

Abstract
There are a number of conflicting reports describing the clinical outcomes of using N-acetylcysteine for the treatment of idiopathic pulmonary fibrosis. We have, therefore, performed a meta-analysis to evaluate the efficacy of N-acetylcysteine, compared with control, for the treatment of idiopathic pulmonary fibrosis.
Original controlled clinical trials evaluating the efficacy of N-acetylcysteine for the treatment of idiopathic pulmonary fibrosis were included in the analysis. Searches for relevant articles were carried out in July 2014 by 2 independent researchers using PubMed, Embase, Cochrane Central, and Google Scholar. Change in forced vital capacity, change in percentage of predicted vital capacity, change in percentage of predicted carbon monoxide diffusing capacity, changes in 6 minutes walking test distance, rate of adverse events, and rate of death were expressed as outcomes using RevMan 5.0.1.
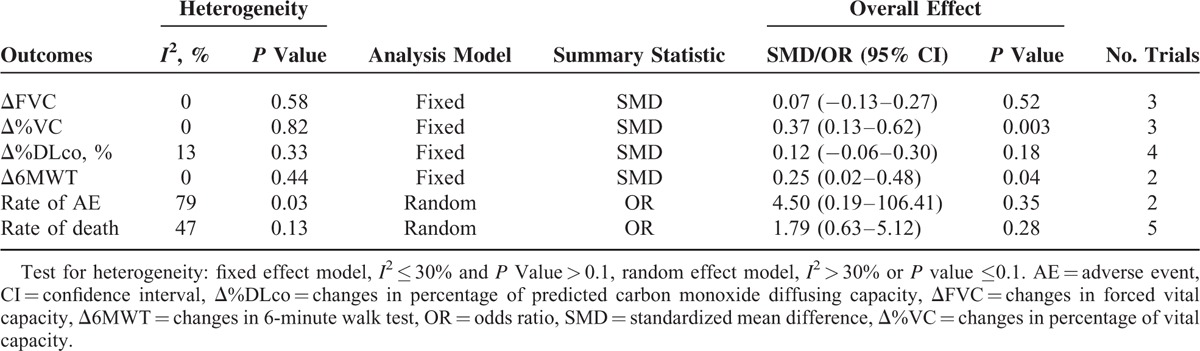
Five trials, with a total of 564 patients, were included in this meta-analysis. The meta-analysis showed that the control group had significant decreases in percentage of predicted vital capacity (standardized mean difference [SMD] = 0.37; 95% confidence interval [CI]: 0.13 to −0.62; P = 0.003) and 6 minutes walking test distance (SMD = 0.25; 95% CI: 0.02–0.48; P = 0.04). There were no statistically significant differences in forced vital capacity (SMD = 0.07; 95% CI: −0.13–0.27; P = 0.52), percentage of predicted carbon monoxide diffusing capacity (SMD = 0.12; 95% CI: −0.06–0.30; P = 0.18), rates of adverse events (odd ratio = 4.50; 95% CI: 0.19–106.41; P = 0.35), or death rates (odd ratio = 1.79; 95% CI: 0.3–5.12; P = 0.28) between the N-acetylcysteine group and the control group.
N-Acetylcysteine was found to have a significant effect only on decreases in percentage of predicted vital capacity and 6 minutes walking test distance. N-acetylcysteine showed no beneficial effect on changes in forced vital capacity, changes in predicted carbon monoxide diffusing capacity, rates of adverse events, or death rates.
INTRODUCTION
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, interstitial lung disease of unknown etiology. Since there are few effective therapies and the mortality rate is high, new treatments for IPF are urgently needed.1–6 Antiinflammatory therapy with corticosteroids or immunosuppressants fails to significantly improve the survival time of patients with IPF.7–10 Other pharmacological interventions, which include nintedanib,11 etanercept, warfarin, gleevec, and bosentan, remain controversial. Pirfenidone was approved by the European Medicines Agency in 2011 for the treatment of IPF.2,12–14
There have been a number of clinical studies to evaluate the antioxidant N-acetylcysteine for the treatment of IPF, but these have produced conflicting results.1–6,15–17 Bando et al1 found no significant differences in survival curves between IPF patients who were treated with N-acetylcysteine and those who received no treatment. The study was, however, an open case–control study in a single institute and the number of cases was small. Demedts et al3 showed that N-acetylcysteine (600 mg 3 times daily), added to standard therapy with prednisone and azathioprine, improved lung function in IPF patients compared with standard therapy alone. Homma et al4 indicated that N-acetylcysteine monotherapy may have beneficial effects in patients with early-stage IPF, and Tomioka et al6 demonstrated that N-acetylcysteine may delay disease progression.5 Significantly, however, the IPF Clinical Research Network found that N-acetylcysteine offered no significant benefit compared with placebo in IPF patients with mild-to-moderate impairment in lung function.
