

A genome-wide protein-protein interaction network is computed by combining three-dimensional structure with functional information in Arabidopsis.
Abstract
Protein-protein interactions (PPIs) are essential to almost all cellular processes. To better understand the relationships of proteins in Arabidopsis (Arabidopsis thaliana), we have developed a genome-wide protein interaction network (AraPPINet) that is inferred from both three-dimensional structures and functional evidence and that encompasses 316,747 high-confidence interactions among 12,574 proteins. AraPPINet exhibited high predictive power for discovering protein interactions at a 50% true positive rate and for discriminating positive interactions from similar protein pairs at a 70% true positive rate. Experimental evaluation of a set of predicted PPIs demonstrated the ability of AraPPINet to identify novel protein interactions involved in a specific process at an approximately 100-fold greater accuracy than random protein-protein pairs in a test case of abscisic acid (ABA) signaling. Genetic analysis of an experimentally validated, predicted interaction between ARR1 and PYL1 uncovered cross talk between ABA and cytokinin signaling in the control of root growth. Therefore, we demonstrate the power of AraPPINet (http://netbio.sjtu.edu.cn/arappinet/) as a resource for discovering gene function in converging signaling pathways and complex traits in plants.
Protein-protein interactions (PPIs) are essential to almost all cellular processes, including DNA replication and transcription, signal transduction, and metabolic cycles. Because proteins function through coordinated interaction, it is important to characterize the nature of these relationships. Deciphering the nature of PPIs not only advances our understanding of gene function at the molecular level but also provides insight into complex cellular processes.
The importance of understanding PPIs has prompted the development of various computational approaches, such as gene fusion and Rosetta stone (Marcotte et al., 1999), phylogenetic profiling (Pellegrini et al., 1999), correlated expression of gene pairs (Grigoriev, 2001), gene neighbor (Overbeek et al., 1999), interolog (Matthews et al., 2001), and combining multiple sources of biological data (Rhodes et al., 2005), for uncovering protein interactions over the past decade. Recently, computational methods using structural information for PPI prediction have gained much attention due to the rapid growth of the Protein Data Bank (PDB; Rose et al., 2013). Protein docking is a promising method for discovering protein interactions that is based on three-dimensional structural information, but it remains a challenging and computationally demanding task to predict PPIs on a genome-wide scale (Wass et al., 2011). Alternatively, knowledge-based methods that utilize the structural similarity of protein pairs to interface a known protein complex have been used in PPI prediction (Aytuna et al., 2005; Zhang et al., 2012; Mosca et al., 2013). The common strategy of these structure-based methods is to find a suitable template complex for the two query protein structures; the prediction is then based on the structural similarity of the two protein models to the template complex. An important advantage of structure-based approaches is their ability to identify the putative interface; that is, they are capable of providing more information than any non-structure-based method.
Arabidopsis (Arabidopsis thaliana), a small flowering plant, is widely used as a model organism in plant biology. In recent years, several large-scale experimental PPI studies have begun to unravel the complex cellular networks present in Arabidopsis (Arabidopsis Interactome Mapping Consortium, 2011; Jones et al., 2014). According to an empirical estimation of the size of the protein interactome, experimentally determined interactions represent a small fraction (approximately 6%) of the entire protein interactome, and the relationships among most proteins remain to be discovered (Arabidopsis Interactome Mapping Consortium, 2011). To complement existing experimental resources, genome-wide plant PPI networks have been developed by computational approaches (Cui et al., 2008; Lee et al., 2010; Lin et al., 2011; Wang et al., 2012, 2014). However, these methods attempt to predict PPIs using only nonstructural information.
Here, we developed a computational approach combining three-dimensional structure with functional information to construct genome-wide PPI networks in Arabidopsis. Comparison of the predicted interactions with experimentally validated data revealed its power for discovering protein interactions and for discriminating positive interactions from similar protein pairs. Experimental evaluation of the predicted PPIs demonstrated the ability of AraPPINet to discover novel protein interactions involved in specific processes at 100-fold greater accuracy than screens of random protein-protein pairs for the test case of abscisic acid (ABA) signaling. Using AraPPINet guilt-by-association and genetic analysis, we identified and validated a regulator, ARR1, involved in cross talk in ABA signaling mediated by PYL1. Our findings suggest that AraPPINet not only could advance our understanding of gene function but also could provide insight into the molecular basis of the complex interplay between signaling pathways in plants.
RESULTS
Construction of PPI Networks in Arabidopsis
We integrated three-dimensional structure and functional information using a machine-learning method for genome-wide PPI prediction in Arabidopsis. The PPI model contained four structural and seven nonstructural features. The structural features included structural similarity, structural distance, preserved interface size, and fraction of the preserved interface that had been derived from structural alignments of protein structure homology models to template complexes. Given a PPI model with multiple values for a structural feature, the data with the highest structural similarity were assigned to this interaction model. This approach resulted in approximately 40 million protein interaction models containing structural information extracted from approximately 5.3 billion structural alignments (Supplemental Table S1). The seven nonstructural features of the interaction model were three gene ontologies, gene coexpression, interolog, Rosetta stone protein, and phylogenetic profile similarity, of which the proportion ranged from 0.2% to 100% of expectations from all possible protein pairs (Supplemental Table S1). Approximately 343 million protein pairs with available data for these 11 features were then classified using a random forests algorithm for PPI predictions, resulting in an Arabidopsis PPI network (AraPPINet) that contained 316,747 high-confidence interacting pairs among 12,574 (48%) of the 26,207 Arabidopsis proteins (Supplemental Fig. S1). Almost one-third of the interacting protein pairs predicted by AraPPINet were supported by structural evidence (Supplemental Fig. S2). AraPPINet exhibited small-world properties similar to those of other biological networks (Supplemental Fig. S3A), and high-clustering coefficient values indicated that the topological structure of AraPPINet was highly modular (Supplemental Fig. S3B). Network analysis revealed that AraPPINet was composed of 1,005 functional modules clustered by a Markov clustering algorithm with an inflation parameter of 1.8 (Van Dongen, 2008), in which the largest module was preferentially associated with transcriptional regulation (Supplemental Fig. S1).
