

Abstract
Karyomegalic interstitial nephritis is a rare cause of hereditary chronic interstitial nephritis, described for the first time over 40 years ago.
A 36-year-old woman, of Turkish origin, presented with chronic kidney disease and high blood pressure. She had a history of recurrent upper respiratory tract infections but no familial history of nephropathy. Physical examination was unremarkable. Laboratory tests showed serum creatinine at 2.3 mg/dL with an estimated glomerular filtration rate of 26 mL/min/1.73m2, and gamma-glutamyl transpeptidase and alkaline phosphatase at 3 and 1.5 times the upper normal limit. Urinalysis showed 0.8 g/day of nonselective proteinuria, microscopic hematuria, and aseptic leukocyturia. Immunological tests and tests for human immunodeficiency and hepatitis B and C viruses were negative. Complement level and serum proteins electrophoresis were normal. Analysis of the renal biopsy showed severe interstitial fibrosis and tubular atrophy. Numerous tubular cells had nuclear enlargement with irregular outlines, hyperchromatic aspect, and prominent nucleoli. These findings were highly suggestive of karyomegalic interstitial nephritis, which was further confirmed by exome sequencing of FAN1 gene showing an identified homozygous frameshift mutation due to a one-base-pair deletion in exon 12 (c.2616delA).
The present case illustrates a rare but severe cause of hereditary interstitial nephritis, sometimes accompanied by subtle extrarenal manifestations. Identification of mutations in FAN1 gene underscores recent insights linking inadequate DNA repair and susceptibility to chronic kidney disease.
INTRODUCTION
Karyomegalic interstitial nephritis (KIN) is an uncommon hereditary cause of chronic interstitial nephritis described for the first time over 40 years ago.1–3 In 1974, Burry et al1 reported a young woman dying from hepatocellular carcinoma and who displayed dysplastic nuclei in the renal epithelium. The term of “KIN” was introduced by Mihatsch et al3 in 1979 who described 3 cases of systemic karyomegaly associated with chronic interstitial nephritis. The disease presents as a slowly progressive chronic kidney disease, eventually leading to end-stage renal disease before the age of 50 years. Extrarenal features are absent or mild, consisting of recurrent upper respiratory tract infections and abnormal liver function tests. The presence of karyomegalic tubular epithelial cells on the renal biopsy specimen is the disease hallmark that makes it distinguishable from other common causes of chronic tubulointerstitial nephritis. More recently, the disease has been linked to mutations in the FAN1 (FANCD2/FANCI-Associated Nuclease 1) gene, a gene involved in the DNA damage response pathway, particularly in the kidney, shedding new lights on the potential link between defective DNA repair and chronic kidney disease progression.
CASE REPORT
A 36-year-old woman, of Turkish origin, was referred to our nephrology department because of impaired renal function. The patient was born in France and had a history of recurrent upper respiratory tract infections. She had no familial history of nephropathy, and reported no exposure to nephrotoxic medications, heavy metals, environmental or agricultural toxins, and no consumption of Chinese herbal medicine. Physical examination was unremarkable except for high blood pressure (164/94 mmHg). Serum creatinine was at 2.3 mg/dL with an estimated glomerular filtration rate of 26 mL/min/1.73m2. Serum electrolytes, calcium, and albumin levels were normal. Hemoglobin level was 10 g/dL (normal range [12–15]). The platelet and leukocyte counts were normal. Liver function tests revealed mild cholestasis with gamma-glutamyl transpeptidase and alkaline phosphatase at 3 and 1.5 times the upper normal limit. Urinalysis showed proteinuria (0.8 g/day), hematuria (15 red blood cells/mm3), and aseptic leukocyturia. No glucosuria was detected on repeated urinary dipsticks. Immunological tests were negative, including antinuclear, antineutrophilic cytoplasmic, antismooth muscle, antiliver/kidney microsomal, and antimitochondrial antibodies. Complement level and serum proteins electrophoresis were normal. Tests for human immunodeficiency and hepatitis B and C viruses were negative. Chest CT scan showed evidence of mild bronchiectasis and interstitial infiltrates of lung bases, consistent with the past history of recurrent respiratory tract infections (Figure 1A). Abdominal ultrasonography revealed no hepatic abnormality but showed small kidneys with an atrophic right kidney measured at 7 cm against 9.5 cm for the left one.
