

Abstract
For inherited cardiomyopathies, abnormal sensitivity to intracellular calcium (Ca2+), incurred from genetic mutations, initiates subsequent molecular events leading to pathological remodeling. Here, we characterized the effect of β‐adrenergic stress in familial dilated cardiomyopathy (DCM) using human‐induced pluripotent stem cell (hiPSC)‐derived cardiomyocytes (CMs) from a patient with RBM20 DCM. Our findings suggest that β‐adrenergic stimulation accelerated defective Ca2+ homeostasis, apoptotic changes, and sarcomeric disarray in familial DCM hiPSC‐CMs. Furthermore, pharmacological modulation of abnormal Ca2+ handling by pretreatment with β‐blocker, carvedilol, or Ca2+‐channel blocker, verapamil, significantly decreased the area under curve, reduced percentage of disorganized cells, and decreased terminal deoxynucleotidyl transferase‐mediated deoxyuridine triphosphate nick‐end labeling (TUNEL)‐positive apoptotic loci in familial DCM hiPSC‐CMs after β‐adrenergic stimulation. These translational data provide patient‐based in vitro analysis of β‐adrenergic stress in RBM20‐deficient familial DCM hiPSC‐CMs and evaluation of therapeutic interventions to modify heart disease progression, which may be personalized, but more importantly generalized in the clinic.
Study Highlights
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
✓ In the United States, idiopathic DCM is the most common indication for cardiac transplantation because of progressive heart failure. Despite numerous clinical reports, cardiogenic stress response mechanisms linked to clinical DCM remain elusive.
WHAT QUESTION DID THIS STUDY ADDRESS?
✓ The central hypothesis underpinning our study is that β‐adrenergic stress will promote phenotypic changes including sarcomeric disarray and Ca2+ handling abnormalities in familial DCM hiPSC‐CMs and conversely, that protection from β‐adrenergic stress by pharmacological interventions will attenuate the extent of phenotypic damage.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE?
✓ We found that RBM20‐deficient familial DCM hiPSC‐CMs could be studied in vitro, specifically demonstrating increased susceptibility to stress and therapeutic rescue by beta‐blocker, carvedilol, and Ca2+ channel blocker, verapamil.
HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY AND THERAPEUTICS?
✓ This study provides the first comprehensive in vitro analysis of β‐adrenergic stress in RBM20‐deficient familial DCM hiPSC‐CMs, allowing for further development of therapeutic interventions that modify heart disease progression.
Heart muscle diseases, or cardiomyopathies, are associated with significant morbidity and mortality because of progressive deterioration of cardiac function and refractory heart failure.1, 2, 3 Dilated cardiomyopathy (DCM) is characterized by enlargement of one or both ventricular chambers with impaired systolic function, affecting about 36.5 in 100,000 people, of whom about 25−30% are familial cases.4, 5, 6 Despite the heritability of DCM, development of heart failure is unpredictable and the lack of effective therapeutic management frequently leads to heart transplantation.7 Recent advancements in regenerative biology potentiate cardiovascular disease modeling and drug screening applications using the bioengineered stem cell platform.8, 9 Although ongoing studies highlight the accuracy of human‐induced pluripotent stem cell (hiPSC)‐derived cardiomyocytes (CMs) to model familial DCM phenotypes,9 the utility of hiPSC‐CMs to assess pharmacological compounds that modulate abnormal calcium (Ca2+) handling properties has yet to be validated. Such proof‐of‐principle studies for drug targeting applications require an in vitro stress test to calibrate the phenotypic onset of cardiogenic defects. Herein, we utilized the previously established hiPSC‐based familial DCM model10 to evaluate β‐adrenergic norepinephrine (NE) stress response and characterize therapeutic agents that preserve normal structural and functional phenotype during in vitro stress.
Among the numerous mutations linked to familial DCM, including genes encoding sarcomeric, cytoskeletal, mitochondrial, and nuclear membrane proteins,11, 12 we studied the functionally distinct RBM20 gene involved in regulating alternative splicing. Indeed, RBM20 mutations, which encode an RNA‐binding motif, were recently discovered in individuals with severe cases of familial DCM.13, 14, 15 Importantly, the subset of proteins aberrantly spliced in human RBM20‐ and rat Rbm20‐mutant hearts includes molecules involved in sarcomere structure and function, ion transport, and Ca2+ handling.16 Disruption of normal splicing therefore results in anomalous isoforms of a whole network of cardiac proteins and prevents the heart from effectively responding to both extrinsic and intrinsic stressors.17 Recent reports have shown that RBM20 familial DCM hiPSC‐CMs have an increased susceptibility to β‐adrenergic stimulation and exhibit stress‐induced phenotypic change in sarcomeric structure.10
The central hypothesis underpinning our study is that β‐adrenergic stress will promote phenotypic changes, including sarcomeric disarray and Ca2+ handling abnormalities in familial DCM hiPSC‐CMs, and, conversely, that protection from β‐adrenergic stress by pharmacological interventions will attenuate the extent of phenotypic damage. We first performed a detailed characterization of intracellular Ca2+ handling properties after β‐agonist treatment in familial DCM hiPSC‐CMs. Although greater sensitivity to β‐adrenergic stimulation in DCM has been suggested,9, 18 the effects of in vitro stress on disease severity have not been evaluated. To determine the detrimental effects of enhanced cardiac contractile activity, control and familial DCM hiPSC‐CMs were subjected to NE stress. We hypothesized that β‐adrenergic stimulation would accelerate defective Ca2+ homeostasis, apoptotic changes, and sarcomeric disarray in familial DCM hiPSC‐CMs derived from a patient with mutations in RBM20.
Evidence suggests that early pharmacological intervention in DCM may delay or even prevent development of heart failure.19, 20 β‐Adrenergic receptor‐blocking drugs reduce myocardial chronotropic and inotropic activity and are widely used in the treatment of symptomatic heart failure.21 Individuals with DCM caused by RBM20 mutations are generally treated with standard heart‐failure therapies, including β‐blockers. Additionally, L‐type Ca2+ channel‐blocking drugs, which show vasospasm prevention and restoration of impaired Ca2+ handling, are promising with respect to treatment of heart failure secondary to DCM.22, 23, 24 Although genotype‐positive family members can be identified preclinically, no preventative interventions have been studied. To determine whether β‐blocker and Ca2+‐channel blocker therapy would mitigate against β‐adrenergic stress, we also evaluated the effects of carvedilol and verapamil in familial DCM hiPSC‐CMs. These data provide the first comprehensive in vitro analysis of β‐adrenergic stress in RBM20‐deficient familial DCM hiPSC‐CMs and the first evaluation of therapeutic interventions to modify disease progression, which may be personalized, but, more importantly, generalized in the clinic.
