

Abstract
Primary Sjögren syndrome (SS) is an autoimmune disease mainly affecting the exocrine glands causing a sicca syndrome. Neurological manifestations are rarely seen in SS although they are debilitating. Peripheral neuropathies namely sensory axonal neuropathy and painful small fiber neuropathy are the most frequent neurological manifestations. Sensory neuronopathy (SN) is less frequently seen although leading to more severe handicap.
The aim of the study was to analyze the clinical presentation and treatment efficacy in a series of SS-related SN.
We retrospectively studied patients with SS fulfilling the American–European Classification Criteria and SN according to recent criteria. Studied variables were neurological findings, associated autoimmune diseases, biological profiles, nerve conduction and sensory/motor amplitudes study, treatments received, and outcomes. Handicap scores were studied at beginning and end of each treatment using the modified Rankin Scale (mRS).
Thirteen patients were included (12 women, 1 man; median age 55 years at SN diagnosis) presenting with SN with a median follow-up of 3 years (range 2–17). In 11 patients, SN preceded or coincided with SS diagnosis. Most common neurological findings were ataxia and areflexia followed by paresthesia and pain. Lower limbs were more affected than upper limbs, neurological deficits were often symmetric and cranial nerves were affected in 3 patients. Seven patients were treated with corticosteroids, 7 with mycophenolate mofetil, 6 with hydroxychloroquine, 5 with intravenous immunoglobulins, 4 with cyclophosphamide, and 2 patients received other immunosuppressive drugs. At the beginning and at the end of follow-up, average mRS was 2.15 (median 2) and 2.38 (median 2), respectively.
SS-related SN progression is heterogeneous but tends to be chronic, insidious, and debilitating despite treatment. From these data concerning a small number of patients, treatment strategies with corticosteroids in association with immunosuppressive drugs, namely mycophenolate mofetil, had positive results. In contrast, intravenous immunoglobulins had disappointing results.
INTRODUCTION
Sensory neuronopathies (SN) or ganglionopathies are pure sensory neuropathies caused by dorsal root ganglia neuronal destruction.1 This process results in a multifocal pattern of sensory deficits, contrasting with the usual length-dependent pattern found in axonal neuropathies. SN is rare when compared to other peripheral neuropathies, but its true prevalence is not known. Several pathogenic mechanisms have been proposed, such as genetic susceptibility, drug-related toxicity, infectious agents, and autoimmune damage. Acquired SN may be secondary to paraneoplastic syndromes (often associated with anti-Hu antibody positivity), inflammatory/autoimmune disease, neurotropic viral infections, B12 insufficiency, chemotherapy agents, and pyridoxine toxicity.1 Among autoimmune diseases, primary Sjögren syndrome (SS) is frequently involved, as well as celiac disease.1–4
SN is characterized by paresthesia, ataxia, difficulty with fine motor movements due to impaired proprioception, reduced vibration sense, and reduced or absent reflexes and Romberg sign. Muscular strength is usually preserved. Autonomic dysfunction may also be found.5–7 Electrophysiological studies reveal a widespread reduction of sensory potential amplitudes, without a distal worsening gradient toward the legs. Asymmetric responses may be observed.3,8 Most of the time, motor nerve conduction studies and distal motor amplitudes are normal.8 Somatosensory evoked potentials may reveal abnormal central conduction times, which are probably due to the degeneration of dorsal root columns in the spinal cord. Magnetic resonance imaging (MRI) has been used as a sensitive technique especially in those patients with long disease duration, showing a hyperintense T2-weighted lesion at posterior columns and volumetric reduction in cervical area resulting from dorsal root degeneration of their projections in the gracile and cuneate fasciculi.9 Although excisional biopsy of dorsal root ganglion with histological analysis is the gold standard for diagnosis of SN, it is rarely performed due to the possible side effects. Sural nerve biopsy usually shows a massive axonal loss and it is not helpful in the diagnosis.10
Primary SS is an autoimmune disease affecting about 1% of the population and more frequently seen in women.11 It is a systemic disorder characterized by sicca symptomatology of mucosal surfaces. Xerophthalmy and xerostomy are the most frequent symptoms although pulmonary and neurological involvement may also occur. Biologically, patients typically present with hypergammaglobulinemia and positive antinuclear antibodies (ANA) of which anti-SSA and anti-SSB are more specific. Histological main characteristic is a focal lymphocytic infiltration of exocrine glands. Neurological involvement in SS is rare and affects the central and peripheral nervous system. Some series have reported a prevalence of peripheral neuropathy in >50% of patients with SS. The peripheral nervous system involvement occurs in several forms.12–15 In a series of 92 patients with SS-related neuropathies, 39% had SN, 20% small fiber neuropathy, 16% trigeminal neuropathy, 12% multiple mononeuropathies, 5% had multiple cranial neuropathies, 4% had polyradiculoneuropathies, and 3% had autonomic neuropathies.14 Some authors estimate that among all SS patients 5% have SN and 5% to 10% have a small fiber neuropathy.16 SN is probably less frequent than painful axonal neuropathy. Although less frequent than other forms of peripheral neuropathies, SN causes greater handicap.