FIGURE 1.
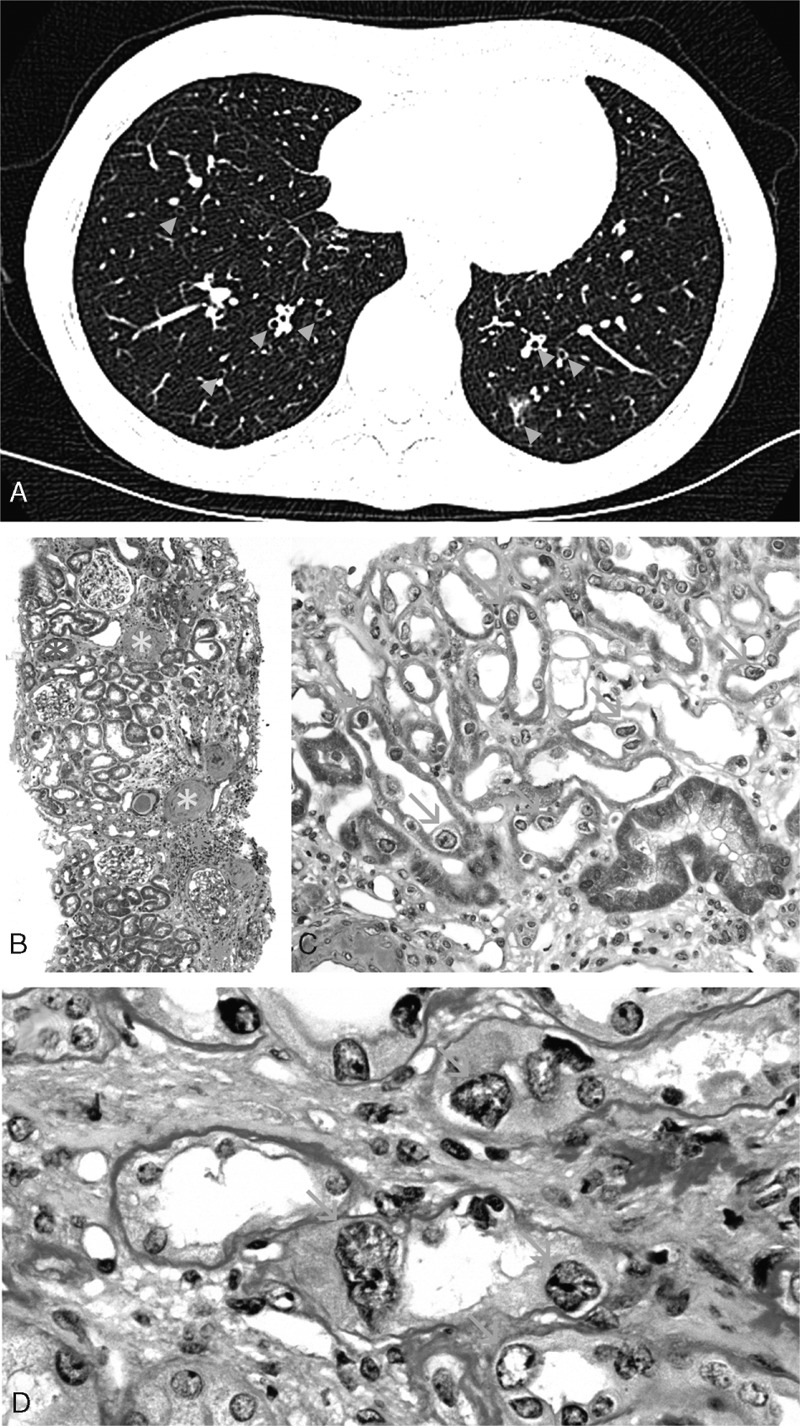
(A) Chest CT-scan showing bilateral bronchectasis with bronchial wall thickening (blue arrowheads). (B) Light microscopy at low magnification using Masson's trichrome staining showed chronic tubulointerstitial nephritis (green asters) with severe fibrosis, tubular atrophy, and inflammatory interstitial infiltrates. Globally sclerotic glomeruli (yellow asters) and severe vascular lesions (red asters) were also observed (original magnification x50). (C) Remarkably, numerous tubular cells in both the cortex and medulla showed nuclear enlargement with irregular outlines (orange arrows, original magnification x200). (D) Periodic acid-Schiff staining showing typical karyomegalic tubular epithelial cells (orange arrows) characterized by markedly enlarged nuclei with irregular outlines, and hyperchromatic and prominent nucleoli (original magnification x600).