Evaluating the Performance of AraPPINet
To compare the accuracy of this combined approach with approaches utilizing either structural or nonstructural information, a 10-fold cross-validation was carried out using the reference data sets. This evaluation showed that the true positive rate (TPR) of this method reached 49.8% (Supplemental Fig. S4A), while the false positive rate (FPR) remained at the low level of 0.09% (Supplemental Fig. S4B). Furthermore, receiver operating characteristic (ROC) curves showed that the combined method had a higher area under the curve (AUC) value than methods relying on only structural or nonstructural information (Fig. 1A). This approach was comparable in performance to other methods over the entire range of the FPR values (Supplemental Fig. S5), which tended to produce fewer false positives. This result suggested that the combined approach was more efficient than others at separating positives from a large number of negatives at the genome-wide scale.
Figure 1.

Evaluation of AraPPINet performance. A, ROC curves for PPIs inferred from different data sources. AUC values are reported in parentheses. B, Venn diagram of predicted overlapping PPIs with experimentally determined interactions. C, Comparison of AraPPINet with other methods based on the high-throughput PPI data set. D, Comparison of AraPPINet with other methods based on newly reported interactions. The random PPI networks are generated by randomly rewiring edges while preserving the original degree distribution of AraPPINet. The average TPR is calculated from 10 randomized networks.
Although cross-validation indicated that the performance of the combined method was superior to those of other PPI predictive models reliant on only structural or nonstructural information, these results could not be applied to estimate the combined model’s ability to discover novel PPIs in practice. Thus, the performance of AraPPINet was tested on an independent data set of protein interactions determined by high-throughput experiments from public databases. After discarding 190 positive reference interactions, a total of 11,669 protein interactions remained as the independent data set for the performance evaluation. Among these interactions, 1,401 PPIs were successfully predicted by this method (Fig. 1B). The evaluation indicated that our predictive method was powerful in novel PPI discovery at the genome-wide scale.
Next, we used two independent data sets to evaluate the performance of AraPPINet compared with that of three other publicly available PPI prediction methods: AtPID (Cui et al., 2008), AtPIN (Brandão et al., 2009), and PAIR (Lin et al., 2011). Among the 11,669 protein interactions from high-throughput experiments, approximately 12% of protein interactions could be successfully recognized by AraPPINet, which significantly outperformed the AtPID, AtPIN, and PAIR methods (Fig. 1C). As further validation of AraPPINet, we tested its performance on a data set comprising 4,533 newly reported protein interactions in Arabidopsis after March 2014. Of these protein interactions, 443 (9.8%) were predicted by AraPPINet (Fig. 1D), which exhibited better TPR than AtPID, AtPIN, and the most recently developed PPI prediction method, PAIR. Furthermore, we also compared the accuracy of different prediction methods using F-measure. From Table I, AraPPINet apparently showed great improvement over all three published PPI prediction methods based on the F1 score.
Table I. Comparison of different prediction approaches with F1 score.
The average true positive of the random network is calculated from 10 randomized networks. Precision represents predicted PPIs with experimental evidence/all predicted PPIs, and recall represents predicted PPIs with experimental evidence/all experimentally determined PPIs.
| Method | Predicted PPIs with Experimental Evidence | All Predicted PPIs | All Experimentally Determined PPIs | Precision | Recall | F1 Score |
|---|---|---|---|---|---|---|
| Random network | 348 | 316,747 | 21,282 | 0.001 | 0.016 | 0.002 |
| AtPID | 386 | 24,418 | 21,282 | 0.016 | 0.018 | 0.017 |
| AtPIN | 449 | 90,043 | 21,282 | 0.005 | 0.021 | 0.008 |
| PAIR | 1,995 | 145,355 | 21,282 | 0.014 | 0.094 | 0.024 |
| AraPPINet | 6,040 | 316,747 | 21,282 | 0.019 | 0.284 | 0.036 |
It is reasonable to estimate the performance of AraPPINet by its ability to discriminate between interacting and noninteracting pairs of proteins with similar sequences. A total of 1,663 interactions between similar proteins in 56 families were used for a performance evaluation. As shown in Figure 2A, only 0.3% of the mean TPR was expected by chance alone, and the three PPI prediction methods, AtPIN, AtPID, and PAIR, had mean TPRs ranging from 11.8% to 32.1% for the tested data set. Compared with the performance of the other three methods, AraPPINet showed high predictive power for discovering interactions between protein family members with a mean TPR of 69.5%. In addition, interactions between members of three specific transcription factor families identified by high-throughput experiments were used to evaluate the performance of AraPPINet for individual cases, including 255 interactions among 72 MADS, 25 interactions among 17 bZIP, and 418 interactions among 49 ARF/IAA transcription factors (de Folter et al., 2005; Ehlert et al., 2006; Vernoux et al., 2011). As shown in Figure 2B, AraPPINet had AUC values ranging from 0.65 for the MADS family to 0.77 for the bZIP family. The precision of PPI prediction reached 22.1% for MADS and 50.6% for ARF/IAA. A number of the interactions between similar members predicted in AraPPINet overlapped highly with those determined through experimental assays (Fig. 2C), suggesting that AraPPINet is a powerful tool for discriminating positive interactions from similar protein pairs within gene families in Arabidopsis.
Figure 2.

Evaluation of AraPPINet for predicting interactions between similar protein pairs. A, Comparison of performance for predicting interactions between similar protein pairs. The box plots indicate interquartile ranges of these data. The bar in each box plot indicates the median. B, ROC curves for AraPPINet-based predictions for the MADS, bZIP, and ARF/IAA families. AUC values are reported in parentheses. C, Venn diagrams of the predicted protein interactions overlapping with the experimentally determined interactions of the MADS, bZIP, and ARF/IAA families.
ABA Signaling Network in AraPPINet
The ABA signaling pathway is a gene network that plays important roles in numerous biological processes in plants. Although a linear core ABA signal transduction pathway has been established, the complexity of gene regulation suggests that ABA functions through an intricate network that is interwoven with additional cellular processes. Using the core ABA components as query proteins, including 14 ABA receptors, nine type 2C protein phosphatases, 10 SNF1-related protein kinases 2, and 10 bZIP transcription factors, we identified 1,912 proteins in AraPPINet predicted to interact with the 43 core components in the ABA signaling pathway. The inferred ABA signaling network contains 7,272 interactions, of which 197 connections have been experimentally proven (Fig. 3A). Among these node proteins, ABI5, ABI1, and ABI2 have the top three connections in the ABA signaling network, consistent with data indicating that these three genes play the most important roles in the signal transduction pathway.
Figure 3.