METHODS
Detailed methods used for human iPSC derivation, culture, and cardiac differentiation, as well as MatLab analysis, flow cytometry, and terminal deoxynucleotidyl transferase‐mediated deoxyuridine triphosphate nick‐end labeling (TUNEL) staining is in the Supplementary Material.
Fluo‐4AM Ca2+ imaging
Intracellular calcium (Ca2+) dynamics in control and familial DCM hiPSC‐CMs at day 20 were assessed.25 Briefly, cells were dissociated into small clumps using collagenase type II (480 U/mL for 15 min; Worthington, Lakewood, NJ) and plated on geltrex‐coated 35‐mm glass‐bottom dishes (Thermo Scientific, Waltham, MA) overnight. After attachment, cells were loaded with the Ca2+‐fluorescent probe Fluo‐4 AM (Invitrogen) for 2 h in Tyrode's solution containing 140 mM NaCl, 5.4 mM KCl, 1 mM MgCl2, 10 mM glucose, 1.5 mM CaCl2, and 10 mM HEPES (pH 7.4) and imaged with a Zeiss LSM Live 5 laser confocal microscope (Zeiss, Oberkochen, Germany). Spontaneous Ca2+ transients were recorded at 37°C.26, 27
Norepinephrine stress
Control and familial DCM hiPSC‐CMs were dissociated using type II collagenase and plated on 35 mm glass‐bottom dishes. After attachment, cells were treated with 10 μM NE (Sigma‐Aldrich) for 48 h. Cells were stained with 1:1000 dilution α‐actinin (Sigma‐Aldrich) to characterize sarcomeric disorganization defined as ≥25% punctate appearance.9 All images were acquired with a Zeiss LSM 510 confocal microscope (Zeiss). Image J software was utilized to measure the length and the width of the sarcomere.
Carvedilol and verapamil pretreatment
For pharmacological modulation of Ca2+ handling, sarcomere disarray, and apoptosis analyses, control and familial DCM hiPSC‐CMs at day 20 were treated with either 10 μM carvedilol (Tocris, Bristol, UK) or 1 nM verapamil (Tocris) for 24 h (Supplementary Table S1). Cells were washed with CDM3 media before in vitro stress testing with 10 μM NE.
Statistical analysis
Results are presented as means ± SEM. Paired group analysis was performed using Student's t test. Two‐way repeated‐measures analysis of variance was used for comparison between groups (JMP 9; SAS Institute, Cary, NC). Kaplan‐Meier analysis was applied with log‐rank testing. Any p < 0.05 was predetermined as significant.
RESULTS
Regulation of Β‐adrenergic stress in familial DCM hiPSC‐CMs
In vitro stress testing of hiPSC‐CMs with β‐adrenergic agonist, NE, has led to increased cardiac contractility by the G‐protein coupled second messenger system.9 Familial DCM has been associated with increased intracellular Ca2+ overload as well as an increased susceptibility to chronotropic stress from NE.10 To modulate this Ca2+ overload and minimize chronotropic stress response, we utilized pretreatment with carvedilol, β‐blocker, and verapamil, L‐type Ca2+ channel blocker (Figure 1 a). Specifically, we compared the effects of carvedilol, which indirectly decreases intracellular Ca2+, to verapamil, which directly regulates intracellular Ca2+ through the L‐type Ca2+ channel (Figure 1 b). In this study, hiPSC‐CMs from controls and patients with familial DCM were pretreated with 10 μM carvedilol or 1 nM verapamil for 24 h and then induced in vitro stress testing with 10 μM NE for 48 h (Figure 1 c). Previously, we found that 10 μM NE was sufficient to induce a positive chronotropic effect in RBM20‐deficient familial DCM hiPSC‐CMs.10 Here, we determined the carvedilol concentration based on studies utilizing β‐adrenergic blocker, metoprolol (10 μM), in familial DCM hiPSC‐CMs.9 Similarly, the verapamil concentration was established based on studies measuring cardiotoxic responses in familial DCM hiPSC‐CMs.28 Furthermore, dose‐dependent analysis using real‐time cell analyzer recordings showed that concentrations ≥10 nM yielded a decreased beating activity in control hiPSC‐CMs (Supplementary Figure S1). Indeed, pretreatment with 10 μM verapamil elicited a cardiotoxic response (data not shown). Outcome measures in this study included Ca2+ handling, sarcomere disarray, and apoptosis analyses.
Figure 1.
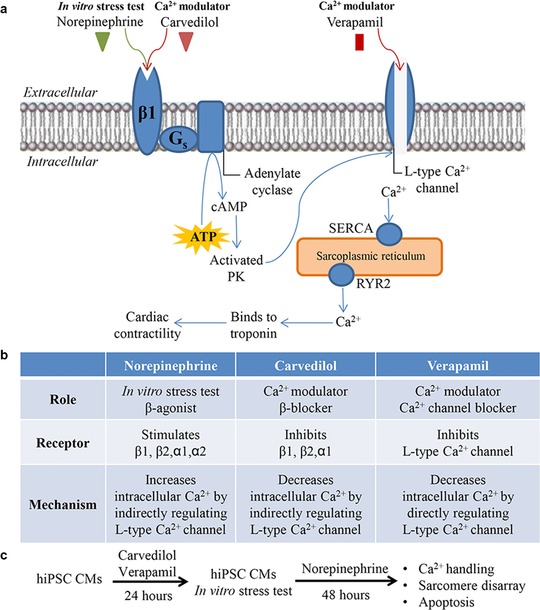
Overview of in vitro cardiac stress with norepinephrine (NE) and calcium (Ca2+) modulation with carvedilol and verapamil. (a) Schematic representation of stimulating (NE) and repressing (carvedilol) the β1 receptor situated on the cardiac sarcolemma. Both NE and carvedilol have an indirect effect on the L‐type Ca2+ channel by intracellular G‐protein coupled second messenger activity. L‐type Ca2+ channel blocker (verapamil) directly inhibits the opening of this channel. (b) Summary of the drug role, receptor/channel binding, and mechanism of NE, carvedilol, and verapamil. (c) Experimental design of this study illustrating control and familial dilated cardiomyopathy (DCM) human‐induced pluripotent stem cell (hiPSC)‐cardiomyocytes (CMs) that were pretreated with carvedilol or verapamil for 24 h and then subjected to in vitro cardiac stress test using NE for 48 h. Outcome measures included Ca2+ handling, sarcomere disarray, and apoptosis analyses. PK, protein kinase; ATP, adenosine triphosphate.
