

Abstract
Background
Concerted hormone secretion is essential for glucose homeostasis and growth. The oocyte testis gene 1 (Otg1) has limited information in mammals before. Human OTG1 has been identified as an antigen associated with cutaneous T cell lymphoma, while worm Otg1 is recently reported to be a vesicle trafficking regulator in neurons. To understand the physiological role of Otg1 and its potential relation to hormone secretion, we characterized a mutation caused by the piggyBac transposon (PB) insertion in mice.
Results
Oocyte testis gene 1 encodes a Golgi localized protein that is expressed with a broad tissue distribution in mice. The PB insertion effectively blocks Otg1 expression, which results in postnatal lethality, growth retardation, hypoglycemia and improved insulin sensitivity in mice. Otg1 mutants exhibit decreased levels of insulin, leptin and growth hormone in the circulation and reduced hepatic IGF-1 expression. Decreased expression of Otg1 in pituitary GH3 cells causes reduced grow hormone expression and secretion, as well as the traffic of the VSVG protein marker.
Conclusions
Our data support the hypothesis that Otg1 impacts hormone secretion by regulating vesicle trafficking. These results revealed a previously unknown and important role of Otg1 in hormone secretion and glucose homeostasis in mammals.
Electronic supplementary material
The online version of this article (doi:10.1186/s13578-016-0108-4) contains supplementary material, which is available to authorized users.
Keywords: Otg1, Hypoglycemia, Hypoinsulinemia, Vesicle trafficking
Background
Glucose is the key source for energy production in mammals. Under normal physiological conditions, the blood glucose level is well regulated by concerted actions of the pancreas, liver, adipose tissue, muscle and brain [1, 2]. Abnormal glucose homeostasis would result in hyperglycemia or hypoglycemia. Hyperglycemia is the characteristic condition of diabetes, which has becoming a rapidly growing health threat in modern society [3]. Chronic hyperglycemia causes glycation of proteins or lipids, which causes many of the long-term complications in diabetic patients [4]. In contrast, since glucose supplies almost all the energy for the brain, hypoglycemia may quickly cause loss of consciousness or even death.
Peptide hormones such as insulin, glucagon, growth hormone and IGF-1 play critical roles in regulating glucose homeostasis [5–9]. Being expressed, peptide hormones are packaged in vesicles at the trans-Golgi network (TGN), transported on microtubules toward the plasma membrane and loaded onto an actin/myosin system for distal transport through the actin cortex to just below the plasma membrane. After tethered there, a subpopulation of vesicles are docked and primed to become the readily-releasable pools [10, 11]. Upon stimulation, these vesicles in the readily-releasable pool would immediately fuse to the plasma membrane to release the contents. This process is essential for activity-dependent hormone secretion to mediate various endocrinological functions. Despite of many identified proteins involved in vesicle budding, trafficking, tethering/docking and cargo secretion, the molecular mechanisms and molecules participating peptide hormone secretion remain to be explored.
The oocyte-testis gene 1 (Otg1) was originally identified in the RIKEN Mouse Gene Encyclopedia Project [12]. The full-length transcript has 16 exons that encode a 917-amino acid peptide. The OTG1 protein has several coiled-coil domains that occupy almost half of the peptide. Other than these, no functional motifs have been predicted in OTG1 [13]. The human homologue of Otg1 encodes the protein that has been recognized as a cutaneous T-cell lymphoma (CTCL) associated antigen [14]. Recently, the C. elegans homologue of Otg1 has been reported as a vesicle trafficking regulator in neurons [15]. Here we report that mouse Otg1 encodes a protein with prominent Golgi localization. Loss of Otg1 results in postnatal lethality, aberrant glucose homeostasis and defective hormone secretion in mice. These results revealed an unknown role of Otg1 in participating hormone secretion and metabolic regulation in mammals.
Results
Disruption of Otg1 results in postnatal lethality and growth retardation in mice
We identified an Otg1 mutant in a screen for mice bearing metabolic defects [16]. The mutant carries a piggyBac transposon (PB) insertion in the eighth exon of Otg1 (Otg1PB) that effectively disrupts gene expression (Fig. 1a). In wild-type animals, Otg1 proteins can be readily detected in various organs such as the brain, heart, lung, stomach, liver, kidney, pancreas and gut. In tissues from homozygous mutants, Otg1 expression is no longer detectable (Fig. 1b). Similar changes were also observed by immunofluorescence staining in pancreatic cells and embryonic fibroblasts (MEFs) with different genotypes (Fig. 1c and Additional file 1: Figure S1). Similar as that of the C. elegans homologue, immunofluorescence staining in wild-type pancreatic cells and MEFs revealed co-localization of OTG1 proteins with the Golgi compartment marker Giantin, confirming a Golgi localization of OTG1 in mice (Fig. 1c) [15, 17].
Fig. 1.
