heated at reflux for 14 h then allowed to cool to rt. The mixture was filtered over Celite, and the filter cake was washed with EtOAc. The filtrate was concentrated under reduced pressure, and the resulting residue was purified by flash column chromatography (Isco CombiFlash Rf unit, 40 g Gold RediSep column, 0%-15% EtOAc in hexanes) to give methyl 3-methyl-6-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)-hexahydrocyclopenta[c]pyrrol-2(1H)-yl)picolinate as a white solid (0.366 g, 66%): MS (ESI+) m/z 405 [M + H]+.
Step B
A mixture of methyl 3-methyl-6-((3aR,5r,6aS)-5-(2-(trifluo-romethyl)phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)picolinate (0.366 g, 0.905 mmol) and LiOH·H2O (0.114 g, 2.71 mmol) in THF (4 mL), CH3OH (4 mL), and H2O (4 mL) was stirred at rt for 72 h. The mixture was acidified to pH 6 via the addition of 2 N aq HCl and then extracted with CH2Cl2 (3 × 50 mL). The combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by flash column chromatography (Isco CombiFlash Rf unit, 40 g RediSep column, 0%-8% CH3OH in CH2Cl2) to give 3-methyl-6-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydrocyclopenta[c]-pyrrol-2(1H)-yl)picolinic acid (50) as a white solid (0.337 g, 95%): mp 148–150 °C; 1H NMR (500 MHz, CDCl3) δ 11.97 (bs, 1H), 7.67 (s, 1H), 7.59–7.50 (m, 3H), 7.27 (m, 1H), 6.65 (s, 1H), 3.70–3.44 (m, 5H), 2.94 (m, 2H), 2.61 (s, 3H), 1.70 (m, 2H), 1.50 (m, 2H); MS (ESI+) m/z 391 [M + H]+; HPLC >99% (AUC), (Method A), tR = 11.8 min.
5-((3aR,5r,6aS)-5-(2-(Trifluoromethyl)phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)nicotinic Acid (51)
Step A
To a mixture of (3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)octahydro-cyclopenta[c]pyrrole hydrochloride (26, 0.153 g, 0.524 mmol), methyl 5-bromonicotinate (0.103 g, 0.477 mmol), XantPhos (0.083 g, 0.140 mmol), and Cs2CO3 (0.465 g, 1.43 mmol) in 1,4-dioxane (8 mL) was added Pd2(dba)3 (0.043 g, 0.047 mmol). The mixture was heated at reflux for 16 h, then allowed to cool to rt. The mixture was concentrated under reduced pressure, and the resulting residue was purified by flash column chromatography (Isco CombiFlash Rf unit, 40 g Gold RediSep column, 0%-25% CH3OH in CH2Cl2) to give methyl 5-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)-nicotinate as a yellow film (0.077 g, 41%): 1H NMR (500 MHz, CDCl3) δ 8.59 (s, 1H), 7.62 (s, 1H), 7.53 (m, 1H), 7.48 (m, 3H), 7.29 (m, 1H), 3.94 (s, 3H), 3.52 (m, 1H), 3.43 (m, 4H), 2.98 (m, 2H), 2.45 (m, 2H), 1.67 (m, 2H); MS (ESI+) m/z 391 [M + H]+.
Step B
A mixture of methyl 5-((3aR,5r,6aS)-5-(2-(trifluoromethyl)-phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)nicotinate (0.076 g, 0.194 mmol) and LiOH·H2O (0.081 g, 1.94 mmol) in THF (4 mL), CH3OH (2 mL), and H2O (1 mL) was stirred at rt for 4 h. The mixture was acidified to pH 2 via the addition of 2 N aq HCl and then diluted with H2O (40 mL). The aqueous mixture was extracted with CH2Cl2 (3 × 50 mL), and the combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure to provide 5-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)-hexahydrocyclopenta[c]pyrrol-2(1H)-yl)nicotinic acid (51) as a yellow solid (0.063 g, 88%): mp 302–304 °C; 1H NMR (500 MHz, DMSO-d6) δ 13.17 (bs, 1H), 8.39 (s, 1H), 8.23 (s, 1H), 7.72–7.59 (m, 3H), 7.38 (m, 2H), 3.44 (m, 5H), 2.94 (m, 2H), 2.30 (m, 2H), 1.65 (m, 2H); MS (ESI+) m/z 377 [M + H]+; HPLC >99% (AUC), (Method B), tR = 7.8 min.
3-((3aR,5r,6aS)-5-(2-(Trifluoromethyl)phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)benzoic Acid (52)
Step A
To a mixture of (3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)octahydrocyclopenta[c]-pyrrole hydrochloride (26, 0.250 g, 0.86 mmol), NaOt-Bu (0.180 g, 0.89 mmol), and BINAP (0.105 g, 0.17 mmol) in toluene (10 mL) was added Pd(OAc)2 (0.020 g, 0.089 mmol). The mixture was heated at 110 °C for 16 h, then allowed to cool to rt. The mixture was filtered over Celite, and the filter cake was washed with EtOAc. The filtrate was concentrated under reduced pressure, and the resulting residue was purified by flash column chromatography (Isco CombiFlash Rf unit, 40 g Gold RediSep column, 0%-15% EtOAc in hexanes) to give methyl 3-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydro-cyclopenta[c]pyrrol-2(1H)-yl)benzoate as a white solid (0.132 g, 63%): MS (ESI+) m/z 390 [M + H]+.
Step B
A mixture of methyl 3-((3aR,5r,6aS)-5-(2-(trifluoromethyl)-phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)benzoate (0.396 g, 0.95 mmol) and LiOH·H2O (0.400 g, 9.5 mmol) in THF (4 mL), CH3OH (2 mL), and H2O (1 mL) stirred at rt for 16 h. The mixture was acidified to pH 2 via the addition of 2 N aq HCl and then diluted with H2O (40 mL). The aqueous mixture was extracted with CH2Cl2 (3 × 50 mL), and the combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure to provide 3-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)-hexahydrocyclopenta[c]pyrrol-2(1H)-yl)benzoic acid (52) as a yellow solid (0.026 g, 18%): 1H NMR (300 MHz, DMSO-d6) δ 12.72 (bs, 1H), 7.72–7.63 (m, 3H), 7.41 (m, 1H), 7.30–7.20 (m, 3H), 6.89 (m, 1H), 3.40 (m, 5H), 2.91 (m, 2H), 2.31 (m, 2H), 1.64 (m, 2H); MS (ESI+) m/z 376 [M + H]+; HPLC 98.8% (AUC), (Method B), tR = 18.6 min.
