

Abstract
Previous NMR studies demonstrated that lonidamine (LND) selectively diminishes the intracellular pH (pHi) of DB-1 melanoma and mouse xenografts of a variety of other prevalent human cancers while decreasing their bioenergetic status (tumor βNTP/Pi ratio) and enhancing the activities of melphalan and doxorubicin in these cancer models. Since melphalan and doxorubicin are highly toxic agents, we have examined three other nitrogen (N)-mustards, chlorambucil, cyclophosphamide and bendamustine, to determine if they exhibit similar potentiation by LND. As single agents LND, melphalan and these N-mustards exhibited the following activities in DB-1 melanoma xenografts; LND: 100% tumor surviving fraction (SF); chlorambucil: 100% SF; cyclophosphamide: 100% SF; bendamustine: 79% SF; melphalan: 41% SF. When combined with LND administered 40 min prior to administration of the N-mustard (to maximize intracellular acidification) the following responses were obtained; chlorambucil: 62% SF; cyclophosphamide: 42% SF; bendamustine: 36% SF; melphalan: 10% SF. The effect of LND on the activities of these N-mustards is generally attributed to acid stabilization of the aziridinium active intermediate, acid inhibition of glutathione-S-transferase, which acts as a scavenger of aziridinium, and acid inhibition of DNA repair by O6-alkyltransferase. Depletion of ATP by LND may also decrease multidrug resistance and increase tumor response. At similar maximum tolerated doses, our data indicate that melphalan is the most effective N-mustard in combination with LND when treating DB-1 melanoma in mice, but the choice of N-mustard for coadministration with LND will also depend on the relative toxicities of these agents, and remains to be determined.
Introduction
Melanoma, the most deadly of all skin cancers [1], is primarily treated by surgical excision, which is curative in about 80% of patients if the tumor is detected in its early stages. However, with recurrence and distant metastases, the prognoses is very poor since effective methods for treating systemic disease are still under development and are among the most active fields of current pharmacological research. About 40–60% of melanoma patients exhibit a BRAF mutation occurring with a ninety percent frequency substitution of glutamate for valine at amino acid 600 (i.e., V600E mutation) [2]. Agents targeting this mutation exhibited considerable initial success, but response was variable and of limited duration [2]. An effort to deal with melanoma resistance to these agents has utilized MEK, RAS and other signal-transduction inhibitors used in combination [3]. Immunotherapy with Ipilumimab [4] as well as PD-1, PD-L1 checkpoint inhibitor therapy [5] is currently under clinical evaluation. Adoptive cell transfer therapy and vaccine development are also under development with some anecdotal success, but a consistent result of these approaches with any solid tumors is yet to be achieved [6, 7]. The role of melanin in therapeutic outcome of melanomas has also been studied [8–12]. As promising as these novel targeted therapies are, a curative treatment of systemic melanoma remains elusive. The most promising approach for systemic treatment of this disease will probably be the development of multiple therapeutic approaches based on different mechanisms of action that could be administered simultaneously or sequentially in order to overcome the inherent heterogeneity of melanoma and its ability to resist almost any agent based on a single mechanism of action. Towards this end, we have been exploring the use of an existing drug, LND, to selectively sensitize melanoma and other tumors to treatment with nitrogen (N)-mustards and anthracyclines [13–15]. Conventional cytotoxic chemotherapy with refined methods to increase melanoma-targeted specificity, drug delivery and minimize systemic toxicity will probably play a crucial role in a broadly based approach to the treatment of this disease. Melphalan is currently used in hyperthermic isolated limb perfusion for treatment of melanoma in transit or soft-tissue sarcomas of the limbs, and is also used in the treatment of multiple myeloma, whereas doxorubicin remains in prominent use in the treatment of a wide range of malignancies. Therefore, increasing the activities of these agents by coadministration with LND may have considerable impact on treatment of neoplastic disease. While we have previously demonstrated that LND potentiates melphalan response, we now compare this N-mustard with three additional N-mustards to determine the relative efficacy of the LND-N-mustard combination for treating melanoma.
As a consequence of high levels of aerobic glycolysis [16], tumors generally exhibit a slightly acidic extracellular pH (pHe) and a neutral to alkaline intracellular pH (pHi) leading to a plasmalemmal pH gradient, that is slightly acidic on the outside and neutral to slightly alkaline on the inside. Since normal cells do not grow well in an acidic environment, this gradient enables tumor cells to successfully compete with stromal cells during invasive tumor growth [17]. Manipulation of pHe and/or pHi of tumors impacts tumor growth, metastasis and response to therapy [18, 19]. The microenvironment of tumors can be modified by administering sodium bicarbonate in order to increase the pHe and thereby reduce tumor invasiveness [17, 20–25]. In contrast, our aim was to decrease the pHi in order to increase the intracellular activity of N-mustards. We accomplished this by administering lonidamine (LND, 100 mg/kg, intraperitoneally; i.p.), an inhibitor, of the monocarboxylate transporter (MCT) that blocks cellular export of lactic acid and also impedes mitochondrial metabolism [13–15], which would otherwise prevent accumulation of lactate in the cytosol. Phosphorus-31 magnetic resonance spectroscopy (MRS) measurements indicate that LND decreases the pHi of the tumor to a minimum of 6.33 ± 0.10 in about 80 min following i.p. administration; these lower levels of pHi are sustained for at least 3hr, whereas the bioenergetic status of the tumor (βNTP/Pi) decreases monotonically after LND treatment, falling by over 66.8 ± 5.7% in 3 hr while having no effect on pHi or bioenergetic status of muscle or brain, and only a small transient effect on pHi and bioenergetic status of liver [15]. Since acidification has been reported to enhance the activity of N-mustards [26–28], we have evaluated the effect of LND-induced acidification on three representative N-mustards, chlorambucil, cyclophosphamide, and bendamustine. These findings point to the potential utility of N-mustards when administered after LND in the systemic treatment of disseminated melanoma and perhaps also other malignancies.
Materials and Methods
Materials
LND was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz CA, USA). The drug (LND; 5 mg) was dissolved in 227 μL of tris/glycine buffer (22.0 mg/mL), vortexed until the solution was clear, and administered i.p. at a dose of 100 mg/kg. The buffer consisted of trizma base (1.2 g) and glycine (5.76 g) in 100 mL sterile water (final pH = 8.3). Chlorambucil was purchased from Sigma Aldrich (St. Louis, MO, USA) and was dissolved by solubilization in 70% acid ethanol followed by 10-fold dilution with PBS immediately prior to i.v. administration (20 mg/kg). Cyclophosphamide was purchased from Cayman Chemical Company, Inc. (MI, USA), dissolved in PBS and administered i.v. (40 mg/kg). Bendamustine was purchased from TCI (Tokyo Chemical Industry Co., Ltd., Tokyo, Japan), dissolved in PBS and administered i.v. (25 mg/kg).