A transjugular biopsy of the right kidney was performed. Analysis of the renal biopsy revealed 60% of sclerotic glomeruli, the remainder appearing normal with no deposits by immunofluorescence study. Predominant lesions included patchy but severe interstitial fibrosis, tubular atrophy, and arteriosclerosis (Figure 1B). Remarkably, numerous tubular cells in the cortex and medulla showed nuclear enlargement with irregular outlines, hyperchromatic aspect, and prominent nucleoli (Figure 1C, D). No intranuclear inclusions or virus-like particles were observed. Altogether, these findings were highly suggestive of karyomegalic interstitial nephritis (KIN), a hereditary cause of chronic interstitial nephritis. Consistently, genetic analysis by exome sequencing identified a homozygous frameshift mutation due to a one-base-pair deletion in exon 12 of FAN1 gene (c.2616delA), resulting in the appearance of a premature STOP codon. After 2 years of follow-up, the patient had experienced 3 episodes of lung infection, including 1 requiring hospitalization. Serum creatinine level was 2.7 mg/dL with an estimated glomerular filtration rate of 21 mL/min/1.73m2. Urinalysis showed an increase in proteinuria level up to 1.2 g/day and liver function tests still revealed mild cholestasis.
DISCUSSION
Chronic interstitial nephritis is a heterogeneous condition that may be secondary to a broad spectrum of causes, including drugs and toxins, infections, and immunological conditions, and other hereditary disorders. Frequently, no etiology can be identified, leading to the diagnosis of the so-called “idiopathic” forms. The clinical and biological setting of interstitial nephritis, including mild to moderate renal dysfunction, absence or mild degree of proteinuria, and/or urinary sediment abnormalities, often make physicians reluctant to perform a renal biopsy. Even if done, renal lesions are most often nonspecific, and definite etiological diagnosis may remain difficult to establish.
KIN is an uncommon hereditary cause of chronic interstitial nephritis described for the first time over 40 years ago.1–3 In 1974, Burry et al1 reported the case of a young woman dying from hepatocellular carcinoma and who displayed dysplastic nuclei in the renal epithelium. The term of “KIN” was then introduced by Mihatsch et al3 in 1979 who described 3 cases of systemic karyomegaly associated with chronic interstitial nephritis. Since then, <50 cases of KIN have been reported in the English literature (Table 1 ).1–22 KIN usually presents as a slowly progressive chronic kidney disease, eventually leading to end-stage renal disease in the early adulthood.8 Patients display mild proteinuria, usually of <1 g/day. More than 75% also have glucosuria, whereas less than one-third present with urinary sediment abnormalities, mostly consisting of hematuria.11 Analysis of the renal biopsy specimen usually shows nonspecific but severe chronic interstitial fibrosis and tubular changes, associated with nonspecific glomerulosclerosis and vascular lesions. The presence of karyomegalic tubular epithelial cells, lining the proximal and distal tubules, and characterized by markedly enlarged and hyperchromatic nuclei, represent the disease hallmark.8
TABLE 1.