6-Methyl-2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)-hexahydrocyclopenta[c]pyrrol-2(1H)-yl)nicotinic Acid (53)
Step A
To a mixture of (3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)octahydro-cyclopenta[c]pyrrole hydrochloride (26, 0.250 g, 0.857 mmol), methyl 2-chloro-6-methylnicotinate (0.146 g, 0.787 mmol), XantPhos (0.135 g, 0.233 mmol), and Cs2CO3 (0.647 g, 1.98 mmol) in toluene (25 mL) was added Pd2(dba)3 (0.071 g, 0.077 mmol). The mixture was heated at 100 °C for 16 h, then allowed to cool to rt. The mixture was concentrated under reduced pressure, and the resulting residue was purified by flash column chromatography (Isco CombiFlash Rf unit, 24 g Gold RediSep column, 0%-25% CH3OH in CH2Cl2) to give methyl 6-methyl-2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydro-cyclopenta[c]pyrrol-2(1H)-yl)nicotinate as a crude yellow film (0.036 g).
Step B
A mixture of methyl 6-methyl-2-((3aR,5r,6aS)-5-(2-(trifluo-romethyl)phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)nicotinate (0.036 g, 0.089 mmol) and LiOHH2O (0.037 g, 0.89 mmol) in THF (4 mL), CH3OH (4 mL), and H2O (1 mL) stirred at 50 °C for 30 min. The mixture was acidified to pH 2 via the addition of 2 N aq HCl and then diluted with H2O (40 mL). The aqueous mixture was extracted with CH2Cl2 (3 × 20 mL), and the combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by reversed phase column chromatography (Isco RediSep 12 g Gold C18 reverse phase column, 0%-100% CH3CN in H2O) to provide 6-methyl-2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)-hexahydrocyclopenta[c]pyrrol-2(1H)-yl)nicotinic acid (53) as a white solid (0.007 g, 20% over two steps): mp 156–157 °C; 1H NMR (500 MHz, DMSO-d6) δ 13.28 (bs, 1H), 7.71–7.59 (m, 3H), 7.38 (m, 1H), 6.87 (s, 1H), 6.74 (s, 1H), 3.55 (m, 4H), 3.41 (m, 1H), 2.89 (m, 2H), 2.35 (s, 3H), 2.27 (m, 2H), 1.65 (m, 2H); MS (ESI+) m/z 391 [M + H]+; HPLC 98.6% (AUC), (Method A), tR = 8.4 min.
2-((3aR,5r,6aS)-5-(2-(Trifluoromethyl)phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)nicotinic Acid (54)
Step A
A mixture of (3aR,5R,6aS)-5-(2-(trifluoromethyl)phenyl)octahydrocyclopenta[c]-pyrrole hydrochloride (26, 0.160 g, 0.55 mmol), methyl 2-chloronicotinate (0.071 g, 0.41 mmol), Pd(OAc)2 (0.011 g 0.05 mmol), Xantphos (0.011 g), and Cs2CO3 (0.050 g, 0.15 mmol) in toluene (10 mL) was stirred at 110 °C for 16 h. The reaction was diluted with H2O (200 mL) and extracted with EtOAc (3 × 100 mL). The combined organic extracts were washed with H2O (3 × 100 mL), brine (100 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0% to 30% EtOAc in hexanes) to give methyl 2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydrocyclopenta[c]-pyrrol-2(1H)-yl)nicotinate as an off-white solid (0.07 g, 33%).
Step B
A solution of 2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)-phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)nicotinate (0.70 g, 0.18 mmol) and 2 N aq NaOH (10 mL) in a 1:1 mixture of CH3OH/THF (20 mL) was stirred at room temperature for 16 h. The mixture was carefully neutralized at 0 °C with 2 N HCl and extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0% to 10% CH3OH in CH2Cl2) to give 2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydrocyclopenta-[c]pyrrol-2(1H)-yl)nicotinic acid (54) as an off-white solid (0.057 g, 85%): mp 158–160 °C; 1H NMR (500 MHz, DMSO-d6) δ 8.20 (s, 1H), 7.79 (m, 1H), 7.63 (m, 3H), 7.39 (m, 1H), 6.71 (s, 1H), 3.53 (m, 4H), 3.48 (m, 1H), 2.80 (m, 2H), 2.24 (m, 2H), 1.61 (m, 2H); MS (ESI+) m/z 377 [M + H]+; HPLC >99% (AUC), (Method A), tR = 10.4 min.
6-((3aR,5r,6aS)-5-(2-(Trifluoromethyl)phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)pyrazine-2-carboxylic Acid (55)
Step A
A mixture of 5-(2-(trifluoromethyl)phenyl)octahydrocyclopenta[c]-pyrrole hydrochloride (26, 0.057 g, 0.29 mmol), methyl 2-chloro-pyrazine-6-carboxylate (0.051 g, 0.29 mmol), and i-Pr2NEt (0.14 mL, 1.09 mmol) in DMF (10 mL) was heated at 60 °C for 16 h. The mixture was allowed to cool to rt and poured into brine (50 mL). The aqueous mixture was extracted with EtOAc (3 × 50 mL), and the combined organic extracts were washed with a saturated NaHCO3 (3 × 50 mL), dried over Na2SO4, and concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0–50% EtOAc in hexanes) to afford methyl 6-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)-pyrazine-2-carboxylate (66 mg, 57%): 1H NMR (300 MHz, CDCl3) δ 8.53 (s, 1H), 8.10 (s, 1H), 7.66–7.56 (m, 1H), 7.56–7.42 (m, 2H), 3.99 (s, 3H), 3.81–3.48 (m, 5H) 3.07–2.92 (m, 2H), 2.51–2.34 (m, 2H), 1.75–1.61 (m, 2H), aromatic CH obscured by a residual peak; ESI MS m/z 392 [M + H]+.
