

Abstract
Carvedilol is the current β-blocker of choice for suppressing ventricular tachyarrhythmia (VT). However, carvedilol’s benefits are dose-limited, attributable to its potent β-blocking activity that can lead to bradycardia and hypotension. The clinically used carvedilol is a racemic mixture of β-blocking S-carvedilol and non-β-blocking R-carvedilol. We recently reported that novel non-β-blocking carvedilol analogues are effective in suppressing arrhythmogenic Ca2+ waves and stress-induced VT without causing bradycardia. Thus, the non-β-blocking R-carvedilol enantiomer may also possess this favourable anti-arrhythmic property. To test this possibility, we synthesized R-carvedilol and assessed its effect on Ca2+ release and VT. Like racemic carvedilol, R-carvedilol directly reduces the open duration of the cardiac ryanodine receptor (RyR2), suppresses spontaneous Ca2+ oscillations in human embryonic kidney (HEK) 293 cells, Ca2+ waves in cardiomyocytes in intact hearts and stress-induced VT in mice harbouring a catecholaminergic polymorphic ventricular tachycardia (CPVT)-causing RyR2 mutation. Importantly, R-carvedilol did not significantly alter heart rate or blood pressure. Therefore, the non-β-blocking R-carvedilol enantiomer represents a very promising prophylactic treatment for Ca2+-triggered arrhythmia without the bradycardia and hypotension often associated with racemic carvedilol. Systematic clinical assessments of R-carvedilol as a new anti-arrhythmic agent may be warranted.
Keywords: β-blockers, carvedilol enantiomers, Ca2+ waves, Ca2+ -triggered arrhythmias, ryanodine receptor, sarcoplasmic reticulum
INTRODUCTION
Ventricular tachyarrhythmia (VT) is a leading cause of sudden cardiac death in patients with heart failure (HF). Despite an intense effort to suppress VT in HF, there are only a few anti-VT treatments that bring a significant survival benefit to HF patients [1–3]. Large clinical trials have shown that carvedilol, a non-selective β-blocker with α-blocking and antioxidant activities, substantially reduces VT and sudden death risk in HF patients [4–8]. However, the molecular mechanism underlying carvedilol’s favourable anti-arrhythmic effect is unclear.
A major cause of VT in HF is abnormal diastolic intracellular Ca2+ handling [9,10]. It is well known that cardiac stress can lead to sarcoplasmic reticulum (SR) Ca2+ overload. Elevated SR Ca2+ load promotes cardiac ryanodine receptor (RyR2) opening, resulting in spontaneous SR Ca2+ release in the form of Ca2+ waves [9–11]. These Ca2+ waves can evoke delayed after depolarizations (DADs), triggered arrhythmia and sudden death [12–18]. Importantly, Ca2+ waves and DADs frequently occur in failing hearts [9,10]. Furthermore, naturally occurring mutations in RyR2 cause catecholaminergic polymorphic ventricular tachycardia (CPVT) by enhancing the propensity for Ca2+ waves and DADs [19–26]. Thus, spontaneous Ca2+ waves are a common cause of CPVT as well as VT in HF.
Suppressing abnormal RyR2-mediated Ca2+ waves would thus represent an effective strategy for preventing Ca2+ -triggered arrhythmias in both CPVT and HF. In support of the present view, we have recently shown that carvedilol is the only β-blocker tested that effectively suppresses spontaneous Ca2+ waves by directly modifying RyR2 gating, an action independent of its well-described β-blocking, α-blocking and antioxidant activities [27,28]. Racemic carvedilol, but not metoprolol, is also able to suppress CPVT in a RyR2 mutant mouse model [27]. This unique inhibitory action of racemic carvedilol on Ca2+ waves probably contributes to its favourable anti-arrhythmic benefits.
Racemic carvedilol is a potent β-blocker with an IC50 of ∼1 nM [29]. Unfortunately, a substantially higher carvedilol concentration (∼1 μM) is required to optimally suppress Ca2+ waves [27] and these higher doses would result in excessive β-blockade and the accompanying adverse effects, such as bradycardia and hypotension [30,31]. In other words, the anti-Ca2+ wave benefit of racemic carvedilol is dose-limited by the agent’s potent β-blocking activity. This is indeed the case, as clinical studies show that high doses of racemic carvedilol produced better outcomes [32], but were associated with bradycardia and hypotension [31]. We have recently isolated the RyR2-targeted Ca2+ wave inhibition of carvedilol by synthesizing several carvedilol analogues that possess minimal β-blockade, but still inhibit Ca2+ waves [27,28]. These novel carvedilol analogues suppress CPVT in mice without causing bradycardia [27]. Further development of these non-β-blocking carvedilol analogues with anti-Ca2+ wave action is likely to lead to a new class of RyR2-targeted anti-arrhythmic agents that can be used to specifically limit Ca2+ -triggered arrhythmias.
Today, the clinically used carvedilol is a racemic mixture composed of R- and S-carvedilol enantiomers [33–35]. Interestingly, unlike S-carvedilol, the R-carvedilol enantiomer does not have β-blocking activity [35–38]. This raises the exciting possibility that, like our novel non-β-blocking carvedilol analogues, the R-carvedilol enantiomer may provide carvedilol’s favourable RyR2-targeted anti-arrhythmic action without the adverse effects of excessive β-blockade. To test this possibility, we synthesized R-carvedilol and assessed its effect on single RyR2 channels, Ca2+ waves in cells and stress-induced VT in mice. R-carvedilol inhibited Ca2+ waves in intact hearts by directly modifying the gating of RyR2 and suppressed VT in mice harbouring a CPVT-causing RyR2 mutation (R4496C). Importantly, R-carvedilol, unlike racemic carvedilol, did not cause bradycardia or hypotension in mice. These results indicate that R-carvedilol could be an effective anti-arrhythmic agent for limiting fatal Ca2+-triggered arrhythmias without the bradycardia or hypotension associated with racemic carvedilol.
MATERIALS AND METHODS
Animal studies
All animal studies were approved by the Institutional Animal Care and Use Committees at Rush University Medical Center or the University of Calgary and were performed in accordance with NIH (National Institutes of Health) guidelines. The knockin mouse model harbouring the CPVT RyR2 mutation (R4496C) [27] and its wild-type littermates in the 129/SvImJ background were used in the present research.
Synthesis of the (R)-(+)-carvedilol enantiomer
Commercially available (R)-glycidol was converted into its (R)-o-nitrobenzenesulfonate (nosylate) by the method of Shiratsuchi et al. [39]. The (R)-glycidyl nosylate (9.60 g, 37.0 mmol) in 35 ml of N,N-dimethylformamide (DMF) was added drop-wise to a cooled (0°C) solution of 4-hydroxycarbazole (6.90 g, 37.7 mmol) and sodium hydroxide (1.55 g, 38.7 mmol) in 100 ml of DMF and 1 ml of water. Stirring was continued for 5 h at 0°C and then at room temperature overnight. The mixture was diluted with brine, extracted with ethyl acetate and the combined organic layers were washed with saturated aqueous sodium bicarbonate, 1 M sodium hydroxide and brine. The resulting solution was dried over anhydrous sodium sulfate, concentrated under vacuum and subjected to flash chromatography over silica gel (elution with 2–4% ethyl acetate/toluene) to afford 7.95 g (90%) of the corresponding 4-[(R)-1-oxiranylmethoxy]-9H-carbazole as a white solid, melting point (mp) 159–160°C, with 1H and 13C NMR spectra identical to those of the racemic material.
2-(2-Methoxyphenoxy)ethylamine (7.00 g, 41.9 mmol) in 15 ml of propan-2-ol was added drop-wise to the above product (5.49 g, 22.9 mmol) in 35 ml of propan-2-ol. The mixture was refluxed for 1.5 h. The solvent was evaporated and the product was purified by flash chromatography over silica gel (elution with 3–7% of methanol–dichloromethane) to provide 6.20 g (67%) of (R)-(+)-carvedilol as a white solid foam, mp 115–116°C; [α]D21 + 17.3°C (c 1.0, acetic acid); lliterature mp 121–123°C; [α]D20 + 18.4° (c 1, acetic acid) [40]. Elemental analysis calculated for C24H26N2O4: C 70.93, H 6.45, N 6.89; found: C 70.75, H 6.67, N 6.85. The product gave IR, 1H and 13C NMR spectra identical to those of authentic racemic carvedilol.