Pharmacological modulation of Ca2+ handling decreases Ca2+ overload due to β‐adrenergic stress in familial DCM hiPSC‐CMs
To assess Ca2+ handling properties, we recorded the fluorescent activity of Fluo‐4AM labeled control and familial DCM hiPSC‐CMs and analyzed spontaneous Ca2+ transients. Intracellular Ca2+ activity during diastole and systole revealed amplified Ca2+ transients in familial DCM hiPSC‐CMs at baseline and after NE stress test, whereas decreased fluorescent response was noted in the carvedilol and verapamil pretreated cohorts (Figure 2 a). Comparatively, Ca2+ transients from control hiPSC‐CMs showed an increased frequency and amplitude after NE stress alone compared with decreased frequency and amplitude with carvedilol and verapamil pretreatment (Figure 2 b). Interestingly, Ca2+ transients from familial DCM hiPSC‐CMs also displayed increased frequency and amplitude as well as an irregular transient pattern after NE stress test (Figure 2 c, top). Indeed, pretreatment with carvedilol and verapamil reduced both frequency and amplitude, whereas verapamil additionally contributed to ablating the irregular transient pattern associated with in vitro stress test (Figure 2 c, bottom). To further characterize this irregular transient pattern in familial DCM hiPSC‐CMs because of NE stress test, we assessed in vitro beating activity. β‐Adrenergic stimulation in familial DCM hiPSC‐CMs resulted in increased and arrhythmic beating, which was reduced to slower beating activity by carvedilol pretreatment and arrhythmia‐free beating activity by verapamil pretreatment (Supplementary Figure S2). Hence, spontaneous Ca2+ tracing denotes defective Ca2+ machinery in familial DCM hiPSC‐CM population compared with control, which can be modulated by pharmacologically targeting the L‐type Ca2+ channel either indirectly with carvedilol or directly with verapamil.
Figure 2.

Pharmacological modulation of calcium handling with carvedilol and verapamil decreases calcium overload in familial dilated cardiomyopathy (DCM) human‐induced pluripotent stem cell (hiPSC)‐cardiomyocytes (CMs). (a) Diastolic and systolic patterns of calcium (Ca2+) fluorescence as measured by Fluo4‐AM intracellular Ca2+ dye at baseline, norepinephrine (NE) stress, and carvedilol or verapamil pretreatment. Calcium modulation is highlighted in red. (b) Control hiPSC‐CMs increase the frequency and amplitude of calcium transients after NE stress. This response is muted after carvedilol or verapamil pretreatment. (c) Familial DCM hiPSC‐CMs increase the frequency, amplitude, and display irregular calcium transients after NE stress. Verapamil pretreatment ablates the irregular calcium transient pattern after NE stress.
Quantification of hiPSC‐CM Ca2+ transients after β‐adrenergic stimulation
We next quantified the area under the curve, time between peaks, characteristic time of Ca2+ extrusion, and Ca2+ spike amplitude in control and familial DCM hiPSC‐CMs. Notably, compared with healthy controls, familial DCM hiPSC‐CMs exhibited greater area under the curve at baseline and after NE stress (DCM baseline: 472.568 ± 15.912 arbitrary units (AU) [n = 3]; DCM post‐NE: 992.757 ± 172.931AU [n = 3] vs. control baseline: 397.648 ± 17.248AU [n = 3]; and control post‐NE: 682.703 ± 41.393AU [n = 3]). Pretreatment with carvedilol and verapamil significantly decreased the area under curve in familial DCM hiPSC‐CMs (DCM carvedilol: 327.252 ± 23.494AU [n = 3; p < 0.001]; DCM verapamil: 293.213 ± 60.049AU [n = 3; p < 0.0001] vs. control carvedilol: 101.620 ± 11.654AU [n = 3]; and control verapamil: 202.796 ± 80.809AU [n = 3]), corresponding with reduced relative Ca2+ levels in the cytoplasm (Figure 3 a).
Figure 3.
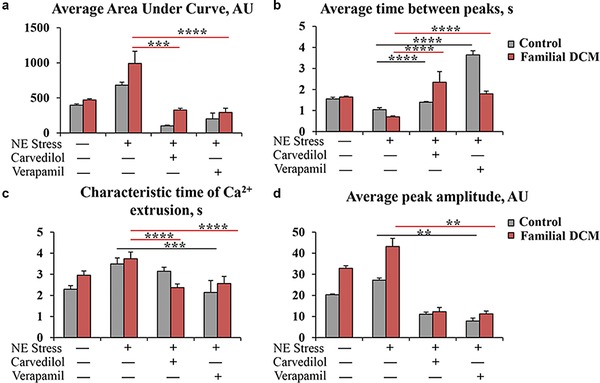
Quantification of calcium (Ca2+) transients after pharmacological pretreatment with carvedilol and verapamil in familial dilated cardiomyopathy (DCM) human‐induced pluripotent stem cell (hiPSC)‐cardiomyocytes (CMs). (a) Carvedilol and verapamil decrease average area under the curve (arbitrary units [AUs]) after norepinephrine (NE) stress. (b) Average time between peaks (s) is increased with carvedilol and verapamil, suggesting slowed calcium transients. (c) Characteristic time of Ca2+ extrusion (seconds [s]) is moderately decreased in both control and DCM hiPSC‐CMs with carvedilol and verapamil treatment. (d) Average peak amplitude (AU) is decreased with pharmacological modulation of intracellular Ca2+ using carvedilol and verapamil. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Furthermore, individual Ca2+ transients suggest that the average time between peaks was decreased after an NE stress test in both control and familial DCM hiPSC‐CMs compared with baseline (DCM baseline: 1.644 ± 0.043 s [n = 3]; DCM post‐NE: 0.707 ± 0.042 s [n = 3] vs. control baseline: 1.555 ± 0.076 s [n = 3]; and control post‐NE: 1.044 ± 0.094 s [n = 3]) suggesting an increased frequency with chronotropic stress. Indeed, pretreatment with carvedilol and verapamil significantly elongated the time between peaks in familial DCM hiPSC‐CMs (DCM carvedilol: 2.351 ± 0.502 s [n = 3; p < 0.001]; DCM verapamil: 1.799 ± 0.125 s [n = 3; p < 0.0001) vs. control carvedilol: 1.399 ± 0.314 s [n = 3; p < 0.001]; control verapamil: 3.646 ± 0.197 s [n = 3; p < 0.001]) indicating a slower beating activity and reduced frequency (Figure 3 b).