Human Melanoma Xenografts in Nude Mice
Male athymic nude mice (01B74) 4–6 weeks of age obtained from the National Cancer Institute, (Frederick, MD, USA) were housed in microisolator cages with access to water and autoclaved mouse chow ad libitum. DB-1 melanoma cells were early passage human melanoma cells derived from a lymph node biopsy of a human patient with metastatic melanoma that was excised before administration of any treatment by Dr. David Berd (Thomas Jefferson University Hospital, Philadelphia, PA). The tissue harvest protocol was reviewed and approved by the institutional review board at Thomas Jefferson University Hospital in order to protect the rights and anonymity of the patient. At that time, the committee concluded that this study was exempt from informed consent due to the samples being collected absent of patient identifiers. A melanoma lung metastasis from a previously untreated patient was excised for therapeutic purposes associated with immunotherapy in 1989. The explant was cooled on ice immediately after surgery, as described by Hill LL et al. [29]. A portion of the excised tissue was disaggregated to a single cell suspension by collagenase and DNAase. The presence of melanoma cells was verified by melanoma antigens [30]. Cell suspensions were then frozen in 10% DMSO and then stored in liquid nitrogen. For experiments, cell suspensions were rapidly thawed, counted in an electronic counter (Coulter Diagnostics, Hialeah, FL) and kept on ice in aliquots of 25x106 cells in 2 ml before inoculation subcutaneously for growth as xenografts. The DB-1 line was serially transplanted twice as subcutaneous xenografts in SCID mice. After the second transfer, cells were disaggregated and grown in monolayer culture at 37°C and pHe 7.3 in 5% CO2 in α-MEM supplemented with 10 FBS, 12 mM glucose, 10 ml/L nonessential amino acids solution and 2.9 g/L glutamine. Culture medium contained penicillin-streptomycin antibiotics for the first three weeks, and then cells were cultured in antibiotic free medium. Cells were maintained in exponential growth and passaged twice a week for 16 passages. The doubling time of the cells was approximately 36 hrs, and the plating efficiency (PE) was approximately 60%. These cyro-preserved cells were the source of inoculum used for our experiments. Cultures were not propagated longer than 2 months before being discarded and replaced by new outgrowth of frozen cells. Periodically, frozen cell stocks had to be replenished by another 16th passage amplification. Cells were monitored periodically to be free of mycoplasma contamination. Over the years using this process, the cell cycle time, PE and morphology were unchanged. During this period, the doubling time for DB-1 xenografts has remained unchanged at about 5 days. DB-1 melanoma cell preparation and inoculation in male athymic nude mice and tumor volume measurement were performed as described previously [15].
Chemotherapy with Chlorambucil, Cyclophosphamide and Bendamustine and in DB-1 Human Melanoma Xenografts
When tumors of human melanoma xenografts (DB-1) reached 20.2 ± 3.0 mm3, 8 cohorts of age- and weight-matched animals were randomized to the following treatment groups: Cohort 1 (sham-treated control) administered sham i.p. injections of tris/glycine buffer and was infused intravenously (i.v.) with PBS; Cohort 2 was infused i.v. with PBS 40 min after LND administration i.p. (100 mg/kg); Cohort 3 was injected i.p. with tris/glycine buffer and infused i.v. with chlorambucil (20 mg/kg); Cohort 4 was injected i.p. with tris/glycine buffer and infused i.v. with cyclophosphamide (40 mg/kg). Cohort 5 was injected i.p. with tris/glycine buffer and infused i.v. with bendamustine (25 mg/kg). Cohort 6 was infused i.v. with chlorambucil (20 mg/kg) 40 min after LND administration i.p. (100 mg/kg). Cohort 7 was infused i.v. with cyclophosphamide (40 mg/kg) 40 min after LND administration i.p. (100 mg/kg). Cohort 8 was infused i.v. with bendamustine (25 mg/kg) 40 min after LND administration i.p. (100 mg/kg). Values shown are means ± SEM; n = 5 animals for controls, LND, chlorambucil, cyclophosphamide, bendamustine, LND + chlorambucil, LND + cyclophosphamide, and LND + bendamustine treated animals.
During the treatment and sham-treatment procedures, all animals were anesthetized with ketamine hydrochloride (10 mg/mL) and acepromazine (1 mg/mL) with additional anesthesia being re-administered approximately every 45–60 min to maintain sedation. Animals were placed on a water pad heater (Gaymar T-Pump, Gaymar Industries, Inc., Orchard Park, NJ, USA) to maintain body temperature during anesthesia. Tumor dimensions were measured as well as body weight. To prevent blood clotting, tail vein catheters (I.V. Catheters FEP, Tyco Healthcare) filled with heparin (100 USP Units/mL) were placed using a restrainer (MTI Braintree Scientific, Braintree Scientific Inc., Braintree, MA, USA). After treatment, catheters were removed and animals were allowed to recover in cages. For the first five days post-treatment, tumor volume and animal weight were measured daily with calipers (Scienceware, Bel-Art Products, Wayne, NJ, USA) and scale (Acculab PP401, H & C Weighing Systems, Columbia, MD, USA), respectively. Afterwards, these measurements were repeated every other day. The tumor dimensions were measured with a caliper in three orthogonal directions, and the volume was calculated using the equation, V = π(a × b × c)/6, where a, b, and c are the length, width, and depth of the tumor. All animal studies were performed in accordance with a protocol approved by the University of Pennsylvania Institutional Animal Care and Use Committee (IACUC).
DNA Purification, Library Preparation, and Sequencing
DNA purification was performed using the DNeasy Blood & Tissue Kit (Qiagen). Five hundred ng of genomic DNA was sheared randomly into 200 bp fragments with the CovarisTM S200 UltraSonicator (Covaris®). Sheared DNA was A-tailed and ligated with adaptor-embedded indices using the NEBNext® UltraTM DNA Library Prep Kit for Illumina® (New England BioLabs, Inc.). DNA quality, fragment size, and concentration of library preps were measured using Agilent’s DNA 1000 chips in conjunction with the 2100 Bioanalyzer (Agilent Technologies). Samples were equimolarly pooled prior to capture with a 2.2 Mbp SureSelectXT Custom Target Enrichment Kit (Agilent Technologies) targeting 108 genes previously implicated in melanomagenesis. Paired-end sequencing was performed on the HiSeqTM 2000 sequencing system (Illumina) at the Perelman School of Medicine Next-Generation Sequencing Core Facility.
Mutational Analysis
Short-sequenced reads were aligned to the hg19 human reference genome using the Burrows-Wheeler Alignment (BWA) tool [31]. Duplicate reads were removed, as well as reads that map to more than one location, off-target reads, and variants annotated with the incorrect transcript. The Genome Analysis Toolkit (GATK) was used for data quality assurance as well as for Single Nucleotide Variant (SNV) and small insertion and deletion (indel) calling [32, 33]. After down-sampling by GATK, a mean target coverage of 197X was achieved. Variants were annotated with wANNOVAR [34].
Copy Number Variation Prediction
Prediction of copy number variation from sequencing data was performed using CODEX [35]. This algorithm normalizes the data using a Poisson latent factor model that removes biases due to GC content, exon capture, amplification efficiency, and latent systemic artifacts. Six latent factors were used for the normalization of the dataset in this study. Segmentation was restricted to exons only for all genes. Only homozygous loss and high amplification calls are reported. Log2 ratio thresholds used for high amplification and homozygous loss were 1.33 (copy number five) and -1.2, respectively. Visual confirmation of SNV calls was accomplished with Nexus 7.5 (BioDiscovery, Inc.) software.