Single-cell Ca2+ imaging of HEK293 cells
Stable, inducible human embryonic kidney (HEK) 293 cells expressing the CPVT-causing RyR2 mutant, R4496C, display robust spontaneous Ca2+ oscillations when perfused with 1 mM Ca2+ in Krebs–Ringer–HEPES (KRH) buffer (125 mM NaCl, 5 mM KCl, 1.2 mM KH2PO4, 6 mM glucose, 1.2 mM MgCl2 and 25 mM HEPES, pH 7.4). These RyR2-R4496C cells were used to assess the impact of R-carvedilol on spontaneous Ca2+ oscillations. Ca2+ oscillations were measured using single-cell Ca2+ imaging and the fluorescent Ca2+ indicator dye fura 2 acetoxymethyl ester (fura 2/AM) (Invitrogen) as described previously [27]. Briefly, cells grown on glass coverslips for 20–24 h after induction by 1 μg/ml tetracycline were loaded with 5 μM fura 2/AM in KRH buffer plus 0.02% pluronic F-127 and 0.1 mg/ml BSA for 20 min at room temperature (23°C). The coverslips were then mounted in a perfusion chamber on an inverted microscope (Nikon TE2000-S) and perfused with KRH buffer containing CaCl2 stepped from 0.1 mM, 0.5 mM to 1 mM for 5 min each. The cells were then continuously perfused with KRH buffer containing 1 mM CaCl2 and R-carvedilol or DMSO (control) stepped from 0 μM to 3 μM, 10 μM and 30 μM for 8 min each. Caffeine (15 mM) was applied at the end of each experiment to verify the presence of functional RyR2 channels. Time-lapse images (0.25 frame/s) were captured and analysed with Nikon NIS-Elements software. Fluorescence intensities were measured from regions of interest centred on individual cells that responded to caffeine. All chemicals were obtained from Sigma unless otherwise specified.
Confocal Ca2+ imaging of intact hearts
Excised hearts were loaded with Rhod-2 AM (rhodamine 2 acetoxymethyl ester; 3–4 μM; Biotium) in Krebs–Henseleit (KH) solution (120 mM NaCl, 24 mM NaHCO3, 11.1 mM glucose, 5.4 mM KCl, 1 mM MgCl2, 0.42 mM NaH2PO4, 10 mM taurine and 5 mM creatine, oxygenated with 95% O2 and 5% CO2) at room temperature for 40 min via a retrograde Langendorff perfusion system, as described previously [41]. After Rhod-2 AM loading, hearts were attached to a confocal microscope system and perfused sequentially with 5 ml of KH solution containing 0 mM, 0.25 mM, 0.5 mM, 1 mM and 2 mM Ca2+ and then continuously with 6 mM Ca2+ (37°C) to induce Ca2+ waves. In situ confocal line-scan imaging of Ca2+ signals arising from epicardial myocytes was performed and acquired at a rate of 1.93 ms per line. To minimize motion artefacts during Ca2+ imaging, blebbistatin (5–10 μM) was added to the perfusion solution. To eliminate the intrinsic sinus rhythm interrupting Ca2+ wave measurements, as well as to help standardize SR Ca2+ load, the arterioventricular (AV) node was ablated by electro-cautery. The AV-node-ablated hearts were then paced at 6 Hz for at least 30 s before being switched to 1 Hz to promote Ca2+ waves [42]. The frequency of Ca2+ waves per 100 μm line was measured.
Single-channel recordings in planar lipid bilayers
Heavy SR microsomes were prepared from rat ventricular muscle using the method described by Chamberlain et al. [43]. Planar lipid bilayers were composed of a 5:4:1 mixture (50 mg/ml in n-decane) of bovine brain phosphatidylethanolamine, phosphatidylserine and phosphatidylcholine. Bilayers were formed across a 100-μm diameter hole in a 10-μm thick teflon partition separating two compartments. The solution on one side of the bilayer (cis) was virtually grounded and initially contained 250 mM HEPES and 120 mM Tris/HCl (pH 7.4). The solution on the other side initially contained 250 mM HEPES and 10 mM CaCl2 (pH 7.4). Heavy SR microsomes (5–15 μg) were added to the cis solution along with 500 mM CsCl and 2 mM CaCl2 to promote microsome–bilayer fusion. SR vesicles were pre-incubated for 1 h with the tested drugs (1 μM) before their addition to the cis solution. After microsome fusion, the cytosolic side of the RyR2 channel faced the cis compartment [44,45] and solutions in both compartments were exchanged for cell-relevant solutions. The cell-relevant cytosolic solution contained 120 mM HEPES/Tris/HCl (pH 7.2), 50 μM free Ca2+ (0.5 mM EGTA), 1 mM free Mg2+ and 5 mM total ATP. The cell-relevant luminal solution contained 200 mM Cs-HEPES (pH 7.2), 1 mM free Mg2+ and 1 mM free Ca2+. Solutions on both sides of the bilayer (before and after SR microsome fusion) contained 1 μM of the tested drug. Recipes for the Ca2+ buffer solutions were generated using WinMAXC 2.05 and verified by a Ca2+ electrode. Single RyR2 recordings were made at room temperature (20–22°C) with net current in the luminal-to-cytosolic direction. Analysis was done using pCLAMP9 software (Molecular Devices). Currents were sampled at 50 μs/pt and filtered (4-pole Bessel) at 1 kHz for display.
ECG recordings and induction of VTs in anaesthetized mice
RyR2-R4496C heterozygous mutant mice (RC) readily display polymorphic VT as well as bidirectional VT when challenged by an intraperitoneal injection of epinephrine (adrenaline) (1.2 mg/kg of body weight) and caffeine (100 mg/kg of body weight) cocktail (referred to as epi/caff) [27]. A 3–10 min baseline ECG (electrocardiogram; heart rate stable) was established before epi/caff injection. Following epi/caff injection, the ECG was recorded for 30 min (continuously). Briefly, mice were lightly anaesthetized with isoflurane vapour (0.5%) and 95% O2. Anaesthetized mice were placed on a heating pad (27°C) and needle electrodes (BIOPAC MP System) were inserted subcutaneously into the right-upper limb and left-lower abdomen. The VT duration (as percentages of time) in each of ten consecutive 3-min periods or over the entire 30-min period post-injection was determined. VT was defined as three or more consecutive ectopic beats.
Drug treatments of RyR2-R4496C+/− mice
RyR2-R4496C+/− mice were treated with different doses of R-carvedilol (0.4, 0.8, 1.6 or 3.2 mg/kg/day) or DMSO of the same volume according to the body weight via intraperitoneal injection for five consecutive days. ECG recordings were performed on day five as described previously [27]. Heart rate before epi/caff injection and the VT duration after epi/caff challenge in control and drug-treated mice were determined.
Measurement of isoproterenol-stimulated heart rates
Drug (racemic carvedilol, R-carvedilol or DMSO) action on isoprenaline (also known as isoproterenol) (Iso)-stimulated heart rate was determined in anaesthetized wild-type mice [27]. Briefly, mice were lightly anaesthetized with isoflurane vapour (0.5%) and then placed on a heating pad (27°C) where needle electrodes were applied subcutaneously into the right-upper limb and left-lower abdomen for ECG recordings (BIOPAC MP System). The animals’ ECGs were continuously monitored under anaesthesia until the heart rate stabilized. The heart rates were continuously monitored via ECG recordings. Iso (0.8 mg/kg) was then intraperitoneally injected to increase the heart rate. Three minutes later, various drugs were intraperitoneally injected to evaluate their action on the Iso-stimulated heart rate, which were continuously monitored via ECG recordings.