The ABA signaling network in AraPPINet. A, ABA signaling network inferred from AraPPINet. Node size represents a node degree. Core ABA signaling proteins are represented in red nodes, and their interacting partners are represented in green nodes. The predicted protein interactions are shown in gray lines, and experimentally demonstrated interactions are shown in red lines. B, Comparison of AraPPINet with other methods for discovering PPIs in ABA signaling based on the high-throughput PPI data set. C, Comparison of AraPPINet with other methods for discovering PPIs in ABA signaling based on newly reported interactions. D and E, Enriched Gene Ontology (GO) functional categories (D) and enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (E) of proteins interacting with core ABA signaling proteins. F, Enriched ABA-responsive genes within the ABA signaling network.
The accuracy of the inferred ABA signaling network was validated using two sets of experimentally verified interactions. Among the 31 proteins interacting with the core ABA components determined by high-throughput experiments, seven (22.6%) proteins were successfully predicted by AraPPINet (Fig. 3B). In addition, among 131 newly reported protein interactions involved in ABA signaling, only two interactions were predicted by the published methods AtPIN, AtPID, and PAIR. In contrast, AraPPINet recognized 39 (29.8%) novel protein interactions in the data set, approximately 100-fold more than screens of random protein-protein pairs involved in ABA signaling (Fig. 3C). These results suggest that AraPPINet is very useful for identifying novel genes using known genes in this pathway as bait.
The functional representation of proteins in the ABA signaling network was performed using plant GO Slim categories (Ashburner et al., 2000). Candidate genes were significantly enriched in specific biological processes such as cellular response to stimulus, regulation of cellular process, and signal transduction (Fig. 3D). The full functional enrichment data can be found in Supplemental Table S2. It is worth noting that genes involved in ABA signaling were highly enriched in the protein-binding GO molecular function category, implying that these proteins preferentially interact with core ABA components to perform their functions. Similarly, functional analysis carried out using the KEGG database showed that these proteins are significantly enriched in several specific pathways (Supplemental Table S3). In addition to the expected plant hormone signal transduction pathway, pathways involved in plant-pathogen interaction and plant circadian rhythm were identified (Fig. 3E). In addition, genes interacting with core ABA signaling components were altered significantly in response to ABA treatments (Fig. 3F), and some of them were induced by other plant hormones (Supplemental Fig. S6), suggesting that these proteins may act as interacting nodes between ABA and other hormone signaling pathways.
Validating Novel Interactions in the ABA Signaling Network
Given that AraPPINet successfully discovered novel PPIs involved in ABA signaling, we evaluated the hypothesized interaction-mediated cross talk between ABA and other signaling pathways. Among the 1,912 proteins interacting with ABA signaling, 29 proteins predicted to interact with the ABA receptors PYL1 (AT5G46790) or ABI5 (AT2G36270) were selected for the validation of their physical interactions by yeast two-hybrid assay (yeast strain AH109) (Supplemental Table S4). As shown in Figure 4A, AT3G16857, which encodes a regulator of the cytokinin response, was found to interact with PYL1 by screening eight candidate partners. In addition, ABI5, another core component in ABA signaling, was selected as bait to identify new binding partners. We found that four novel proteins (AT5G06960, AT3G22840, AT1G12610, and AT1G13260) were determined to interact with ABI5 by screening 21 candidates (Fig. 4A). AT5G06960, better known as TGA5, is a core component in salicylic acid signaling (Zhang et al., 2003), while AT3G22840 is involved in early light response and seed germination (Harari-Steinberg et al., 2001; Rizza et al., 2011). The proteins AT1G12610 and AT1G13260 (RAV1) belong to the B3-AP2 transcription factor family; the former is involved in GA signaling and response to abiotic stresses (Magome et al., 2008; Kang et al., 2011). These results indicated that the accuracy of AraPPINet is more than 17% in the test case of ABA signaling.
Figure 4.

Yeast two-hybrid and bimolecular fluorescence complementation (BiFC) assays for detecting interacting partners of PYL1 or ABI5. A, Two-hybrid validation in yeast of the interactions between PYL1 and ARR1 (AT3G16857) as well as between ABI5 and TGA5 (AT5G06960), ELIP (AT3G22840), RAV1 (AT1G13260), and DDF1 (AT1G12610). BD-protein indicates the fusion protein with the Gal4 binding domain, and AD-protein indicates the fusion protein with the Gal4 activation domain. AD or BD was used as a negative control. +His indicates synthetic dropout medium deficient in Leu and Trp, and −His indicates synthetic dropout medium deficient in Leu, Trp, and His. B, BiFC assay for ABI5 and RAV1. 35S:ABI5-YFPN and 35S:RAV1-YFPC constructs were coinfiltrated into N. benthamiana leaves. YFP signal intensity was detected from 48 to 60 h after infiltration.
A recent study showed that RAV1 coexpression with ABI5 enhanced plant resistance to imposed drought stress (Mittal et al., 2014). Thus, BiFC assays were performed to validate the interaction between the proteins RAV1 and ABI5 in plant cells. Constructs encoding ABI5-YFPN and RAV1-YFPC were coinfiltrated into Nicotiana benthamiana leaves, resulting in a visible yellow fluorescent protein (YFP) fluorescence signal in nuclei (Fig. 4B). As a control, empty pEG201YN vector was coinfiltrated with RAV1-YFPC, and no fluorescence signal was observed (Fig. 4B). These results, coupled with the phenotype of transgenic cotton (Gossypium hirsutum), suggested that RAV1 interacted physically with ABI5 to increase resistance to drought stress in plants (Mittal et al., 2014).
The structural interaction model of ARR1 and PYL1 was produced by superimposing homology models on the template of the contact-dependent growth inhibition toxin/immunity complex from Burkholderia pseudomallei (Fig. 5A). The homology model of ARR1 was structurally similar to chain A of the template complex, while the PYL1 model was structurally aligned to chain B. The interaction model has a high interacting probability score of 0.788, arising from both structural similarity and a conserved interface. Similarly, homology models of ABI5 and its four interacting partners were superimposed with their respective homologous template complexes. These structural interaction models showed a significant level of conservation in the interfaces between ABI5 and the four interacting partners (Fig. 5, B–E), which resulted in the interaction of protein pairs with different global structures via similar interface architectures. The structural details of these interactions revealed that conserved interfaces could effectively improve the accuracy of discovering PPIs by computational approaches.
Figure 5.