Most of the literature on SS and neuropathies focus on axonal neuropathy so that there is limited data on long-term outcome and therapeutic response for SS-related SN. Due to the rarity of this affection, there are no controlled randomized trials and conclusions are based on individual observations or small series. In the literature, there are some papers about the efficacy of immunosuppressive therapy, plasma exchange,17 intravenous immunoglobulins,18,19 anti-CD20 therapies,20 and anti-TNF drugs21 with controversial results.
The aim of this study was to review clinical presentation of SS-associated SN as well as treatment efficacy and long-term outcome.
METHODS
The study was completed retrospectively between 1995 and 2013.
We searched through the database of the Internal Medicine Department and the Neurophysiology Department of the Pitié-Salpêtrière Hospital for the terms “ganglionopathy,” “sensory neuronopathy,” and “Sjögren syndrome.”
Inclusion criteria were
Primary SS as defined by the American–European Consensus Group22 (see Table 1).
Probable SN defined by Camdessanché criteria23 (see Table 2).
TABLE 1.
American–European Consensus Group Revised International Classification Criteria for Sjögren Syndrome22

TABLE 2.
Camdessanché Criteria for the Diagnosis of Sensory Neuronopathy

Exclusion criteria were
Insufficient data
Past history of cisplatine use
Active neoplasm
Celiac disease
B12 insufficiency.
Data were extracted from medical records. The following variables were studied: patients’ signs and symptoms, neurological examinations, associated autoimmune diseases, biological profiles (antinuclear antibodies [ANA], precipitating antibodies to extractable nuclear antigens Ro/SSA and La/SSB, rheumatoid factor, complement factors, immunoglobulins, monoclonal immunoglobulin component, and cryoglobulinemia). As we collected the data retrospectively and anonymously, this study was not considered as a biomedical research.
Electrophysiological studies were noted, including motor and sensory amplitudes and nerve conduction velocities, at the beginning and end of each treatment.
All treatments given were noted. Handicap was evaluated using modified Rankin Scale (mRS) at the beginning and the end of each treatment. Side effects of the treatments were also recorded.
Treatment response was classified as improvement, stability, or degradation for each treatment given for at least 6 months. Improvement was defined by a decrease of 1 or more points in the mRS or increase of 5 microVolts in at least 2 sensory nerve amplitudes. Worsening was defined by an increase of 1 or more points in the MRS or decrease of 5 microvolts in at least 2 sensory nerve amplitudes. Patients presenting other outcomes were classified as presenting disease stability. A positive treatment response was defined as improvement or stability.
RESULTS
Patients’ Characteristics
Thirteen patients were included, 12 female and 1 male. Median age at onset of SS was 55 years old (range 20–72). Eleven patients (85%) had xerostomy, 9 (69%) had a positive sugar test, and 9 (69%) patients had a Chisholm grade 3 or 4 on accessory salivary gland biopsy. Ten patients had xerophthalmy (77%) and 9 (69%) had a Schirmer test < 5 mm (69%). Three patients had articular signs (23%) and Raynaud phenomenon was present in 2 patients (15%). Two patients had pulmonary involvement with nonspecific interstitial pneumonitis (NSIP). Three patients had associated autoimmune thyroiditis (23%).