Statistical Analyses
The approach to the primary analysis followed methods described by Demidenko [36]. The analysis plan was designed to address the question of whether each alkylating agent delays tumor growth, and whether the addition of LND further delays tumor growth relative to the alkylating agent alone. The data were first visualized using individual growth curves and smoothed using locally weighted scatterplot smoothing (loess). For each animal, we determined the time from treatment until the tumor reached a volume of 150 mm3, or approximately 3 doublings from its initial volume at the time treatment began. The treatment delay was estimated from the difference in the mean time to 150 mm3 between the (T) treated and (C) control arm and p-values determined using a Wald test. To determine the proportion of tumor cells surviving, we first computed the slope of the log tumor volume curve for each animal. This slope was computed using pre-treatment data for the treated animals and both the pre-treatment data and the data up to day 20 post-treatment in the controls, where the log tumor volume curve for the controls remained linear. We then computed the mean slope of the growth curves (B) across both arms of the study and estimated the proportion of the tumor cells surviving treatment as exp(−(T−C) × B). This estimate was truncated at a maximum of 1.0 as surviving cells cannot exceed 100%. Standard errors of the estimates were computed using the bootstrap percentile method and used to construct Wald tests. We further compared the data from these experiments to our previous work on melphalan in the absence of glucose [14]. While these data were analyzed using the same methods described here, the initial tumor volume in the earlier experiment was larger; thus, the target volume was four times the initial volume, and the control data were terminated at day 6 post-treatment. We used one-sided tests and a type I error rate of 0.05. To be consistent with the one-sided p-value, we present two-sided 90% confidence intervals obtained by the bootstrap percentile method.
We used weight loss as a measure of toxicity. The minimum weight for each animal in the first 21 days post-treatment was normalized to its median pre-treatment weight. The log of this ratio was the outcome in a series of linear models where the predictor was the alkylating agent with or without LND. P-values were one-sided as it is of scientific interest to determine whether the alkylating agents increased weight loss relative to the control, and whether the combination caused increased weight loss relative to the individual agent. Analyses were conducted in R Studio (V 99.484) using R (Version 3.2.1; R Foundation for Statistical Computing, Vienna, Austria) [37].
Results
The effects of treatment with LND in combination with chlorambucil, cyclophosphamide and bendamustine (Fig 1) were evaluated using tumor growth delay experiments. For these experiments, mean tumor-doubling times ranged from 4.6 days (LND alone) to 11.0 days (bendamustine alone). These rates are comparable to those observed in our earlier experiences with melphalan (doubling times of 5.3 to 5.9 days).
Fig 1. Growth Delay Experiments on DB-1 Melanoma Xenografts.

Growth delay experiments performed on DB-1 human melanoma xenografts in nude mice treated with 20 mg/kg chlorambucil, 40 mg/kg cyclophosphamide or 25 mg/kg bendamustine. Mice were treated on Day 0 as follows: control (sham i.p. tris/glycine buffer + sham i.v. PBS), LND (lonidamine; 100 mg/kg; i.p.), chlorambucil, LND + chlorambucil, cyclophosphamide and LND + cyclophosphamide, or bendamustine, LND + bendamustine. Each grey line is the trajectory for an individual animal. The trend for each group is illustrated using the loess smoothed curve (dark line) and its standard error (shaded area). The time at which each animal’s tumor achieved a volume of 150 mm3 (dashed line) was used to estimate the mean tumor growth delay (T-C) shown in Table 1. P-values for the difference in mean growth delay for each pair of plots are shown. Values labeled NS did not achieve the threshold of 0.05. P-values for the mean difference in cell survival rates appear in Table 1. Abbreviations are as follows: Ctl, Control; Chl, Chlorambucil; Cyc, Cyclophosphamide; Ben, Bendamustine; NS, Not Significant.
Compared to control (Cohort 1), Table 1 shows that LND alone neither significantly delayed tumor growth nor altered cell survival. Similarly, each of the alkylating agents, when administered as single agents (Cohorts 3–5), yielded no statistically significant differences relative to control for either mean tumor growth delay or cell surviving fraction (Table 1). Among the three alkylating agents administered individually, the largest mean growth delay of 3.8 days occurred for bendamustine (p = 0.11) which also exhibited the most reduced cell surviving fraction of 79% (p = 0.28).
Table 1. Summary of estimated mean growth delay (T-C), and surviving fraction as a percent (100×exp(−(T−C)×B), with bootstrap 95% CI), log cell kill method, by experiment and treatment arm in DB-1 human melanoma xenografts.
Individual growth curve data appear in Fig 1.
| Experimental Groups | Growth Delay (T-C; days) | Surviving fraction (%) Estimates (90% CI) | ||||
|---|---|---|---|---|---|---|
| Estimate (90% CI) | p value | Estimates (90% CI) | p value | |||
| Vs Control | Vs Single Agent | Vs Control | Vs Single Agent | |||
| Lonidamine (LND) | -2.1* | 0.73 | NA | 100 | 0.67 | NA |
| (-7.4, 2.9) | (59, 100) | |||||
| Chlorambucil | -4.1* | 0.96 | NA | 100 | 0.81 | NA |
| (-8.1, 0.2) | (98, 100) | |||||
| Cyclophosphamide | -1.3 | 0.65 | NA | 100 | 0.63 | NA |
| (-6.1, 3.6) | (62, 100) | |||||
| Bendamustine | 3.8 | 0.11 | NA | 79 | 0.28 | NA |
| (-1.8, 9.8) | (42, 100) | |||||
| Melphalan | 7.6 | 0.007 | NA | 41 | < 0.001 | NA |
| (2.3, 14.6) | (16, 75) | |||||
| LND + Chlorambucil | 7.7 | 0.008 | 0.002 | 62 | 0.004 | 0.090 |
| (2.5, 13.1) | (35, 86) | |||||
| LND + Cyclophosphamide | 5.9 | 0.018 | 0.050 | 42 | 0.001 | 0.068 |
| (0.6, 10.8) | (19, 92) | |||||
| LND + Bendamustine | 10.6 | <0.001 | 0.052 | 36 | < 0.001 | 0.13 |
| (4.9, 16.0) | (20, 62) | |||||
| LND + Melphalan | 17.8 | <0.001 | 0.015 | 10 | < 0.001 | 0.247 |
| (10.8, 23.8) | (5, 24) | |||||
Note: Mean growth delay and survival estimates for LND (lonidamine) alone, and for each of the three alkylating agents, either alone or in combination with LND. All p values are one-sided. Doubling times are assumed identical across all treatments. Compared to control, LND alone did not yield significant differences in either growth delay (p = 0.73) or percent survival (p = 0.67). NA is not applicable.
*Negative growth delay indicates return to tumor volume at an estimated rate that was slightly faster than the control (Fig 1). The 90% CI in all cases where the estimate was negative covered zero, suggesting that these differences represent random variation among animals.
In sharp contrast, each of the alkylating agents in combination with LND (Cohorts 6–8) showed both statistically significant growth delays and statistically significant reductions in cell surviving fraction relative to the control group (Cohort 1). Specifically, compared to control, the mean growth delay for each of the combinations of LND with an alkylating agent ranged from 5.9 to 10.6 day (p < 0.05 for each agent in combination with LND, see Table 1 or Fig 1 for specific individual p-values). Similarly, compared to control, the mean surviving fraction for each of the combinations ranged from 41 to 62% (p < 0.01 for each agent in combination with LND, see Table 1 for specific p-values).
We next compared the response to the combination of LND plus an alkylating agent to the alkylating agent alone. Here we found that the mean growth delay for each of the alkylating agents, combined with LND, yielded a mean growth delay that achieved or approached statistical significance. Specifically the mean growth delay for LND + chlorambucil (Cohort 6) exceeded chlorambucil alone Cohort 3 (p = 0.002) and the mean growth delay for LND + cyclophosphamide (Cohort 7) exceeded cyclophosphamide alone (p = 0.050). The mean growth delay for LND + bendamustine (10.6 days, Cohort 8) exceeded bendamustine alone (3.8 days, Cohort 5,) but this result did not achieve strict statistical significance (p = 0.052) (Table 1).