In view of the conflicting evidence, we have undertaken the 1st systematic review and meta-analysis to evaluate the efficacy of N-acetylcysteine for the treatment of patients with IPF.
MATERIALS AND METHODS
Searching Strategy
PubMed, EMBASE, Cochrane Central, and Google Scholar were searched in July 2014 by 2 independent researchers using the free text terms “acetylcysteine,” “N-acetylcysteine,” “NAC,” “idiopathic pulmonary fibrosis,” and “IPF.” All studies that included potentially relevant information about N-acetylcysteine and the treatment of IPF were retrieved.
Inclusion criteria were as follows: studies that compared an N-acetylcysteine-treated group with a control group for the treatment of IPF; studies that reported outcome measures, including changes in pulmonary function tests (change in forced vital capacity [ΔFVC], change in percentage of predicted vital capacity [Δ%VC], and change in percentage of predicted carbon monoxide diffusing capacity [Δ%DLco]), changes in 6 minutes walking test distance (Δ6MWT), rates of adverse events, and rates of death; and articles published in English.
Quality Score
Whether the studies were of sufficiently high quality to be included in the analysis was evaluated by 2 independent researchers (CY and GY) and disagreements were resolved by a 3rd researcher. The quality of each study included in the analysis was assessed using the Cochrane Risk of Bias Tool for systematic reviews of interventions (version 5.0.1)18 and also using the Jadad scale.
Data Extraction
Data were independently extracted by 2 researchers and differences were resolved by discussion with a 3rd researcher. For all eligible articles, the following data were extracted from the original publication: year of publication, number of patients, study design, and outcomes.
Data Analysis
Statistical analysis for dichotomized outcomes was performed using odds ratio (OR) and 95% confidence interval (95% CI). Standardized mean difference (SMD) and 95% CI were used for statistical analysis of continuous variables. Outcomes were calculated using the reported P values; P < 0.05 was considered to be statistically significant. The I2 statistic was applied to estimate heterogeneity. Statistical heterogeneity was present when I2 > 30% or P ≤ 0.1; in this case, a random-effects model was used. A fixed-effect model was used when I2 ≤ 30% and P > 0.1.18 Sensitivity analysis was performed using an exchanging effect model. All statistical analyses were conducted using Review Manager Version 5.0.1.18
RESULTS
Search Identification and Selection
The database searches initially yielded 66 results. Duplicates (26) and animal studies (4) were excluded. Twenty-eight further studies were deemed irrelevant, based on the title or abstract, and were also excluded. Of the remaining 8 reports, 2 described the same clinical study, 1 lacked important outcomes, and 1 lacked a control group, leaving only 5 studies that met the predetermined inclusion criteria (Figure 1). All of the included studies were published in English.
FIGURE 1.
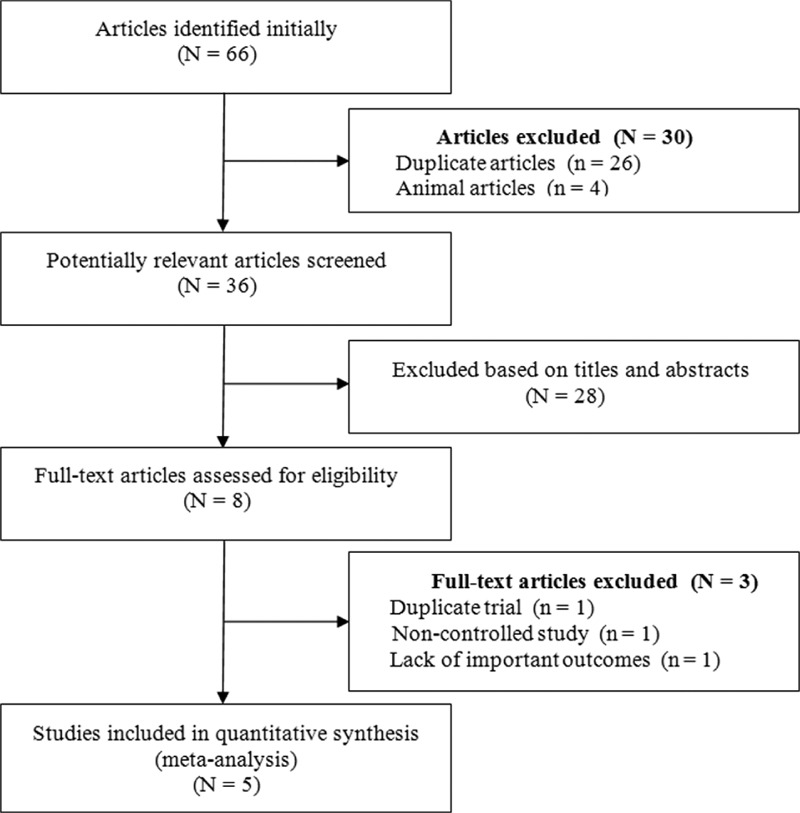
Study selection flow diagram of this meta-analysis.