Structural models of PPIs. A, Structural interaction model for ARR1 and PYL1. Considering the structural template (PDB structure 4G6V) of the contact-dependent growth inhibition toxin/immunity complex (chain A and B), the homology models of ARR1 and PYL1 were structurally superimposed. B, Structural interaction model for DDF1 and ABI5 based on the template complex (PDB structure 1RB6). C, Structural interaction model for RAV1 and ABI5 based on the template complex (PDB structure 1IJ2). D, Structural interaction model for ELIP and ABI5 based on the template complex (PDB structure 2WUK). E, Structural interaction model for TGA5 and ABI5 based on the template complex (PDB structure 2Z5I). The homology models of PYL1 and ABI5 are shown in blue, the models for the interacting protein partners are shown in red, and the template complexes are shown in gray. The conserved interfaces in the van der Waals representation are shown in matching colors.
Identifying Cross Talk in ABA Signaling
The PYL1-interacting partner ARR1, a type B response regulator, is an important regulator involved in the cytokinin signaling pathway (Sakai et al., 2001; Wang et al., 2011). Based on the physical interaction between ARR1 and PYL1, we hypothesized that ARR1 might be involved in regulating the ABA signaling pathway mediated by PYL1. To validate this hypothesis, the response of the type B arr1,11,12 triple mutant (CS6986) to ABA was tested by the primary root elongation assay to determine whether the cytokinin signaling core regulator ARR1 also was involved in the regulation of ABA signaling (Leung et al., 1994; Cary et al., 1995). The arr1,11,12 triple mutant and wild-type seedlings were grown on Murashige and Skoog (MS) medium, and they showed similar primary root elongation phenotypes (Fig. 6, A and B). Compared with wild-type seedlings, the arr1,11,12 mutant seedlings displayed insensitivity to the cytokinin-mediated inhibition of primary root growth on MS medium containing 10 µm 6-BA cytokinin (Fig. 6, A and B). On MS medium supplemented with 10 µm ABA, primary root elongation was inhibited in wild-type seedlings (Fig. 6, A and B). In contrast, primary root elongation in arr1,11,12 mutant seedlings was not inhibited by ABA. These data indicated that arr1,11,12 mutant seedlings were insensitive to cytokinin and ABA, suggesting that type B ARRs, including ARR1, might be involved in ABA signaling regulated by the ABA receptor PYL1 in addition to cytokinin signaling (Fig. 6C).
Figure 6.

arr1,11,12 mutant seedlings are insensitive to the inhibition of cytokinin and ABA on primary root growth. A, Representative examples of wild-type and arr1,11,12 seedlings on MS medium plates or plates with either 10 µm 6-benzyladenine (6-BA) or 10 µm ABA. Five-day-old seedlings grown on MS medium were transferred to MS plates or plates supplemented with either 6-BA or ABA. The photographs were taken 5 d after the transfer. Bars = 10 mm. B, Primary root lengths of wild-type and arr1,11,12 seedlings at 5 d after transfer to MS medium plates or plates with either 6-BA or ABA. The data represent mean values ± se (n > 15). Asterisks indicate that the primary root length of arr1,11,12 is significantly longer than that of the wild type on MS medium plates with either 6-BA or ABA (Student’s t test, P < 0.05). C, Schematic representation of the interactions between ABA and cytokinin signaling mediated by ARR1 and PYL1.
DISCUSSION
In this study, we developed a computational approach through combining three-dimensional structures with functional evidence to infer the genome-wide PPI network in Arabidopsis. The power of this method for discovering protein interactions as well as predicting interactions between protein family members was demonstrated by a comparison with other publicly available PPI prediction methods as well as by experimentally determined PPI data sets. The predicted AraPPINet network contains 316,747 interactions covering nearly half of the proteome (Arabidopsis Genome Initiative, 2000), which is in agreement with the estimated size of the protein interactome in Arabidopsis (Arabidopsis Interactome Mapping Consortium, 2011). Furthermore, AraPPINet includes many interactions (85.6% of our predicted PPIs) beyond those found by simple interolog prediction from reference model organisms, which suggested that our predictive method was powerful in novel PPI discovery.
Structural information has been used successfully to validate or improve large-scale PPI networks (Prieto and De Las Rivas, 2010; Zhang et al., 2012). Our analysis demonstrated that structural evidence could increase the reliability of predicted PPI networks by reducing false positives from the large amount of data regarding noninteracting protein pairs at the genome-wide scale (Supplemental Fig. S4B). It is critical that only very low FPR can produce a small enough number of false positives to be used effectively in practice. In addition to structural evidence, functional clues preferentially contributed to the increased coverage of positive interaction prediction (Supplemental Fig. S4A). These two factors combined to create a balance between accuracy and coverage in PPI prediction using AraPPINet.
AraPPINet represents a reliable PPI network and was useful for identifying new proteins involved in specific processes using a set of known genes as bait. For example, using network guilt-by-association, we defined an ABA signaling network encompassing more than 7,000 interactions involving approximately 1,950 proteins in Arabidopsis. This protein interaction map greatly expanded the known ABA signaling network in plants. An evaluation based on two independent data sets showed that nearly 30% of the known protein interactions involved in ABA signaling were recognized successfully by AraPPINet, indicating that our computational approach for discovering novel PPIs in ABA signaling is comparable in accuracy to high-throughput experiments (von Mering et al., 2002). Using yeast two-hybrid tests on a set of predicted PPIs linked to the ABA and other signaling pathways, five proteins were validated as newly interacting partners of core components in the ABA signaling pathway. Interestingly, ARR1 was predicted to interact with the ABA receptor PYL1 by AraPPINet and also was detected by experimental screening. By genetic analysis, we validated ARR1 involvement in the regulation of cytokinin and ABA signaling through its interaction with PYL1. Our findings suggested that AraPPINet not only could advance our understanding of new gene functions but also provided new insight into the cross talk between pathways such as ABA signaling with respect to diverse biological processes.
AraPPINet represents a step toward the goal of computationally reconstructing reliable PPI networks at a genome-wide scale, and this global protein interaction map could facilitate the understanding of the biological organization of genes for specific functions in plants.
MATERIALS AND METHODS
Gold Standard Data Set
The experimentally determined PPIs for Arabidopsis (Arabidopsis thaliana) were extracted from five databases, BioGRID (Chatr-Aryamontri et al., 2015), IntAct (Orchard et al., 2014), DIP (Salwinski et al., 2004), MINT (Licata et al., 2012), and BIND (Isserlin et al., 2011). Proteins without a valid The Arabidopsis Information Resource (TAIR) locus or that were undefined in the Arabidopsis genome were removed, resulting in a total of 17,569 PPIs among 6,725 proteins. The PPIs available in different databases are listed in Supplemental Table S5.