For each of the three alkylating agents, the estimate of the cell surviving fraction for the combination with LND was smaller than for the alkylating agent alone. However, in no case did the differences achieve strict statistical significance. We note that the surviving cell fraction for LND + cyclophosphamide was 42% versus 100% for cyclophosphamide alone, and that the surviving cell fraction for LND + chlorambucil was 62% versus 100% for chlorambucil alone, with p < 0.10 for both contrasts (Table 1).
In previous work we showed that the mean growth delay for LND + melphalan (17.8 days, previously published results) exceeded melphalan alone (7.6 days, previously published results, p = 0.015). The mean difference in cell surviving fraction for LND + melphalan (10%, previously published results) versus melphalan alone (41%, previously published results) did not achieve significance [14, 15].
LND + chlorambucil (Cohort 6), LND + cyclophosphamide (Cohort 7) and LND + bendamustine (Cohort 8) resulted in a 38%, 58% and 64% cell kill, respectively. In our previous study LND + melphalan yield 90% cell kill, respectively, [14, 15] (Table 1).
Weight Loss
For each cohort, Fig 2 displays boxplots of the maximum weight loss, relative to the pre-treatment value. The mean pre-treatment weight across all animals was 30.1 g (95% CI of 29.3, 30.3). Without LND, the average weight of the controls post- versus pre-treatment was 97.0% (90% CI 96.1, 97.9%, p < 0.001). With LND, the average weights of the controls post- versus pre-treatment was 91% (90% CI 90.2, 92.0) of control, and significantly lower than in the absence of LND (p < 0.001). Fig 2 and Table 2 show that for the alkylating agents alone, the mean post-treatment weight declined to levels that significantly exceeded that of the weight loss observed in the control without LND. The pre-treatment: post-treatment weight reduction ranged from 89.4% (chlorambucil, Cohort 3, p < 0.001 versus control without LND) to 96.6% (cyclophosphamide, Cohort 4, p = 0.030 vs control without LND) (Table 2).
Fig 2. Weight Loss Experiments on DB-1 Melanoma Xenografts.

Box plot of weight loss relative to median pretreatment weight for the control and each alkylating agent with (+) or without (-) LND (lonidamine). Bold horizontal lines indicate median, boxes indicate interquartile range, (IQR) whiskers extend to the smaller of either the range of the data or the box +/- 1.5 IQR. Outliers appear as dots. Abbreviations are as follows: Ctl, Control; NS, Not Significant.
Table 2. Weight loss of DB-1 human melanoma xenografts following treatment with nitrogen-mustards alone or in combination with LND.
| Experimental Groups | Ratio of Post-treatment: Pretreatment Weight (% Relative to Control) | ||
|---|---|---|---|
| Estimate (90% CI) | p value | ||
| Vs Control | Vs Single Agent | ||
| Alkylating Agent Alone | |||
| Chlorambucil | 89.4 | < 0.001 | NA |
| (86.1, 92.8) | |||
| Cyclophosphamide | 96.6 | 0.030 | NA |
| (93.8, 99.5) | |||
| Bendamustine | 93.2 | < 0.001 | NA |
| (91.3, 95.1) | |||
| Melphalan* | 92.0 | 0.011 | NA |
| (86.8, 97.4) | |||
| Alkylating Agent in Combination with LND | |||
| LND + Chlorambucil | 90.5 | < 0.001 | 0.627 |
| (87.1, 93.9) | |||
| LND + Cyclophosphamide | 92.6 | 0.001 | 0.015 |
| (89.7, 95.5) | |||
| LND+ Bendamustine | 84.7 | < 0.001 | < 0.001 |
| (83.0, 86.4) | |||
| LND + Melphalan* | 87.7 | 0.001 | 0.082 |
| (83.0, 92.6) | |||
Note: Ratio of minimum post-treatment to pre-treatment weight for each group, normalized to the ratio in the control group. P-values are for the comparison to control, and for the comparison of the combined LND (lonidamine)-alkylating agent to the alkylating agent alone. The mean weight at the start of treatment for animals in the current experiment was 30.1 g (95% CI 29.3, 30.3). For the control group the relative post- to pre-treatment weight was 97.0% (90% CI of 96.1, 97.9%).
For the animals treated with the alkylating agent in combination with LND, post-treatment relative to pre-treatment weight ranged from 84.7% (LND + bendamustine, Cohort 8) to 92.6% (LND + cyclophosphamide, Cohort 7) (Table 2). For chlorambucil, the weight loss was similar with or without LND. In contrast, for cyclophosphamide and bendamustine, the estimated weight loss was significantly lower than for the alkylating agent without LND (p < 0.015 and p < 0.001 respectively) (Table 2).
We note that, the minimum post versus pre-treatment weight of animals receiving melphalan (92%, p = 0.011) or LND + melphalan (87.7%, p = 0.001) was significantly lower than control, but that differences between the two groups were not significant (p = 0.082) [15] (Table 2).
Fig 3 shows in vitro, in vivo and ex vivo characteristics of human DB-1 melanoma cells. S1 Spreadsheet shows tumor growth delay and body weight data.
Fig 3. In vitro, In vivo and Ex vivo Characteristics of Human DB-1 Melanoma Cells.
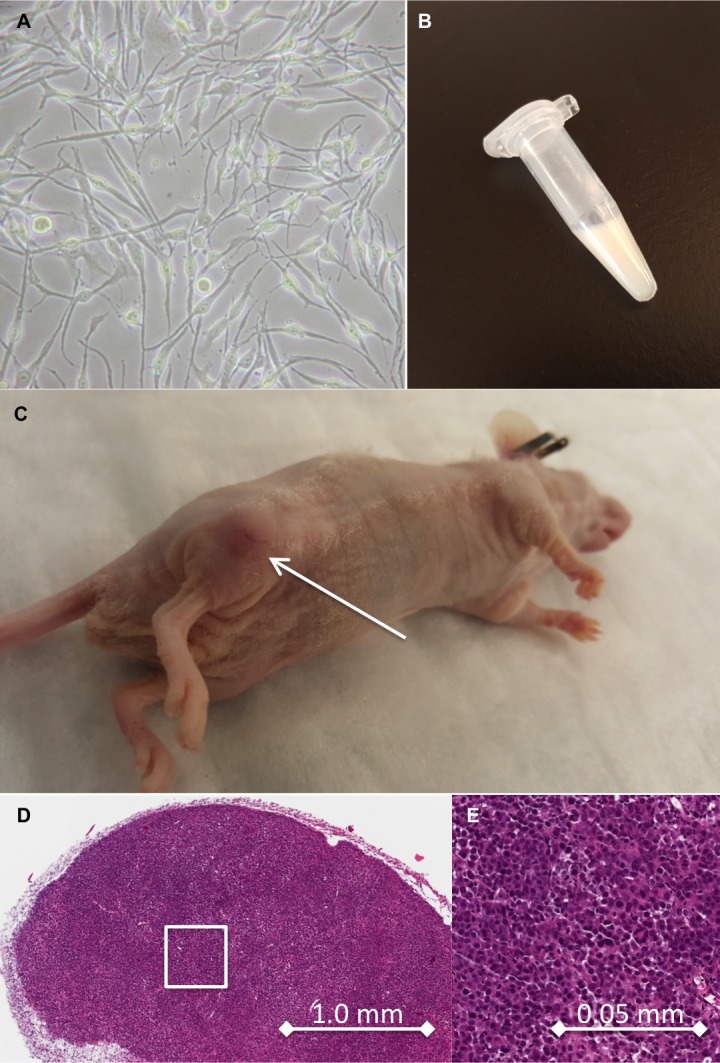
(A) DB-1 cells in culture at 40X magnification. (B) Cell pellet demonstrating amelanotic characteristic of cells. (C) Typical xenograft tumor on flank of an athymic nude mouse. Arrow pointing towards lesion. (D) H&E stain at 42X magnification of section through harvested tumor. Inset represents region shown in panel E at 840X magnification.