Blood pressure measurements
R-carvedilol (1.6 mg/kg/day), racemic carvedilol (1.6 mg/kg/day) or DMSO of the same volume according to the body weight was administered to 129/SvImJ wild-type mice by intraperitoneal injection for five consecutive days. Blood pressure before and after the 5-day treatment was measured by using the CODA™ 8-Channel High Throughput Non-Invasive tail-cuff blood pressure system (Kent Scientific) following the manufacturer’s instructions. Briefly, mice were allowed to enter holders freely. The holders were placed on an infrared warming platform (25°C). A blood pressure cuff was then placed on the mouse’s tail. Recordings were performed with five acclimation cycles, 20 test cycles, 5 s between cycles, a deflation time of 20 s, a maximum occlusion pressure of 250 mmHg and a minimum volume of 15 μl. Systolic, diastolic and mean blood pressures were determined using the CODA program.
Statistical analysis
All values shown are means ± S.E.M. unless indicated otherwise. To test for differences between groups, we used Student’s t test (two-tailed) or one-way ANOVA with post-hoc test. The paired t test was used to compare blood pressures before and after drug treatment. Statistical analyses were performed using the SPSS V.15.0 (SPSS). A P-value <0.05 was considered statistically significant. To make a priori statistical power calculations, we used G*Power software to predict the needed number of experiments to detect a significant difference.
RESULTS
R-carvedilol suppresses spontaneous Ca2+ oscillations in HEK293 cells
To determine whether the non-β-blocking R-carvedilol enantiomer is able to suppress spontaneous Ca2+ release, we synthesized R-carvedilol (Figure 1A) and assessed its effect on spontaneous Ca2+ oscillations in HEK293 cells expressing a CPVT-causing RyR2 mutation (R4496C). As shown in Figure 1(B), RyR2-R4496C-expressing HEK293 cells displayed spontaneous Ca2+ oscillations in the absence of R-carvedilol (control). Application of R-carvedilol inhibited these Ca2+ oscillations in a concentration-dependent manner (Figures 1C and 1D), similar to that of racemic carvedilol [27]. Thus, like racemic carvedilol, R-carvedilol is also able to suppress spontaneous RyR2-mediated Ca2+ release in HEK293 cells.
Figure 1. R-carvedilol inhibits spontaneous Ca2+ release in HEK293 cells.
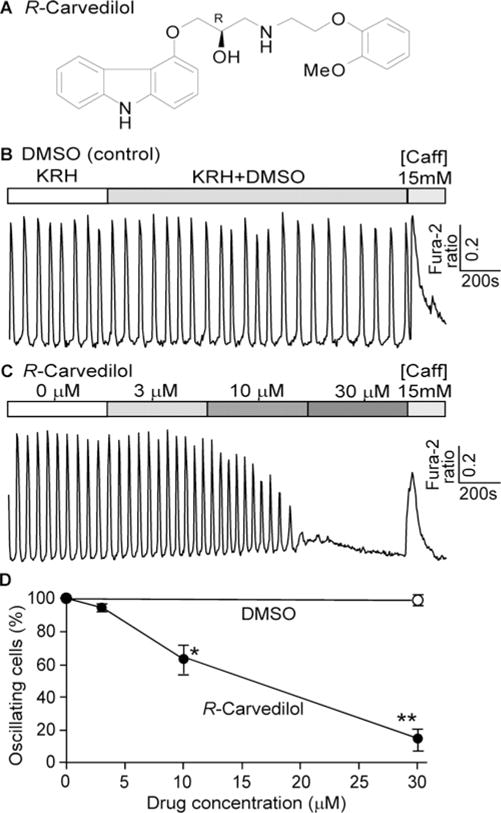
(A) Chemical structure of R-carvedilol. (B and C) Stable inducible HEK293 cells expressing the RyR2-R4496C mutant were loaded with 5 μM fura2/AM in KRH buffer. Representative traces of fura 2 ratios in HEK293 cells (∼197–419) perfused with 1 mM extracellular Ca2+ in KRH buffer containing DMSO (B) or various concentrations of R-carvedilol (0, 3, 10 and 30 μM) are shown. (C) Percentage of cells showing spontaneous Ca2+ oscillations in cells treated with DMSO (control) or R-carvedilol. Data shown are means ± S.E.M. (n=5–7; * P < 0.05, **P < 0.001 compared with DMSO).
R-carvedilol directly modifies the gating of the RyR2 channel
We have shown that racemic carvedilol suppresses spontaneous Ca2+ release by directly altering RyR2 channel function [27]. To determine whether R-carvedilol directly modifies the function of the RyR2 channel, we assessed the effect of R-carvedilol on single RyR2 channels in lipid bilayers. We have previously shown that with longer drug incubation time, lower carvedilol concentrations were able to modify the gating of single RyR2 channels and suppress spontaneous Ca2+ release in cardiomyocytes [27]. Hence, we pre-incubated SR microsomes with R-carvedilol (1 μM) for 1 h prior to their incorporation into lipid bilayers. We found that R-carvedilol (1 μM) significantly reduced the mean open time (OT; 40.8 ± 12.9–9.1 ± 3.6 ms; P < 0.05) and increased the event frequency (133 ± 15–257 ± 6 s−1; P < 0.001) of single RyR2 channels (Figure 2). The open probability (Po; 0.84 ± 0.06 compared with 0.57 ± 0.16) and mean closed time (CT; 3.7 ± 0.8 compared with 9.1 ± 6.1 ms) were not significantly altered by R-carvedilol (Figure 2).
Figure 2. R-carvedilol modifies the gating of single RyR2 channels.
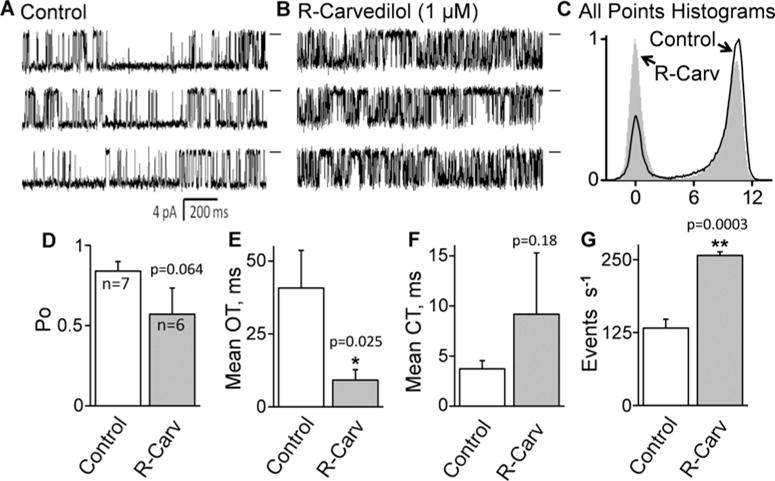
Single rat RyR2 channels were recorded in quasi-physiological salt solutions. SR microsomes were pre-incubated for 1 h without (control) (A) or with (1 μM) R-carvedilol (B) before they were fused into the lipid bilayer with the drug in both the cytosolic and luminal solutions. Single control (n=7) and R-carvedilol-treated (n = 6) channels were recorded at −40 mV. Openings are downward. Baselines are indicated (short bars). All-point histograms (C), Po (D), OT (E), CT (F) and event frequency (s−1) (G) are shown (*P < 0.05; **P < 0.01 compared with control).
The possibility that the drug-treated channels are displaying an increased frequency of sub-conductance states was evaluated by plotting amplitude frequency (all points) histograms (Figure 2). The control and drug-treated histograms each had two distinct peaks (closed and full open). There was no distinct peak associated with a sub-conductance state in the drug-treated case. Note that the full-open peak is skewed but this skewing is equal in both the control and drug-treated histograms. Thus, there was no evidence that R-carvedilol evokes a sub-conductance state.
We have also analysed the open and closed time distribution (Supplementary Figure S1). The control and R-carvedilol-treated open and closed time histograms were best fitted by assuming three open-state components. R-carvedilol reduced OT and narrowed the overall open time range. This smaller open time range is consistent with the smaller (compared with control) error level for the R-carvedilol-treated OT bar in (Figure 2). In contrast, R-carvedilol increased CT and widened the overall closed time range. This larger closed time range is consistent with the larger (compared with control) error level for the R-carvedilol-treated CT bar in Figure 2. These results demonstrate that, like racemic carvedilol [27], R-carvedilol directly modifies the gating of RyR2.