A data set derived from those small-scale or high-throughput experiments with at least two supporting publications was used as a primary positive reference. After removing protein self-interactions or interacting proteins encoded by mitochondria or chloroplast genes, 5,080 interacting protein pairs remained as the gold standard data set (Supplemental Table S6). Simultaneously, a number of protein pairs were randomly chosen to form the negative reference data set. The negative data set created by this method may contain several potentially interacting protein pairs; however, given the empirically estimated ratio of interacting protein pairs of five to ten interactions per 10,000 protein pairs in the Arabidopsis genome (Arabidopsis Interactome Mapping Consortium, 2011), this level of contamination is likely acceptable. In this way, 508,000 protein pairs not overlapping with 17,569 experimentally determined interactions were randomly generated as the negative reference data set for training the protein interaction classifier.
Collection of Structural Information
The structure homology models of Arabidopsis proteins were taken from the ModBase database (Pieper et al., 2014). A protein structure was considered to be reliable if it met at least one of following model evaluation criteria: (1) an E value less than 10−5; (2) a ModPipe quality score of 0.5 or greater; (3) a GA341 score of 0.5 or greater; or (4) a z-DOPE score less than 0. When multiple structures were available for a target protein, we chose the representative model with the highest ModPipe quality score. Based on the above criteria, a total of 17,391 structure homology models were collected for Arabidopsis.
A total of 90,424 and 88,999 structures involving multiple organisms were available in the PDB (Rose et al., 2013) and Proteins, Interfaces, Structures, and Assemblies (Xu et al., 2008) databases, respectively. The chain-chain binary interface and the binding sites of protein complexes were generated in the PIBASE software package with an interatomic distance cutoff of 6.05 Å (Davis and Sali, 2005). A total of 91,652 protein complexes with 368,306 interacting chain pairs among 317,112 chains were identified for use as templates for protein interaction predictions.
Protein structure homology models were aligned to template complexes using TM-align (Zhang and Skolnick, 2005). Approximately 5.3 billion structural alignments were created between the protein structure homology models and the chains of template complexes. With a normalized TM score cutoff of 0.4 for structural similarity, a total of 50,001,762 structural alignments were generated that involved 16,679 protein structure models.
Structural features were derived from the structural alignments of protein structure homology models to template complexes. Because two structural alignments are obtained for each protein pair (i.e. protein structure homology model i is aligned to template chain i′, while protein model j is aligned to chain j′; TM-Scorei is the score between protein structure model i and template chain i′, and TM-Scorej is the score between protein structure model j and template chain j′), structural similarity is calculated as the square root of the product of TM-Scorei and TM-Scorej. Similarly, structural distance is calculated as the square root of the product of root-mean-square deviations RMSDi and RMSDj. The preserved interface size is defined as the number of interacting residue pairs in the potential protein-protein model preserved in the template complex. The fraction of the preserved interface is the proportion of the conserved interface of the protein-protein model relative to the corresponding interface in the template complex. When multiple values were available for a protein pair or a structural feature, the data with the highest structural similarity to the protein pair were chosen to create an interaction model.
Similarity Analysis of Gene Function
The GO data used were gene_ontology.1_2.obo (Ashburner et al., 2000). The functional similarity of a gene pair was defined as log(n/N)/log(2/N), where n is the number of genes in the lowest GO category that contained both genes and N is the total number of genes annotated for the organism. This formula normalizes the functional similarity range from 0 to 1.
Interolog Analysis
Interolog analysis was performed similarly to a strategy proposed by Jonsson and Bates (2006). A confidence score S for each PPI prediction was assigned. Given a protein pair A and B, S could be defined as follows:
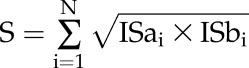 |
where A′i and B′i are the corresponding orthologs of A and B, which show an interaction in one model organism (i.e. the protein pair A′i and B′i is called an interolog of protein pair A and B). ISai is the InParanoid score between A′i and A, while ISbi is the InParanoid score between B′i and B. The orthologs of Arabidopsis proteins in Escherichia coli, Saccharomyces cerevisae, Caenorhabditis elegans, Drosophila melanogaster, Mus musculus, and Homo sapiens were identified by InParanoid with default settings. N is the total number of interologs of protein pair A and B identified in the six model organisms. The experimentally determined PPI data sets used for interolog analysis was obtained from the BioGRID, IntAct, DIP, MINT, and BIND databases (Supplemental Table S7).
Coexpression Analysis
Arabidopsis GeneChip probe-gene mapping was performed as described previously (Harb et al., 2010). The oligonucleotide sequences of probes were mapped to genes from the TAIR 10 database using BLASTN with E < 10−5 (Altschul et al., 1997). If two or more probe sets mapped to a single gene, the expression value for that gene was determined by averaging the signals across the probe sets. In this way, a total of 21,122 genes remained after disregarding probe sets that mapped to more than one gene. A total of 1,388 Affymetrix Arabidopsis arrays were obtained from the TAIR Gene Expression Omnibus (http://www.arabidopsis.org). These slides were normalized using RMAExpress software (Bolstad et al., 2003). The correlations between genes were determined by the Pearson correlation coefficient.
Phylogenetic Profile Analysis
As described previously by Pellegrini et al. (1999), phylogenetic profiles for all Arabidopsis protein sequences were constructed using BLASTP searches (E < 10−10) against a collection of 30 eukaryotic and 660 prokaryotic genomes after applying an evolutionary filter for similar genomes. This calculation across collected genomes yielded a 690-dimensional vector representing the presence or absence of homologs of a query protein in these genomes. The probability of two coevolved proteins is given by P as follows:
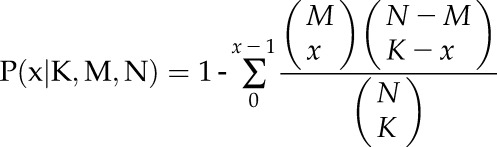 |
where x is the number of the cooccurrence homologs of proteins A and B in the genomes; K and M are the numbers of homologs of proteins A and B, respectively; and N is the total number of collected genomes. The negative log P value of phylogenetic profile similarity is used for prediction.