Mutations and Copy Number Aberrations in the DB-1 Human Melanoma Cell Line
The DB-1 cell line contained the following known pathogenic mutations: BRAF p. V600E; TP53 p.L145R; CDK4 p.R24C and TERT promoter region Chr5:1295228–1295229 GG>AA. Additional variants identified were PTEN p.T176A, JAK3 p.P693L, EGFR p.P753S, and APC p.140G. The cell line also contained homozygous loss of CDKN2B.
Discussion
The Warburg effect has been exploited for the detection and treatment of cancer for many years. PET (Positron Emission Tomography) imaging with FDG (fluorodeoxyglucose) has utilized it for the minimally invasive and sensitive detection of cancer. We reasoned that if we could trap the lactate produced by tumor cells inside the cytosol, we would have a method to selectively acidify cancer cells and make them susceptible to alkylating agents whose activity is enhanced by acid [13–15, 38] and whose lethal effect is also enhanced under acidic conditions. The goal of the present study was to determine whether intracellular tumor acidification and deenergization could increase the efficacy of bendamustine, cyclophosphamide and chlorambucil in DB-1 melanoma xenografts following LND administration. Previous studies had indicated that i.p. injection of LND at 100 mg/kg produced the optimal effects on tumor acidification and bioenergetic decline without significant toxicity to normal tissues. N-mustards are generally administered by i.v. infusion, and were administered at levels comparable to those used by other investigators in studies of mouse xenografts [15]. Compared to the alkylating agent alone, we observed significant, or near-significant increases in growth delays for all three alkylating agents when combined with LND. Additionally, compared to control each alkylating agent in combination with LND showed statistically significant cell kill, with rates ranging from 38 (Chlorambucil) to 64% (Bendamustine). In contrast, the alkylating agents alone demonstrated very little cell kill with rates ranging from 0 to 21%. We note that cell kill rates for the combination of alkylating agent plus LND compared to the alkylating agent alone did not achieve strict statistical significance (p values ranging from 0.068 to 0.13). We chose to report the experiment as planned, rather than repeating it, given the statistical comparison to the control, and the consistent pattern in both growth delay and cell kill across all three agents in combination with LND. In our experiment relative cytotoxicities of these agents in DB-1 melanoma xenografts after LND administration were bendamustine (cell kill, 64%) > cyclophosphamide (cell kill, 58%) > chlorambucil (cell kill, 38%). These differences in cytotoxicities are small and conclusions regarding the relative toxicity of these agents would require a substantially larger number of animals than was used in the current study. We have previously reported cytotoxicity of melphalan (cell kill, 90%) following LND administration in DB-1 melanoma xenograft [14], suggesting that among the N-mustards, melphalan may exhibit the highest level of potentiation following LND treatment. This conclusion must be taken with some caution as (1) confidence intervals were broadly overlapping and (2) the experiments were not conducted concurrently, and (3) we cannot strictly rule out differences between experimental conditions for the two sets of results. Lastly, we note that the three alkylating agents examined here were associated with a 3.4% to 10.6% increase in post-treatment weight loss relative to control, while the combination of agents with LND were associated with a 7.4% to 15.3% increased weight loss relative to control, values that for cyclophosphamide and bendamustine were significantly larger than for the alkylating agent alone. The effect on weight loss is similar to those observed for melphalan.
In preparation for studies combining LND with alkylating agents, we have already performed ex vivo investigations of the effects of a number of candidate platinum and alkylating agents on cultured DB-1 melanoma cells [15]. Melphalan exhibited the greatest cytotoxicity as a single agent, followed by cisplatin, bendamustine and chlorambucil. LND also exhibited a low level of cytotoxicity as a single agent in these experiments. This most likely occurred due to the presence of serum in the medium, thus limiting the bioavailability of LND to exhibit an effect. Cyclophosphamide was not included in ex vivo study at that time. Recently, we have demonstrated by 31P MRS (Phosphorus Magnetic Resonance Spectroscopy) that the treatment of DB-1 melanoma xenografts with LND (100 mg/kg, i.p.) selectively acidifies and de-energizes tumors [15].
The activity of N-mustards increases with increasing acidification of tumors [26–28, 39–42]. In the case of N-mustards, this could be caused by three effects: (i) increased concentrations of the active intermediate cyclic aziridinium ion; (ii) decreased concentrations of competing nucleophiles, such as hydroxide and glutathione, whose production is diminished by decreased activity of gluthathione-S-transferase under acidic conditions; and (iii) decreased DNA repair as a result of acid inhibition of O6-alkyltransferase [40, 42]. This is probably largely because acid shifts the equilibrium between the various forms of these agents towards more active forms. In the case of N-mustards, the active species is the cyclic aziridinium ion. In cells, the reactivity of these agents will also be affected by considerations of transport into the cell and, eventually, into the nucleus, where DNA alkylation occurs. Elimination of these agents by active transport via multidrug resistant pumps will also modify the activities of melphalan [43] and other N-mustards. Since multidrug resistance is an energy consuming process, deenergization of the tumor should increase the retention and, hence, also the activities of these alkylating agents in tumor cells.
Differences were observed between the three groups but did not achieve statistical significance. This may reflect the sample size within each group, which was chosen with the primary goal of determining differences between the individual agent and the combination with LND. With this limitation in mind, it is of interest that bendamustine exhibited the longest growth delay and largest cell kill. Previous reports indicated that increased efficacy of bendamustine might be due to inhibition of mitotic checkpoints, inefficient DNA repair, and initiation of p53-dependent DNA-damage stress response, all of which lead to mitotic catastrophe and apoptosis [44]. Moreover, the presence of a benzimidazole ring in addition to the N-mustard group may influence the way bendamustine interacts with DNA and/or its antimetabolite properties [44]. The benzimidazole central ring system is unique to bendamustine; the intent of adding this structure to the N-mustard was to include the antimetabolite properties of benzimidazole. This heterocyclic ring structure may contribute to the unique antitumor activity of bendamustine and distinguish it from conventional 2-chloroethylamine alkylators-cyclophosphamide, chlorambucil and melphalan [44].
Studies have shown that melanotic melanomas exhibited significantly shorter disease-free survival and overall survival than those with amelanotic lesions. Similarly, melanin-producing lymph node metastases were linked to shorter overall survival and disease-free survival, which was confirmed by a significantly longer mean/median disease-free survival for amelanotic versus melanotic metastases [9]. Bryoanza et al. have shown that melanin content also acts as a radioprotector and scavenger resulting in changing of properties of melanoma cells [10]. Melanogenesis is a metabolic pathway characteristic of normal and malignant melanocytes that can affect the behavior of melanoma cells or their surrounding environment, and the inhibition of melanogenesis might represent a valid therapeutic target for the management of advanced melanotic melanomas [11]. However, in the current study, with the DB-1 melanoma cell pellet indicating a very low content of melanin, based on a lack of colorization (amelanotic), DB-1 is probably an ideal cell line to examine these therapeutic effects independent of pH interaction with melanin. A possible future direction would be to examine LND effects on pH in a xenograft model that is more highly expressive of melanin.