R-carvedilol suppresses spontaneous Ca2+ waves in intact hearts
To determine whether R-carvedilol suppresses spontaneous Ca2+ release in the context of cardiac cells and intact hearts, we assessed the effect of R-carvedilol on spontaneous Ca2+ waves in ventricular myocytes in intact hearts expressing the wave-promoting RyR2 mutation (R4496C). Spontaneous Ca2+ waves were induced by elevating the extracellular Ca2+ concentration to 6 mM and monitored by using confocal line-scan Ca2+ imaging of intact hearts (ex vivo) [42]. To eliminate the influence of the intrinsic sinus rhythm on Ca2+ wave, we ablated the AV node by electro-cautery [42]. AV-node-ablated hearts were electrically paced at 6 Hz to standardize SR Ca2+ loading before the pace was reduced to 1.0 Hz. As shown in Figure 3, spontaneous Ca2+ waves occurred in 89.3 ± 4.7% of cells with a frequency of 3.3 ± 0.4 Hz/100 μm during the 1.0 Hz pacing period (Figure 3A) under control conditions (DMSO). As in our single RyR2 channel studies, these intact hearts were pre-incubated with R-carvedilol (1 μM) for 1 h, as this would allow carvedilol to accumulate in the tissue [46–51]. We found that R-carvedilol (1 μM) significantly reduced the occurrence (51.1 ± 7.9%) and frequency (0.6 ± 0.1 Hz/100 μm) of Ca2+ waves (i.e. the Ca2+ release that occurred between electrical stimulations), as compared with control (P < 0.001) (Figures 3B and 3C). We previously showed that racemic carvedilol also inhibits spontaneous Ca2+ waves in cardiomyocytes [27]. Thus, like racemic carvedilol, R-carvedilol suppresses spontaneous Ca2+ waves in cardiomyocytes.
Figure 3. R-carvedilol suppresses spontaneous Ca2+ waves in intact hearts.
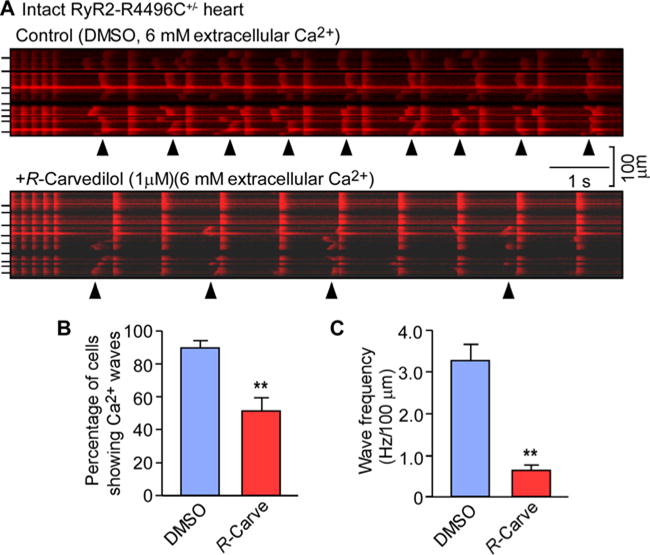
Intact hearts isolated from RyR2-R4496C+/− mice were loaded with Rhod-2 AM and Langendorff-perfused with KH solution containing 6 mM extracellular Ca2+ to induce SR Ca2+ overload. Blebbistatin (10 μM) was used to inhibit muscle contraction. The AV node was ablated by electro-cautery and the hearts were paced at 6 Hz first and then the pace was switched to 1 Hz. Intracellular Ca2+ dynamics in epicardial ventricular myocytes in intact hearts was monitored by using line-scan confocal Ca2+ imaging. (A) Representative line-scan images of Ca2+ dynamics in hearts treated with DMSO or R-carvedilol (1 μM). (B) Percentage of cells showing Ca2+ waves. (C) Frequency of spontaneous Ca2+ waves. Arrowheads show the occurrence of Ca2+ waves. Short bars to the left indicate cell boundaries within the intact heart. Data shown are means ± S.E.M. from 27–34 areas of four hearts for each group (**P <0.001 compared with DMSO).
R-carvedilol does not alter depolarization-induced Ca2+ transients in intact hearts
We also examined the action of R-carvedilol on depolarization-induced Ca2+ transients. The amplitude and dynamics of Ca2+ transients evoked by depolarization were measured using confocal line-scan Ca2+ imaging of intact hearts ex vivo. We found that R-carvedilol (1 μM) had no significant impact on the amplitude (ΔF/F0) (R-carvedilol: 1.64 ± 0.09 compared with DMSO: 1.86 ± 0.19), time to peak (R-carvedilol: 36.0 ± 0.7 ms compared with DMSO: 35.4 ± 1.0 ms) or time to 50% decay (T50); R-carvedilol: 58.0 ± 1.4 ms compared with DMSO: 60.0 ± 2.8 ms) of depolarization-evoked Ca2+ transients (Figure 4). Thus, R-carvedilol suppressed spontaneous Ca2+ waves that occur between electrical stimulations (Figure 3), but did not significantly alter the depolarization-induced Ca2+ transients.
Figure 4. R-carvedilol does not markedly affect depolarization-induced Ca2+ transients in intact hearts.
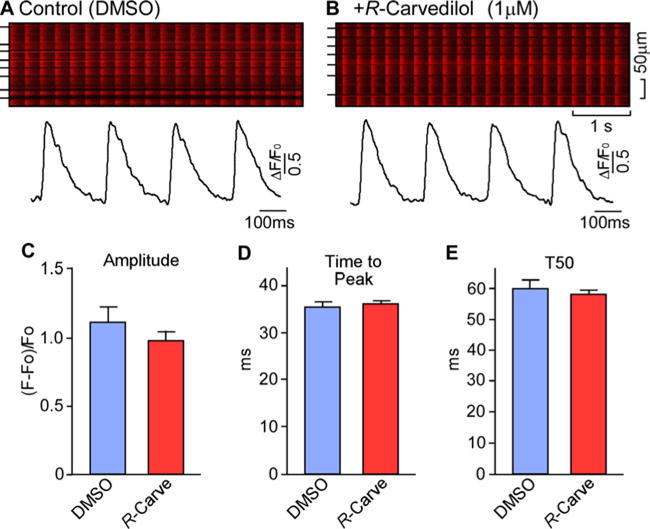
Intact hearts isolated from RyR2-R4496C+/− mice were loaded with Rhod-2 AM in KH solution and Langendorff-perfused with blebbistatin (10 μM) to inhibit muscle contraction. The perfused heart was paced at 4 Hz and Ca2+ transients were monitored by line-scan confocal Ca2+ imaging. Representative images/traces in hearts treated with DMSO (A) or with R-carvedilol (1 μM) (B). Short bars to the left indicate cell boundaries within the intact heart. The amplitude (C), time to peak (D) and time to 50% decay (T50) (E) of Ca2+ transients in hearts treated with DMSO or R-carvedilol are shown. Data shown are means ± S.E.M. from six or seven areas of three or four hearts for each group.
R-carvedilol suppresses CPVT in RyR2-R4496C mutant mice
CPVT is caused by spontaneous Ca2+ waves. Our finding that R-carvedilol suppresses Ca2+ waves would suggest that R-carvedilol may also limit CPVT. To test this idea, we employed RyR2-R4496C mutant mice that are known to develop Ca2+ wave-evoked CPVT [27]. As shown in Figure 5, long-lasting VTs (66.4 ± 4.4%) including bidirectional VTs were readily induced in R4496C mice by the injection of caffeine and epinephrine (Figures 5A and 5C). Pre-treating R4496C mice with R-carvedilol daily for 5 days suppressed CPVT in a dose-dependent manner. At a dose of 1.6 mg kg/day, R-carvedilol reduced VT duration by ∼70% (to 20.8 ± 4.2%; P < 0.001) (Figures 5B and 5D). Since R-carvedilol was given daily for 5 days, it is possible that metabolites of R-carvedilol produced during the 5-day treatment may contribute to the observed effects of R-carvedilol, either by directly interacting with the RyR2 channel or via potential long-term effects on, e.g., gene expression or membrane properties. We have previously shown that racemic carvedilol (1.6 mg/kg/day for 5 days) reduces VT duration in RyR2-R4496C mice by ∼80% [27]. Thus, like racemic carvedilol, Ca2+ -wave-inhibiting R-carvedilol is also effective at suppressing CPVT.