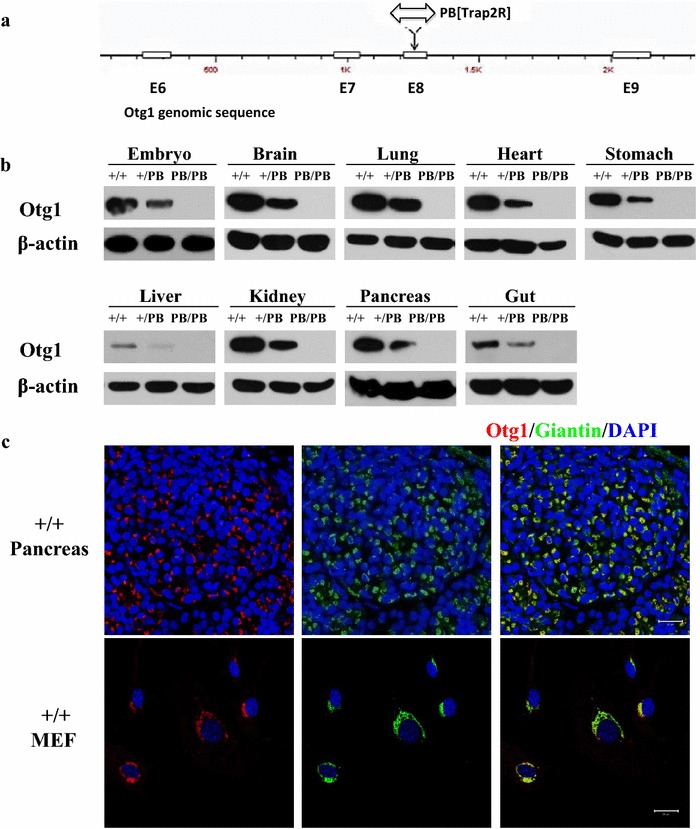
A PB insertion effectively abolished Otg1 expression. a Schematic representation of the genomic sequence flanking PB insertion site in Otg1. White box: exon. b Western blot did not detect OTG1 expression in either E13.5 Otg1 PB/PB embryos or tissues of P1 Otg1 PB/PB mice. c Immunofluorescence staining showed co-localization of OTG1 protein and Giantin in both pancreatic tissues of MEFs from wild-type mice
Oocyte testis gene 1PB/PB animals were born in consistent with a Mendelian pattern of inheritance (Additional file 2: Table S1). However, 46.5 % (53/114) homozygous Otg1 mutants could not survive throughout the first day after birth (P1), while others gradually died within the next 30 days. In contrast, 96.4 % (108/112) wild-type and 94.2 % (196/208) heterozygous littermates kept alive at the age of one month (Fig. 2a). The external morphology of mutants died at P1 was apparently normal. In contrast, the most obvious morphological changes of other dead mutants were their small sizes (Fig. 2c). Further analysis showed that Otg1PB/PB individuals had comparable body weight with their littermates both at the end of fetal development (embryonic day 18.5, Additional file 3: Figure S2C) and at birth (Fig. 2b). Soon after that, the survivors suffered from severe growth retardation. The body weight of Otg1PB/PB mice increased much slower than that of the wild-type and heterozygous littermates. In fact, homozygous Otg1 mutants always exhibit lipohypotrophy and usually died before the body weight reaches 5 grams (Fig. 2b, d).
Fig. 2.
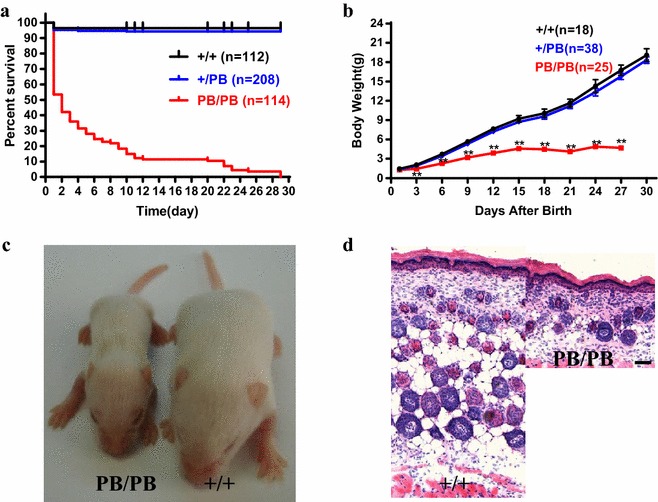
Postnatal lethality and growth retardation in Otg1 mutant mice. a Survival curves of Otg1 PB/PB (n = 114), Otg1 PB/+ (n = 208) and wild-type (n = 114) littermates. b Body weight curves of Otg1 PB/PB (n = 25), Otg1 PB/+ (n = 38) and wild-type (n = 18) littermates. c Representative image showing the body size difference between an Otg1 PB/PB and a wild-type mouse at P11. d Representative image of the hypodermis histological section of P4 Otg1 PB/PB and Otg1 +/+ mice, showing severe lipodystrophy of the mutant
Otg1 mutation leads to impaired glucose homeostasis
We then explored pathophysiological alterations that may lead to postnatal lethality and growth retardation in Otg1PB/PB mice. Alcian blue-alizarin red staining revealed normal skeleton structures in Otg1PB/PB mutants (Additional file 3: Figure S2A). This result, along with the normal body weight of newborn animals, suggests that severe embryonic developmental defects, such as abnormal pattern formation, shall not be accounted for postnatal lethality and growth retardation. We often observed milk in the stomach of Otg1PB/PB pups, suggesting that feeding failure is unlikely the reason for growth retardation and lethality (Additional file 3: Figure S2B).