Reported Cases of Karyomegalic Interstitial Nephritis in the English Literature
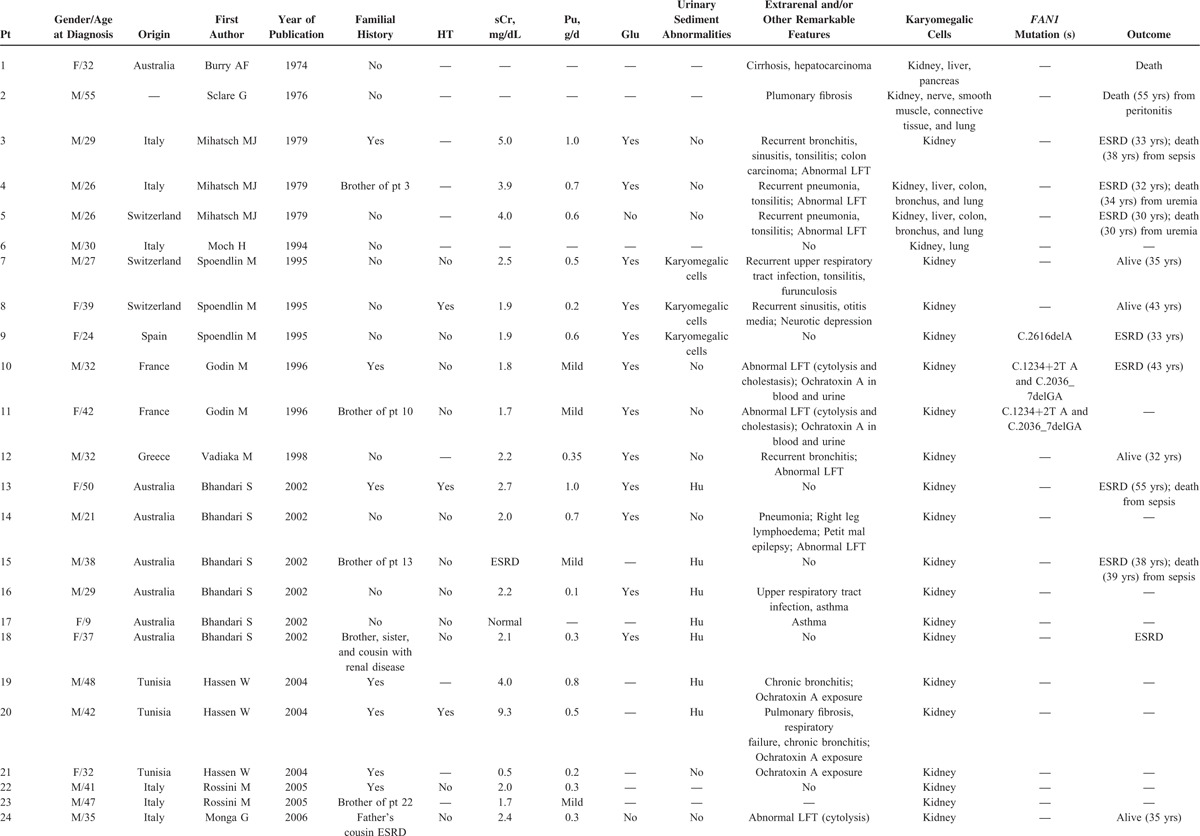
TABLE 1 (Continued).
Reported Cases of Karyomegalic Interstitial Nephritis in the English Literature
Almost half of patients display a past medical history of recurrent upper respiratory tract infections and abnormal liver function tests.8,11 Consistently, karyomegaly has been described in the liver and lung, but also in other organs, including the brain, skin, and digestive tissues.3,11 Nevertheless, and in sharp contrast to the kidney, extrarenal karyomegaly is usually associated with only subtle clinical and biological changes. Only 1 case of systemic karyomegaly with primary pulmonary manifestations and without significant kidney impairment has been reported in the literature.19 Karyomegalic cells can also be detected in the urine samples.11 Renal karyomegaly may be related to a few alternative causes, including viral infections, immunosuppressive therapy such as alkylating agents, and exposure to heavy metals and mycotoxins, particularly ochratoxin A.9,23 In the present case, no exposure to the above-mentioned agents and no evidence for viral infections were identified.