Step B
A mixture of methyl 6-((3aR,5r,6aS)-5-(2-(trifluoromethyl)-phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)pyrazine-2-carboxy-late (66.0 mg, 0.169 mmol) and LiOH·H2O (0.027 g, 0.64 mmol) in THF (4 mL), CH3OH (4 mL), and H2O (1 mL) was stirred at rt for 30 min. The mixture was acidified to pH 6 using 2 N aq HCl and extracted with CH2Cl2 (3 × 20 mL), and the combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by reversed phase column chromatography (Isco RediSep 12 g Gold C18 reverse phase column, 0%-100% CH3CN in H2O) to provide 2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydro-cyclopenta[c]pyrrol-2(1H)-yl)nicotinic acid (55) as a white solid (0.015 g, 24%). mp 182–185 °C; 1H NMR: (300 MHz, CDCl3) δ 8.60 (s, 1H), 8.29–8.00 (br s, 1H), 7.74–7.55 (m, 1H), 7.55–7.39 (m, 2H), 3.93–3.40 (m, 5H), 2.79–2.54 (m, 2H) 2.50–2.22 (m, 2H), 1.80–1.44 (m, 2H); ESI MS m/z: 378 [M + H]+; HPLC 98.7% (AUC), (Method B), tR = 14.0 min.
6-((3aR,5r,6aS)-5-(2-(Trifluoromethyl)phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)pyridazine-4-carboxylic Acid (56)
Step A
A mixture of (3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)octahydro-cyclopenta[c]pyrrole hydrochloride (26, 0.500 g, 1.71 mmol), methyl 3-chloropyridazine-5-carboxylate (0.295 g, 1.71 mmol), and Et3N (0.71 mL, 5.15 mmol) in DMF (10 mL) was heated at 60 °C for 16 h. The mixture was then allowed to cool to rt and diluted with H2O (50 mL). The aqueous mixture was extracted with EtOAc (3 × 50 mL), and the combined organic extracts were washed with H2O (3 × 50 mL) and brine. The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by flash column chromatography (Isco CombiFlash Rf unit, 40 g RediSep column, 0%-50% EtOAc in hexanes) to give methyl 6-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydrocyclopenta[c]-pyrrol-2(1H)-yl)pyridazine-4-carboxylate as an oil (0.069 g, 10%): MS (ESI+) m/z 392 [M + H]+.
Step B
A mixture of methyl 6-((3aR,5r,6aS)-5-(2-(trifluoromethyl)-phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)pyridazine-4-carboxy-late (0.060 g, 0.153 mmol) and LiOH·H2O (0.020 g, 0.477 mmol) in THF (5 mL) and H2O (5 mL) was stirred at rt for 16 h. The mixture was acidified to pH 6 via the addition of 2 N aq HCl and then extracted with CH2Cl2 (3 × 20 mL). The combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure to provide 6-((3aR,5r,6aS)-5-(2-(trifluoro-methyl)phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)pyridazine-4-carboxylic acid (56) as a light-yellow solid (0.054 g, 95%): 1H NMR (300 MHz, DMSO-d6) δ 13.92 (bs, 1H), 8.82 (s, 1H), 7.75–7.62 (m, 3H), 7.38 (m, 1H), 7.22, (s, 1H), 3.72–3.66 (m, 4H), 3.44 (m, 1H), 2.94 (m, 2H), 2.29 (m, 2H), 1.67 (m, 2H); MS (ESI+) m/z 378 [M + H]+; HPLC 97.4% (AUC), (Method A), tR = 12.8 min.
5-Methyl-2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)-hexahydrocyclopenta[c]pyrrol-2(1H)-yl)thiazole-4-carboxylic Acid (57)
Step A
To a solution of (3aR,5r,6aS)-5-(2-(trifluoromethyl)-phenyl)hexahydrocyclopenta[c]pyrrole-2(1H)-carbonyl chloride (0.300 g, 1.03 mmol) in DMSO (8 mL) and ethyl 2-chloro-5-methyl-thiazole-4-carboxylate (0.211 g, 1.03 mmol) was added i-Pr2NEt (0.40 g, 3.09 mmol). The resulting solution was heated at 110 °C for 24 h. The reaction was allowed to cooled to rt and diluted with H2O (30 mL) and extracted with EtOAc (3 × 30 mL). The combined organic extracts were washed with brine (2 × 30 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was chro-matographed over silica gel (Isco CombiFlash Rf unit, 12 g Redisep column, 0–30% EtOAc in hexanes) to give ethyl 5-methyl-2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydrocyclopenta[c]-pyrrol-2(1H)-yl)thiazole-4-carboxylate as an off-white solid (0.042 g, 9%): 1H NMR (300 MHz, DMSO-d6) δ 7.76–7.70 (m, 1H), 7.68–7.54 (m, 2H), 7.42–7.35 (m, 1H), 4.22 (q, J = 6.9 Hz, 2H), 3.55–3.42 (m, 2H), 3.42–3.35 (m, 2H), 2.97–2.83 (m, J = 2H), 2.55 (s, 3H), 2.32–2.18 (m, 3H), 1.74–1.53 (m, 2H), 1.27 (t, J = 6.9 Hz, 3H); MS (ESI+) m/z 425 [M + H]+.
Step B
A mixture of ethyl 5-methyl-2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)-thiazole-4-carboxylate (0.040 g, 0.094 mmol) and LiOH·H2O (0.039 g, 0.94 mmol) in THF (2 mL), CH3OH (1 mL), and H2O (0.5 mL) was stirred at rt for 16 h. The mixture was then acidified to pH 5 with 2 N aq HCl and diluted with H2O (50 mL). The resulting solids were collected by filtration to provide 5-methyl-2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)-thiazole-4-carboxylic acid (57) as a white solid (0.030 g, 81%): mp 166–170 °C; 1H NMR (500 MHz, DMSO-d6) δ 12.41 (br s, 1H), 7.72 (d, J = 8.0 Hz, 1H), 7.67–7.58 (m, 2H), 7.42–7.36 (m, 1H), 3.51–3.45 (m, 2H), 3.42–3.32 (m, 3H), 2.95–2.85 (m, 2H), 2.53 (s, 3H), 2.30–2.22 (m, 2H), 1.66–1.57 (m, 2H); MS (ESI+) m/z 397 [M + H]+; HPLC >99% (AUC), (Method B), tR = 14.5 min.