Study Characteristics and Quality
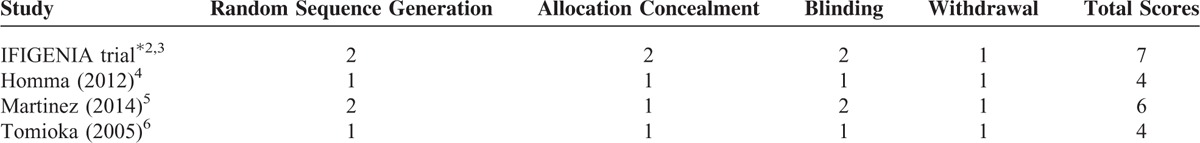
The characteristics of the 5 selected studies are presented in Table 1. The dataset includes 564 patients with a diagnosis of IPF, 286 patients in the N-acetylcysteine group and 278 patients in the control group. The quality scores of each study included in the analysis were assessed using the Cochrane Risk of Bias Tool18 and are described in Table 2. The quality scores attributed to each study using a modified Jadad scale are described in Table 3. The quality scores of the studies ranged from 2 to 7, with a mean of 3.9. A score of 1 to 3 is defined as low quality and a score of 4 to 7 is defined as high quality.
TABLE 1.
Main Characteristics of the Trials Included in the Meta-Analysis
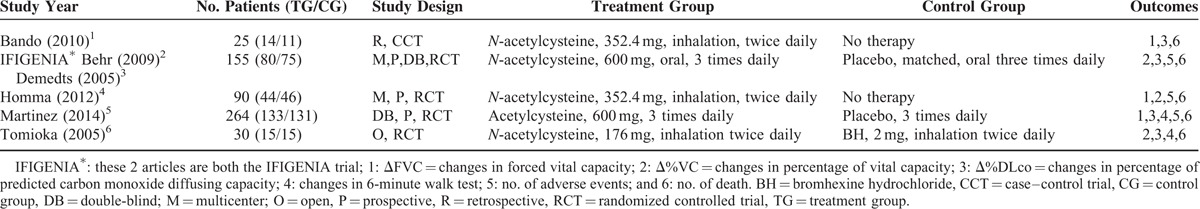
TABLE 2.
Risk of Bias Assessment for the RCTs Included in This Meta-Analysis
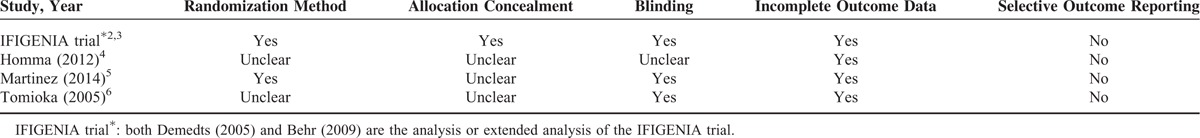
TABLE 3.
Quality Scores by Modified Jaded Scale
Conflicting Evidence
N-acetylcysteine has recently received increased attention as a novel treatment for IPF. A number of studies have evaluated the efficacy and safety of N-acetylcysteine for the treatment of IPF, but with conflicting results.1–6 Two studies found that N-acetylcysteine provided no benefit and did not significantly improve the lung function of patients with IPF.1,6 Demedts et al,3 however, showed that N-acetylcysteine had a beneficial effect on lung function in IPF patients and significantly delayed disease progression. They also found that triple therapy with prednisone, azathioprine, and high-dose N-acetylcysteine (600 mg 3 times daily) was more effective than N-acetylcysteine therapy alone. Remarkably, Tomioka et al6 suggested that although long-term administration of N-acetylcysteine as an aerosol may delay disease progression, it did not improve pulmonary function or quality of life. A study by the IPF Clinical Research Network showed that N-acetylcysteine has some beneficial effects in patients with early-stage IPF.5
The results of all of these trials should, however, be interpreted with caution since the low statistical power limits the interpretability of the findings.