Given the critical role of glucose in supporting growth, we examined glucose homeostasis in mutant mice. We observed progressively developed hypoglycemia in Otg1 mutants. The blood glucose level in free–fed Otg1PB/PB mice was similar as that in the wild-type and heterozygous littermates at birth, then decreased to approximately 25 % blow normal within two days and further dropped to 58 % of that in the wild-type at the age of 11 days (P11) (Fig. 3a, b). Fasted blood glucose levels of P11 Otg1PB/PB mice were only 47 % of that in the wild-type mice. In intraperitoneal glucose tolerance test (IPGTT), the blood glucose level of P11 Otg1PB/PB mice changed with the same tendency as that of the wild-type or heterozygous mice, but kept to be approximately 60 % lower at each time point (Fig. 3c). In addition to hypoglycemia, we recorded extremely low level of serum insulin and elevated insulin sensitivity in the mutants. Compared with 0.66 and 0.57 ng/ml of serum insulin detected in wild-type and heterozygous littermates, respectively, ELISA analysis revealed an average insulin concentration of 0.02 ng/ml in P11 homozygotes (Fig. 3d). In the insulin tolerance test (ITT), the blood glucose level of P11 Otg1PB/PB mice dropped more rapidly than that of the wild-type and heterozygous littermates. In fact, it decreased to a level too low to be detected within 30 min (Fig. 3e). Finally, we observed hypoleptinemia in Otg1 mutants. Consistent with lipodystrophy, Otg1PB/PB mice had circulating leptin below detectable level (<0.2 ng/ml) at P11 (Fig. 3f).
Fig. 3.

Impaired glucose homeostasis in Otg1 mutant mice. a Blood glucose levels of newborn Otg1 PB/PB (P1: n = 13; P2: n = 10), Otg1 PB/+ (P1: n = 30; P2: n = 30) and wild-type (P1: n = 16; P2: n = 11) littermates. b Blood glucose levels of free fed or 2-hour starved Otg1 PB/PB (n = 10), Otg1 PB/+ (n = 19) and wild-type (n = 10) littermates at P11. c Intraperitoneal glucose tolerance test (IPGTT) performance of Otg1 PB/PB (n = 4), Otg1 PB/+ (n = 6) and wild-type (n = 3) littermates at P11. d Serum insulin levels of Otg1 PB/PB (n = 10), Otg1 PB/+ (n = 19) and wild-type (n = 10) littermates at P11. e Insulin tolerance test (ITT) results of Otg1 PB/PB (n = 4), Otg1 PB/+ (n = 8) and wild-type (n = 5) littermates at P11. f Serum leptin levels of Otg1 PB/PB (n = 7), Otg1 PB/+ (n = 5) and wild-type (n = 4) littermates. g Serum growth hormone levels of Otg1 PB/PB (n = 17) and wild-type (n = 15) littermates. h Relative IGF-1 expression in Otg1 PB/PB (P1: n = 5; P2: n = 4) and wild-type (P1: n = 3; P11: n = 5) littermates. *p < 0.05, **p < 0.01, ***p < 0.005
Growth retardation, hypoinslulinemia, hypoglycemia and increased insulin sensitivity have been reported in mice with defective growth hormone receptors (GHRs) [7, 9]. This raises the possibility that growth hormone (GH) signaling is aberrant in Otg1PB/PB mice. We measured the expressions of GH and its downstream mediator IGF-1. Compared with those of the wild-type littermates, ELISA revealed approximately 35 % reduction of serum GH levels in P11 Otg1PB/PB mice, while real-time RT-PCR detected 50 and 87 % decrease of hepatic IGF-1 expression in P1 and P11 mutants, respectively (Fig. 3g, h). Taken together, the results above suggest that disruption of Otg1 leads to impaired glucose homeostasis in mice.