Historically, KIN was thought to be a hereditary disorder because almost half of patients had a familial history of nephropathy. Consistently, the disease has been recently ascribed to autosomal recessive mutations in the FAN1 gene, which encodes Fanconi anemia-associated nuclease 1.17 This nuclease belongs to the Fanconi anemia DNA damage response pathway and is required for the repair of DNA interstrand crosslinks (Figure 2). The recognition of DNA interstrand crosslink by Fanconi Anemia Complementation group M (FANCM) and associated proteins leads to the recruitment of the Fanconi anemia core complex and monoubiquitylation of Fanconi Anemia Complementation group D2 (FANCD2) and Fanconi Anemia Complementation group I (FANCI). FAN1 is then recruited at sites of DNA damage by monoubiquitinated FANCD2 and cleaves DNA successively, allowing to excise an interstrand crosslink from 1 strand through flanking incisions. FAN1 is more specifically involved in the repair of interstrand crosslinks-induced DNA breaks by being required for efficient homologous recombination, and probably acts in the resolution of homologous recombination intermediates.24 Patients with Fanconi anemia usually display developmental abnormalities, bone marrow failure, and predisposition to cancers.25 Strikingly, contrary to other FAN genes, FAN1 mutations have not been associated with a Fanconi anemia phenotype, likely because of a predominant expression of the gene in the kidney, liver, and neuronal tissue.17 Recently, Segui et al26 linked colorectal cancer predisposition to monoallelic germline mutations in the FAN1 gene. Interestingly, in case of biallelic loss of FAN1, as observed in patients with KIN, development of cancer at early ages has also been reported in 2 cases.1,3 Of note, contrary to KIN-associated biallelic mutations in FAN1, which localize toward the C-terminus of the protein, monoallelic mutations associated with hereditary colorectal cancer do not show a preferential gene location.26 Interstrand crosslink lesions could be actually processed differentially depending on the organ affected, eventually leading to different clinical phenotypes.17 In particular, FAN1 could be more specifically engaged in the repair of a possible “renal-specific” DNA damage. Godin et al6 reported 2 related cases of KIN associated with a homozygous FAN1 mutation who also displayed detectable ochratoxin A in the urine and blood samples. Exposure to ochratoxin A has also been identified in a few other patients with KIN, suggesting that FAN1 mutations could render tubular cells more susceptible to environmental genotoxin-induced renal DNA damage (Figure 2). In this line, Chaki et al27 also identified mutations in 2 other genes of the DNA damage response pathway in patients with renal ciliopathies, reinforcing the concept of a potential link between defective DNA damage repair and the pathogenesis of chronic kidney diseases.
FIGURE 2.
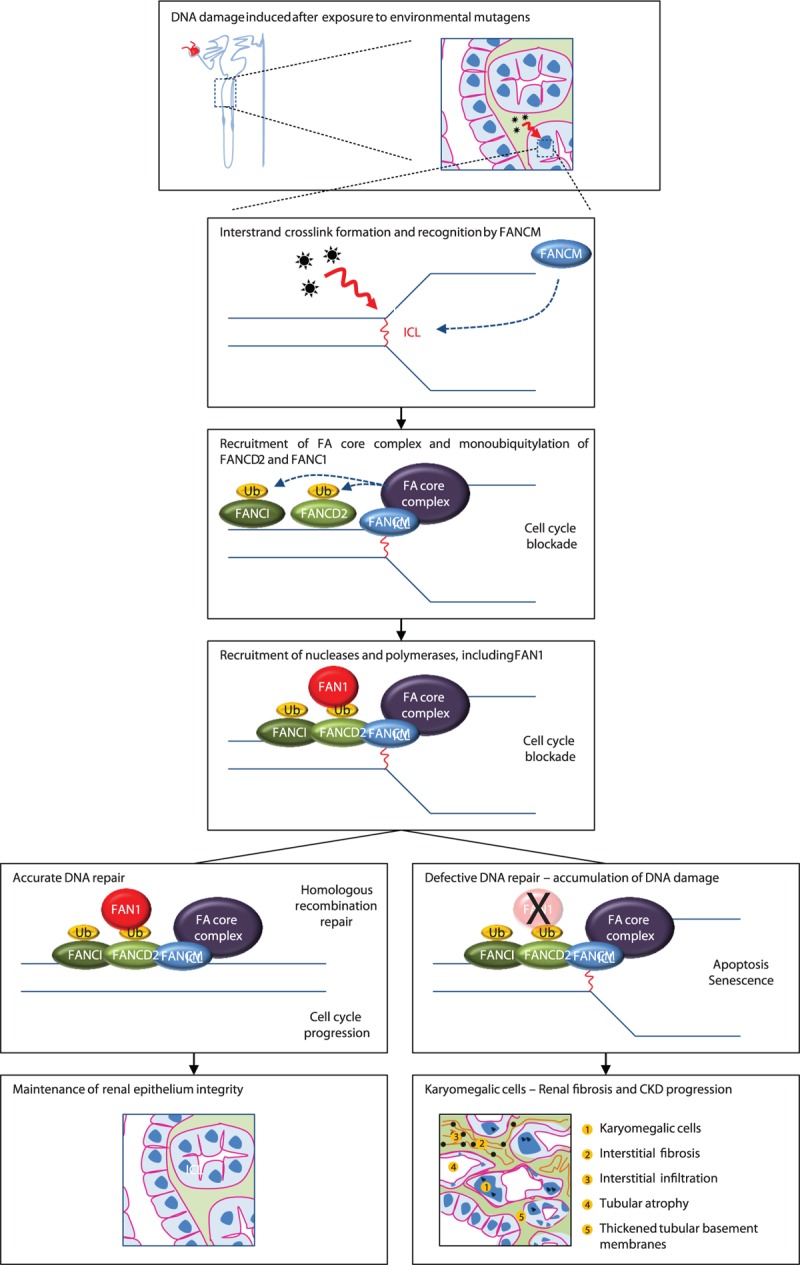