Outcomes
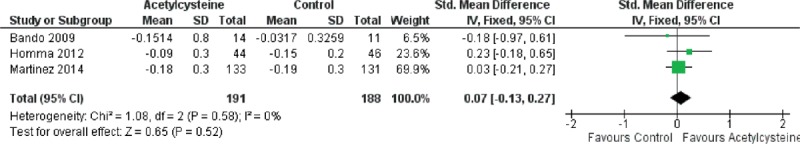
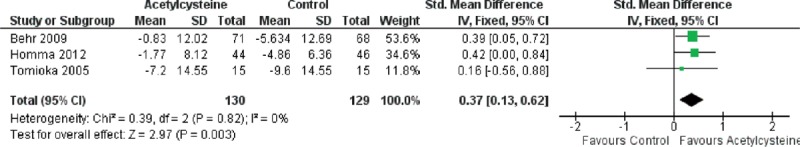
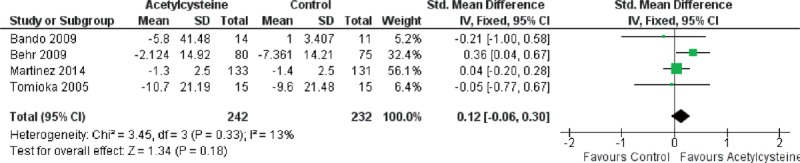
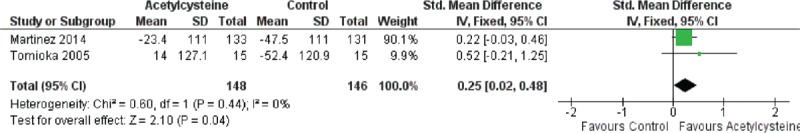
Because of the presence of heterogeneity, we used a random-effects model for the analysis of adverse events and death. We used a fixed-effects model for the analysis of ΔFVC, Δ%VC, Δ%DLco, and Δ6MWT. The decrease in %VC was significantly less in the N-acetylcysteine group than in the control group (SMD = 0.37, 95% CI: 0.13 to −0.62; P = 0.003) (Figure 2). The decline in 6MWT was significantly less in the N-acetylcysteine group (SMD = 0.25, 95% CI: 0.02–0.48; P = 0.04) (Figure 3). There were no statistically significant differences in ΔFVC (SMD = 0.07, 95% CI: −0.13–0.27; P = 0.52) (Figure 4) or Δ%DLco (SMD = 0.12, 95% CI: −0.06–0.30; P = 0.18) (Figure 5) between the treatment and control groups. There were also no statistically significant differences in the occurrence of adverse events (OR = 4.50, 95% CI: 0.19–106.41; P = 0.35) (Figure 6) or death (OR = 1.79, 95% CI: 0.3–5.12; P = 0.28) (Figure 7). The results of the meta-analysis (Table 4) show no difference after sensitivity analysis using an exchanging effect model.
FIGURE 2.
Forest plot evaluating effects of acetylcysteine group on Δ%VC compared with control group. (Fixed model: 130 of acetylcysteine, 129 of control, SMD = 0.37, 95% CI 0.13–0.62, P = 0.003). CI = confidence interval, SMD = standardized mean difference, Δ%VC = changes in percentage of vital capacity.
FIGURE 3.
Forest plot evaluating effects of acetylcysteine group on 6MWT compared with control group. (Fixed model: 148 of acetylcysteine, 146 of control, SMD = 0.25, 95% CI 0.02–0.48, P = 0.04). CI = confidence interval, 6MWT = 6-minute walk test, SMD = standardized mean difference.
FIGURE 4.
Forest plot evaluating effects of acetylcysteine group on ΔFVC compared with control group. (Fixed model: 191 of acetylcysteine, 188 of control, SMD = 0.07, 95% CI −0.13–0.27, P = 0.52). CI = confidence interval, ΔFVC = changes in forced vital capacity, SMD = standardized mean difference.