Otg1 mutation leads to aberrant vesicle trafficking
Given its Golgi localization and the reported role of the C. elegans homologue in neurons [15], Otg1 is likely to be involved in vesicle trafficking, a process that is critical for protein hormone secretion in mammals. Consistent with this predicted role, we found Otg1PB/PB mice had islet cells 48 % of the sizes of their wild-type littermates (Fig. 4a, b). This is likely the consequence of defective vesicle trafficking rather than the result of smaller body size, since Otg1PB/PB hepatocytes are of similar sizes as those of the Otg1+/PB animals (Additional file 4: Figure S3). Electron microscopy also revealed a greatly reduced number of insulin granules in the cytoplasm of mutant β cells (Fig. 4c). To mask the possible effect on insulin secretion from reduced GH signaling in vivo, we isolated islets from newborn mutants and examined their response to glucose challenge by measuring secreted insulin in the culture medium. There was no significant difference between Otg1PB/PB and the wild-type islets when they were provided with basal level (3 mM) of glucose. However, when challenged by 25 mM of glucose for 1 h, the insulin concentration in the culture medium of Otg1PB/PB islets was only 25 % of that of the wild-type islets (Fig. 4d). Altogether, these results suggest that the smaller islet cells are likely the result of defective vesicle trafficking caused by the Otg1 mutation.
Fig. 4.

Decreased Otg1 expression leads to impaired hormone secretion. a H&E staining of pancreas from P11 Otg1 PB/PB and wild-type littermates. b Relative pancreas islet cell sizes of P11 Otg1 PB/PB (n = 31) and wild-type (n = 20) littermates. c Scanning EM of islet cells of P6 Otg1 PB/PB and wild-type littermates. White arrows indicate insulin granules. N: nucleus. G: Golgi. d Insulin levels of glucose stimulated insulin secretion (GSIS) after isolation of islets isolated from Otg1 PB/PB (n = 7) and wild-type (n = 8) littermates at P1. e Western blot showed the decreased Otg1 expression in GH3 cells transfected with small hairpin RNAs, but not scramble shRNAs. GAPHD serves as the loading control. f Released growth hormone levels in Otg1 knockdown cells compared with cells transfected with scramble shRNAs, which is arbitrary set as 1. g Growth hormone levels in Otg1 knockdown cells compared with cells transfected with scramble shRNAs, which is arbitrary set as 1. *p < 0.05, **p < 0.01, ***p < 0.005
The role of Otg1 in hormone secretion was further confirmed in rat pituitary GH3 somatolactotropes, a popular model to study GH secretion [18]. We first knocked down Otg1 expression with small hairpin RNAs (shRNAs), then measured GH released into the medium within a period of two hours. Compared with cells transfected with scramble shRNAs, shRNA-1 and shRNA-2 transfected cells produced 55 and 58 % less Otg1 proteins, respectively (Fig. 4e). As expected, shRNA-1 transfection resulted in a reduction of GH content and secretion by 52 and 58 %, respectively, while shRNA-2 transfection resulted in a reduction of GH content and secretion by 43 and 56 %, respectively (Fig. 4f, g).
Decreased hormone secretion in both mouse islet and rat GH3 suggested a critical role of Otg1 in hormone secretion in mammals. To monitor the effect of Otg1 on intracellular transport, we used VSVG-mEmerald, a fluorescent reporter that translocates from endoplasmic reticulum to the plasma membrane via the Golgi apparatus at 37 °C [19]. Live-cell imaging showed that the transportation of VSVG-mEmerald was significantly blocked in Otg1 knockdown GH3 cells (Additional file 5: Video S1 and Additional file 6: Video S2). The average transport speed of VSVG-mEmerald vesicles (n = 110) was 0.068 μm/sec in shRNA-1 treated cells, but 0.186 μm/sec in scramble shRNA treated cells (Fig. 5a). Standard deviations of the transport speed in each shRNA-1 treated cell (n = 11) were also decreased (Fig. 5b and Additional file 7: Figure S4). These results suggest that Otg1 is required for vesicle trafficking in mammalian cells.
Fig. 5.