2-((3aR,5r,6aS)-5-(5-Fluoro-2-(trifluoromethyl)phenyl)-hexahydrocyclopenta[c]pyrrol-2(1H)-yl)-6-methylpyrimidine-4-carboxylic Acid (61)
Step A
To a N2 degassed mixture of (3aS,6aS)-tert-butyl 5-(((trifluoromethyl)sulfonyl)oxy)-3,3a,6,6a–tetrahydro-cyclopenta[c]pyrrole-2(1H)-carboxylate ((±)-23, 5.0 g, 14.0 mmol), (5-fluoro-2-(trifluoromethyl)phenyl)boronic acid (2.91 g, 14.0 mmol), and aqueous 2 M Na2CO3 (100 mL) in DME (200 mL) was added Pd(PPh3)4 (0.500 g, 1.4 mmol). The mixture was heated at 80 °C for 6 h, then allowed to cool to rt and diluted with H2O (500 mL). The aqueous mixture was extracted with EtOAc (2 × 200 mL), and the combined organic extracts were washed with H2O (200 mL) and brine (200 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography (Isco CombiFlash Rf unit, 330 g Redisep column, 0% to 10% EtOAc in hexanes) to give (3aR,6aS)-tert-butyl 5-(5-fluoro-2-(trifluoromethyl)phenyl)-3,3a,6,6a–tetrahydrocyclopenta[c]pyrrole-2(1H)-carboxylate (58a) as a clear, viscous oil (4.88 g, 94%).
Step B
A mixture of 5-(5-fluoro-2-(trifluoromethyl)phenyl)-3,3a,6,6a–tetrahydrocyclopenta[c]pyrrole-2(1H)-carboxylate (4.8 g, 12.9 mmol) and 10% Pd/C (1.57 g wet, 10% w/w) in CH3OH (50 mL) was subjected to an atmosphere of H2 gas (40 psi) using a Parr Shaker apparatus for 16 h at rt. The mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure. The resulting residue was purified by flash column chromatography (Isco CombiFlash Rf unit, 40 g Redisep column, 0% to 30% EtOAc in hexanes) to give (3aR,5r,6aS)-tert-butyl 5-(5-fluoro-2-(trifluoromethyl)phenyl)hexahydro-cyclopenta[c]pyrrole-2(1H)-carboxylate (59a) as a clear, viscous oil (4.5 g 95%).
Step C
To a 0 °C cooled solution of (3aR,5r,6aS)-tert-butyl 5-(5-fluoro-2-(trifluoromethyl)phenyl)hexahydrocyclopenta[c]pyrrole-2(1H)-carboxylate (4.50 g, 12.1 mmol) in CH2Cl2 (60 mL) was added a 2.0 M HCl solution in Et2O (100 mL), and the mixture was allowed to stir at rt for 24 h. The mixture was diluted with Et2O (200 mL), and the precipitated product was filtered to give (3aR,5r,6aS)-5-(5-fluoro-2-(trifluoromethyl)phenyl)octahydrocyclopenta[c]pyrrole hydrochloride (60a) as a white solid (3.45 g, 92%): MS (ESI+) m/z 274 [M + H]+.
Step D
To a solution of (3aR,5r,6aS)-5-(5-fluoro-2-(trifluoromethyl)phenyl)octahydrocyclopenta[c]pyrrole hydrochloride (1.0 g, 3.23 mmol) and Et3N (1.43 mL, 10.29 mmol) in DMF (50 mL) was added a methyl 2-chloropyrimidine-4-carboxylate (0.641 g, 3.43 mmol), and the resulting solution was stirred at 60 °C for 16 h. The reaction was diluted with H2O (200 mL) and extracted with EtOAc (3 × 100 mL). The combined organic extracts were washed with H2O (3 × 100 mL) and brine (100 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0% to 30% EtOAc in hexanes) to give methyl 2-((3aR,5r,6aS)-5-(5-fluoro-2-(trifluoromethyl)phenyl)-hexahydrocyclopenta[c]pyrrol-2(1H)-yl)-6-methylpyrimidine-4-carboxy-late as an off-white solid (1.14 g, 84%): MS (ESI+) m/z 424 [M + H]+.
Step E
A solution of methyl 2-((3aR,5r,6aS)-5-(5-fluoro-2-(trifluoromethyl)phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)-6-methylpyrimidine-4-carboxylate (1.0 g, 2.36 mmol) and 2 N aq NaOH (20 mL) in a 1:1 mixture of CH3OH/THF (40 mL) was stirred at room temperature for 16 h. The mixture was carefully neutralized at 0 °C with 2 N aq HCl and extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0% to 10% CH3OH in CH2Cl2) to give 2-((3aR,5r,6aS)-5-(5-fluoro-2-(trifluoromethyl)-phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)-6-methylpyrimidine-4-carboxylic acid (61) as an off-white solid (0.831 g, 86%): 1H NMR (500 MHz, DMSO-d6) δ 7.74 (m, 1H), 7.54 (m, 1H), 7.24 (m, 1H), 6.63 (s, 1H), 3.63 (m, 4H), 3.58 (m, 1H), 2.82 (m, 2H), 2.36 (m, 2H), 2.27 (s, 3H), 1.61 (m, 2H); MS (ESI+) m/z 410 [M + H]+; HPLC >99% (AUC) (Method B), tR = 10.8 min.
2-((3aR,5r,6aS)-5-(3-Fluoro-2-(trifluoromethyl)phenyl)-hexahydrocyclopenta[c]pyrrol-2(1H)-yl)-6-methylpyrimidine-4-carboxylic Acid (62)
Compound 62 was prepared according to a similar procedure described for the synthesis of 61. 1H NMR (300 MHz, DMSO-d6) δ 7.52–7.44 (m, 1H), 7.02–6.99 (d, J = 8.4 Hz, 1H), 6.89–6.84 (m, 2H), 3.80–3.73 (m, 2H), 3.46–3.42 (m, 4H), 3.14–3.04 (m, 4H); ESI MS m/z 411 [M + H]+; HPLC >99.0% purity (Method B), tR = 16.8 min.