FIGURE 5.
Forest plot evaluating effects of acetylcysteine group on Δ%DLco compared with control group. (Fixed model: 242 of acetylcysteine, 232 of control, SMD = 0.12, 95% CI −0.06–0.30, P = 0.18). CI = confidence interval, Δ%DLco = changes in percentage of predicted carbon monoxide diffusing capacity, SMD = standardized mean difference.
FIGURE 6.
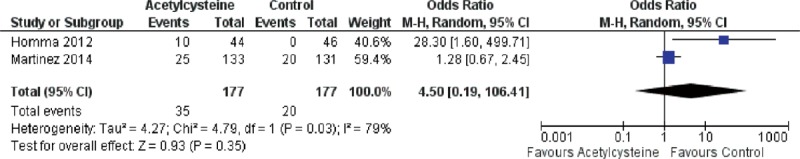
Forest plot evaluating effects of acetylcysteine group on rate of adverse events compared with control group. (Random model: 177 of acetylcysteine, 177 of control, OR = 4.5, 95% CI 0.19–106.41, P = 0.35). CI = confidence interval, OR = odds ratio.
FIGURE 7.
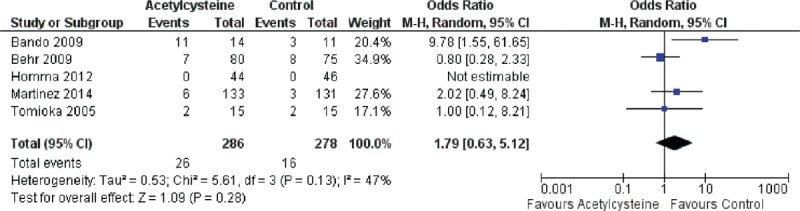
Forest plot evaluating effects of acetylcysteine group on rate of death compared with control group. (Random model: 286 of acetylcysteine, 278 of control, OR = 1.79, 95% CI 0.63–5.12, P = 0.28). CI = confidence interval, OR = odds ratio.
TABLE 4.
Clinical Outcomes of Acetylcysteine Group Compared With Control Group
Publication Bias
Publication bias among the studies was assessed using funnel plots. The small number of studies, together with the low statistical power of these 5 studies, has limited the interpretability of our results and a relatively high publication bias is therefore present in this meta-analysis.
DISCUSSION
The pathogenesis of IPF has been suggested to be linked to abnormal fibroblast response mechanisms.19 In our opinion, however, the etiology of IPF remains ill-defined and there are likely a number of risk factors, including heredity, environment, autoimmunity, viral infections, and gastroesophageal reflux.20 The 1st step in treating patients with IPF is to establish a definite diagnosis; multidisciplinary assessment is recommended to increase the diagnostic accuracy. Several clinical tests, such as high-resolution computed axial tomography, transbronchial biopsy, and histopathological tests, help to increase diagnostic accuracy.
Before treating patients with IPF, a number of factors need to be considered. These include prognostic factors, stage of IPF, complications, and comorbidities. Several therapeutic strategies should be combined. First, and most importantly, the patient should receive treatment for the IPF; second, risk factors that may aggravate IPF should be avoided; and third, attention should be paid to symptomatic and palliative treatment.21 It has been reported that patients with IPF may benefit from nonpharmacological treatments, such as home oxygen therapy, respiratory rehabilitation, lung transplantation, cell and gene therapy, and palliative care.19,20 Pharmacological interventions for IPF are also receiving increased attention.1–6,12,13,15–17,19–25
We have, we believe, carried out the 1st meta-analysis to evaluate the efficacy and safety of N-acetylcysteine for the treatment of patients with IPF. Meta-analysis is an ideal statistical tool for increasing statistical power and is an important component of a systematic review, which provides more powerful evidence for clinical decision-making than a single clinical trial. The present meta-analysis seems to contradict the results of several previous clinical trials that demonstrated that IPF patients benefit from treatment with N-acetylcysteine. In our meta-analysis, treatment with N-acetylcysteine failed to provide benefits in terms of ΔFVC, Δ%DLco, adverse events, or death. ΔFVC and Δ%DLco are considered to be important indices for the evaluation of N-acetylcysteine treatment in patients with IPF. Whether some of the adverse events reported in the studies, including cough, fever, abdominal pain, respiratory failure, edema, increased C-reactive protein, increased blood glucose, dyspnea, and asthenia, are really drug-related “adverse events” or are, in fact, complications and comorbidities of IPF needs clarification in future studies. N-Acetylcysteine treatment was associated with a significantly smaller decrease in %VC and a smaller decline in 6MWT, compared with the control group. Our meta-analysis found no significant difference in adverse events or mortality between patients receiving N-acetylcysteine and the control group. This indicates that N-acetylcysteine is relatively safe for patients with IPF but, because of the small number of studies included, our results need to be validated by further studies.