Figure 5. R-Carvedilol suppresses CPVT in RyR2-R4496C+/− mutant mice.
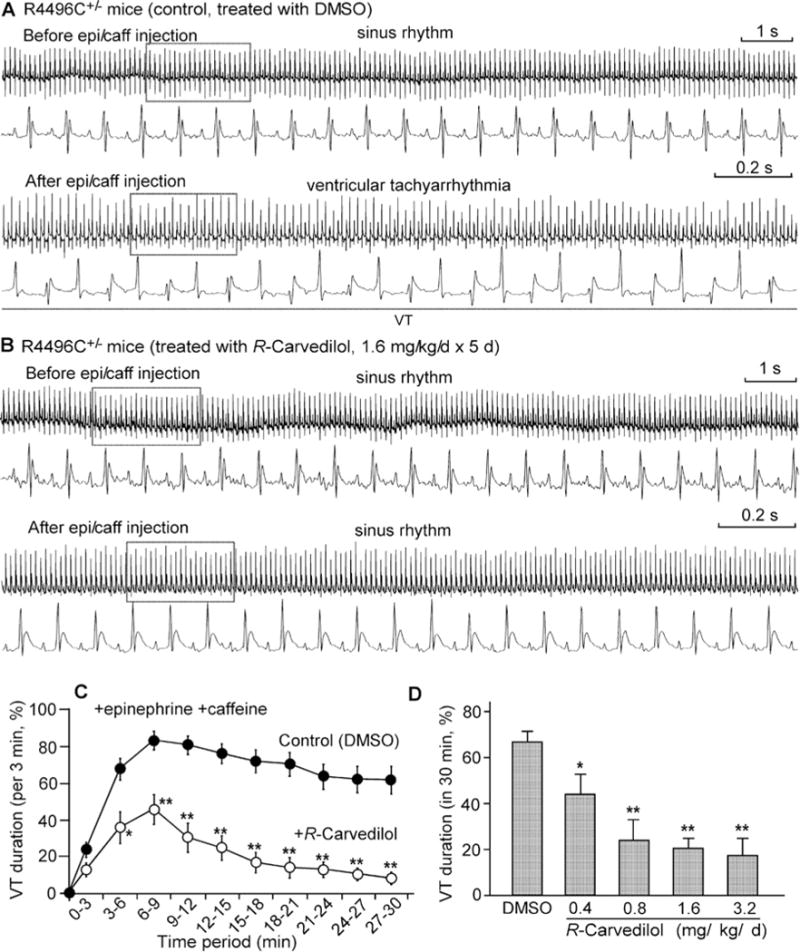
Representative ECG recordings (15 s-trace at top, 2 s-trace at bottom) of RyR2-R4496C+/− mice treated with DMSO (control) (A) or with R-carvedilol (1.6 mg/kg/day for 5 days) (B) before (top panels) and 3–6 min after (bottom panels) intraperitoneal injection of epinephrine (1.2 mg/kg) and caffeine (100 mg/kg). VT occurred intermittently and percentage time in VT (VT duration) in mice treated with DMSO (control) or R -carvedilol (1.6 mg/kg/day) was measured in sequential 3-min periods post-injection (C). Average time in VT (VT duration) in mice treated with DMSO or various doses of R-carvedilol (0.4, 0.8, 1.6 and 3.2 mg/kg/day) was measured (post-injection) over the entire 30-min recording period (D). The time scale bar sets for ECG traces before and after injection of epinephrine and caffeine are the same. Data shown are means ± S.E.M. from 12–38 mice for each group (*P <0.05, **P <0.001 compared with DMSO).
R-carvedilol does not alter heart rate or blood pressure
Racemic carvedilol is a potent β-blocker that lowers both heart rate and blood pressure [35–38]. Since R-carvedilol has little β-blocking activity, R-carvedilol would be expected to have minimal impact on heart rate or blood pressure. Indeed, we found that R-carvedilol (1.6 mg/kg) had no effect on Iso-induced heart rate increase, whereas racemic carvedilol significantly suppressed it (Figure 6A). Pre-treating mice with racemic carvedilol (1.6 mg/kg/day) for 5 days also decreased heart rate [racemic carvedilol: 420 ± 8 beats per minute (bpm) compared with DMSO: 659 ± 9 bpm; P < 0.001], but pretreatment with R-carvedilol (1.6 mg/kg/day) for 5 days did not (639 ± 9 bpm; Figure 6B). We also assessed the action of R-carvedilol on blood pressure. Pre-treating mice with racemic carvedilol (1.6 mg/kg/day) for 5 days significantly reduced the systolic, diastolic and mean blood pressure, whereas pre-treatment with R-carvedilol (1.6 mg/kg/day) for 5 days had no significant effect on any of these blood pressure measurements (only mean blood pressure shown; R-carvedilol: from 98.6 ± 4.5 mmHg to 96.4 ± 3.6 mmHg; compared with DMSO: from 98.4 ± 3.1 mmHg to 95.6 ± 3.0 mmHg; compared with racemic carvedilol: from 100.1 ± 2.6 mmHg to 83.3 ± 2.5 mmHg; Figure 6C). This is consistent with previous reports [35–38]. Taken together, our data indicate that, unlike racemic carvedilol, the non-β-blocking R-carvedilol enantiomer suppresses stress-induced VT without lowering heart rate or blood pressure.
Figure 6. Effect of R-carvedilol on heart rate and blood pressure.
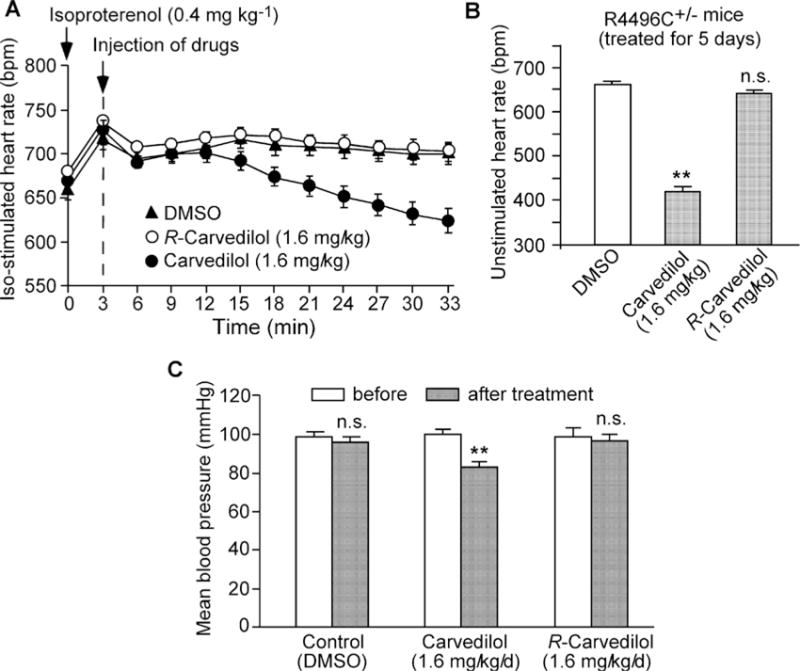
(A) Iso-stimulated heart rate was monitored by ECG recordings in R4496C+/− mice (n=6 for each group) after the injection of Iso (0.8 mg/kg), followed by the treatment with DMSO (control), R-carvedilol (1.6 mg/kg) or racemic carvedilol (1.6 mg/kg). (B) Unstimulated heart rate was determined by ECG recordings in R4496C+/− mice (n = 18–38 for each group) after the treatment with DMSO (control), R-carvedilol (1.6 mg/kg/day) or racemic carvedilol (1.6 mg/kg/day) for 5 days. (C) Mean blood pressure was determined by measuring the systolic and diastolic blood pressure using the CODA™ high-throughput non-invasive tail-cuff blood pressure system in wild-type mice (n=7–11 for each group) treated with DMSO (control), R-carvedilol (1.6 mg/kg/day) or racemic carvedilol (1.6 mg/kg/day) for 5 days. Data shown are means ± S.E.M. (**P <0.001; compared with DMSO).