Recent insights in the pathophysiology of karyomegalic interstitial nephritis. Tubular epithelial cells are exposed to various types of aggressions, potentially leading to cellular injury. In case of DNA damage with ICL, which may be favored by environmental genotoxins, FANCM is recruited at the site of ICL and initiates the recruitment of the Fanconi anemia core complex leading to the monoubiquitylation of FANCD2 and FANCI, and cell cycle blockade to allow DNA repair. FAN1 is then recruited by monoubiquitinated FANCD2 and cleaves DNA successively, allowing to excise an interstrand crosslink from 1 strand through flanking incisions. FAN1 has been reported to be more specifically involved in the repair of ICL-induced DNA breaks by being required for efficient homologous recombination, and probably acts in the resolution of homologous recombination intermediates. In the absence of FAN1, DNA repair is impaired resulting in the accumulation of DNA damage and impairment of cell proliferation. In particular, FAN1 could be more specifically engaged in the repair of a possible “renal-specific” DNA damage. FAN1 = FANCD2/FANCI-Associated Nuclease 1; FANCI = Fanconi Anemia Complementation group I, FANCD2 = Fanconi Anemia Complementation group D2, FANCM = Fanconi Anemia Complementation group M, ICL = interstrand crosslinks.
In summary, KIN is a rare but severe cause of chronic interstitial nephritis, sometimes accompanied by subtle extrarenal manifestations. Identification of mutations in FAN1 gene underscores recent insights linking inadequate DNA repair and susceptibility to chronic kidney disease.
Footnotes
Abbreviations: FAN1 = FANCD2/FANCI-Associated Nuclease 1, FANCD2 = Fanconi Anemia Complementation group D2, FANCI = Fanconi Anemia Complementation group I, FANCM = Fanconi Anemia Complementation group M, ICL = interstrand crosslinks, KIN = Karyomegalic interstitial nephritis.
MR and JL, and BK and MZ contributed equally to this work. PI, JL, BK, and MZ were involved in the management of the patient. MR analyzed the kidney biopsy specimen. CA performed the genetic analysis. PI, BK, and MZ wrote the article, which was approved by all co-authors. Informed consent to publish was obtained from the patient. The authors report no conflicts of interest.
REFERENCES
- 1.Burry AF. Extreme dysplasia in renal epithelium of a young woman dying from hepatocarcinoma. J Pathol 1974; 113:147–150. [DOI] [PubMed] [Google Scholar]
- 2.Sclare G. A case of unexplained karyomegaly. Beiträge Zur Pathol 1976; 157:301–306. [DOI] [PubMed] [Google Scholar]
- 3.Mihatsch MJ, Gudat F, Zollinger HU, et al. Systemic karyomegaly associated with chronic interstitial nephritis. A new disease entity? Clin Nephrol 1979; 12:54–62. [PubMed] [Google Scholar]
- 4.Moch H, Spöndlin M, Schmassmann A, et al. Systemic karyomegaly with chronic interstitial nephritis. Discussion of the disease picture based on an autopsy case. Pathologe 1994; 15:44–48. [DOI] [PubMed] [Google Scholar]
- 5.Spoendlin M, Moch H, Brunner F, et al. Karyomegalic interstitial nephritis: further support for a distinct entity and evidence for a genetic defect. Am J Kidney Dis 1995; 25:242–252. [DOI] [PubMed] [Google Scholar]
- 6.Godin M, Francois A, Le Roy F, et al. Karyomegalic interstitial nephritis. Am J Kidney Dis 1996; 27:166. [DOI] [PubMed] [Google Scholar]
- 7.Vadiaka M, Sotsiou F, Koufos C. A case of systemic karyomegaly associated with interstitial nephritis. Ann Med Interne 1998; 149:291–294. [PubMed] [Google Scholar]
- 8.Bhandari S, Kalowski S, Collett P, et al. Karyomegalic nephropathy: an uncommon cause of progressive renal failure. Nephrol Dial Transplant 2002; 17:1914–1920. [DOI] [PubMed] [Google Scholar]