Rosetta Stone Analysis
Rosetta stone proteins were detected as described previously (Marcotte et al., 1999; Bowers et al., 2004). All protein sequences in the Arabidopsis genome were BLASTPed against the nonredundant database with E < 10−10. Two nonhomologous proteins were aligned over at least 70% of their sequences to different portions of a third protein, and the third protein was referred to as a candidate Rosetta stone protein.
Although proteins containing highly conserved domains may not be fused, they are often linked to each other by the Rosetta stone method. To eliminate these confounding Rosetta stone proteins, the probability that two proteins are linked was computed by chance alone. The probability is given by P as follows:
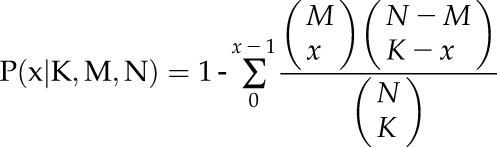 |
where x is the number of candidate Rosetta stone proteins; K and M are the numbers of homologs of proteins A and B, respectively; and N is the total number of sequences in the nonredundant database. The negative log P value of the Rosetta stone protein is used for prediction.
Random Forest Classifier
The positive and negative reference sets with 11 features were used to train the random forest classifier in R with the setting 500 trees (Liaw and Wiener, 2002). All protein pairs were then classified by the trained random forest model for PPI prediction. The predicted PPI network was drawn using Cytoscape (Cline et al., 2007).
Measurement of Classification Performance
The positive and negative reference sets were randomly divided into 10 subsets of equal size. Each time, nine subsets were used for classifier training, and the remaining subset was used to test the trained classifier. This procedure was repeated 10 times using different subsets for training and testing, and a prediction for each interaction was finally generated. The number of TP (true positive), FP (false positive), TN (true negative), and FN (false negative) predictions were counted. TPR (recall) = TP/(TP +FN), FPR = FP/(FP +TN), precision = TP/(TP +FP), F1 score = 2 × precision × recall/(precision + recall), and the AUC values were calculated against the ROC curves.
Comparison of AraPPINet with Other Predicted Interactomes
The performance of AraPPINet was compared with those of three published PPI prediction methods. The AtPID data set was retrieved from the AtPID database (version 4; Cui et al., 2008). The AtPIN data set was downloaded from the AtPIN database (Brandão et al., 2009). The latest PAIR data set (version 3) was provided by Dr. X. Chen (Lin et al., 2011). The random PPI networks were generated based on AraPPINet using an edge-shuffling method, which randomly rewired edges while preserving the original degree distribution of AraPPINet.
Functional Enrichment Analysis
We used plant GO Slim categories to characterize the functions of interacting proteins. The statistical enrichment of proteins in a GO category was obtained by comparing the proteins with those in AraPPINet as well as the Arabidopsis genome using Fisher’s exact test in Blast2GO (Conesa and Götz, 2008).
Similarly, KEGG pathway enrichment analysis of proteins was performed using Fisher’s exact test implemented in a Perl script against a reference data set of AraPPINet and the Arabidopsis genome.
Identification of Hormone-Responsive Genes
The identification of hormone-responsive genes was based on the normalized expression profiling data (submission nos. ME00333, ME00334, ME00344, ME00337, ME00336, ME00343, and ME00335) of Arabidopsis seedlings from the TAIR Gene Expression Omnibus. The average signal intensities of biological replicates for each sample were used to calculate the fold change of gene expression, and differentially expressed genes were identified as having a minimum fold change of 2.
Yeast Two-Hybrid Assay
Plasmids were constructed using Gateway Cloning Technology (Invitrogen). Insertions of PYL1, ABI5, and the coding sequences of candidate genes were prepared using pENTR/D-TOPO. The bait vector pDEST32 and the prey vector pDEST22 were used for the yeast two-hybrid assay. Sets of bait and prey constructs were cotransformed into the AH109 yeast strain. The transformed yeast cells were selected on synthetic minimal double dropout medium deficient in Trp and Leu. Protein interaction tests were assessed on triple dropout medium deficient in Trp, Leu, and His. At least six clones were analyzed with three repeats and generated similar results.
BiFC Analysis
The BiFC vectors pEarleyagte201-YN and pEarleygate202-YC were kindly provided by Steven J. Rothstein for analysis. RAV1 was fused to the C-terminal end (amino acids 175–239) of the YFP protein (YFPC) in the vector pEG202YC, which generated the RAV1-YFPC protein. ABI5 was fused to the N-terminal end (amino acids 1–174) of the YFP protein (YFPN) in the vector pEG201YN, which generated the ABI5-YFPN protein. The ABI5-YFPN, RAV1-YFPC, and empty pEG201-YFPN constructs were introduced into Agrobacterium tumefaciens strain GV3101. Pairs were coinfiltrated into Nicotiana benthamiana. YFP signal was observed after infiltration from 48 to 60 h by confocal laser scanning microscopy (Leica TCS SP5-II). Each observation was repeated at least three times.
Plant Materials and Root Growth Assay
Arabidopsis Columbia was used as the wild-type plant. The transfer DNA insertion mutant line arr1,11,12 (CS6986) was kindly provided by Dr. Jiawei Wang. For the root growth assay, seeds of different genotypes were surface sterilized and then maintained in the dark for 3 d at 4°C to disrupt dormancy. The seeds were sown onto MS medium containing 1.5% agar. To investigate the inhibition of root growth by cytokinin and ABA, 5-d-old seedlings were transferred onto plates supplemented with 6-BA and ABA (Sigma). The primary root growth of seedlings was measured 5 d after transfer to the plates.
Supplemental Data
The following supplemental materials are available.
Supplemental Figure S1. Global view of AraPPINet with the top six assigned modules containing at least 200 node proteins.
Supplemental Figure S2. Coverage of features for interactions in AraPPINet.
Supplemental Figure S3. Topological properties of AraPPINet.
Supplemental Figure S4. Comparisons of performance for methods using different sources of information.
Supplemental Figure S5. Partial ROC curves for PPIs predicted based on different sources of information.
Supplemental Figure S6. Heat map of genes within the ABA signaling network in response to plant hormones at different times.
Supplemental Table S1. Features of protein-protein pairs in Arabidopsis.
Supplemental Table S2. Enriched GO functional categories of interacting proteins within the ABA signaling network.
Supplemental Table S3. Enriched KEGG pathways of interacting proteins within the ABA signaling network.
Supplemental Table S4. Predicted PPIs were screened with the yeast two-hybrid assay.