DISCUSSION
The major finding of the present study is that the non-β-blocking R-carvedilol enantiomer suppresses arrhythmogenic spontaneous Ca2+ waves and stress-induced VT in mice without significantly lowering heart rate or blood pressure. We have shown that the clinically used racemic carvedilol also suppresses Ca2+ waves and stress-induced VT, but also causes bradycardia [27]. Indeed, bradycardia and hypotension are two major adverse effects of carvedilol attributable to this agent’s potent β-blocking activity [30,31]. Therefore, R-carvedilol’s capacity to suppress Ca2+ waves provides a promising new anti-arrhythmic prophylactic option for treating Ca2+ -triggered arrhythmias without the adverse effects of bradycardia and hypotension currently associated with racemic carvedilol.
Spontaneous SR Ca2+ release in the form of Ca2+ waves in cardiac cells can lead to DADs, which in turn can cause triggered activities, cardiac arrhythmias and sudden death [9–11]. These arrhythmogenic Ca2+ waves result from abnormal opening of RyR2 channels by elevated SR luminal Ca2+ during SR Ca2+ overload [21,22,26,52–56]. Many conditions such as excessive β-adrenergic receptor stimulation (as during emotional or physical stress) can lead to SR Ca2+ overload and store-overload-induced Ca2+ waves [12–18]. These Ca2+ waves and subsequent DADs frequently occur in diseased hearts [9,10]. Spontaneous Ca2+ waves are thus a common pathological entity. Therapeutically targeting spontaneous Ca2+ waves and especially the RyR2 channel may represent an effective and attractive approach to suppressing dangerous Ca2+ -wave-evoked VT in various pathological settings. Indeed, we have demonstrated that racemic carvedilol directly modifies the gating of single RyR2 channels and effectively suppresses Ca2+ wave and wave-evoked CPVT [27]. Additionally, we reported that a carvedilol analogue (VK-II-86) with minimal β-blocking activity also alters RyR2 gating and suppresses Ca2+ waves and CPVT [27]. In the present study, we demonstrate that the non-β-blocking R-carvedilol enantiomer reduces the duration of RyR2 channel openings and inhibits spontaneous Ca2+ waves and CPVT. Taken together, these observations support the practicality and effectiveness of this therapeutic strategy for preventing Ca2+ -mediated arrhythmias by modifying the gating of the RyR2 channel [27,56].
Our studies showed that R-carvedilol and novel carvedilol analogues (e.g. VK-II-86) have limited β-blocking activity, but still suppress Ca2+ waves and wave-evoked VT. This indicates that β-blockade is not required for inhibiting Ca2+ waves and wave-evoked VT in our experimental setting. However, blocking β-adrenergic receptor signalling does suppress the stress-induced SR Ca2+ overload that promotes Ca2+ waves. Hence, β-blockade will reduce the likelihood of spontaneous Ca2+ waves. Indeed, clinical studies have consistently demonstrated the benefit of β-blockade in reducing the occurrence of VT and risk of sudden death [57–59]. Therefore, although excessive β-blockade can lead to adverse effects, adequate (well-managed) β-blockade is clearly valuable and beneficial. In this regard, combining the benefit of the non-β-blocking R-carvedilol with the benefit of a well-managed β-blockade regimen might provide a promising new approach to preventing Ca2+ -triggered arrhythmia with optimal control of heart rate and blood pressure.
We have recently produced and characterized a large number of carvedilol analogues that are capable of suppressing RyR2-mediated spontaneous Ca2+ release [27,28]. Developing these novel compounds into clinically useful anti-arrhythmic agents will require years of efforts. However, the R-carvedilol enantiomer is already present in racemic carvedilol that is being used clinically today. In other words, R-carvedilol is currently being used in humans along with the S-carvedilol enantiomer. More encouragingly, R-carvedilol alone has already been applied to healthy human volunteers without major adverse effects [35]. Thus, an important and exciting future effort is to systematically assess the safety and efficacy of R-carvedilol in human patients. Given that abnormal intracellular Ca2+ handling commonly occurs in many cardiac settings, R-carvedilol with its anti-Ca2+ wave action may provide a promising and effective treatment for CPVT as well as for VT in HF and other heart conditions associated with SR Ca2+ mishandling.
In summary, the clinically used carvedilol is a racemic mixture of the β-blocking S-carvedilol and non-β-blocking R-carvedilol. We show that R-carvedilol inhibits spontaneous Ca2+ waves by directly modifying the gating of RyR2 and suppresses Ca2+ wave-evoked VT without significantly lowering heart rate or blood pressure. Thus, R-carvedilol can limit Ca2+ -triggered arrhythmias without the unwanted bradycardia and hypotension that are typically associated with racemic carvedilol. Given our results in the present study and this agent’s current clinical application, large-scale clinical assessments of R-carvedilol as a novel anti-arrhythmic agent may be warranted.
Supplementary Material
Acknowledgments
FUNDING
This work was supported by the U.S. National Institutes of Health [grant numbers R01HL075210 (to M.F., T.G.B. and S.R.W.C.), R01HL057832 (to M.F.) and R01HL090905 (to L.S.S.)]; the Canadian Institutes of Health Research the Heart and Stroke Foundation of Alberta, the Canada Foundation for Innovation; and the Heart and Stroke Foundation/Libin Cardiovascular Institute Professorship in Cardiovascular Research (to S.R.W.C.).
Abbreviations
- AV
arterioventricular
- bpm
beats per minute
- CPVT
catecholaminergic polymorphic ventricular tachycardia
- CT
mean closed time
- DAD
delayed after depolarization
- DMF
dimethylformamide
- ECG
electrocardiogram
- epi/caff
epinephrine (adrenaline) and caffeine
- fura 2/AM
fura 2 acetoxymethyl ester
- HEK
human embryonic kidney
- HF
heart failure
- Iso
isoprenaline
- KH
Krebs–Henseleit
- KRH
Krebs–Ringer–HEPES
- mp
melting point
- OT
mean open time
- Po
open probability
- Rhod-2 AM
rhodamine 2 acetoxymethyl ester
- RyR2
cardiac ryanodine receptor
- SR
sarcoplasmic reticulum
- VT
ventricular tachyarrhythmia
Footnotes
AUTHOR CONTRIBUTION
Jingqun Zhang, Qiang Zhou, Chris Smith, Long-Sheng Song, Michael Fill, Thomas Back and Wayne Chen designed the research. Jingqun Zhang, Qiang Zhou, Chris Smith, Haiyan Chen, Zhen Tan, Biyi Chen, Alma Nani and Guogen Wu performed the research. Jingqun Zhang, Qiang Zhou, Chris Smith, Thomas Back and Wayne Chen analysed the data. Jingqun Zhang, Chris Smith, Long-Sheng Song, Michael Fill, Thomas Back and Wayne Chen wrote the paper.