Unfortunately, we have no clinical history of the response of the primary tumor in donor from whom the DB-1 cell line originated. Nor do we have any information regarding the treatment response of the donor’s metastases. The DB-1 cell line, besides being a cancer cell model, is a model of metastatic disease. It is hoped that the results we observe with DB-1 xenografts, investigating the tumor sensitizing effects of LND, are a guide to the response to treatment with alkylating agents of patients with metastatic melanoma.
Earlier studies have supported the mechanism of MCT inhibition by LND [15, 45–47]. MCT-1 and MCT-4, the other isoform of MCT present in DB-1 melanoma [48], have Km values of about 4.5 mM and 22 mM for lactate, respectively [49]. Recent LC-MS (Liquid chromatography-mass spectrometry) analysis of the effects of LND on DB-1 melanoma cells indicates that this agent also inhibits complex II at the ubiquinone reduction step [50]. Furthermore, our recent data demonstrates that LND inhibits the MPC of isolated liver and heart mitochondria with an IC50 of 2.5 μM [51]. Thus, the most potent site of activity of LND is the MPC followed by MCT1 and MCT4, and complex II (in that order) at increasing concentrations of LND.
Our group and other researchers have identified the sites of action of LND that are common to both normal tissue and melanoma [50, 51]. Explanation for why melanoma and other cancers demonstrate selectivity for metabolic response to LND compared to normal tissues needs further investigation. Oncogenic mutations of BRAF have been implicated in changes of the metabolic phenotypes seen in melanoma favoring a shift towards more oxidative and less glycolytic metabolism [52, 53]. Prior to treatment the DB-1 tumor derives about equal amounts of its ATP from both of these pathways [54–56]. LND should decrease mitochondrial ATP production to a greater extent than glycolytic energy production since it inhibits mitochondrial uptake and oxidation of pyruvate resulting in a decrease in TCA cycle flux, and it also inhibits electron transport, while the concentration of lactate (and, hence, the extent of glycolysis) in the tumor triples [15]. Therefore, the effect of shifting tumor metabolism in the direction of oxidative metabolism as a result of the BRAF-V600E mutation makes the tumor more responsive to LND and agents that it potentiates such as N-mustards and anthracyclines.
Limited success has been achieved in treating melanomas with agents that target the BRAF-V600E mutation observed in about 50% of melanomas [2] and with ipilumumab [57, 58], but none of these have been curative. Development of resistance following oncogene inhibition is common in the clinic, where tumor cells acquire or develop pro-survival mechanisms to maintain their growth [59, 60]. While BRAF inhibitors have displayed a rapid and dramatic response rate in BRAF mutated melanoma patients, most of these patients eventually relapse [61, 62]. Studies have shown the increased expression of genes involved in TCA cycle, oxidative phosphorylation and ATP generation in melanomas with mutant BRAF treated with vemurafenib, an inhibitor of BRAF-V600E [52]. Combination of an immunotherapeutic agent and a BRAF inhibitor may help overcome oncogene inhibition resistance [63], and, as noted above, LND, should make the mutant BRAF more responsive to N-mustards and anthracyclines. The most promising approach for the systemic treatment of this disease will probably involve the development of multiple therapeutic agents functioning by a variety of independent mechanisms that would be very difficult to simultaneously circumvent. The present study demonstrates one such method that utilizes the Warburg effect and agents that trap lactate in tumors to selectively acidify melanoma and sensitize it to systemic therapy with conventional alkylating agents.
Supporting Information
(XLSX)
Acknowledgments
Jeff Roman and Kevin Muriuki are acknowledged for their help to perform this study. Support for this project was provided in part by NIH grants R01-CA129544 and R01-CA172820.
Abbreviations
- LND
lonidamine
- MRS
magnetic resonance spectroscopy
- MCT
monocarboxylate transporter
- pHi
intracellular pH
- pHe
extracellular pH
- i.p.
intraperitoneal
- i.v.
intravenous
- s.c.
subcutaneous
- NTP
nucleoside triphosphate
- CHC
α-cyano-4-hydroxycinnamic acid
- PD-1
programmed cell death-1
- PD-L1
programmed cell death ligand-1
Data Availability
Regarding the availability of data used in this report, beyond the primary data within the paper, spreadsheets containing additional information have been uploaded as supplementary information.
Funding Statement
This work was supported by NIH grants R01-CA129544 and R01-CA172820.
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, et al. Cancer statistics, 2006. CA Cancer J Clin. 2006;56(2):106–30. Epub 2006/03/04. doi: 56/2/106 [pii]. . [DOI] [PubMed] [Google Scholar]
- 2.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363(9):809–19. Epub 2010/09/08. 10.1056/NEJMoa1002011 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang AX, Qi XY. Targeting RAS/RAF/MEK/ERK signaling in metastatic melanoma. IUBMB Life. 2013;65(9):748–58. Epub 2013/07/31. 10.1002/iub.1193 . [DOI] [PubMed] [Google Scholar]
- 4.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA4 blockade in metastatic melanoma. Science. 2015. Epub 2015/09/12. [pii] 10.1126/science.aad0095 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lutzky J. Checkpoint inhibitors in the treatment of cutaneous malignant melanoma. Chin Clin Oncol. 2014;3(3):30 Epub 2015/04/07. 10.3978/j.issn.2304-3865.2014.05.13 . [DOI] [PubMed] [Google Scholar]
- 6.Liu S, Riley J, Rosenberg S, Parkhurst M. Comparison of common gamma-chain cytokines, interleukin-2, interleukin-7, and interleukin-15 for the in vitro generation of human tumor-reactive T lymphocytes for adoptive cell transfer therapy. J Immunother. 2006;29(3):284–93. Epub 2006/05/16. 10.1097/01.cji.0000190168.53793.6b 00002371-200605000-00006 [pii]. . [DOI] [PubMed] [Google Scholar]
- 7.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–9. Epub 2006/09/02. doi: 1129003 [pii] 10.1126/science.1129003 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Slominski A, Tobin DJ, Shibahara S, Wortsman J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol Rev. 2004;84(4):1155–228. Epub 2004/09/24. 10.1152/physrev.00044.2003 84/4/1155 [pii]. . [DOI] [PubMed] [Google Scholar]