The most frequent biological feature was the presence of positive ANA in 9 patients (69%), followed by a positive anti-SSA in 5 patients (38%) and hypergammaglobulinemia in 3 patients (23%). Other disease characteristics presented by these patients are displayed in Table 3.
TABLE 3.
Patients Characteristics

Clinical and Electrophysiologial Presentation of SN
Median age at the onset of SN was 55 years (range 24–69). In 4 patients, neurological symptoms of SN occurred before SS diagnosis (with a median delay of 3 years, range 1–10 years). On the other side, 2 patients were diagnosed with SN after SS diagnosis, respectively, 4 and 9 years later. In 7 patients, both entities were diagnosed simultaneously.
Most common neurological findings included upper limb and gait ataxia (n=9 [69%] and n = 5 [38%], respectively), paresthesia (n = 7 [54%]), upper or lower members areflexia (n = 7 [54%] and 4 [31%], respectively) and superior and inferior members pain (n = 4 [31%] and 3 [23%], respectively). In 10 (77%) patients these signs affected both sides in a symmetrical pattern. Ataxia and areflexia were more common in the lower limbs. Other neurological manifestations included cranial nerves (III or V) involvement in 3 patients (23%). No patient presented sphincterian signs or autonomic dysfunction. Median mRS at SN diagnosis was 2.15 (range 1–5). Detailed neurological findings are summarized in Table 4.
TABLE 4.
Neurological Findings

Treatments and Follow-Up
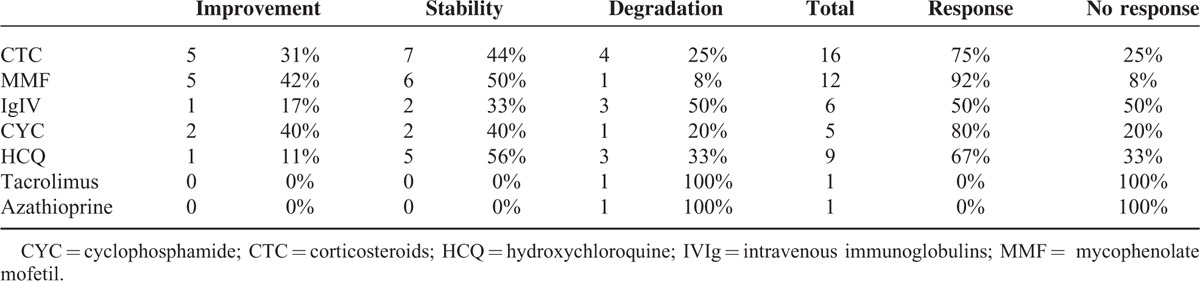
Median follow-up period of the patients was 3 years (range 2–17 years). Two patients did not receive specific treatment for SN or SS because they presented minor and not incapacitating symptoms. The 11 remaining patients received several treatments, which are detailed in Table 5. Seven patients were treated with corticosteroids (54%), 7 with MMF (54%), 6 with hydroxychloroquine (HCQ) (46%), 5 with intravenous immunoglobulins (IVIg) (38%), and 4 with cyclophosphamide (CYC) (31%). Two patients received other immunosuppressive drugs: tacrolimus (n = 1) and azathioprine (AZA) (n = 1). Among the patients who were treated, 7 patients experienced >1 treatment (54%). Two patients were under monotherapy with neuronopathy stability. Table 5 depicts patients’ treatments, their responses to treatments, and the SN course. Further details on treatment responses are depicted in Table 6.
TABLE 5.