In addition, the characteristic time of Ca2+ extrusion, which evaluates the duration to remove Ca2+ from the cytoplasm, was longer in familial DCM hiPSC‐CMs than control at baseline and after NE stress (DCM baseline: 2.957 ± 0.201 s [n = 3]; DCM post‐NE: 3.741 ± 0.313 s [n = 3] vs. control baseline: 2.294 ± 0.170 s [n = 3]; and control post‐NE: 3.492 ± 0.288 s [n = 3]). Pretreatment with carvedilol and verapamil significantly shortened the characteristic time of Ca2+ extrusion in familial DCM hiPSC‐CMs (DCM carvedilol: 2.375 ± 0.169 s [n = 3; p < 0.001]; DCM verapamil: 2.569 ± 0.329 s [n = 3; p < 0.0001] vs. control carvedilol: 3.147 ± 0.191 s [n = 3]; and control verapamil: 2.142 ± 0.561 s [n = 3; p < 0.001]; (Figure 3 c).
Finally, the average peak amplitude was greater in familial DCM hiPSC‐CMs at baseline and post‐NE stress test (DCM baseline: 32.870 ± 1.207AU [n = 3]; DCM post‐NE: 43.198 ± 3.811AU [n = 3] vs. control baseline: 20.334 ± 0.370AU [n = 3]; and control post‐NE: 27.227 ± 1.028AU [n = 3]), correlating with a higher concentration of Ca2+ release into the cytoplasm. Pretreatment with carvedilol and verapamil significantly reduced the Ca2+ spike amplitude in familial DCM hiPSC‐CMs (DCM carvedilol: 12.276 ± 2.036AU [n = 3]; DCM verapamil: 11.279 ± 1.297AU [n = 3; p < 0.01] vs. control carvedilol: 11.068 ± 1.007AU [n = 3]; and control verapamil: 7.862 ± 1.393AU [n = 3; p < 0.01]) corresponding with reduced intracellular Ca2+ (Figure 3 d). Hence, both carvedilol and verapamil contribute to restoring the defective Ca2+ machinery in familial DCM hiPSC‐CMs.
Pre‐administration of carvedilol and verapamil reduces the extent of β‐adrenergic stress‐induced sarcomeric disarray in familial DCM hiPSC‐CMs
To examine whether treatment with β‐adrenergic agonist, a positive inotropic reagent, can induce phenotypic stress response, we analyzed both control and familial DCM hiPSC‐CMs with α‐actinin immunostaining. Indeed, in vitro treatment with 10 μM NE markedly increased the number of CMs with punctate sarcomeric α‐actinin distribution from familial DCM iPSC clones compared with baseline (Figure 4 a, left). Additionally, familial DCM hiPSC‐CMs showed complete myofilament degeneration after NE treatment, which was not present in control hiPSC‐CMs. Notably, carvedilol and verapamil pretreated hiPSC‐CMs displayed reduced disease‐relevant phenotypic response with decreased sarcomeric disarray (Figure 4 a, right). Disorganized sarcomeric pattern, defined as ≥25% punctate appearance, was quantified and more evident in familial DCM hiPSC‐CMs (DCM baseline: 27.5 ± 4.8%; DCM post‐NE: 62.4 ± 3.9% vs. control baseline: 15.3 ± 2.4%; control post‐NE: 35.6 ± 3.3%; Figures 4 b,c). In agreement with α‐actinin structural changes, pretreatment with carvedilol and verapamil significantly reduced percentage of disorganized cells in familial DCM hiPSC‐CMs after in vitro stress test (DCM carvedilol: 35.6 ± 3.7%; DCM verapamil: 29.5 ± 2.0% [p < 0.05] vs. control carvedilol: 27.8 ± 2.9%; and control verapamil: 29.5 ± 3.5%; Figure 4 b,c). These findings suggest that carvedilol and verapamil pretreatment limits susceptibility to stress induced by β‐adrenergic stimulation in familial DCM hiPSC‐CMs.
Figure 4.
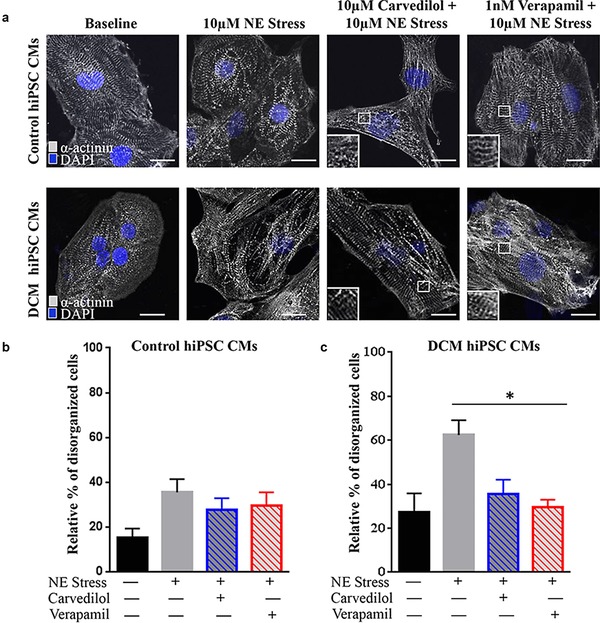
Pretreatment with carvedilol and verapamil reduces the extent of norepinephrine (NE)‐induced sarcomeric disarray. (a) Sarcomeric disarray defined as ≥25% punctate appearance in α‐actinin staining suggests an increased number of punctate cells in control and familial dilated cardiomyopathy (DCM) human‐induced pluripotent stem cell (hiPSC)‐cardiomyocytes (CMs) after NE stress. Carvedilol and verapamil reduce this disease‐relevant phenotypic response. Scale bar: 20μm. (b) Control hiPSC‐CMs exhibit a lower percentage of disorganized cells after NE treatment compared with familial DCM hiPSC‐CMs. Carvedilol and verapamil pretreatment additionally reduced this percent disarray. (c) Familial DCM hiPSC‐CMs had an increased percentage of disorganized cells after NE stress, which was reduced by carvedilol and significantly by verapamil pretreatment. *p < 0.05.
Carvedilol and verapamil pretreatment decreases NE‐induced apoptosis in familial DCM hiPSC‐CMs
Previous studies have shown that activation of the β‐adrenergic pathway by NE stimulates apoptotic response in adult rat cardiac myocytes in vitro.29 To assess localization of apoptotic DNA fragmentation and cell death in control and familial DCM hiPSC‐CMs after NE stress, we utilized TUNEL and flow cytometry with annexin V fluorescein isothiocyanate and propidium iodide assays. Representative TUNEL stained images corresponded with increased TUNEL‐positive cells after NE stress in both control and familial DCM hiPSC‐CMs (Figure 5 a, left). Carvedilol and verapamil pretreatment reduced this in vitro stress‐induced apoptotic response (Figure 5 a, right). Furthermore, quantification of TUNEL‐positive loci exhibited a lower percentage of apoptotic cells after NE treatment in control than familial DCM hiPSC‐CMs (DCM baseline: 7.1 ± 4.4%; DCM post‐NE: 40.3 ± 3.7% vs. control baseline: 2.8 ± 0.7%; and control post‐NE: 30.1 ± 0.7%; Figure 5 b,c). In agreement with TUNEL‐positive staining, pretreatment with carvedilol and verapamil significantly reduced percentage of TUNEL‐positive loci in familial DCM hiPSC‐CMs after in vitro stress test (DCM carvedilol: 23.8 ± 4.5% [p < 0.01]; DCM verapamil: 15.8 ± 5.6% [p < 0.01] vs. control carvedilol: 30.9 ± 5.6%; and control verapamil: 26.1 ± 3.4%; Figure 5 b,c).