The mechanism of action of N-acetylcysteine in the therapy of IPF is uncertain. One hypothesis, developed using animal models of pulmonary fibrosis, is that N-acetylcysteine increases the synthesis of glutathione, a potent antioxidant, and decreases the fibrotic response.20 A second hypothesis is that N-acetylcysteine-mediated downregulation of lysyl oxidase activity alleviates bleomycin-induced pulmonary fibrosis in rats.26 A 3rd suggestion27 is that N-acetylcysteine slows progression of IPF by inhibiting epithelial-mesenchymal transition.
There are several potential limitations of this meta-analysis, these include the use of different drug doses, low statistical power, high dropout rates, and significant clinical heterogeneity among the studies. The results of additional high-quality randomized controlled trials (RCTs) are thus needed to add weight to the analysis. We also failed to assess some meaningful end points because necessary data were lacking in the studies included in this meta-analysis. Only 5 trials were included in this meta-analysis, suggesting that a relatively high publication bias may exist. The control groups included no therapy, placebo, and bromhexine hydrochloride treatment, which introduces clinical heterogeneity into the studies included in this meta-analysis. In light of these considerations, additional high-quality RCTs are awaited to verify our findings and to provide the best clinical recommendations. Further studies should focus on a number of important issues, including disease stage, prognostic factors, drug dose, duration of dosing, drug combinations, statistical power of the study, and monitoring complications and comorbidities, as well as the cost of N-acetylcysteine therapy for patients with IPF.
CONCLUSIONS
The limited evidence currently available suggests that N-acetylcysteine has a significant effect only on decreases in VC and 6MWT and fails to significantly reduce changes in FVC, changes in DLco, adverse events, or death. N-acetylcysteine should, therefore, not be recommended for routine treatment of patients with IPF until additional high-quality RCTs have been performed. Our results need to be interpreted with caution because of the heterogeneity and low statistical power of the studies included in this meta-analysis.
Footnotes
Abbreviations: CI = confidence interval, IPF = idiopathic pulmonary fibrosis, OR = odds ratio, RCT = randomized controlled trial, SMD = standardized mean difference.
We are grateful to all the previous study authors and the study participants. TS and DWZ conceived the study; TS, JL, and DWZ collected data and provided data analysis; DWZ submitted the final manuscript; and all authors read and approved the final manuscript, and designed the study and participated in writing the paper.
The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Bando M, Hosono T, Mato N, et al. Long-term efficacy of inhaled N-acetylcysteine in patients with idiopathic pulmonary fibrosis. Intern Med 2010; 49:2289–2296. [DOI] [PubMed] [Google Scholar]
- 2.Behr J, Demedts M, Buhl R, et al. Lung function in idiopathic pulmonary fibrosis-extended analyses of the IFIGENIA trial. Respir Res 2009; 10:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Demedts M, Behr J, Buhl R, et al. High-dose acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med 2005; 353:2229–2242. [DOI] [PubMed] [Google Scholar]
- 4.Homma S, Azuma A, Taniguchi H, et al. Efficacy of inhaled N-acetylcysteine monotherapy in patients with early stage idiopathic pulmonary fibrosis. Respirology 2012; 17:467–477. [DOI] [PubMed] [Google Scholar]
- 5.Martinez FJ, de Andrade JA, Anstrom KJ, et al. Idiopathic Pulmonary Fibrosis Clinical Research Network. Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370:2093–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tomioka H, Kuwata Y, Imanaka K, et al. A pilot study of aerosolized N-acetylcysteine for idiopathic pulmonary fibrosis. Respirology 2005; 10:449–455. [DOI] [PubMed] [Google Scholar]