Treatments and Follow-Up of Sensory Neuropathy in Patients With SS
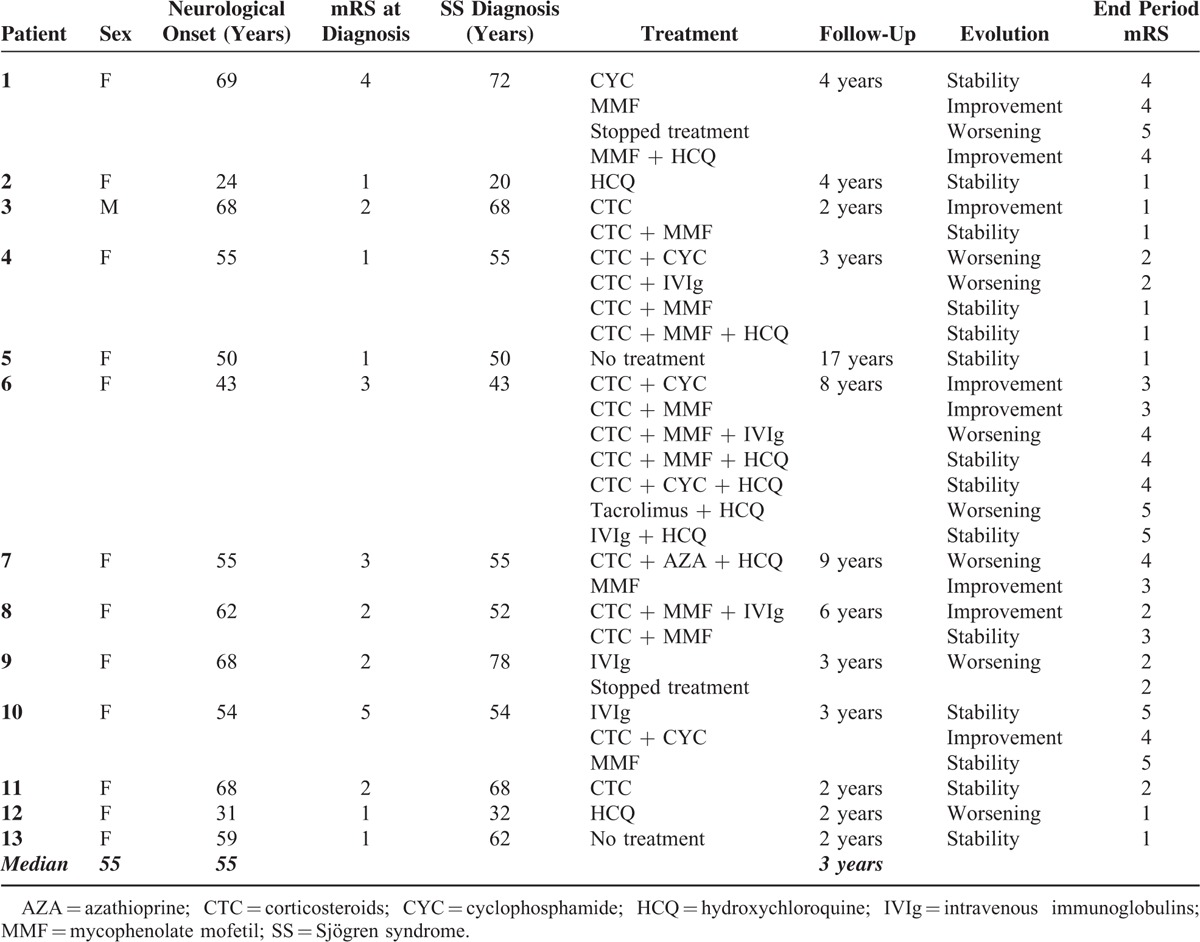
TABLE 6.
Treatment Response
At the end of follow-up, median mRS score was 2.38 (range: 1–5).
Treatments were well tolerated except for 1 patient who presented a pulmonary infection during MMF therapy.
DISCUSSION
SN is a rare form of peripheral neurological involvement in SS patients with no validated treatment strategy. Evolution and response to treatment remain unclear due to the rarity of long-term studies.