Figure 5.
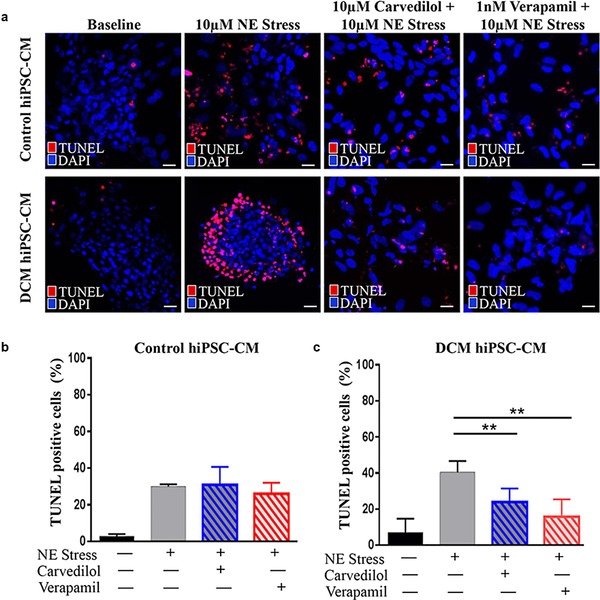
Pretreatment with carvedilol and verapamil reduces the extent of norepinephrine (NE)‐induced apoptosis. (a) Representative terminal deoxynucleotidyl transferase‐mediated deoxyuridine triphosphate nick‐end labeling (TUNEL) stained images of control and familial dilated cardiomyopathy (DCM) human‐induced pluripotent stem cell (hiPSC)‐cardiomyocytes (CMs) after NE stress. Carvedilol and verapamil reduce this in vitro stress‐induced apoptotic response. Scale bar: 20 μm. (b) Quantification of TUNEL‐positive loci in control hiPSC‐CMs exhibit a lower percentage of apoptotic cells after NE treatment compared with familial DCM hiPSC‐CMs. Carvedilol and verapamil pretreatment additionally reduced this percent disarray. (c) Familial DCM hiPSC‐CMs had a moderately increased percentage of apoptotic cells after NE stress, which was significantly reduced by carvedilol and verapamil pretreatment. **p < 0.01.
To confirm and characterize apoptosis, flow cytometry with annexin V fluorescein isothiocyanate (apoptosis marker) and propidium iodide (cell death marker) was performed. Familial DCM hiPSC‐CMs exhibited an increased percentage of apoptotic and dead cells after NE stress (DCM post‐NE dead cells: 10.71%; DCM post‐NE apoptotic cells: 10.42% vs. control post‐NE dead cells: 6.50%; and control post‐NE apoptotic cells: 3.51%; Supplementary Figure S3a, Supplementary Table S2). Indeed, pretreatment with carvedilol and verapamil increased the percent of live cells after NE stress, and reduced the percent of apoptosis in familial DCM hiPSC‐CMs (DCM carvedilol dead cells: 10.22%; DCM carvedilol apoptotic cells: 8.31% vs. DCM verapamil dead cells: 7.65%; and DCM verapamil apoptotic cells: 4.25%; Supplementary Figure S3a, Supplementary Table S2). In accordance with the TUNEL data, total annexin and propidium iodide‐positive cell count determined that pharmacological pretreatment with carvedilol and verapamil modestly reduced chronotropic stress‐induced apoptosis and cell death rate in familial DCM hiPSC‐CMs (Supplementary Figure S3c,d).
DISCUSSION
Endstage heart failure is a major burden for the individual patient and a universal challenge for health care systems. In the United States, idiopathic DCM is the most common indication for cardiac transplantation because of progressive heart failure.30 Affected individuals with familial DCM have a poor prognosis because of the high frequency of arrhythmias and sudden death. RBM20‐related DCM represents at least 1.9–3% of idiopathic DCM encountered in clinical practice.13 Since the first description of isolated DCM related to RBM20 mutations by Brauch et al.30 in 2009, more than 30 RBM20‐dependent alternatively spliced cardiac genes have been reported.31 Given its global post‐transcriptional regulation in the heart, RBM20 familial DCM is associated with excitation‐contraction coupling abnormalities as well as an increased propensity to ventricular tachycardia.
Here, we utilized familial DCM hiPSC‐CMs and compared them to healthy control hiPSC‐CMs to demonstrate increased susceptibility to β‐adrenergic stress test with NE and therapeutic rescue by pretreatment with β‐antagonist, carvedilol, and L‐type Ca2+ channel blocker, verapamil (Table 1). In vitro stress test resulted in abnormal Ca2+ handling, including increased average area under the curve, peak amplitude, characteristic time of Ca2+ extrusion, and decreased time between peaks, as well as irregular Ca2+ transient rhythm, beating activity, sarcomeric disarray, and increased apoptotic and cell death response. Pretreatment with either carvedilol or verapamil muted this response to β‐adrenergic stress, contributing to the restoration of defective Ca2+ machinery in familial DCM hiPSC‐CMs.
Table 1.
Summary of calcium handling, sarcomere disarray, and apoptosis analyses in control and familial DCM hiPSC‐CMs after pharmacological modulation
| In vitro stress test | Ca2+ modulation | ||
|---|---|---|---|
| Norepinephrine | Carvedilol | Verapamil | |
| Ca2+ handling: average area under the curve | ↑↑↑↑ | ↓↓ | ↓↓ |
| Ca2+ handling: peak amplitude | ↑↑↑↑ | ↓↓ | ↓↓ |
| Ca2+ handling: average time between peaks | ↓ | ↑↑ | ↑↑ |
| Ca2+ handling: characteristic time of extrusion | ↑↑↑ | ↓ | ↓ |
| Ca2+ handling: transient rhythm | Irregular | Irregular | Regular |
| Arrhythmia (beating activity) | Irregular | Irregular | Regular |
| Sarcomeric disarray (α‐actinin immunostaining) | ↑↑↑↑ | ↑↑ | ↑↑ |
| Cell death (TUNEL and flow cytometry) | ↑↑↑↑ | ↑↑ | ↑↑ |
| Apoptosis (flow cytometry) | ↑↑↑ | ↑↑ | ↑ |
Ca2+, calcium; TUNEL, terminal deoxynucleotidyl transferase‐mediated deoxyuridine triphosphate nick‐end labeling.