- 7.Xaubet A, Ancochea J, Blanquer R, et al. Diagnosis and treatment of diffuse interstitial lung diseases. Arch Bronconeumol 2003; 39:580–600. [DOI] [PubMed] [Google Scholar]
- 8.Pinheiro GA, Antao VC, Wood JM, et al. Occupational risks for idiopathic pulmonary fibrosis mortality in the United States. Int J Occup Environ Health 2008; 14:117–123. [DOI] [PubMed] [Google Scholar]
- 9.Taskar VS, Coultas DB. Is idiopathic pulmonary fibrosis an environmental disease? Proc Am Thorac Soc 2006; 3:293–298. [DOI] [PubMed] [Google Scholar]
- 10.Lee JS, Ryu JH, Elicker BM, et al. Gastroesophageal reflux therapy is associated with longer survival in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011; 184:1390–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luppi F, Spagnolo P, Cerri S, et al. The big clinical trials in idiopathic pulmonary fibrosis. Curr Opin Pulm Med 2012; 18:428–432. [DOI] [PubMed] [Google Scholar]
- 12.Taniguchi H, Ebina M, Kondoh Y, et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J 2010; 35:821–829. [DOI] [PubMed] [Google Scholar]
- 13.Jiang C, Huang H, Liu J, et al. Adverse events of pirfenidone for the treatment of pulmonary fibrosis: a meta-analysis of randomized controlled trials. PLoS One 2012; 7:e47024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Behr J, Richeldi L. Recommendations on treatment for IPF. Respir Res 2013; 14 Suppl 1:S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wells AU. Antioxidant therapy in idiopathic pulmonary fibrosis: hope is kindled. Eur Respir J 2006; 27:664–666. [DOI] [PubMed] [Google Scholar]
- 16.Meyer A, Buhl R, Kampf S, et al. Intravenous N-acetylcysteine and lung glutathione of patients with pulmonary fibrosis and normals. Am J Respir Crit Care Med 1995; 152:1055–1060. [DOI] [PubMed] [Google Scholar]
- 17.Meyer A, Buhl R, Magnussen H. The effect of oral N-acetylcysteine on lung glutathione levels in idiopathic pulmonary fibrosis. Eur Respir J 1994; 7:431–436. [DOI] [PubMed] [Google Scholar]
- 18.Higgins JPT, Green S, eds. 2008. Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.1 [updated September 2008]. Chichester, United Kingdom: The Cochrane Collaboration; Available at: http://www.cochrane-handbook.org. [Google Scholar]
- 19.Chan AL, Rafii R, Louie S, et al. Therapeutic update in idiopathic pulmonary fibrosis. Clin Rev Allergy Immunol 2013; 44:65–74. [DOI] [PubMed] [Google Scholar]
- 20.Xaubet A, Ancochea J, Bollo E, et al. Guidelines for the diagnosis and treatment of idiopathic pulmonary fibrosis. Sociedad Española de Neumología y Cirugía Torácica (SEPAR) Research Group on Diffuse Pulmonary Diseases. Arch Bronconeumol 2013; 49:343–353. [DOI] [PubMed] [Google Scholar]
- 21.Richeldi L, Davies HR, Ferrara G, et al. Corticosteroids for idiopathic pulmonary fibrosis. Cochrane Database Syst Rev 2003; CD002880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wells AU, Behr J, Costabel U, et al. Triple therapy in idiopathic pulmonary fibrosis: an alarming press release. Eur Respir J 2012; 9:805–806. [DOI] [PubMed] [Google Scholar]
- 23.McGrath EE, Millar AB. Hot off the breath: triple therapy for idiopathic pulmonary fibrosis – hear the PANTHER roar. Thorax 2012; 67:97–98. [DOI] [PubMed] [Google Scholar]
- 24.Davies HR, Richeldi L. Idiopathic pulmonary fibrosis: current and future treatment options. Am J Respir Med 2002; 1:211–224. [DOI] [PubMed] [Google Scholar]
- 25.Lota HK, Wells AU. The evolving pharmacotherapy of pulmonary fibrosis. Expert Opin Pharmacother 2013; 14:79–89. [DOI] [PubMed] [Google Scholar]
- 26.Li S, Yang X, Li W, et al. N-acetylcysteine downregulation of lysyl oxidase activity alleviating bleomycin-induced pulmonary fibrosis in rats. Respiration 2012; 84:509–517. [DOI] [PubMed] [Google Scholar]
- 27.Wolters PJ. Could N-acetylcysteine slow progression of idiopathic pulmonary fibrosis by inhibiting EMT? Am J Physiol Lung Cell Mol Physiol 2009; 297:L803–L804. [DOI] [PubMed] [Google Scholar]