2-((3aR,5r,6aS)-5-(2-Chloro-5-fluorophenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)-6-methylpyrimidine-4-carboxylic Acid (63)
Compound 63 was prepared according to a similar procedure described for the synthesis of 61. Mp 183–185 °C; 1H NMR (500 MHz, DMSO-d6) δ 13.22 (br s, 1H). 7.44 (dd, J = 9.0, 5.5 Hz, 1H), 7.34 (dd, J = 10.5, 3.0 Hz, 1H), 7.10–7.05 (m, 1H), 6.99 (s, 1H), 3.70–3.64 (m, 2H), 3.62–3.57 (m, 2H), 3.50–3.41 (m, 1H), 2.92–2.81 (m, 2H), 2.36 (s, 3H), 2.34–2.25 (m, 2H), 1.57–1.47 (m, 2H); MS (ESI+) m/z 376 [M + H]+; HPLC >99% purity (Method B), tR = 12.7 min.
2-((3aR,5r,6aS)-5-(2-Chloro-3-fluorophenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)-6-methylpyrimidine-4-carboxylic Acid (64)
Compound 64 was prepared according to a similar procedure described for the synthesis of 61. 1H NMR (300 MHz, DMSO-d6) δ 7.35–7.28 (m, 2H), 7.28–7.19 (m, 1H), 6.68 (s, 1H), 3.62–3.54 (m, 4H), 3.53–3.41 (m, 1H), 2.91–2.76 (m, 2H), 2.40–2.26 (m, 2H), 2.33 (s, 3H), 1.56–1.41 (m, 2H); MS (ESI+) m/z 376 [M + H]+; HPLC >99% purity (Method B), tR = 10.9 min.
2-((3aR,5r,6aS)-5-(2-Chloro-3-fluorophenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)-6-methylpyrimidine-4-carboxylic Acid (65)
Compound 65 was prepared according to a similar procedure described for the synthesis of 61. Mp 176–180 °C; 1H NMR (300 MHz, DMSO-d6) δ 13.31 (br s, 1H), 8.99 (s, 1H), 8.65 (d, J = 5.1 Hz, 1H), 7.65 (d, J = 5.1 Hz, 1H), 6.99 (s, 1H), 3.75–3.62 (m, 4H), 3.41–3.35 (m, 1H), 2.91–2.90 (m, 2H), 2.37 (s, 3H), 2.32–2.27 (m, 2H), 1.80–1.74 (m, 2H); MS (ESI+) m/z 393 [M + H]+; HPLC 98.3% purity (Method H), tR = 13.8 min.
2-((3aR,5r,6aS)-5-(2-Chloro-3-fluorophenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)-6-methylpyrimidine-4-carboxylic Acid (66)
Compound 66 was prepared according to a similar procedure described for the synthesis of 61. Mp 161 – 165 °C; 1H NMR (300 MHz, DMSO-d6) δ 13.31 (br s, 1H), 8.55–8.53 (m, 1H), 8.22 (d, J = 8.1 Hz, 1H), 7.67–7.62 (m, 1H), 7.00 (s, 1H), 3.71–3.61 (m, 4H), 3.44–3.38 (m, 1H), 2.91–2.89 (m, 2H), 2.37 (s, 3H), 2.31–2.23 (m, 2H), 1.67–1.57 (m, 2H); MS (ESI+) m/z 393 [M + H]+; HPLC >99.0% purity (Method B), tR = 14.7 min.
Supplementary Material
Acknowledgments
This study was supported by NIH Grants U01 NS074476 (to K.P.), P30 EY019007 (Core Support for Vision Research), and unrestricted funds from Research to Prevent Blindness (New York, NY) to the Department of Ophthalmology, Columbia University. We thank The Burch Family Foundation, the Mary Jaharis-John Catsimatidis Scholarship Fund, the Kaplen Foundation, and the Eye Surgery Fund for gifts supporting this study. This project has also been funded in whole or in part with Federal funds from the National Institute of Neurological Disorders and Stroke, the National Eye Institute, the NIH Blueprint Neurotherapeutics Network, Blueprint for Neuro-science Research Program, National Institutes of Health, Department of Health and Human Services, under Contract No. HHSN271201100013C.
ABBREVIATIONS USED
- AMD
age-related macular degeneration
- A2E
N-retinide-N-retinylidene ethanolamine
- SPA
scintillation proximity assay
- HTRF
homogeneous time-resolved fluorescence assay
- HLM
human liver microsomes
- RLM
rat liver microsomes
- n-BuLi
n-butyl lithium
- THF
tetrahydrofuran
- DMF
N,N-dimethylformamide
- DME
dimethoxyethane
- Et2O
diethyl ether
- EtOAc
ethyl acetate
- CH3OH
methyl alcohol
- EtOH
ethyl alcohol
- Boc2O
di-tert-butyl dicarbonate
- TFA
trifluoroacetic acid
- Pd(OAc)2
palladium(II) acetate
- Pd2(dba)3
tris-(dibenzylideneacetone) dipalladium(0)
- BINAP
2,2′-bis-(diphenylphosphino)-1,1′-binaphthalene
- XantPhos
4,5-bis-(diphenylphosphino)-9,9-dimethylxanthene
- JohnPhos
(2-biphenyl)di-tert-butylphosphine
- Ac2O
acetic anhydride
- NaOAc
sodium acetate
- LiHMDS
lithium bis(trimethylsilyl)amide
- i-Pr2NEt
N,N-diisopropylethylamine
- Et3N
triethylamine
- LiOH·H2O
lithium hydroxide monohydrate
- NaOH
sodium hydroxide
- SOCl2
thionyl chloride
- Pd(PPh3)4
tetrakis(triphenylphosphine)palladium(0)
- PhN(SO2CF3)2
1,1,1-trifluoro-N-phenyl-N-((trifluoromethyl)sulfonyl)-methanesulfonamide
- LiAlH4
lithium aluminum hydride
- Gly
glycine
- Tyr
tyrosine
- Arg
arginine
- Gln
glutamine
- Leu
leucine
- Phe
phenylalanine
- CYP
cytochrome P450
- CYP2C9
cytochrome P450 2C9
- CYP2C19
cytochrome P450 2C19
- CYP2D6
cytochrome P450 2D6
- CYP3A4
cytochrome P450 3A4
- GPCR
G-protein coupled receptor
- hERG
human ether-à-go-go-related gene
- PK
pharmacokinetics
- PD
pharmacodynamics
- iv
intravenous
- po
oral
- qd
once daily
- Cl
clearance
- Vss
volume of distribution at steady state
- AUC
area under the curve
- % F
% bioavailability
- SAR
structure–activity relationship
- SPR
structure–property relationship
- ADME
absorption, distribution, metabolism, elimination
- PPB
plasma protein binding
- THF
tetrahydrofuran
- DME
dimethoxyethane
- CH2Cl2
dichloromethane
- CH3CN
acetonitrile
- aq
aqueous
Footnotes
ASSOCIATED CONTENT
Supporting Information
In vivo protocols and 1H NMR spectral data. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.5b00423.