Previous studies have established the link between Ca2+ handling alterations and cardiomyopathy.9, 16, 25 Beraldi et al.25 demonstrated Rbm20‐dependent dysregulation of Ca2+ homeostasis in knockdown mouse embryoid bodies. Additionally, analysis of Ca2+ transients by fluorescence microscopy in RBM20 familial DCM hiPSC‐CMs revealed abnormalities that indicated anomalous Ca2+ handling within the cytoplasm.10 This study identified that familial DCM hiPSC‐CMs display dysregulation of Ca2+ homeostasis following in vitro stress test with NE. Indeed, changes in Ca2+‐sensitivity and subsequent alteration of Ca2+ homeostasis could be a common and an important underlying mechanism that triggers arrhythmias leading to sudden cardiac death or the development of congestive heart failure.32, 33, 34, 35 To further understand the mechanism of familial DCM and its therapies, we pharmacologically modulated the L‐type Ca2+ channel either indirectly with carvedilol or directly with verapamil.
Currently, familial DCM is clinically managed with carvedilol, a third generation nonselective β‐blocker with ancillary vasodilatory and antioxidant properties.36 Kao et al.37 have shown that in DCM ventricular reverse‐remodeling associated with β‐blockade is driven by changes in myocardial gene expression including Ca2+ handling. This finding correlates with clinical studies that suggest carvedilol not only slows deterioration of cardiac function, but also improves cardiac function.38 Moreover, the protective role of carvedilol in reducing the left ventricular end‐diastolic diameter and left ventricular fractional shortening has been described in the lamin A/C‐deficient familial DCM model after mechanical stress.39 Other studies of familial DCM because of mutations in cardiac troponin T have indicated that treatment with β‐adrenergic blockers has improved function of hiPSC‐derived cardiomyocytes.9 Consistent with these findings, our results show that carvedilol treatment led to a relatively negative chronotropic effect and an improved global Ca2+ transient on familial DCM hiPSC‐CMs. However, β‐adrenergic blockade did not ameliorate the irregular transient rhythm and beating activity after NE stress.
Prior studies have shown that inotropic agents, including isoproterenol, dobutamine, and NE, have induced arrhythmias in rat and hamster hearts because of increased intracellular Ca2+ levels.40 Indeed, Rbm20‐deficient rats and humans with RBM20 missense mutations share cardiomyopathy with arrhythmia and sudden death.16 Verapamil is an L‐type Ca2+ channel blocker that is widely used for the treatment of cardiac arrhythmias.41 Recent findings suggest that verapamil treatment reduced beating frequency by 30–40% as well as decreased the amplitude of contractions in hiPSC‐CMs.42 After β‐adrenergic stimulation, we also observed reduction in beating rate and intracellular Ca2+ relative to Ca2+‐dependent fluorescence transients in the verapamil pretreated cohort. Additionally, irregular Ca2+ transient rhythm resulting from β‐adrenergic stress was absent with verapamil pretreatment. Further studies using patch‐clamping and multiple electrode array technologies could elucidate the anti‐arrhythmogenic mechanism based on transient current and cell‐to‐cell communication.
Insights into the structural sarcomeric changes of RBM20‐related DCM are mainly based on the observations from experimental animal models.14, 16 In Rbm20‐deficient (Rbm20 −/−) and Rbm20‐insufficient (Rbm20 +/−) rat models, animals were born with normal functioning hearts, but eventually developed ventricular enlargement, arrhythmia, increased rate of sudden death, and extensive fibrosis, closely resembling to those observed in humans with heterozygous RBM20 mutations.16 The application of β‐adrenergic stimulation and chronotropic stress to familial DCM hiPSC CMs because of mutations in cardiac troponin T resulted in increased abnormal sarcomeric α‐actinin distribution.9 Our findings in RBM20 hiPSC CMs were consistent. However, while Sun et al.9 reported an increased number of CMs with punctate sarcomeric α‐actinin distribution after 1 week of 10 μM NE treatment, we observed increased number of disorganized cells after 48 h of 10 μM NE treatment. Discerning whether this early onset of phenotypic response is related to greater sensitivity of RBM20 hiPSC CMs to β‐adrenergic stress merits additional studies. Interestingly, in accordance with Ca2+ handling measures, the increased susceptibility to β‐adrenergic stimulation was limited by carvedilol or verapamil pretreatment. Indeed, we investigated the synergistic function between carvedilol and verapamil; however, the combination of both agents hindered the regulation of membrane excitability and did not illicit synchronized calcium dynamics (data not shown). Future investigations could modulate drug concentrations of carvedilol and verapamil, or additional β‐blocker and Ca2+‐channel blocker agents, to identify a possible combination therapy that would be better tolerated by hiPSC‐CMs.
In heart failure, chronic β‐adrenergic system stimulation contributes to a progression of both remodeling and apoptosis.43, 44 Furthermore, NE stimulates apoptosis in adult rat ventricular myocytes, as shown by an increased DNA laddering on agarose gel electrophoresis and increased the percentage of TUNEL‐positive cells.29 In accordance, our findings suggest that familial DCM hiPSC‐CMs have increased susceptibility to apoptosis and cell death after exposure to external chronotropic stress, and that pharmacological pretreatment with carvedilol or verapamil modestly reduced chronotropic stress‐induced apoptosis and cell death rate in familial DCM hiPSC‐CMs. In addition to negating inotropic and chronotropic actions, carvedilol also participates in antioxidant responses by mediating oxidative stress.45, 46 Supplementary studies of the role of oxidative stress and carvedilol in RBM20 hiPSC‐CMs could be informative in modifying apoptotic response and disease progression.