- 9.Hassen W, Abid-Essafi S, Achour A, et al. Karyomegaly of tubular kidney cells in human chronic interstitial nephropathy in Tunisia: respective role of Ochratoxin A and possible genetic predisposition. Hum Exp Toxicol 2004; 23:339–346. [DOI] [PubMed] [Google Scholar]
- 10.Rossini M, Coventry S, Duff DR, et al. Chronic renal failure and abnormal tubular cells in 2 siblings. Am J Kidney Dis 2005; 46:982–987. [DOI] [PubMed] [Google Scholar]
- 11.Monga G, Banfi G, Salvadore M, et al. Karyomegalic interstitial nephritis: report of 3 new cases and review of the literature. Clin Nephrol 2006; 65:349–355. [DOI] [PubMed] [Google Scholar]
- 12.Baba F, Nanovic L, Jaffery JB, et al. Karyomegalic tubulointerstitial nephritis—a case report. Pathol Res Pract 2006; 202:555–559. [DOI] [PubMed] [Google Scholar]
- 13.Palmer D, Lallu S, Matheson P, et al. Karyomegalic interstitial nephritis: a pitfall in urine cytology. Diagn Cytopathol 2007; 35:179–182. [DOI] [PubMed] [Google Scholar]
- 14.Verine J, Reade R, Janin A, et al. Karyomegalic interstitial nephritis: a new French case. Ann Pathol 2010; 30:240–242. [DOI] [PubMed] [Google Scholar]
- 15.Uz E, Bayram Y, Haltas H, et al. Karyomegalic tubulointerstitial nephritis: a rare cause of chronic kidney disease. Nephrourol Mon 2011; 3:201–203. [Google Scholar]
- 16.Mölne J. Slide seminar: Karyomegalic tubulointerstitial nephritis: Case 3. European Society of Pathology. Nephropathology working group. 2011. http://www.nephropathology-esp.org/meeting-lecture/slide-seminar-karyomegalic-tubulointerstitial-nephritis Accessed March 2, 2016. [Google Scholar]
- 17.Zhou W, Otto EA, Cluckey A, et al. FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat Genet 2012; 44:910–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lucisano G, Comi N, Cianfrone P, et al. An uncommon presentation of an uncommon nephropathy: the karyomegalic interstitial nephritis. J Nephrol 2013; 26:1188–1191. [DOI] [PubMed] [Google Scholar]
- 19.Tagliente DJ, Voss JS, Peters SG, et al. Systemic karyomegaly with primary pulmonary presentation. Hum Pathol 2014; 45:180–184. [DOI] [PubMed] [Google Scholar]
- 20.Zschiedrich S, Huber TB, Hildebrandt F, et al. Karyomegalic interstitial nephritis. Lancet Lond Engl 2013; 382:2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Radha S, Tameem A, Rao BS. Karyomegalic interstitial nephritis with focal segmental glomerulosclerosis: a rare association. Indian J Nephrol 2014; 24:117–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakhoul F, Ben Itzhaq O, Farber E. Karyomegalic interstitial nephritis with chronic kidney disease. Isr Med Assoc J IMAJ 2015; 17:581–582. [PubMed] [Google Scholar]
- 23.McCulloch T, Prayle A, Lunn A, et al. Karyomegalic-like nephropathy, Ewing's sarcoma and ifosfamide therapy. Pediatr Nephrol Berl Ger 2011; 26:1163–1166. [DOI] [PubMed] [Google Scholar]
- 24.Wang R, Persky NS, Yoo B, et al. DNA repair. Mechanism of DNA interstrand cross-link processing by repair nuclease FAN1. Science 2014; 346:1127–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Auerbach AD. Fanconi anemia and its diagnosis. Mutat Res 2009; 668:4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seguí N, Mina LB, Lázaro C, et al. Germline mutations in FAN1 cause hereditary colorectal cancer by impairing DNA repair. Gastroenterology 2015; 149:563–566. [DOI] [PubMed] [Google Scholar]
- 27.Chaki M, Airik R, Ghosh AK, et al. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell 2012; 150:533–548. [DOI] [PMC free article] [PubMed] [Google Scholar]