Supplemental Table S5. Presence of PPIs in Arabidopsis from various databases.
Supplemental Table S6. The gold standard PPI data from various databases.
Supplemental Table S7. Availability of PPIs in six model organisms from various databases.
Supplementary Material
Acknowledgments
We thank Dr. Yi Huang (Oil Crops Research Institute, Chinese Academy of Agricultural Sciences) for helpful discussions and for critical reading of the article; Jiawei Wang (Institute of Plant Physiology and Ecology, Chinese Academy of Sciences) for kindly providing arr1,11,12 transfer DNA insertion mutant (CS6986) seeds; Steven J. Rothstein (University of Guelph) for providing the BiFC vectors; and the staff of the computing facility in the PI cluster at Shanghai Jiao Tong University.
Glossary
- PPI
protein-protein interaction
- PDB
Protein Data Bank
- ABA
abscisic acid
- TPR
true positive rate
- FPR
false positive rate
- ROC
receiver operating characteristic
- AUC
area under the curve
- GO
Gene Ontology
- BiFC
bimolecular fluorescence complementation
- MS
Murashige and Skoog
- 6-BA
6-benzyladenine
- TAIR
The Arabidopsis Information Resource
Footnotes
This work was supported by the National Basic Research Program of China (grant no. 2013CB733903–03) and the National Natural Science Foundation of China (grant no. 31371229).
References
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408: 796–815 [DOI] [PubMed] [Google Scholar]
- Arabidopsis Interactome Mapping Consortium (2011) Evidence for network evolution in an Arabidopsis interactome map. Science 333: 601–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000) Gene Ontology: tool for the unification of biology. Nat Genet 25: 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aytuna AS, Gursoy A, Keskin O (2005) Prediction of protein-protein interactions by combining structure and sequence conservation in protein interfaces. Bioinformatics 21: 2850–2855 [DOI] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP (2003) A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19: 185–193 [DOI] [PubMed] [Google Scholar]
- Bowers PM, Pellegrini M, Thompson MJ, Fierro J, Yeates TO, Eisenberg D (2004) Prolinks: a database of protein functional linkages derived from coevolution. Genome Biol 5: R35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandão MM, Dantas LL, Silva-Filho MC (2009) AtPIN: Arabidopsis thaliana protein interaction network. BMC Bioinformatics 10: 454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cary AJ, Liu W, Howell SH (1995) Cytokinin action is coupled to ethylene in its effects on the inhibition of root and hypocotyl elongation in Arabidopsis thaliana seedlings. Plant Physiol 107: 1075–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatr-Aryamontri A, Breitkreutz BJ, Oughtred R, Boucher L, Heinicke S, Chen D, Stark C, Breitkreutz A, Kolas N, O’Donnell L, et al. (2015) The BioGRID interaction database: 2015 update. Nucleic Acids Res 43: D470–D478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline MS, Smoot M, Cerami E, Kuchinsky A, Landys N, Workman C, Christmas R, Avila-Campilo I, Creech M, Gross B, et al. (2007) Integration of biological networks and gene expression data using Cytoscape. Nat Protoc 2: 2366–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, Götz S (2008) Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics 2008: 619832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Li P, Li G, Xu F, Zhao C, Li Y, Yang Z, Wang G, Yu Q, Li Y, et al. (2008) AtPID: Arabidopsis thaliana protein interactome database: an integrative platform for plant systems biology. Nucleic Acids Res 36: D999–D1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis FP, Sali A (2005) PIBASE: a comprehensive database of structurally defined protein interfaces. Bioinformatics 21: 1901–1907 [DOI] [PubMed] [Google Scholar]
- de Folter S, Immink RG, Kieffer M, Parenicová L, Henz SR, Weigel D, Busscher M, Kooiker M, Colombo L, Kater MM, et al. (2005) Comprehensive interaction map of the Arabidopsis MADS box transcription factors. Plant Cell 17: 1424–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlert A, Weltmeier F, Wang X, Mayer CS, Smeekens S, Vicente-Carbajosa J, Dröge-Laser W (2006) Two-hybrid protein-protein interaction analysis in Arabidopsis protoplasts: establishment of a heterodimerization map of group C and group S bZIP transcription factors. Plant J 46: 890–900 [DOI] [PubMed] [Google Scholar]
- Grigoriev A. (2001) A relationship between gene expression and protein interactions on the proteome scale: analysis of the bacteriophage T7 and the yeast Saccharomyces cerevisiae. Nucleic Acids Res 29: 3513–3519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harari-Steinberg O, Ohad I, Chamovitz DA (2001) Dissection of the light signal transduction pathways regulating the two early light-induced protein genes in Arabidopsis. Plant Physiol 127: 986–997 [PMC free article] [PubMed] [Google Scholar]
- Harb A, Krishnan A, Ambavaram MM, Pereira A (2010) Molecular and physiological analysis of drought stress in Arabidopsis reveals early responses leading to acclimation in plant growth. Plant Physiol 154: 1254–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isserlin R, El-Badrawi RA, Bader GD (2011) The Biomolecular Interaction Network Database in PSI-MI 2.5. Database (Oxford) 2011: baq037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones AM, Xuan Y, Xu M, Wang RS, Ho CH, Lalonde S, You CH, Sardi MI, Parsa SA, Smith-Valle E, et al. (2014) Border control: a membrane-linked interactome of Arabidopsis. Science 344: 711–716 [DOI] [PubMed] [Google Scholar]
- Jonsson PF, Bates PA (2006) Global topological features of cancer proteins in the human interactome. Bioinformatics 22: 2291–2297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HG, Kim J, Kim B, Jeong H, Choi SH, Kim EK, Lee HY, Lim PO (2011) Overexpression of FTL1/DDF1, an AP2 transcription factor, enhances tolerance to cold, drought, and heat stresses in Arabidopsis thaliana. Plant Sci 180: 634–641 [DOI] [PubMed] [Google Scholar]
- Lee K, Thorneycroft D, Achuthan P, Hermjakob H, Ideker T (2010) Mapping plant interactomes using literature curated and predicted protein-protein interaction data sets. Plant Cell 22: 997–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung J, Bouvier-Durand M, Morris PC, Guerrier D, Chefdor F, Giraudat J (1994) Arabidopsis ABA response gene ABI1: features of a calcium-modulated protein phosphatase. Science 264: 1448–1452 [DOI] [PubMed] [Google Scholar]