- 9.Brozyna AA, Jozwicki W, Carlson JA, Slominski AT. Melanogenesis affects overall and disease-free survival in patients with stage III and IV melanoma. Hum Pathol. 2013;44(10):2071–4. Epub 2013/06/25. doi: S0046-8177(13)00138-X [pii] 10.1016/j.humpath.2013.02.022 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bro AYAA, Jo AWW, Roszkowski K, Filipiak J, Slominski AT. Melanin content in melanoma metastases affects the outcome of radiotherapy. Oncotarget. 2016. Epub 2016/02/26. doi: 7528 [pii] 10.18632/oncotarget.7528 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brozyna AA, VanMiddlesworth L, Slominski AT. Inhibition of melanogenesis as a radiation sensitizer for melanoma therapy. Int J Cancer. 2008;123(6):1448–56. Epub 2008/06/21. 10.1002/ijc.23664 . [DOI] [PubMed] [Google Scholar]
- 12.Slominski A, Zbytek B, Slominski R. Inhibitors of melanogenesis increase toxicity of cyclophosphamide and lymphocytes against melanoma cells. Int J Cancer. 2009;124(6):1470–7. Epub 2008/12/17. 10.1002/ijc.24005 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nath K, Nelson DS, Heitjan DF, Leeper DB, Zhou R, Glickson JD. Lonidamine induces intracellular tumor acidification and ATP depletion in breast, prostate and ovarian cancer xenografts and potentiates response to doxorubicin. NMR Biomed. 2015;28(3):281–90. Epub 2014/12/17. 10.1002/nbm.3240 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nath K, Nelson DS, Heitjan DF, Zhou R, Leeper DB, Glickson JD. Effects of hyperglycemia on lonidamine-induced acidification and de-energization of human melanoma xenografts and sensitization to melphalan. NMR Biomed. 2015;28(3):395–403. Epub 2015/02/24. 10.1002/nbm.3260 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nath K, Nelson DS, Ho AM, Lee SC, Darpolor MM, Pickup S, et al. 31P and 1H MRS of DB-1 melanoma xenografts: lonidamine selectively decreases tumor intracellular pH and energy status and sensitizes tumors to melphalan. NMR Biomed. 2013;26(1):98–105. Epub 2012/06/30. 10.1002/nbm.2824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Warburg. The metabolism of tumors. London: Constable and Co.; 1930. [Google Scholar]
- 17.Gatenby RA, Gillies RJ. A microenvironmental model of carcinogenesis. Nat Rev Cancer. 2008;8(1):56–61. Epub 2007/12/07. doi: nrc2255 [pii] 10.1038/nrc2255 . [DOI] [PubMed] [Google Scholar]
- 18.Gerweck LE, Seetharaman K. Cellular pH gradient in tumor versus normal tissue: potential exploitation for the treatment of cancer. Cancer Res. 1996;56(6):1194–8. Epub 1996/03/15. . [PubMed] [Google Scholar]
- 19.Tannock IF, Rotin D. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res. 1989;49(16):4373–84. Epub 1989/08/15. . [PubMed] [Google Scholar]
- 20.Silva AS, Yunes JA, Gillies RJ, Gatenby RA. The potential role of systemic buffers in reducing intratumoral extracellular pH and acid-mediated invasion. Cancer Res. 2009;69(6):2677–84. Epub 2009/03/12. doi: 0008-5472.CAN-08-2394 [pii] 10.1158/0008-5472.CAN-08-2394 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raghunand N, Mahoney BP, Gillies RJ. Tumor acidity, ion trapping and chemotherapeutics. II. pH-dependent partition coefficients predict importance of ion trapping on pharmacokinetics of weakly basic chemotherapeutic agents. Biochem Pharmacol. 2003;66(7):1219–29. Epub 2003/09/25. doi: S0006295203004684 [pii]. . [DOI] [PubMed] [Google Scholar]
- 22.Raghunand N, Altbach MI, van Sluis R, Baggett B, Taylor CW, Bhujwalla ZM, et al. Plasmalemmal pH-gradients in drug-sensitive and drug-resistant MCF-7 human breast carcinoma xenografts measured by 31P magnetic resonance spectroscopy. Biochem Pharmacol. 1999;57(3):309–12. Epub 1999/01/16. doi: S0006-2952(98)00306-2 [pii]. . [DOI] [PubMed] [Google Scholar]
- 23.Raghunand N, He X, van Sluis R, Mahoney B, Baggett B, Taylor CW, et al. Enhancement of chemotherapy by manipulation of tumour pH. Br J Cancer. 1999;80(7):1005–11. Epub 1999/06/11. 10.1038/sj.bjc.6690455 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robey IF, Baggett BK, Kirkpatrick ND, Roe DJ, Dosescu J, Sloane BF, et al. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res. 2009;69(6):2260–8. Epub 2009/03/12. doi: 0008-5472.CAN-07-5575 [pii] 10.1158/0008-5472.CAN-07-5575 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martinez-Zaguilan R, Raghunand N, Lynch RM, Bellamy W, Martinez GM, Rojas B, et al. pH and drug resistance. I. Functional expression of plasmalemmal V-type H+-ATPase in drug-resistant human breast carcinoma cell lines. Biochem Pharmacol. 1999;57(9):1037–46. Epub 2000/05/05. doi: S0006-2952(99)00022-2 [pii]. . [DOI] [PubMed] [Google Scholar]
- 26.Jahde E, Glusenkamp KH, Klunder I, Hulser DF, Tietze LF, Rajewsky MF. Hydrogen ion-mediated enhancement of cytotoxicity of bis-chloroethylating drugs in rat mammary carcinoma cells in vitro. Cancer Res. 1989;49(11):2965–72. Epub 1989/06/01. . [PubMed] [Google Scholar]
- 27.Jahde E, Glusenkamp KH, Rajewsky MF. Nigericin enhances mafosfamide cytotoxicity at low extracellular pH. Cancer Chemother Pharmacol. 1991;27(6):440–4. Epub 1991/01/01. . [DOI] [PubMed] [Google Scholar]
- 28.Jahde E, Roszinski S, Volk T, Glusenkamp KH, Wiedemann G, Rajewsky MF. Metabolic response of AH13r rat tumours to cyclophosphamide as monitored by pO2 and pH semi-microelectrodes. Eur J Cancer. 1992;29A(1):116–22. Epub 1992/01/01. . [DOI] [PubMed] [Google Scholar]
- 29.Hill LL KR, Jaworsky C, Murphy G, McCue P, Berd D. Growth and metastasis of fresh human melanoma tissue in mice with severe combined immunodeficiency. Cancer Res. 1991;51(18):4937–41. [PubMed] [Google Scholar]
- 30.Berd D, Herlyn M, Koprowski H, Mastrangelo MJ. Flow cytometric determination of the frequency and heterogeneity of expression of human melanoma-associated antigens. Cancer Res. 1989;49(23):6840–4. Epub 1989/12/01. . [PubMed] [Google Scholar]
- 31.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60. Epub 2009/05/20. doi: btp324 [pii] 10.1093/bioinformatics/btp324 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–8. Epub 2011/04/12. doi: ng.806 [pii] 10.1038/ng.806 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303. Epub 2010/07/21. doi: gr.107524.110 [pii] 10.1101/gr.107524.110 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protoc. 2010;10(10):1556–66. Epub 2015/09/18. doi: nprot.2015.105 [pii] 10.1038/nprot.2015.105 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang Y, Oldridge DA, Diskin SJ, Zhang NR. CODEX: a normalization and copy number variation detection method for whole exome sequencing. Nucleic Acids Res. 2015;43(6):e39 Epub 2015/01/27. [pii] 10.1093/nar/gku1363 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Demidenko E. Three endpoints of in vivo tumour radiobiology and their statistical estimation. Int J Radiat Biol. 2010;86(2):164–73. Epub 2010/02/13. 10.3109/09553000903419304 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Efron B, Tibshirani T. Bootstrap method for standard errors, confidence intervals, and other measures of statistical accuracy. Stat Sci. 1986;1(1):54–77. [Google Scholar]
- 38.Zhou R, Bansal N, Leeper DB, Pickup S, Glickson JD. Enhancement of hyperglycemia-induced acidification of human melanoma xenografts with inhibitors of respiration and ion transport. Acad Radiol. 2001;8(7):571–82. Epub 2001/07/14. 10.1016/s1076-6332(03)80681-5 . [DOI] [PubMed] [Google Scholar]