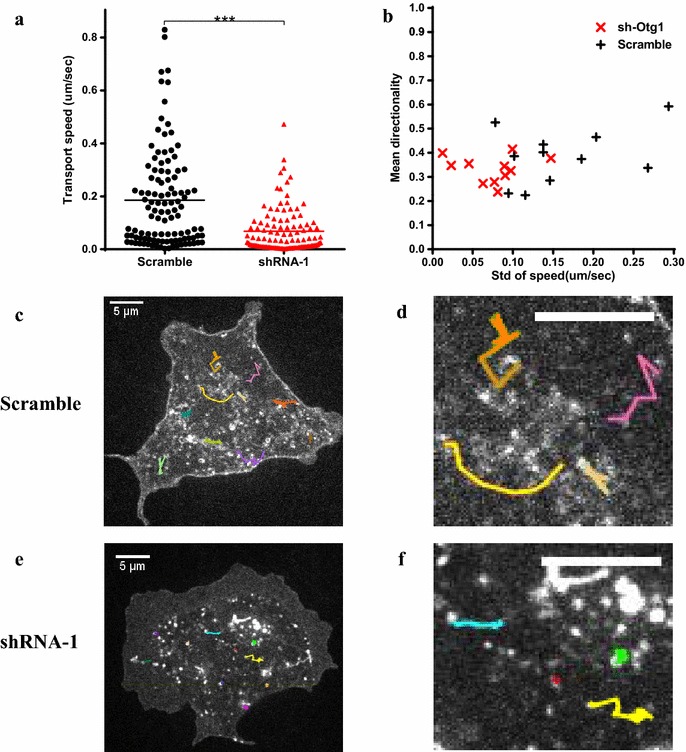
Blocked intracellular transport in Otg1 knockdown cells. GH3 cells were co-transfected with VSVG-mEmerald and Otg1 shRNA or scramble shRNA. a Scatter plot showing decreased transport speed of VSVG-mEmerald vesicles in Otg1 knockdown (red triangles) and scramble shRNA treated (black dots) GH3 cells. Eleven cells were scored for each group. Ten vesicles were randomly picked from each cell. b Scatter plot showing Otg1 knockdown resulted in decreased standard deviation of transport speed (std speed) and unaltered directionality of VSVG-mEmerald vesicles in each cell. Data of vesicles selected in (a) were used for the calculation. c, d A representative image (c) and its close-up (d) showing tracked movement of 10 VSVG-mEmerald vesicles (lines in color) in scramble shRNA treated GH3 cells. (e, f) A representative image (e) and its close-up (f) showing tracked movement of 10 VSVG-mEmerald vesicles (lines in color) in Otg1 knock down GH3 cells. Scale bars 5 μm. *p < 0.05, **p < 0.01, ***p < 0.005
Discussion
In the present study, we have shown that Otg1 encodes a Golgi protein that is required for normal vesicle trafficking in mammalian cells. Disruption of Otg1 results in growth retardation and postnatal lethality in mice. Various abnormalities related to glucose homeostasis, such as hypoinslulinemia, hypoglycemia, increased insulin sensitivity, decreased serum growth hormone level and reduced hepatic IGF-1 expression, could be observed in mutant animals before death. These results revealed an unknown function Otg1 in metabolic regulation.
Oocyte testis gene 1 is ubiquitously expressed with a prominent Golgi localization. Almost half of the peptide sequence is occupied by coiled-coil regions with short interruptions. OTG1 is a conserved protein during evolution and the human homologue has been identified as a tumor antigen. These features are reminiscent of those of the golgin coiled-coil proteins, which are known as membrane and cytoskeleton tethers [20, 21]. Although the C-terminus of OTG1 lacks a transmembrane or a small GTPase interacting signal, which are usually presented in a typical goglin, OTG1 may still be involved in similar intracellular activities by serving as molecular partners of typical golgins. Under this scenario, OTG1 may be involved not only in capturing incoming vesicles, but also in providing specificity to the tethering step.
Hypoglycemia is normal during the first hours of mammalian life. However, prolonged neonatal hypoglycemia would cause long-term neuronal deficits [22]. In contrast to extensively recognized hyperinsulinemic hypoglycemia, hypoinsunlinemic hypoglycemia is an extrememly rare condition in human. Limited cases of hypoinsunlinemic hypoglycemia are usually related to impaired insulin signaling pathway. For example, a hyperactive mutation of AKT2, the gene required for insulin-induced translocation of GLUT4 to the plasma membrane, caused hypoinsunlinemic hypoglycemia in four patients [23, 24]. On the other hand, non-islet cell tumor-induced hypoglycemia (NICTH) is caused by the secretion of incompletely processed precursors of IGF-II, which has an insulin-like hypoglycaemic activity [25]. We have shown that disruption of Otg1 caused hypoinsunlinemic hypoglycemia in mice, which implies a possible role of Otg1 mutations in human patients. The fact that Otg1 mutation blocked vesicle trafficking also suggests a new etiology of this rarely observed disease condition. Examine other regulators of vesicle trafficking in human patients may identify more causative mutations of hypoinsunlinemic hypoglycemia in the future.
The mechanism through which Otg1 modulates vesicle trafficking remains to be investigated. The C. elegans homologue of Otg1 works as a partner of Rab-2 and Rund-1 in regulating neuronal vesicle trafficking [15]. However, mutations of the homologues of Rab-2 (Rab-2a and Rab-2b) or Rund-1 (Rundc-1) showed different phenotypes from that of Otg1 mutants in mice [26, 27]. Unlike the C. elegans mutant, Otg1PB/PB mice did not show gross behavioral defects. The observation that Otg1PB/PB pups are capable of sucking milk suggests they may have normal neuronal functions (Additional file 3: Figure S2B). In addition, the human orthologue of Otg1 is a tumor antigen. Thus, further studies of Otg1 may not only shed light on the mechanisms of vesicle trafficking in mammals, but also contribute to the study of related diseases such as metabolic abnormalities or cancer.