The authors declare the following competing financial interest(s): C.L.C., N.D., E.E.F., M.P.C., P.C., L.Z., G.J., and K.P. are inventors on the patent applications for compounds disclosed in this paper that are assigned to The Trustees of Columbia University in the City of New York and licensed to iCura Vision. K.P., G.J., and P.G.P. are co-founders of iCura Vision.
REFERENCES
- 1.Petrukhin K. New therapeutic targets in atrophic age-related macular degeneration. Expert Opin. Ther. Targets. 2007;11(5):625–639. doi: 10.1517/14728222.11.5.625. [DOI] [PubMed] [Google Scholar]
- 2.Hubschman JP, Reddy S, Schwartz SD. Age-related macular degeneration: current treatments. Clin. Ophthalmol. 2009;3:155–166. doi: 10.2147/opth.s2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. [accessed June 10, 2015];Macular Degeneration Facts and Statistics. http://www.brightfocus.org/macular/about/understanding/facts.html.
- 4.(a) Young RW. Pathophysiology of age-related macular degeneration. Surv. Ophthalmol. 1987;31(5):291–306. doi: 10.1016/0039-6257(87)90115-9. [DOI] [PubMed] [Google Scholar]; (b) Dorey CK, Wu G, Ebenstein D, Garsd A, Weiter JJ. Cell loss in the aging retina. Relationship to lipofuscin accumulation and macular degeneration. Invest. Ophthalmol. Visual Sci. 1989;30(8):1691–1699. [PubMed] [Google Scholar]; (c) Holz FG, Bellman C, Staudt S, Schutt F, Volcker HE. Fundus autofluorescence and development of geographic atrophy in age-related macular degeneration. Invest. Ophthalmol. Visual Sci. 2001;42(5):1051–1056. [PubMed] [Google Scholar]; (d) Holz FG, Bellmann C, Margaritidis M, Schutt F, Otto TP, Volcker HE. Patterns of increased in vivo fundus autofluorescence in the junctional zone of geographic atrophy of the retinal pigment epithelium associated with age-related macular degeneration. Graefe's Arch. Clin. Exp. Ophthalmol. 1999;237(2):145–152. doi: 10.1007/s004170050209. [DOI] [PubMed] [Google Scholar]; (e) Holz FG, Bindewald-Wittich A, Fleckenstein M, Dreyhaupt J, Scholl HP, Schmitz-Valckenberg S. Progression of geographic atrophy and impact of fundus autofluorescence patterns in age-related macular degeneration. Am. J. Ophthalmol. 2007;143(3):463–472. doi: 10.1016/j.ajo.2006.11.041. [DOI] [PubMed] [Google Scholar]; (f) Schmitz-Valckenberg S, Fleckenstein M, Scholl HP, Holz FG. Fundus autofluorescence and progression of age-related macular degeneration. Surv. Ophthalmol. 2009;54(1):96–117. doi: 10.1016/j.survophthal.2008.10.004. [DOI] [PubMed] [Google Scholar]; (g) Finnemann SC, Leung LW, Rodriguez-Boulan E. The lipofuscin component A2E selectively inhibits phagolysosomal degradation of photoreceptor phospholipid by the retinal pigment epithelium. Proc. Natl. Acad. Sci. U. S. A. 2002;99(6):3842–3847. doi: 10.1073/pnas.052025899. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Suter M, Reme C, Grimm C, Wenzel A, Jaattela M, Esser P, Kociok N, Leist M, Richter C. Age-related macular degeneration. The lipofusion component N-retinyl-N-retinylidene ethanolamine detaches proapoptotic proteins from mitochondria and induces apoptosis in mammalian retinal pigment epithelial cells. J. Biol. Chem. 2000;275(50):39625–39630. doi: 10.1074/jbc.M007049200. [DOI] [PubMed] [Google Scholar]; (i) Sparrow JR, Fishkin N, Zhou J, Cai B, Jang YP, Krane S, Itagaki Y, Nakanishi K. A2E, a byproduct of the visual cycle. Vision Res. 2003;43(28):2983–2990. doi: 10.1016/s0042-6989(03)00475-9. [DOI] [PubMed] [Google Scholar]; (j) Sparrow JR, Gregory-Roberts E, Yamamoto K, Blonska A, Ghosh SK, Ueda K, Zhou J. The bisretinoids of retinal pigment epithelium. Prog. Retinal Eye Res. 2012;31(2):121–135. doi: 10.1016/j.preteyeres.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Delori FC. RPE Lipofuscin in Ageing and Age-Related Macular Degeneration. In: Coscas G, Piccolino FC, editors. Retinal Pigment Epithelium and Macular Diseases (Documenta Ophthalmologica) Vol. 62. Dordrecht, The Netherlands: Kluwer Academic Publishers; 1995. pp. 37–45. [Google Scholar]
- 5.Weng J, Mata NL, Azarian SM, Tzekov RT, Birch DG, Travis GH. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt’s disease from the phenotype in abcr knockout mice. Cell (Cambridge, MA, U. S.) 1999;98(1):13–23. doi: 10.1016/S0092-8674(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 6.(a) Sparrow JR, Cai B. Blue light-induced apoptosis of A2E–containing RPE: involvement of caspase-3 and protection by Bcl-2. Invest. Ophthalmol. Visual Sci. 2001;42(6):1356–1362. [PubMed] [Google Scholar]; (b) Bergmann M, Schutt F, Holz FG, Kopitz J. Inhibition of the ATP-driven proton pump in RPE lysosomes by the major lipofuscin fluorophore A2-E may contribute to the pathogenesis of age-related macular degeneration. FASEB J. 2004;18(3):562–564. doi: 10.1096/fj.03-0289fje. [DOI] [PubMed] [Google Scholar]; (c) Sparrow JR, Parish CA, Hashimoto M, Nakanishi K. A2E, a lipofuscin fluorophore, in human retinal pigmented epithelial cells in culture. Invest. Ophthalmol. Visual Sci. 1999;40(12):2988–2995. [PubMed] [Google Scholar]; (d) De S, Sakmar TP. Interaction of A2E with model membranes. Implications to the pathogenesis of age-related macular degeneration. J. Gen. Physiol. 