- 39.Atema A, Buurman KJ, Noteboom E, Smets LA. Potentiation of DNA-adduct formation and cytotoxicity of platinum-containing drugs by low pH. Int J Cancer. 1993;54(1):166–72. Epub 1993/04/22. . [DOI] [PubMed] [Google Scholar]
- 40.Kuin A, Aalders M, Lamfers M, van Zuidam DJ, Essers M, Beijnen JH, et al. Potentiation of anti-cancer drug activity at low intratumoral pH induced by the mitochondrial inhibitor m-iodobenzylguanidine (MIBG) and its analogue benzylguanidine (BG). Br J Cancer. 1999;79(5–6):793–801. Epub 1999/03/10. 10.1038/sj.bjc.6690127 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Skarsgard LD, Skwarchuk MW, Vinczan A, Kristl J, Chaplin DJ. The cytotoxicity of melphalan and its relationship to pH, hypoxia and drug uptake. Anticancer Res. 1995;15(1):219–23. Epub 1995/01/01. . [PubMed] [Google Scholar]
- 42.Wong P, Lee C, Tannock IF. Reduction of intracellular pH as a strategy to enhance the pH-dependent cytotoxic effects of melphalan for human breast cancer cells. Clin Cancer Res. 2005;11(9):3553–7. Epub 2005/05/04. doi: 11/9/3553 [pii] 10.1158/1078-0432.CCR-04-2472 . [DOI] [PubMed] [Google Scholar]
- 43.Barnouin K, Leier I, Jedlitschky G, Pourtier-Manzanedo A, Konig J, Lehmann WD, et al. Multidrug resistance protein-mediated transport of chlorambucil and melphalan conjugated to glutathione. Br J Cancer. 1998;77(2):201–9. Epub 1998/02/14. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leoni LM, Bailey B, Reifert J, Bendall HH, Zeller RW, Corbeil J, et al. Bendamustine (Treanda) displays a distinct pattern of cytotoxicity and unique mechanistic features compared with other alkylating agents. Clin Cancer Res. 2008;14(1):309–17. Epub 2008/01/04. doi: 14/1/309 [pii] 10.1158/1078-0432.CCR-07-1061 . [DOI] [PubMed] [Google Scholar]
- 45.Ben-Horin H, Tassini M, Vivi A, Navon G, Kaplan O. Mechanism of action of the antineoplastic drug lonidamine: 31P and 13C nuclear magnetic resonance studies. Cancer Res. 1995;55(13):2814–21. Epub 1995/07/01. . [PubMed] [Google Scholar]
- 46.Ben-Yoseph O, Lyons JC, Song CW, Ross BD. Mechanism of action of lonidamine in the 9L brain tumor model involves inhibition of lactate efflux and intracellular acidification. J Neurooncol. 1998;36(2):149–57. Epub 1998/04/03. . [DOI] [PubMed] [Google Scholar]
- 47.Mardor Y, Kaplan O, Sterin M, Ruiz-Cabello J, Ash E, Roth Y, et al. Noninvasive real-time monitoring of intracellular cancer cell metabolism and response to lonidamine treatment using diffusion weighted proton magnetic resonance spectroscopy. Cancer Res. 2000;60(18):5179–86. Epub 2000/10/04. . [PubMed] [Google Scholar]
- 48.Wahl ML, Owen JA, Burd R, Herlands RA, Nogami SS, Rodeck U, et al. Regulation of intracellular pH in human melanoma: Potential therapeutic implications. Molecular Cancer Therapeutics. 2002;1(8):617–28. . [PubMed] [Google Scholar]
- 49.Halestrap AP, Price NT. The proton-linked monocarboxylate transporter (MCT) family: structure, function and regulation. Biochem J. 1999;343 Pt 2:281–99. Epub 1999/10/08. . [PMC free article] [PubMed] [Google Scholar]
- 50.Guo L, Shestov AA, Worth AJ, Nath K, Nelson DS, Leeper DB, et al. Inhibition of mitochondrial complex II by the anti-cancer agent lonidamine. J Biol Chem. 2015. Epub 2015/11/02. [pii] 10.1074/jbc.M115.697516 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nancolas B, Guo L, Zhou R, Nath K, Nelson DS, Leeper DB, et al. The anti-tumour agent lonidamine is a potent inhibitor of the mitochondrial pyruvate carrier and plasma membrane monocarboxylate transporters. Biochem J. 2016. Epub 2016/02/03. doi: BJ20151120 [pii] 10.1042/BJ20151120 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, et al. Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell. 2013;23(3):302–15. Epub 2013/03/13. doi: S1535-6108(13)00067-6 [pii] 10.1016/j.ccr.2013.02.003 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parmenter TJ, Kleinschmidt M, Kinross KM, Bond ST, Li J, Kaadige MR, et al. Response of BRAF-mutant melanoma to BRAF inhibition is mediated by a network of transcriptional regulators of glycolysis. Cancer Discov. 2014;4(4):423–33. Epub 2014/01/29. doi: 2159-8290.CD-13-0440 [pii] 10.1158/2159-8290.CD-13-0440 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shestov AA, Mancuso A, Lee SC, Guo L, Nelson DS, Roman JC, et al. Bonded Cumomer Analysis of Human Melanoma Metabolism Monitored by 13C NMR Spectroscopy of Perfused Tumor Cells. J Biol Chem. 2016;291(10):5157–71. Epub 2015/12/26. doi: M115.701862 [pii] 10.1074/jbc.M115.701862 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shestov AA, Mancuso A, Leeper DB, Glickson JD. Metabolic network analysis of DB1 melanoma cells: how much energy is derived from aerobic glycolysis? Adv Exp Med Biol. 2013;765:265–71. Epub 2012/08/11. 10.1007/978-1-4614-4989-8_37 . [DOI] [PubMed] [Google Scholar]
- 56.Shestov AA, Lee SC, Nath K, Guo L, Nelson DS, Roman JC, et al. 13C MRS and LC-MS Flux Analysis of Tumor Intermediary Metabolism. Front Oncol. 2016;6:135 Epub 10.3389/fonc.2016.00135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23. Epub 2010/06/08. doi: NEJMoa1003466 [pii] 10.1056/NEJMoa1003466 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thompson JA, Hamid O, Minor D, Amin A, Ron IG, Ridolfi R, et al. Ipilimumab in treatment-naive and previously treated patients with metastatic melanoma: retrospective analysis of efficacy and safety data from a phase II trial. J Immunother. 2012;35(1):73–7. Epub 2011/12/02. 10.1097/CJI.0b013e31823735d6 . [DOI] [PubMed] [Google Scholar]
- 59.Holderfield M, Deuker MM, McCormick F, McMahon M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer. 2014;14(7):455–67. Epub 2014/06/25. doi: nrc3760 [pii] 10.1038/nrc3760 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sun C, Wang L, Huang S, Heynen GJ, Prahallad A, Robert C, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508(7494):118–22. Epub 2014/03/29. [pii] 10.1038/nature13121 . [DOI] [PubMed] [Google Scholar]
- 61.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–16. Epub 2011/06/07. 10.1056/NEJMoa1103782 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366(8):707–14. Epub 2012/02/24. 10.1056/NEJMoa1112302 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ngiow SF, Meeth KM, Stannard K, Barkauskas DS, Bollag G, Bosenberg M, et al. Co-inhibition of colony stimulating factor-1 receptor and BRAF oncogene in mouse models of BRAF melanoma. Oncoimmunology. 2015;5(3):e1089381 Epub 2016/05/04. 10.1080/2162402X.2015.10893811089381 [pii]. . [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLSX)
Data Availability Statement
Regarding the availability of data used in this report, beyond the primary data within the paper, spreadsheets containing additional information have been uploaded as supplementary information.