Conclusions
Our results revealed an essential role of Otg1 in vesicle trafficking, which is critical for peptide hormone secretion, metabolic regulation and postnatal survival in mice.
Methods
Mice
All animal experiments were performed in accordance with protocols approved by the Animal Care and Use Committee of the Institute of Developmental Biology and Molecular Medicine (IDM), Fudan University. The Otg1 mutant strain (H66eR12) was generated on the FVB/NJ background and maintained on 12/12-hour light/dark cycles. The Otg1 mutation carried by H66eR12 was induced with a piggyBac transposon (PB) insertion in the eighth exon. Mapping information of the PB insertion in Otg1 and the mutant genotyping protocol could be found from the PBmice database [16]. All assays were performed in a mixed population of both males and females.
Metabolic assays
Blood glucose levels were analyzed with Glucometer Elite (LifeScan). For glucose tolerant tests (GTTs), animals were fasted two hours before receiving intraperitoneal injection of 20 % glucose saline solutions (2 g glucose per kg body weight). Tail vein blood was then sampled at 0, 15, 30, 60, 90 and 120 min after injection for blood glucose tests. For insulin tolerance tests (ITTs), 2-hour fasted mice received an intraperitoneal injection of insulin (Humulin, Lilly) (0.75 U/kg body weight), then had tail vein blood glucose levels measured at 0, 15, 30, 45 and 60 min later. ELISA was performed following the manufacturer’s protocol to measure serum insulin (Crystal Chem Inc.), leptin (Crystal Chem Inc.) and growth hormone (Millipore) concentrations. All samples were collected from female mice at the age of P11.
Islet culture and glucose stimulated insulin secretion (GSIS)
Pancreatic islets were isolated by collagenase perfusion in situ, digested for 28 min and then purified by single layer histopaque (Sigma). Isolated islets from different mice were mixed and cultured in RPMI 1640 medium supplemented with 11 mM glucose, 7.5 % FCS and 10 mM HEPES (Sigma). For GSIS assay, islets were washed in PBS and incubated in a 96-well plate with glucose-free Krebs–Ringer bicarbonate (KRB) medium (125 mM NaCl, 4.74 mM KCl, 1 mM CaCl2, 1.2 mM KH2PO4, 1.2 mM MgSO4, 5 mM NaHCO3, 25 mM HEPES, pH 7.4, with 0.1 % BSA) at 37 °C for 30 min, then incubated in KRB containing 3 mM or 25 mM glucose at 37 °C for 30 min with 5 islets per well. The amount of insulin released into the incubation medium in each well was assayed using ELISA (Crystal Chem Inc.). At least 5 wells were examined for each genotype with each glucose concentration.
Western blot
Protein extraction was prepared with the RIPA lysis buffer and quantified with the BCA Protein Assay Kit (Pierce). Equal amounts of samples were separated by SDS/PAGE, transferred onto PVDF membranes (Millipore) and immunoblotted following standard protocols. The antibodies used were: rabbit anti-OTG1 (Sigma HPA018019, 1:1000), mouse anti-GAPDH (KangCheng Biotech KC-5G4, 1:10,000) rabbit anti-β-actin (Santa Cruz sc-1616-R, 1:2000), goat anti-mouse IgG-HRP (Santa Cruz sc-2005, 1:5000) and goat anti-rabbit IgG-HRP (Santa Cruz sc-2004, 1:5000).
Histology and immunohistochemistry
Frozen sections were prepared by fixing tissues overnight in 4 % paraformaldehyde, followed by cryoprotection in 30 % sucrose at 4 °C for two days and sectioning with given thickness for histological and immunofluorescence analysis. For morphological analysis, Section (5 μm) were stained with hematoxylin and eosin to have images acquired with a Leica DMRXA2 microscope. For immunofluorescence analysis, Section (6–8 μm) were treated following the standard protocol with following antibodies: rabbit anti-OTG1 (Sigma HPA018019, 1:1000), Alexa 488 conjugated rabbit-anti-Giantin (Covance A488-114L, 1:1000), rabbit anti-insulin (Santa Cruz sc-9168, 1:1000), goat anti-glucagon (Santa Cruz sc-7780, 1:1000), donkey anti-goat IgG-FITC (Chemicon AP180F, 1:2000), donkey anti-rabbit IgG-Cy3 (Millipore AP182C, 1:2000).
Electron microscopy
Pancreas tissues were fixed in a fresh fixative solution consisting of 2 % glutaraldehyde and 4 % paraformaldehyde, postfixed with 1 % osmium tetroxide in phosphate buffer at 4 °C and dehydrated in ascending concentrations of methanol and propylenoxide before being embedded in Epoxy resin. Ultra-thin sections were prepared using a Reichert ultramicrotome, contrasted with uranyl acetate and lead citrate and examined under a Philips CM120 electron microscope.