- Liaw A, Wiener M (2002) Classification and regression by randomForest. R News 2: 18–22 [Google Scholar]
- Licata L, Briganti L, Peluso D, Perfetto L, Iannuccelli M, Galeota E, Sacco F, Palma A, Nardozza AP, Santonico E, et al. (2012) MINT, the molecular interaction database: 2012 update. Nucleic Acids Res 40: D857–D861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M, Zhou X, Shen X, Mao C, Chen X (2011) The predicted Arabidopsis interactome resource and network topology-based systems biology analyses. Plant Cell 23: 911–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magome H, Yamaguchi S, Hanada A, Kamiya Y, Oda K (2008) The DDF1 transcriptional activator upregulates expression of a gibberellin-deactivating gene, GA2ox7, under high-salinity stress in Arabidopsis. Plant J 56: 613–626 [DOI] [PubMed] [Google Scholar]
- Marcotte EM, Pellegrini M, Ng HL, Rice DW, Yeates TO, Eisenberg D (1999) Detecting protein function and protein-protein interactions from genome sequences. Science 285: 751–753 [DOI] [PubMed] [Google Scholar]
- Matthews LR, Vaglio P, Reboul J, Ge H, Davis BP, Garrels J, Vincent S, Vidal M (2001) Identification of potential interaction networks using sequence-based searches for conserved protein-protein interactions or “interologs.” Genome Res 11: 2120–2126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal A, Gampala SS, Ritchie GL, Payton P, Burke JJ, Rock CD (2014) Related to ABA-Insensitive3(ABI3)/Viviparous1 and AtABI5 transcription factor coexpression in cotton enhances drought stress adaptation. Plant Biotechnol J 12: 578–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosca R, Céol A, Aloy P (2013) Interactome3D: adding structural details to protein networks. Nat Methods 10: 47–53 [DOI] [PubMed] [Google Scholar]
- Orchard S, Ammari M, Aranda B, Breuza L, Briganti L, Broackes-Carter F, Campbell NH, Chavali G, Chen C, del-Toro N, et al. (2014) The MIntAct project: IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res 42: D358–D363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek R, Fonstein M, D’Souza M, Pusch GD, Maltsev N (1999) The use of gene clusters to infer functional coupling. Proc Natl Acad Sci USA 96: 2896–2901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO (1999) Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc Natl Acad Sci USA 96: 4285–4288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieper U, Webb BM, Dong GQ, Schneidman-Duhovny D, Fan H, Kim SJ, Khuri N, Spill YG, Weinkam P, Hammel M, et al. (2014) ModBase, a database of annotated comparative protein structure models and associated resources. Nucleic Acids Res 42: D336–D346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto C, De Las Rivas J (2010) Structural domain-domain interactions: assessment and comparison with protein-protein interaction data to improve the interactome. Proteins 78: 109–117 [DOI] [PubMed] [Google Scholar]
- Rhodes DR, Tomlins SA, Varambally S, Mahavisno V, Barrette T, Kalyana-Sundaram S, Ghosh D, Pandey A, Chinnaiyan AM (2005) Probabilistic model of the human protein-protein interaction network. Nat Biotechnol 23: 951–959 [DOI] [PubMed] [Google Scholar]
- Rizza A, Boccaccini A, Lopez-Vidriero I, Costantino P, Vittorioso P (2011) Inactivation of the ELIP1 and ELIP2 genes affects Arabidopsis seed germination. New Phytol 190: 896–905 [DOI] [PubMed] [Google Scholar]
- Rose PW, Bi C, Bluhm WF, Christie CH, Dimitropoulos D, Dutta S, Green RK, Goodsell DS, Prlic A, Quesada M, et al. (2013) The RCSB Protein Data Bank: new resources for research and education. Nucleic Acids Res 41: D475–D482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai H, Honma T, Aoyama T, Sato S, Kato T, Tabata S, Oka A (2001) ARR1, a transcription factor for genes immediately responsive to cytokinins. Science 294: 1519–1521 [DOI] [PubMed] [Google Scholar]
- Salwinski L, Miller CS, Smith AJ, Pettit FK, Bowie JU, Eisenberg D (2004) The Database of Interacting Proteins: 2004 update. Nucleic Acids Res 32: D449–D451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dongen S. (2008) Graph clustering via a discrete uncoupling process. SIAM J Matrix Anal Appl 30: 121–141 [Google Scholar]
- Vernoux T, Brunoud G, Farcot E, Morin V, Van den Daele H, Legrand J, Oliva M, Das P, Larrieu A, Wells D, et al. (2011) The auxin signalling network translates dynamic input into robust patterning at the shoot apex. Mol Syst Biol 7: 508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Mering C, Krause R, Snel B, Cornell M, Oliver SG, Fields S, Bork P (2002) Comparative assessment of large-scale data sets of protein-protein interactions. Nature 417: 399–403 [DOI] [PubMed] [Google Scholar]
- Wang C, Marshall A, Zhang D, Wilson ZA (2012) ANAP: an integrated knowledge base for Arabidopsis protein interaction network analysis. Plant Physiol 158: 1523–1533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li L, Ye T, Zhao S, Liu Z, Feng YQ, Wu Y (2011) Cytokinin antagonizes ABA suppression to seed germination of Arabidopsis by downregulating ABI5 expression. Plant J 68: 249–261 [DOI] [PubMed] [Google Scholar]
- Wang Y, Thilmony R, Zhao Y, Chen G, Gu YQ (2014) AIM: a comprehensive Arabidopsis interactome module database and related interologs in plants. Database (Oxford) 2014: bau117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wass MN, Fuentes G, Pons C, Pazos F, Valencia A (2011) Towards the prediction of protein interaction partners using physical docking. Mol Syst Biol 7: 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Canutescu AA, Wang G, Shapovalov M, Obradovic Z, Dunbrack RL Jr (2008) Statistical analysis of interface similarity in crystals of homologous proteins. J Mol Biol 381: 487–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang QC, Petrey D, Deng L, Qiang L, Shi Y, Thu CA, Bisikirska B, Lefebvre C, Accili D, Hunter T, et al. (2012) Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature 490: 556–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Skolnick J (2005) TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res 33: 2302–2309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Tessaro MJ, Lassner M, Li X (2003) Knockout analysis of Arabidopsis transcription factors TGA2, TGA5, and TGA6 reveals their redundant and essential roles in systemic acquired resistance. Plant Cell 15: 2647–2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.