2002;120(2):147–157. doi: 10.1085/jgp.20028566. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Vives-Bauza C, Anand M, Shirazi AK, Magrane J, Gao J, Vollmer-Snarr HR, Manfredi G, Finnemann SC. The age lipid A2E and mitochondrial dysfunction synergistically impair phagocytosis by retinal pigment epithelial cells. J. Biol. Chem. 2008;283(36):24770–24780. doi: 10.1074/jbc.M800706200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Zhou J, Jang YP, Kim SR, Sparrow JR. Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc. Natl. Acad. Sci. U. S. A. 2006;103(44):16182–16187. doi: 10.1073/pnas.0604255103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Radu RA, Hu J, Yuan Q, Welch DL, Makshanoff J, Lloyd M, McMullen S, Travis GH, Bok D. Complement system dysregulation and inflammation in the retinal pigment epithelium of a mouse model for Stargardt macular degeneration. J. Biol. Chem. 2011;286(21):18593–18601. doi: 10.1074/jbc.M110.191866. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Ben-Shabat S, Parish CA, Vollmer HR, Itagaki Y, Fishkin N, Nakanishi K, Sparrow JR. Biosynthetic studies of A2E, a major fluorophore of retinal pigment epithelial lipofuscin. J. Biol. Chem. 2002;277(9):7183–7190. doi: 10.1074/jbc.M108981200. [DOI] [PubMed] [Google Scholar]; (i) Rozanowska M, Jarvis-Evans J, Korytowski W, Boulton ME, Burke JM, Sarna T. Blue light-induced reactivity of retinal age pigment. In vitro generation of oxygen-reactive species. J. Biol. Chem. 1995;270(32):18825–18830. doi: 10.1074/jbc.270.32.18825. [DOI] [PubMed] [Google Scholar]; (j) Sparrow JR, Zhou J, Ben-Shabat S, Vollmer H, Itagaki Y, Nakanishi K. Involvement of oxidative mechanisms in blue-light-induced damage to A2E–laden RPE. Invest. Ophthalmol. Visual Sci. 2002;43(4):1222–1227. [PubMed] [Google Scholar]; (k) Dontsov AE, Sakina NL, Golubkov AM, Ostrovsky MA. Light-induced release of A2E photooxidation toxic products from lipofuscin granules of human retinal pigment epithelium. Dokl. Biochem. Biophys. 2009;425:98–101. doi: 10.1134/s1607672909020112. [DOI] [PubMed] [Google Scholar]
- 7.(a) Radu RA, Han Y, Bui TV, Nusinowitz S, Bok D, Lichter J, Widder K, Travis GH, Mata NL. Reductions in serum vitamin A arrest accumulation of toxic retinal fluorophores: a potential therapy for treatment of lipofuscin-based retinal diseases. Invest. Ophthalmol. Visual Sci. 2005;46(12):4393–4401. doi: 10.1167/iovs.05-0820. [DOI] [PubMed] [Google Scholar]; (b) Radu RA, Mata NL, Nusinowitz S, Liu X, Sieving PA, Travis GH. Treatment with isotretinoin inhibits lipofuscin accumulation in a mouse model of recessive Stargardt’s macular degeneration. Proc. Natl. Acad. Sci. U. S. A. 2003;100(8):4742–4747. doi: 10.1073/pnas.0737855100. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Maeda A, Maeda T, Golczak M, Imanishi Y, Leahy P, Kubota R, Palczewski K. Effects of potent inhibitors of the retinoid cycle on visual function and photoreceptor protection from light damage in mice. Mol. Pharmacol. 2006;70(4):1220–1229. doi: 10.1124/mol.106.026823. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Palczewski K. Retinoids for treatment of retinal diseases. Trends Pharmacol. Sci. 2010;31(6):284–295. doi: 10.1016/j.tips.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petrukhin K. Pharmacological inhibition of lipofuscin accumulation in the retina as a therapeutic strategy for dry AMD treatment. Drug Discovery Today: Ther. Strategies. 2013;10(1):e11–e20. doi: 10.1016/j.ddstr.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cioffi CL, Dobri N, Freeman EE, Conlon MP, Chen P, Stafford DG, Schwarz DMC, Golden KC, Zhu L, Kitchen DB, Barnes KD, Racz B, Qin Q, Michelotti E, Cywin CL, Martin WH, Pearson PG, Johnson G, Petrukhin K. Design, Synthesis, and Evaluation of Nonretinoid Retinol Binding Protein 4 Antagonists for the Potential Treatment of Atrophic Age-Related Macular Degeneration and Stargardt Disease. J. Med. Chem. 2014;57(18):7731–7757. doi: 10.1021/jm5010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Kanai M, Raz A, Goodman DS. Retinol-binding protein: the transport protein for vitamin A in human plasma. J. Clin. Invest. 1968;47(9):2025–2044. doi: 10.1172/JCI105889. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Naylor HM, Newcomer ME. The structure of human retinol-binding protein (RBP) with its carrier protein transthyretin reveals an interaction with the carboxy terminus of RBP. Biochemistry. 