Cell culture and live imaging
Rat pituitary GH3 cells were cultured at 37 °C in a humidified atmosphere containing 95 % air and 5 % CO2. The culture medium was DMEM supplemented with 10 % fetal bovine serum, 100 U/ml penicillin and 100 mg/ml streptomycin. GH3 cells were transfected with small hairpin RNA (shRNA) constructs (Sigma-Aldrich) or the VSVG-mEmerald plasmid (modified from Addgene plasmid #31947) with Lipofectamine 2000 (Invitrogen). For growth hormone secretion assay, cells were transferred to serum free medium 48 h after transfection and incubated for 2 h before ELISA. Live cell confocal images were acquired 48 h after transfection, using spinning disk confocal scan head (CSU-X/M2 N, Yokogawa) attached to an inverted microscope (IX-81, Olympus) and an EMCCD camera (DU897BV, Andor) controlled by Micro-Manager software. Images (512 × 512 pixels, voxel size 0.0946 μm/pixel) were taken every 0.5 s for 400 frames. Live images were analyzed in NIH ImageJ with the MTrackJ plugin. VSVG containing vesicles (10/cell) were randomly selected in 11 Otg1 knockdown cells and 11 scramble shRNAs treated cells. Directionality of each vesicle was defined as its real transport distance divided by linear distance between the start and end positions.
Statistics
GSIS data were compared by two-way ANOVA analysis. All other data were compared by unpaired two-tailed Student’s t test. Results were shown as mean ± SEM. P < 0.05 was considered statistically significant.
Authors’ contributions
GW characterized the physiological and metabolic alterations of the animals, participated in islet and cell culture experiments. RL participated in physiological and metabolic phenotyping, carried out molecular genetic experiments. YY and LC characterized vesicle trafficking alterations in GH3 cells. SD mapped the mutations and initially identified the lethality phenotype. MH and TX participated in the design of the study and helped to analyze the results. XW conceived of the study and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We thank Dr. Lei Xue for stimulating discussions, Xiaoping Huang, Guoying Ma, Yanfeng Tan, Guicheng Wang and Yanqian Xia for technical assistance.
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
All animal procedures applied in this article have been approved by the Animal Care and Use Committee of the Institute of Developmental Biology and Molecular Medicine, Fudan University, Shanghai, China.
Sources of funding
This work was supported in part by grants from Chinese Hi-tech Research and Development Project (863) [2014AA021104], Natural Science Foundation of China (NSFC) [81170789, 81570756] and Shanghai Municipal Government (15XD1500500 and 12431900100). YY has been supported by the Fudan Undergraduate Research Opportunities Program (FDUROP) and the National Top Talent Undergraduate Training Program (NTTUTP). MH and TX are Howard Hughes Medical Institute investigators.
Availability of data and materials
Not applicable,because we do not have any readily reproducible materials in the manuscript, no new software, no databases and relevant raw data are already included in the text and figures for the reviewers.
Abbreviations
- CTCL
cutaneous T-cell lymphoma
- GH
growth hormone
- GHR
growth hormone receptor
- GSIS
islet culture and glucose stimulated insulin secretion
- GTT
glucose tolerant test
- IDM
The Institute of Developmental Biology and Molecular Medicine
- IPGTT
intraperitoneal glucose tolerance test
- ITT
insulin tolerance test
- MEF
embryonic fibroblast
- NICTH
non-islet cell tumor-induced hypoglycemia
- Otg1
oocyte testis gene 1
- PB
piggyBac
- shRNA
small hairpin RNA
- TGN
trans-Golgi network
Additional files
10.1186/s13578-016-0108-4 Otg1 expression is disrupted by the PB insertion. In contrast to Fig. 1c, immunofluorescence staining showed Giantin (green) but not Otg1 (red) signals in pancreatic tissues and MEFs from Otg1 PB/PB mice.
10.1186/s13578-016-0108-4 Genotypes of new born animals.
10.1186/s13578-016-0108-4 Otg1 mutant mice have no severe developmental defects. (A) Alcian blue-alizarin red staining of P6 Otg1 +/PB and Otg1 PB/PB mice. (B) Representative image showing P6 wild-type and Otg1 PB/PB littermates, with sucked milk in the stomach. (C) Average body weight of Otg1 PB/PB (n = 6), Otg1 PB/+ (n = 15) and wild-type (n = 15) littermates at E18.5.
10.1186/s13578-016-0108-4 Otg1 mutation does not affect hepatocyte size. (A) H&E staining of liver sections from P11 Otg1 PB/PB and Otg1PB/+ littermates. (B) Relative hepatocytes size of P11 Otg1 PB/PB (n = 3) and Otg1 PB/+ (n = 4) littermates.
10.1186/s13578-016-0108-4 GH3 cells co-transfected with VSVG-mEmerald and scramble shRNA.
10.1186/s13578-016-0108-4 GH3 cells co-transfected with VSVG-mEmerald and Otg1 shRNA-1.