References
- 1.Waldo AL, Camm AJ, deRuyter H, Friedman PL, MacNeil DJ, Pauls JF, Pitt B, Pratt CM, Schwartz PJ, Veltri EP. Effect of d-sotalol on mortality in patients with left ventricular dysfunction after recent and remote myocardial infarction. The SWORD investigators Survival with oral d-sotalol. Lancet. 1996;348:7–12. doi: 10.1016/s0140-6736(96)02149-6. [DOI] [PubMed] [Google Scholar]
- 2.Miller L. Limitations of current medical therapies for the treatment of heart failure. Rev Cardiovasc Med. 2003;4(Suppl 2):S21–S29. [PubMed] [Google Scholar]
- 3.Kamath GS, Mittal S. The role of antiarrhythmic drug therapy for the prevention of sudden cardiac death. Prog Cardiovasc Dis. 2008;50:439–448. doi: 10.1016/j.pcad.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 4.Poole-Wilson PA, Swedberg K, Cleland JG, Di Lenarda A, Hanrath P, Komajda M, Lubsen J, Lutiger B, Metra M, Remme WJ, et al. Comparison of carvedilol and metoprolol on clinical outcomes in patients with chronic heart failure in the carvedilol or metoprolol european trial (COMET): randomised controlled trial. Lancet. 2003;362:7–13. doi: 10.1016/S0140-6736(03)13800-7. [DOI] [PubMed] [Google Scholar]
- 5.Stroe AF, Gheorghiade M. Carvedilol: Beta-blockade and beyond. Rev Cardiovasc Med. 2004;5(Suppl 1):S18–S27. [PubMed] [Google Scholar]
- 6.Greenberg B. Nonselective versus selective beta-blockers in the management of chronic heart failure: clinical implications of the carvedilol or metoprolol european trial. Rev Cardiovasc Med. 2004;5(Suppl 1):S10–S17. [PubMed] [Google Scholar]
- 7.Remme WJ. Which beta-blocker is most effective in heart failure? Cardiovasc Drugs Ther. 2010;24:351–358. doi: 10.1007/s10557-010-6247-7. [DOI] [PubMed] [Google Scholar]
- 8.DiNicolantonio JJ, Hackam DG. Carvedilol: A third-generation beta-blocker should be a first-choice beta-blocker. Expert Rev Cardiovasc Ther. 2012;10:13–25. doi: 10.1586/erc.11.166. [DOI] [PubMed] [Google Scholar]
- 9.Bers DM. Calcium and cardiac rhythms: Physiological and pathophysiological. Circ Res. 2002;90:14–17. [PubMed] [Google Scholar]
- 10.Pogwizd SM, Bers DM. Cellular basis of triggered arrhythmias in heart failure. Trends Cardiovasc Med. 2004;14:61–66. doi: 10.1016/j.tcm.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 11.Engelhardt S, Hein L, Dyachenkow V, Kranias EG, Isenberg G, Lohse MJ. Altered calcium handling is critically involved in the cardiotoxic effects of chronic beta-adrenergic stimulation. Circulation. 2004;109:1154–1160. doi: 10.1161/01.CIR.0000117254.68497.39. [DOI] [PubMed] [Google Scholar]
- 12.Kass RS, Tsien RW. Fluctuations in membrane current driven by intracellular calcium in cardiac Purkinje fibers. Biophys J. 1982;38:259–269. doi: 10.1016/S0006-3495(82)84557-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orchard C, Eisner D, Allen D. Oscillations of intracellular Ca2+ in mammalian cardiac muscle. Nature. 1983;304:735–738. doi: 10.1038/304735a0. [DOI] [PubMed] [Google Scholar]
- 14.Stern M, Kort A, Bhatnagar G, Lakatta E. Scattered-light intensity fluctuations in diastolic rat cardiac muscle caused by spontaneous Ca2+ -dependent cellular mechanical oscillations. J Gen Physiol. 1983;82:119–153. doi: 10.1085/jgp.82.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wier W, Kort A, Stern M, Lakatta E, Marban E. Cellular calcium fluctuations in mammalian heart: direct evidence from noise analysis of aequorin signals in Purkinje fibers. Proc Natl Acad Sci USA. 1983;80:7367–7371. doi: 10.1073/pnas.80.23.7367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marban E, Robinson SW, Wier WG. Mechanisms of arrhythmogenic delayed and early afterdepolarizations in ferret ventricular muscle. J Clin Invest. 1986;78:1185–1192. doi: 10.1172/JCI112701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schlotthauer K, Bers DM. Sarcoplasmic reticulum Ca2+ release causes myocyte depolarization. underlying mechanism and threshold for triggered action potentials. Circ Res. 2000;87:774–780. doi: 10.1161/01.res.87.9.774. [DOI] [PubMed] [Google Scholar]
- 18.Xie LH, Weiss JN. Arrhythmogenic consequences of intracellular calcium waves. Am J Physiol Heart Circ Physiol. 2009;297:H997–H1002. doi: 10.1152/ajpheart.00390.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, Sorrentino VV, Danieli GA. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196–200. doi: 10.1161/01.cir.103.2.196. [DOI] [PubMed] [Google Scholar]
- 20.Priori SG, Napolitano C, Memmi M, Colombi B, Drago F, Gasparini M, DeSimone L, Coltorti F, Bloise R, Keegan R, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106:69–74. doi: 10.1161/01.cir.0000020013.73106.d8. [DOI] [PubMed] [Google Scholar]
- 21.Jiang D, Xiao B, Yang D, Wang R, Choi P, Zhang L, Cheng H, Chen SRW. RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR) Proc Natl Acad Sci USA. 2004;101:13062–13067. doi: 10.1073/pnas.0402388101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang D, Wang R, Xiao B, Kong H, Hunt DJ, Choi P, Zhang L, Chen SRW. Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ Res. 2005;97:1173–1181. doi: 10.1161/01.RES.0000192146.85173.4b. [DOI] [PubMed] [Google Scholar]
- 23.Cerrone M, Colombi B, Santoro M, di Barletta MR, Scelsi M, Villani L, Napolitano C, Priori SG. Bidirectional ventricular tachycardia and fibrillation elicited in a knock-in mouse model carrier of a mutation in the cardiac ryanodine receptor. Circ Res. 2005;96:e77–e82. doi: 10.1161/01.RES.0000169067.51055.72. [DOI] [PubMed] [Google Scholar]
- 24.Liu N, Colombi B, Memmi M, Zissimopoulos S, Rizzi N, Negri S, Imbriani M, Napolitano C, Lai FA, Priori SG. Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: Insights from a RyR2 R4496C knock-in mouse model. Circ Res. 2006;99:292–298. doi: 10.1161/01.RES.0000235869.50747.e1. [DOI] [PubMed] [Google Scholar]
- 25.Cerrone M, Noujaim SF, Tolkacheva EG, Talkachou A, O’Connell R, Berenfeld O, Anumonwo J, Pandit SV, Vikstrom K, Napolitano C, Priori SG, Jalife J. Arrhythmogenic mechanisms in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2007;101:1039–1048. doi: 10.1161/CIRCRESAHA.107.148064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Priori SG, Chen SR. Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ Res. 2011;108:871–883. doi: 10.1161/CIRCRESAHA.110.226845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou Q, Xiao J, Jiang D, Wang R, Vembaiyan K, Wang A, Smith CD, Xie C, Chen W, Zhang J, et al. Carvedilol and its new analogs suppress arrhythmogenic store overload-induced Ca2+ release. Nat Med. 2011;17:1003–1009. doi: 10.1038/nm.2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith CD, Wang A, Vembaiyan K, Zhang J, Xie C, Zhou Q, Wu G, Chen SR, Back TG. Novel carvedilol analogues that suppress store-overload-induced Ca2+ release. J Med Chem. 2013;56:8626–8655. doi: 10.1021/jm401090a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yao A, Kohmoto O, Oyama T, Sugishita Y, Shimizu T, Harada K, Matsui H, Komuro I, Nagai R, Matsuo H, Serizawa T, Maruyama T, Takahashi T. Characteristic effects of alpha1-beta1,2-adrenergic blocking agent, carvedilol, on [Ca2+]i in ventricular myocytes compared with those of timolol and atenolol. Circ J. 2003;67:83–90. doi: 10.1253/circj.67.83. [DOI] [PubMed] [Google Scholar]