Patients Characteristics and SN Onset
As in other series, our patients with SS-related SN were mostly females (12/13).2,6,15 Median age of onset of SN was 55 years, similar to other series.2,6,15 The diagnosis of SN preceded that of SS in 4 patients (31%) and was concomitant to SS diagnosis in 7 patients (54%). Previous series reported that about half of the patients were diagnosed with SN before SS diagnosis. In our series, this proportion was only of 1/3 of the patients.6 On the other side, in our series SN diagnosis preceded or was made at the same time than SS diagnosis in 11/13, whereas some series describe a 2/3 ratio.6 Nevertheless, clinicians should consider SS when studying patients presenting with SN. In accordance to other series, our patients presenting with SN had a high prevalence of xerostomy (85%) and xerophtalmy (77%) and high Chisholm grade in accessory salivary glands biopsy (8 patients had Chisholm grade 3). Three patients had associated autoimmune thyroiditis, which is closed to the proportion of SS who have thyroiditis, even without SN.6
Biological Characteristics
Nine patients had positive AAN (69%) or anti-SSA (38%). The role of these antibodies in SS-related SN is unknown.2,6 In immune-mediated SN, available data support the concept of direct inflammatory damage to dorsal root ganglia neurons mediated by CD8 T lymphocytes.24,25 Humoral dysfunction seems to play a minor role in most forms of SN, but anti-GD1b antibodies were associated to sensory neuronopathy in animal-based models.26 None of our patients had antiGD1b antibodies.
SN Presentation and Diagnosis
SN is generally distinguishable from other forms of peripheral neuropathy in SS because it tends to be asymmetrical and to predominate in the upper limbs. In our series, patients presented with symmetrical neurological findings. Furthermore, lower limbs were more frequently affected. These findings contradict previous descriptions in other series.2,6,12 Cranial nerves were affected in 3/13 patients, the same as in other studies that describe a percentage of ∼30%.2,6,12 No patient presented with sphincterian signs or autonomic dysfunction.
Treatments and Follow-Up
Our patients were followed for a median time of 3 years (range 2–17). The overall picture of SS-related SN is a debilitating condition with an insidious chronic progression. Outcomes even after treatment were generally not good with a higher degree of handicap at the end of follow-up (2.15 vs 2.38). In our series, patients’ outcome and disease course were not homogeneous. Two patients did not receive any specific treatment and they remained clinically stable, one of them over a period of 17 years. On the other hand, for patients #4 and #6, handicap worsened despite several lines of treatment. Corticosteroids and HCQ were the most frequently used drugs. Corticosteroids were usually initiated with intravenous methylprednisolone pulses (1 g × 3) followed by 1 mg/kg oral prednisone with progressive reduction over time. In fact, corticosteroids are the most studied drugs in SS-related SN but with various outcome.6 In this series, as in previous, another immunosuppressive drug was often added. MMF was the most used immunosuppressive drug. MMF dose was usually 2 g a day. Interestingly, 2 patients experienced improvement under MMF alone. Except for one, all other patients under MMF alone or in association presented improvement or stability of their disease. We may hypothesize that these positive results obtained with MMF may be explained by the fact that SN pathology is caused by cellular immune-mediated damage to dorsal ganglia root neurons.27 In our patients, intravenous cyclophosphamide was used either alone or in association in with corticosteroids. Cyclophosphamide was usually used as the first immunosuppressive drug and then switched to MMF. Maximum doses of cyclophosphamide were 6 g. Two patients improved after cyclophosphamide, one remained stable and the other worsened despite treatment. The patient who was stable experienced improvement when he started MMF. In the literature, there are almost no data on the efficacy of MMF in SN.28
Two patients received other immunosuppressive drug namely azathioprine and tacrolimus but with disappointing results. Five patients were treated with intravenous immunoglobulins but with unsatisfactory results: 3 patients worsened despite treatment, 1 remained stable, and 1 improved but in association with other immunosuppressive drug. In the literature, there are some studies indicating the benefit role of intravenous immunoglobulins in the treatment of SS-related neuropathies mainly in axonal polyneuropathies.18,29 In our experience, intravenous immunoglobulins in SS-related SN are not useful.
All these data are limited by the retrospective nature of our study and the fact that the treatment-free outcome of SN is not well known.