This study has several limitations. First, due to the lack of specific markers to allow effective sorting of different subset of cardiomyocytes, such as nodal, atrial, and ventricular myocytes, the effects of RBM20 mutation as well as their in vitro response to cell‐specific stimulation remain unclear. Second, the results of this study may not translate to RBM20‐related DCM because of other point mutations. Nevertheless, the ability to reproduce disease phenotypes of arg636‐to‐ser (R636S) substitution in this study suggests that our results should be applicable to point to mutations in the highly conserved RS domain of exon 9 in the RBM20 gene. Third, the variation in carvedilol and verapamil drug concentrations may not accurately reflect in vivo physiological dosing. It is possible that higher doses of β‐adrenergic blockade could contribute to regulating Ca2+ transient rhythm in familial DCM hiPSC‐CMs after β‐adrenergic stress. Indeed, drug screening additional β‐adrenergic blockers and Ca2+‐channel blockers using the familial DCM hiPSC‐CM platform deserves further investigation. Furthermore, establishing hiPSC‐CM maturity remains a high priority for in vitro drug testing investigations. This study utilized day 20 control and DCM hiPSC‐CMs, which were characterized by quantitative reverse transcriptase real‐time polymerase chain reaction (qRT‐PCR) transcriptional profiling data as well as microarray analysis in our recent publication.10 Given that the findings in this study are limited to quality controlled and selected clones based on efficient reprogramming, cardiac differentiation potential, and beating activity, future studies could validate these results in isogenic control hiPSC lines generated by correcting DCM mutations using genome editing tools. Despite these limitations, human pluripotent stem cell‐derived cardiomyocytes in various stages of maturity offer the opportunity to create appropriate human test models with high biological relevance in drug testing, circumventing studies with costly animal models.40, 46, 47, 48, 49
In conclusion, familial DCM hiPSC‐CMs display increased susceptibility to β‐adrenergic stimulation by defective Ca2+ homeostasis, apoptotic changes, and sarcomeric disarray, and pretreatment with carvedilol or verapamil mitigates this stress response. Accordingly, clinical studies indicate that addition of verapamil in patients with nonadvanced DCM improved their Minnesota Quality of Life score and exercise capacity in the 6‐min walk test compared with standard carvedilol‐based therapeutic management.24 Here, we provide the first comprehensive in vitro analysis of β‐adrenergic stress in RBM20‐deficient familial DCM hiPSC‐CMs, allowing for further development of therapeutic interventions that modify heart disease progression.
Author Contributions
S.P.W. and T.J.N. wrote the manuscript; S.P.W. and T.J.N. designed the research; S.P.W. and S.C.H. performed the research; S.P.W., S.C.H., S.R., A.T., T.M.O., and T.J.N. analyzed the data; S.R., A.T., and T.M.O. contributed new reagents/analytical tools.
Conflict of Interest
Mayo Clinic has financial interests and ownership in ReGen Theranostics.
Supporting information
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Supporting Information
Supporting Figure
Supporting Figure
Supporting Figure
Supporting Table
Supporting Table
Supporting Information
Acknowledgments
We are grateful for the assistance and support from Jeanne Theis, Boyd Rasmussen, Matthew Hoplin, Jonathan Nesbitt, Katherine Campbell, Jennifer Miller, and Traci Paulson. We thank Kristin Mantz and James Tarara for help with imaging. Fibroblasts and nuclear reprogramming services were provided by ReGen Theranostics in Rochester, MN. This study was supported by the National Institutes of Health (MSTP/T32/65841, NCATS/TL1/TR000137, and OD007015‐01), Todd and Karen Wanek Family Program for Hypoplastic Left Heart Syndrome, Regenerative Medicine Minnesota, and Mayo Clinic Center for Regenerative Medicine (Graduate Scholar MRM/2015/GSCH/003).
References
- 1. Maron, B.J. et al Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 14, 1807–1816 (2006). [DOI] [PubMed] [Google Scholar]
- 2. Tsirka, A.E. et al Improved outcomes of pediatric dilated cardiomyopathy with utilization of heart transplantation. J. Am. Coll. Cardiol. 44, 391–397 (2004). [DOI] [PubMed] [Google Scholar]
- 3. Hershberger, R.E. , Morales, A. & Siegfried, J.D. Clinical and genetic issues in dilated cardiomyopathy: a review for genetics professionals. Genet. Med. 12, 655–667 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Towbin, J.A. et al Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA 296, 1867–1876 (2006). [DOI] [PubMed] [Google Scholar]
- 5. Hershberger, R.E. , Hedges, D.J. & Morales, A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 10, 531–547 (2013). [DOI] [PubMed] [Google Scholar]
- 6. Fatkin, D. , Otway, R. & Richmond, Z. Genetics of dilated cardiomyopathy. Heart Fail. Clin. 6, 129–140 (2010). [DOI] [PubMed] [Google Scholar]
- 7. Hershberger, R.E. & Morales, A. Dilated cardiomyopathy overview. GeneReviews (Internet) 1993. –2016. [Google Scholar]
- 8. Takahashi, K. et al Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 (2007). [DOI] [PubMed] [Google Scholar]
- 9. Sun, N. et al Patient‐specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci. Transl. Med. 4, 130ra147 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wyles, S.P. et al Modeling structural and functional deficiencies of RBM20 familial dilated cardiomyopathy using human induced pluripotent stem cells. Hum. Mol. Genet. 25, 254–265 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Burkett, E.L. & Hershberger, R.E. Clinical and genetic issues in familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 45, 969–981 (2005). [DOI] [PubMed] [Google Scholar]
- 12. Knöll, R. et al The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell 111, 943–955 (2002). [DOI] [PubMed] [Google Scholar]
- 13. Brauch, K.M. et al Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 54, 930–941 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li, S. , Guo, W. , Dewey, C.N. & Greaser, M.L. Rbm20 regulates titin alternative splicing as a splicing repressor. Nucleic Acids Res. 41, 2659–2672 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wells, Q.S. et al Whole exome sequencing identifies a causal RBM20 mutation in a large pedigree with familial dilated cardiomyopathy. Circ. Cardiovasc. Genet. 6, 317–326 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guo, W. et al RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 18, 766–773 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harvey, P.A. & Leinwand, L.A. The cell biology of disease: cellular mechanisms of cardiomyopathy. J. Cell Biol. 194, 355–365 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harding, S.E. et al Acceleration of contraction by beta‐adrenoceptor stimulation is greater in ventricular myocytes from failing than non‐failing human hearts. Basic Res. Cardiol. 91(Suppl 2), 53–56 (1996). [DOI] [PubMed] [Google Scholar]