- 30.Packer M, Bristow MR, Cohn JN, Colucci WS, Fowler MB, Gilbert EM, Shusterman NH. The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. U.S carvedilol heart failure study group. N Engl J Med. 1996;334:1349–1355. doi: 10.1056/NEJM199605233342101. [DOI] [PubMed] [Google Scholar]
- 31.Ko DT, Hebert PR, Coffey CS, Curtis JP, Foody JM, Sedrakyan A, Krumholz HM. Adverse effects of {beta}-blocker therapy for patients with heart failure: A quantitative overview of randomized trials. Arch Intern Med. 2004;164:1389–1394. doi: 10.1001/archinte.164.13.1389. [DOI] [PubMed] [Google Scholar]
- 32.Bristow MR, Gilbert EM, Abraham WT, Adams KF, Fowler MB, Hershberger RE, Kubo SH, Narahara KA, Ingersoll H, Krueger S, et al. Carvedilol produces dose-related improvements in left ventricular function and survival in subjects with chronic heart failure. Circulation. 1996;94:2807–2816. doi: 10.1161/01.cir.94.11.2807. [DOI] [PubMed] [Google Scholar]
- 33.van Zwieten PA. Pharmacodynamic profile of carvedilol. Cardiology. 1993;82(Suppl 3):19–23. doi: 10.1159/000175939. [DOI] [PubMed] [Google Scholar]
- 34.Frishman WH. Carvedilol. N Engl J Med. 1998;339:1759–1765. doi: 10.1056/NEJM199812103392407. [DOI] [PubMed] [Google Scholar]
- 35.Stoschitzky K, Koshucharova G, Lercher P, Maier R, Sakotnik A, Klein W, Liebmann PM, Lindner W. Stereoselective effects of (R)- and (S)-carvedilol in humans. Chirality. 2001;13:342–346. doi: 10.1002/chir.1042. [DOI] [PubMed] [Google Scholar]
- 36.Nichols AJ, Sulpizio AC, Ashton DJ, Hieble JP, Ruffolo RR., Jr The interaction of the enantiomers of carvedilol with alpha 1- and beta 1-adrenoceptors. Chirality. 1989;1:265–270. doi: 10.1002/chir.530010404. [DOI] [PubMed] [Google Scholar]
- 37.Bartsch W, Sponer G, Strein K, Muller-Beckmann B, Kling L, Bohm E, Martin U, Borbe HO. Pharmacological characteristics of the stereoisomers of carvedilol. Eur J Clin Pharmacol. 1990;38(Suppl 2):S104–S107. doi: 10.1007/BF01409475. [DOI] [PubMed] [Google Scholar]
- 38.Hanada K, Asari K, Saito M, Kawana J, Mita M, Ogata H. Comparison of pharmacodynamics between carvedilol and metoprolol in rats with isoproterenol-induced cardiac hypertrophy: effects of carvedilol enantiomers. Eur J Pharmacol. 2008;589:194–200. doi: 10.1016/j.ejphar.2008.04.055. [DOI] [PubMed] [Google Scholar]
- 39.Shiratsuchi M, Kawamura K, Akashi T, Ishihama H, Nakamura M, Takenaka F. Synthesis and activity of optical isomers of nipradilol. Chem Pharm Bull. 1987;35:3691–3698. doi: 10.1248/cpb.35.3691. [DOI] [PubMed] [Google Scholar]
- 40.Leinert H. Process for the preparation of optically-active carbazole derivatives, new R- and S-carbazole derivatives and pharmaceutical compositions containing these compounds. US Pat 4,697,022 1987
- 41.Wei S, Guo A, Chen B, Kutschke W, Xie YP, Zimmerman K, Weiss RM, Anderson ME, Cheng H, Song LS. T-tubule remodeling during transition from hypertrophy to heart failure. Circ Res. 2010;107:520–531. doi: 10.1161/CIRCRESAHA.109.212324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang J, Chen B, Zhong X, Mi T, Guo A, Zhou Q, Tan Z, Wu G, Chen AW, Fill M, et al. The cardiac ryanodine receptor luminal Ca2+ sensor governs Ca2+ waves, ventricular tachyarrhythmias and cardiac hypertrophy in calsequestrin-null mice. Biochem J. 2014;461:99–106. doi: 10.1042/BJ20140126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chamberlain BK, Volpe P, Fleischer S. Calcium-induced calcium release from purified cardiac sarcoplasmic reticulum vesicles: general characteristics. J Biol Chem. 1984;259:7540–7546. [PubMed] [Google Scholar]
- 44.Tu Q, Velez P, Cortes-Gutierrez M, Fill M. Surface charge potentiates conduction through the cardiac ryanodine receptor channel. J Gen Physiol. 1994;103:853–867. doi: 10.1085/jgp.103.5.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qin J, Valle G, Nani A, Nori A, Rizzi N, Priori SG, Volpe P, Fill M. Luminal Ca2+ regulation of single cardiac ryanodine receptors: Insights provided by calsequestrin and its mutants. J Gen Physiol. 2008;131:325–334. doi: 10.1085/jgp.200709907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hjalmarson A. Cardioprotection with beta-adrenoceptor blockers. does lipophilicity matter? Basic Res Cardiol. 2000;95(Suppl 1):I41–I45. doi: 10.1007/s003950070008. [DOI] [PubMed] [Google Scholar]
- 47.Caron G, Steyaert G, Pagliara A, Reymond F, Crivori P, Gaillard P, Carrupt PA, Avdeef A, Comer J, Box KJ, et al. Structure-lipophilicity relationships of neutral and protonated beta-blockers. Helv Chim Acta. 1999;82:1211–1222. [Google Scholar]
- 48.Neugebauer G, Akpan W, von Mollendorff E, Neubert P, Reiff K. Pharmacokinetics and disposition of carvedilol in humans. J Cardiovasc Pharmacol. 1987;10(Suppl 11):S85–S88. [PubMed] [Google Scholar]
- 49.Doze P, Elsinga PH, Maas B, Van Waarde A, Wegman T, Vaalburg W. Synthesis and evaluation of radiolabeled antagonists for imaging of beta-adrenoceptors in the brain with PET. Neurochem Int. 2002;40:145–155. doi: 10.1016/s0197-0186(01)00081-x. [DOI] [PubMed] [Google Scholar]
- 50.Fujimaki M. Stereoselective disposition and tissue distribution of carvedilol enantiomers in rats. Chirality. 1992;4:148–154. doi: 10.1002/chir.530040304. [DOI] [PubMed] [Google Scholar]
- 51.Stahl E, Mutschler E, Baumgartner U, Spahn-Langguth H. Carvedilol stereopharmacokinetics in rats: affinities to blood constituents and tissues. Arch Pharm. 1993;326:529–533. doi: 10.1002/ardp.19933260907. [DOI] [PubMed] [Google Scholar]
- 52.Bassani JW, Yuan W, Bers DM. Fractional SR Ca release is regulated by trigger Ca and SR Ca content in cardiac myocytes. Am J Physiol. 1995;268:C1313–C1319. doi: 10.1152/ajpcell.1995.268.5.C1313. [DOI] [PubMed] [Google Scholar]
- 53.Shannon TR, Ginsburg KS, Bers DM. Potentiation of fractional sarcoplasmic reticulum calcium release by total and free intra-sarcoplasmic reticulum calcium concentration. Biophys J. 2000;78:334–343. doi: 10.1016/S0006-3495(00)76596-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Diaz ME, Trafford AW, O’Neill SC, Eisner DA. Measurement of sarcoplasmic reticulum Ca2+ content and sarcolemmal Ca2+ fluxes in isolated rat ventricular myocytes during spontaneous Ca2+ release. J Physiol. 1997;501:3–16. doi: 10.1111/j.1469-7793.1997.003bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eisner DA, Kashimura T, Venetucci LA, Trafford AW. From the ryanodine receptor to cardiac arrhythmias. Circ J. 2009;73:1561–1567. doi: 10.1253/circj.cj-09-0478. [DOI] [PubMed] [Google Scholar]
- 56.Chen W, Wang R, Chen B, Zhong X, Kong H, Bai Y, Zhou Q, Xie C, Zhang J, Guo A, et al. The ryanodine receptor store-sensing gate controls Ca2+ waves and Ca2+ -triggered arrhythmias. Nat Med. 2014;20:184–192. doi: 10.1038/nm.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Anh D, Marine JE. Beta blockers as anti-arrhythmic agents. Heart Fail Rev. 2004;9:139–147. doi: 10.1023/B:HREV.0000046369.90417.db. [DOI] [PubMed] [Google Scholar]
- 58.Zicha S, Tsuji Y, Shiroshita-Takeshita A, Nattel S. Beta-blockers as antiarrhythmic agents. Handb Exp Pharmacol. 2006;171:235–266. [PubMed] [Google Scholar]
- 59.Adamson PB, Gilbert EM. Reducing the risk of sudden death in heart failure with beta-blockers. J Card Fail. 2006;12:734–746. doi: 10.1016/j.cardfail.2006.08.213. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.