CONCLUSIONS
SN is a rare type of peripheral neuropathy, which can occur among SS patients. It usually begins before SS diagnosis is made. Its progression is somewhat heterogeneous but tends to be chronic, insidious, and debilitating despite treatment. From the data of our series, treatment strategies with corticoids in association with immunosuppressive drugs, namely MMF, have positive results. In contrast, intravenous immunoglobulins have disappointing results. Randomized trials should be needed, but they may be difficult to perform due to the rarity of this entity.
Footnotes
Abbreviations: ANA = antinuclear antibodies, AZA = azathioprine, CYC = cyclophosphamide, HCQ = hydroxychloroquin, IVIg = intravenous immunoglobulins, MMF = mycophenolate mofetil, mRS = modified Rankin Scale, SN = sensory neuronopathy, SS = Sjogren syndrome, TNF = tumor necrosis factor.
ZA and FCA contributed equally to this study.
The authors have no funding and conflicts of interest to disclose.
REFERENCES
- 1.Sheikh S, Amato A. The dorsal root ganglion under attack: the acquired sensory ganglionopathies. Pract Neurol 2010; 10:326–334. [DOI] [PubMed] [Google Scholar]
- 2.Griffin JW, Cornblath DR, Alexander E, et al. Ataxic sensory neuropathy and dorsal root ganglionitis associated with Sjögren's syndrome. Ann Neurol 1990; 27:304–315. [DOI] [PubMed] [Google Scholar]
- 3.Martinez AR, Nunes MB, Nucci A, et al. Sensory neuronopathy and autoimmune diseases. Autoimmune Dis 2012; 2012:873587.doi: 0.1155/2012/873587. Epub 2012 Jan 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berkowitz A, Samuels M. The neurology of Sjogren's syndrome and the rheumatology of peripheral neuropathy and myelitis. Pract Neurol 2014; 14:14–22.doi: 10.1136/practneurol-2013-000651. Epub 2013 Dec 4. [DOI] [PubMed] [Google Scholar]
- 5.Font J, Valls J, Cervera R, et al. Pure sensory neuropathy in patients with primary Sjögren's syndrome: clinical, immunological and electromyographic findings. Ann Rheum Dis 1990; 49:779–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Font J, Ramos-Casals M, de la Red G, et al. Pure sensory neuropathy in primary Sjogren's syndrome. Longterm prospective followup and review of the literature. J Rheumatol 2003; 30:1552–1557. [PubMed] [Google Scholar]
- 7.Kuntzer T, Antoine JC, Steck AJ. Clinical features and pathophysiological basis of sensory neuronopathies (ganglionopathies). Muscle Nerve 2004; 30:255–268. [DOI] [PubMed] [Google Scholar]
- 8.Lauria G, Pareyson D, Sghirlanzoni A. Neurophysiological diagnosis of acquired sensory ganglionopathies. Eur Neurol 2003; 50:146–152. [DOI] [PubMed] [Google Scholar]
- 9.França MC, Jr, D’Abreu A, Zanardi VA, et al. MRI shows dorsal lesions and spinal cord atrophy in chronic sensory neuronopathies. J Neuroimaging 2008; 18:168–172. [DOI] [PubMed] [Google Scholar]
- 10.Lauia G, Shirlanzoni A, Lombardi R, et al. Epidermal nerve fiber density in sensory ganglionopathies: clinical and neurophysiological correlations. Muscle Nerve 2001; 24:1034–1039. [DOI] [PubMed] [Google Scholar]
- 11.Fox RI. Sjögren's syndrome. Lancet 2005; 366:321–331. [DOI] [PubMed] [Google Scholar]
- 12.Ramos-Casals M, Solans R, Rosas J, et al. Primary Sjögren syndrome in Spain: clinical and immunologic expression in 1010 patients. Medicine 2008; 87:210–219. [DOI] [PubMed] [Google Scholar]
- 13.Lafitte Manifestations neurologiques du syndrome de Gougerot–Sjögren primitif. Rev Neurol (Paris) 1998; 154:658–673. [PubMed] [Google Scholar]