1999;38(9):2647–2653. doi: 10.1021/bi982291i. [DOI] [PubMed] [Google Scholar]; (c) Noy N, Slosberg E, Scarlata S. Interactions of retinol with binding proteins: studies with retinol-binding protein and with transthyretin. Biochemistry. 1992;31(45):11118–11124. doi: 10.1021/bi00160a023. [DOI] [PubMed] [Google Scholar]; (d) Noy N, Xu ZJ. Interactions of retinol with binding proteins: implications for the mechanism of uptake by cells. Biochemistry. 1990;29(16):3878–3883. doi: 10.1021/bi00468a012. [DOI] [PubMed] [Google Scholar]; (e) Monaco HL. The transthyretin-retinol-binding protein complex. Biochim Biophys. Acta, Protein Struct. Mol. Enzymol. 2000;1482(1–2):65–72. doi: 10.1016/s0167-4838(00)00140-0. [DOI] [PubMed] [Google Scholar]; (f) Monaco HL. Three-dimensional structure of the transthyretin-retinol-binding protein complex. Clin. Chem. Lab. Med. 2002;40(12):1229–1236. doi: 10.1515/CCLM.2002.213. [DOI] [PubMed] [Google Scholar]; (g) Monaco HL, Rizzi M, Coda A. Structure of a complex of two plasma proteins: transthyretin and retinol-binding protein. Science. 1995;268(5213):1039–1041. doi: 10.1126/science.7754382. [DOI] [PubMed] [Google Scholar]
- 11.Schlehuber S, Skerra A. Lipocalins in drug discovery: from natural ligand-binding proteins to ″anticalins″. Drug Discovery Today. 2005;10(1):23–33. doi: 10.1016/S1359-6446(04)03294-5. [DOI] [PubMed] [Google Scholar]
- 12.Wolf G. Multiple functions of vitamin A. Physiol. Rev. 1984;64(3):873–937. doi: 10.1152/physrev.1984.64.3.873. [DOI] [PubMed] [Google Scholar]
- 13.(a) Berni R, Formelli F. In vitro interaction of fenretinide with plasma retinol-binding protein and its functional consequences. FEBS Lett. 1992;308(1):43–45. doi: 10.1016/0014-5793(92)81046-o. [DOI] [PubMed] [Google Scholar]; (b) Adams WR, Smith JE, Green MH. Effects of N-(4-hydroxyphenyl)retinamide on vitamin A metabolism in rats. Exp. Biol. Med. 1995;208(2):178–185. doi: 10.3181/00379727-208-43849. [DOI] [PubMed] [Google Scholar]
- 14.Mata NL, Lichter JB, Vogel R, Han Y, Bui TV, Singerman LJ. Investigation of Oral Fenretinide for Treatment of Geographic Atrophy in Age-Related Macular Degeneration. Retina. 2013;33(3):498–507. doi: 10.1097/IAE.0b013e318265801d. [DOI] [PubMed] [Google Scholar]
- 15.Motani A, Wang Z, Conn M, Siegler K, Zhang Y, Liu Q, Johnstone S, Xu H, Thibault S, Wang Y, Fan P, Connors R, Le H, Xu G, Walker N, Shan B, Coward P. Identification and characterization of a non-retinoid ligand for retinol-binding protein 4 which lowers serum retinol-binding protein 4 levels in vivo. J. Biol. Chem. 2009;284(12):7673–7680. doi: 10.1074/jbc.M809654200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dobri N, Qin Q, Kong J, Yamamoto K, Liu Z, Moiseyev G, Ma J-X, Allikmets R, Sparrow JR, Petrukhin K. A1120, a Nonretinoid RBP4 Antagonist, Inhibits Formation of Cytotoxic Bisretinoids in the Animal Model of Enhanced Retinal Lipofuscino-genesis. Invest. Ophthalmol. Visual Sci. 2013;54(1):85–95. doi: 10.1167/iovs.12-10050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Swanson DM, Dubin AE, Shah C, Nasser N, Chang L, Dax SL, Jetter M, Breitenbucher JG, Liu C, Mazur C, Lord B, Gonzales L, Hoey K, Rizzolio M, Bogenstaetter M, Codd EE, Lee DH, Zhang SP, Chaplan SR, Carruthers NI. Identification and biological evaluation of 4-(3-trifluoromethylpyridin-2-yl)-piperazine-1-carboxylic acid (5-trifluoromethylpyridin-2-yl)amide, a high affinity TRPV1 (VR1) vanilloid receptor antagonist. J. Med. Chem. 2005;48(6):1857–1872. doi: 10.1021/jm0495071. [DOI] [PubMed] [Google Scholar]
- 18.(a) Dart MJ, Searle XB, Tietje KR, Toupence RB. Azabicyclic Compounds Are Central Nervous System Active Agents. PCT Int. Appl. WO2004016604 A2. Google Patents. 2004 [Google Scholar]; (b) Lauffer D, Li P, Waal N, Mcginty K, Tang Q, Ronkin S, Farmer L, Shannon D, Jacobs D. C-Met Protein Kinase Inhibitors. PCT Int. Appl. WO2009045992 A2. Google Patents. 2009 [Google Scholar]; (c) Guillemont JEG, Lancois DFA, Motte MMS, Koul A, Balemans WMA, Arnoult EPA. Antibacterial Cyclopenta[c]pyrrole Substituted 3,4-Dihydro-1H-[1,8]naphthyridinones. PCT Int. Appl. WO2013021054 A1. Google Patents. 2013 [Google Scholar]
- 19.Cogan U, Kopelman M, Mokady S, Shinitzky M. Binding affinities of retinol and related compounds to retinol binding proteins. Eur. J. Biochem. 1976;65(1):71–78. doi: 10.1111/j.1432-1033.1976.tb10390.x. [DOI] [PubMed] [Google Scholar]
- 20.Gamble MV, Ramakrishnan R, Palafox NA, Briand K, Berglund L, Blaner WS. Retinol binding protein as a surrogate measure for serum retinol: studies in vitamin A-deficient children from the Republic of the Marshall Islands. Am. J. Clin. Nutr. 2001;73(3):594–601. doi: 10.1093/ajcn/73.3.594. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.