- 19. Yusuf, S. , Sleight, P. , Pogue, J. , Bosch, J. , Davies, R. & Dagenais, G. Effects of an angiotensin‐converting‐enzyme inhibitor, ramipril, on cardiovascular events in high‐risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N. Engl. J. Med. 342, 145–153 (2000). [DOI] [PubMed] [Google Scholar]
- 20. Dahlöf, B. et al The Losartan Intervention For Endpoint reduction (LIFE) in Hypertension study: rationale, design, and methods. The LIFE Study Group. Am. J. Hypertens. 10 (7 Pt 1), 705–713 (1997). [PubMed] [Google Scholar]
- 21. Packer, M. et al The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. U.S. Carvedilol Heart Failure Study Group. N. Engl. J. Med. 21, 1349–1355 (1996). [DOI] [PubMed] [Google Scholar]
- 22. Factor, S.M. , Cho, S.H. , Scheuer, J. , Sonnenblick, E.H. & Malhotra, A. Prevention of hereditary cardiomyopathy in the Syrian hamster with chronic verapamil therapy. J. Am. Coll. Cardiol. 12, 1599–1604 (1988). [DOI] [PubMed] [Google Scholar]
- 23. Sonnenblick, E.H. , Fein, F. , Capasso, J.M. & Factor, S.M. Microvascular spasm as a cause of cardiomyopathies and the calcium‐blocking agent verapamil as potential primary therapy. Am. J. Cardiol. 55, 179B–184B (1985). [DOI] [PubMed] [Google Scholar]
- 24. Wojnicz, R. et al Therapeutic window for calcium‐channel blockers in the management of dilated cardiomyopathy: a prospective, two‐centre study on non‐advanced disease. Cardiology 117, 148–154 (2010). [DOI] [PubMed] [Google Scholar]
- 25. Beraldi, R. et al Rbm20‐deficient cardiogenesis reveals early disruption of RNA processing and sarcomere remodeling establishing a developmental etiology for dilated cardiomyopathy. Hum. Mol. Genet. 23, 3779–3791 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Martinez‐Fernandez, A. et al iPS programmed without c‐MYC yield proficient cardiogenesis for functional heart chimerism. Circ. Res. 105, 648–656 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hodgson, D.M. et al Stable benefit of embryonic stem cell therapy in myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 287, H471–H479 (2004). [DOI] [PubMed] [Google Scholar]
- 28. Liang, P. et al Drug screening using a library of human induced pluripotent stem cell‐derived cardiomyocytes reveals disease‐specific patterns of cardiotoxicity. Circulation 127, 1677–1691 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Communal, C. , Singh, K. , Pimentel, D.R. & Colucci, W.S. Norepinephrine stimulates apoptosis in adult rat ventricular myocytes by activation of the beta‐adrenergic pathway. Circulation 98, 1329–1334 (1998). [DOI] [PubMed] [Google Scholar]
- 30. Refaat, M.M. et al Genetic variation in the alternative splicing regulator RBM20 is associated with dilated cardiomyopathy. Heart Rhythm 9, 390–396 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Maatz, H. et al RNA‐binding protein RBM20 represses splicing to orchestrate cardiac pre‐mRNA processing. J. Clin. Invest. 124, 3419–3430 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Manning, E.P. , Guinto, P.J. & Tardiff, J.C. Correlation of molecular and functional effects of mutations in cardiac troponin T linked to familial hypertrophic cardiomyopathy: an integrative in silico/in vitro approach. J. Biol. Chem. 287, 14515–14523 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Marston, S.B. How do mutations in contractile proteins cause the primary familial cardiomyopathies? J. Cardiovasc. Transl. Res. 4, 245–255 (2011). [DOI] [PubMed] [Google Scholar]
- 34. Liu, B. , Tikunova, S.B. , Kline, K.P. , Siddiqui, J.K. & Davis, J.P. Disease‐related cardiac troponins alter thin filament Ca2+ association and dissociation rates. PLoS One 7, e38259 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schober, T. et al Myofilament Ca sensitization increases cytosolic Ca binding affinity, alters intracellular Ca homeostasis, and causes pause‐dependent Ca‐triggered arrhythmia. Circ. Res. 111, 170–179 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xue, S.R. , Xue, Y. & Xue, R. Carvedilol restore cardiac calcium release channel structure and function in heart failure. Int. J. Cardiol. 116, 231–235 (2007). [DOI] [PubMed] [Google Scholar]
- 37. Kao, D.P. et al Therapeutic molecular phenotype of β‐blocker‐associated reverse‐remodeling in nonischemic dilated cardiomyopathy. Circ. Cardiovasc. Genet. 8, 270–283 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Poole‐Wilson, P.A. et al Comparison of carvedilol and metoprolol on clinical outcomes in patients with chronic heart failure in the Carvedilol Or Metoprolol European Trial (COMET): randomised controlled trial. Lancet 362, 7–13 (2003). [DOI] [PubMed] [Google Scholar]
- 39. Chandar, S. et al Effects of mechanical stress and carvedilol in lamin A/C‐deficient dilated cardiomyopathy. Circ. Res. 106, 573–582 (2010). [DOI] [PubMed] [Google Scholar]
- 40. Stefenelli, T. , Wikman‐Coffelt, J. , Wu, S.T. & Parmley, W.W. Calcium‐dependent fluorescence transients during ventricular fibrillation. Am. Heart J. 120, 590–597 (1990). [DOI] [PubMed] [Google Scholar]
- 41. Vohra, J. Verapamil in cardiac arrhythmias: an overview. Clin. Exp. Pharmacol. Physiol. Suppl. 6, 129–134 (1982). [PubMed] [Google Scholar]
- 42. Mehta, A. et al Pharmacological response of human cardiomyocytes derived from virus‐free induced pluripotent stem cells. Cardiovasc. Res. 91, 577–586 (2011). [DOI] [PubMed] [Google Scholar]
- 43. Goldspink, D.F. , Burniston, J.G. , Ellison, G.M. , Clark, W.A. & Tan, L.B. Catecholamine‐induced apoptosis and necrosis in cardiac and skeletal myocytes of the rat in vivo: the same or separate death pathways? Exp. Physiol. 89, 407–416 (2004). [DOI] [PubMed] [Google Scholar]
- 44. Krishnamurthy, P. , Subramanian, V. , Singh, M. & Singh, K. Beta1 integrins modulate beta‐adrenergic receptor‐stimulated cardiac myocyte apoptosis and myocardial remodeling. Hypertension 49, 865–872 (2007). [DOI] [PubMed] [Google Scholar]
- 45. Hakucho, A. , Liu, J. , Liu, X. & Fujimiya, T. Carvedilol improves ethanol‐induced liver injury via modifying the interaction between oxidative stress and sympathetic hyperactivity in rats. Hepatol. Res. 44, 560–570 (2014). [DOI] [PubMed] [Google Scholar]
- 46. Xu, C. et al β‐Blocker carvedilol protects cardiomyocytes against oxidative stress‐induced apoptosis by up‐regulating miR‐133 expression. J. Mol. Cell. Cardiol. 75, 111–121 (2014). [DOI] [PubMed] [Google Scholar]
- 47. Flora, S.J. & Mehta, A. Monoisoamyl dimercaptosuccinic acid abrogates arsenic‐induced developmental toxicity in human embryonic stem cell‐derived embryoid bodies: comparison with in vivo studies. Biochem. Pharmacol. 78, 1340–1349 (2009). [DOI] [PubMed] [Google Scholar]
- 48. Kola, I. & Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 3, 711–715 (2004). [DOI] [PubMed] [Google Scholar]
- 49. Roden, D.M. Drug‐induced prolongation of the QT interval. N. Engl. J. Med. 350, 1013–1022 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Supporting Information
Supporting Figure
Supporting Figure
Supporting Figure
Supporting Table
Supporting Table
Supporting Information