- 14.Mori K, Iijima M, Koike H, et al. The wide spectrum of clinical manifestations in Sjögren's syndrome-associated neuropathy. Brain 2005; 128:2518–2534. [DOI] [PubMed] [Google Scholar]
- 15.Brito-Zerón P, Akasbi M, Bosch X, et al. Classification and characterization of peripheral neuropathies in 102 patients with primary Sjögren's syndrome. Clin Exp Rheumatol 2013; 31:103–110. [PubMed] [Google Scholar]
- 16.Birnbaum J. Peripheral nervous system manifestations of Sjögren syndrome, clinical patterns, diagnostic paradigms, etiopathogenesis, and therapeutic strategies. Neruologist 2010; 16:287–297. [DOI] [PubMed] [Google Scholar]
- 17.Chen WH, Yeh JH, Chiu HC. Plasmapheresis in the treatment of ataxic sensory neuropathy associated with Sjögren's syndrome. Eur Neurol 2001; 45:270–274. [DOI] [PubMed] [Google Scholar]
- 18.Takahashi Y, Takata T, Hoshino M, et al. Benefit of IVIG for long-standing ataxic sensory neuronopathy with Sjogren's syndrome. Neurology 2003; 60:503–505. [DOI] [PubMed] [Google Scholar]
- 19.Rist S, Sellam, Hachulla E, et al. Experience of intravenous immunoglobulin therapy in neuropathy associated with primary Sjogren's syndrome: a national multicentric retrospective study. Arthritis Care Res 2011; 63:1339–1344. [DOI] [PubMed] [Google Scholar]
- 20.Gorson KC, Natarajan N, Ropper AH, et al. Rituximab treatment in patients with IVIg-dependent immune polyneuropathy: a prospective pilot trial. Muscle Nerve 2007; 35:66–69. [DOI] [PubMed] [Google Scholar]
- 21.Caroyer JM, Manto MU, Steinfeld SD. Severe sensory neuronopathy responsive to infliximab in primary Sjögren's syndrome. Neurology 2002; 59:1113–1114. [DOI] [PubMed] [Google Scholar]
- 22.Vitali C, Bombardieri S, Jonsson R, et al. Classification criteria for Sjögren's syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Ann Rheum Dis 2002; 61:554–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Camdessanché JP, Jousserand G, Ferraud K, et al. The pattern and diagnostic criteria of sensory neuronopathy: a case-control study. Brain 2009; 132:1723–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Colli BO, Carlotti CG, Jr, Assirati JA, et al. Dorsal root ganglionectomy for the diagnosis of sensory neuropathies. Surgical technique and results. Surg Neurol 2008; 69:266–273. [DOI] [PubMed] [Google Scholar]
- 25.Sène D, Jallouli M, Lefaicheur JP, et al. Peripheral neuropathies associated with primary Sjögren syndrome: immunologic profiles of nonataxic sensory neuropathy and sensorimotor neuropathy. Medicine (Baltimore) 2011; 90:133–138. [DOI] [PubMed] [Google Scholar]
- 26.Kusunoki S, Hitoshi S, Kaida KI, et al. Monospecific anti-GD1b IgG is required to induce rabbit ataxic neuropathy”. Ann Neurol 1999; 157:167–170. [PubMed] [Google Scholar]
- 27.Allison AC. Mechanisms of action of mycophenolate mofetil. Lupus 2005; 14 Suppl 1:s2–8. [DOI] [PubMed] [Google Scholar]
- 28.Alix JJ, Hadjivassiliou M, Ali RSl, et al. Sensory ganglionopathy with livedoid vasculopathy controlled by immunotherapy. Muscle Nerve 2015; 51:296–301. [DOI] [PubMed] [Google Scholar]
- 29.Rist S, Sellam J, Hachulla E, et al. On the behalf the club rhumatismes et inflammation: Experience of intravenous immunoglobulins therapy in neuropathy associated with primary Sjögren's syndrome: a multicentric retrospective study. Arthritis Care Res 2011; 63:1339–1344. [DOI] [PubMed] [Google Scholar]