10.1186/s13578-016-0108-4 Otg1 knockdown blocks vesicle transportation in GH3 cells. (A) Unaltered directionality between Otg1 knockdown (red x) and scramble shRNA treated (black crosses) GH3 cells shown in Fig. 5b. (B) Decreased standard deviation of transport speed of cells shown in Fig. 5b. ***p < 0.005.
Footnotes
Guangxue Wang, Rongbo Li and Ying Yang contributed equally to this work
Contributor Information
Guangxue Wang, Email: guangxue_wang@fudan.edu.cn.
Rongbo Li, Email: rongboli@fudan.edu.cn.
Ying Yang, Email: 12307110380@fudan.edu.cn.
Liang Cai, Email: cail@fudan.edu.cn.
Sheng Ding, Email: sheng_ding@fudan.edu.cn.
Tian Xu, Email: tian.xu@yale.edu.
Min Han, Email: mhan@colorado.edu.
Xiaohui Wu, Email: xiaohui_wu@fudan.edu.cn.
References
- 1.Tirone TA, Brunicardi FC. Overview of glucose regulation. World J Surg. 2001;25(4):461–467. doi: 10.1007/s002680020338. [DOI] [PubMed] [Google Scholar]
- 2.Herman MA, Kahn BB. Glucose transport and sensing in the maintenance of glucose homeostasis and metabolic harmony. J Clin Investig. 2006;116(7):1767–1775. doi: 10.1172/JCI29027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roder PV, Wu B, Liu Y, Han W. Pancreatic regulation of glucose homeostasis. Exp Mol Med. 2016;48:e219. doi: 10.1038/emm.2016.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahmed N. Advanced glycation endproducts–role in pathology of diabetic complications. Diabetes Res Clin Pract. 2005;67(1):3–21. doi: 10.1016/j.diabres.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 5.Duvillie B, et al. Phenotypic alterations in insulin-deficient mutant mice. Proc Natl Acad Sci USA. 1997;94(10):5137–5140. doi: 10.1073/pnas.94.10.5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vuguin PM, et al. Ablation of the glucagon receptor gene increases fetal lethality and produces alterations in islet development and maturation. Endocrinology. 2006;147(9):3995–4006. doi: 10.1210/en.2005-1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou Y, et al. A mammalian model for Laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse) Proc Natl Acad Sci USA. 1997;94(24):13215–13220. doi: 10.1073/pnas.94.24.13215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lupu F, Terwilliger JD, Lee K, Segre GV, Efstratiadis A. Roles of growth hormone and insulin-like growth factor 1 in mouse postnatal growth. Dev Biol. 2001;229(1):141–162. doi: 10.1006/dbio.2000.9975. [DOI] [PubMed] [Google Scholar]
- 9.Liu JL, et al. Disruption of growth hormone receptor gene causes diminished pancreatic islet size and increased insulin sensitivity in mice. Am J Physiol Endocrinol Metab. 2004;287(3):E405–E413. doi: 10.1152/ajpendo.00423.2003. [DOI] [PubMed] [Google Scholar]
- 10.Michael DJ, Cai H, Xiong W, Ouyang J, Chow RH. Mechanisms of peptide hormone secretion. Trends Endocrinol Metab TEM. 2006;17(10):408–415. doi: 10.1016/j.tem.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 11.Park JJ, Loh YP. How peptide hormone vesicles are transported to the secretion site for exocytosis. Mol Endocrinol. 2008;22(12):2583–2595. doi: 10.1210/me.2008-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawai J, et al. Functional annotation of a full-length mouse cDNA collection. Nature. 2001;409(6821):685–690. doi: 10.1038/35055500. [DOI] [PubMed] [Google Scholar]
- 13.Oduru S, et al. Gene discovery in the hamster: a comparative genomics approach for gene annotation by sequencing of hamster testis cDNAs. BMC Genom. 2003;4(1):22. doi: 10.1186/1471-2164-4-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartmann TB, Thiel D, Dummer R, Schadendorf D, Eichmuller S. SEREX identification of new tumour-associated antigens in cutaneous T-cell lymphoma. Br J Dermatol. 2004;150(2):252–258. doi: 10.1111/j.1365-2133.2004.05651.x. [DOI] [PubMed] [Google Scholar]
- 15.Ailion M, et al. Two Rab2 interactors regulate dense-core vesicle maturation. Neuron. 2014;82(1):167–180. doi: 10.1016/j.neuron.2014.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.PiggyBac Mutagenesis Information Center. http://www.idmshanghai.cn/PBmice/. Accessed 21 Nov. 2012.
- 17.Linstedt AD, Hauri HP. Giantin, a novel conserved Golgi membrane protein containing a cytoplasmic domain of at least 350 kDa. Mol Biol Cell. 1993;4(7):679–693. doi: 10.1091/mbc.4.7.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamashita H, et al. A rat pituitary tumor cell line (GH3) expresses type I and type II receptors and other cell surface binding protein(s) for transforming growth factor-beta. J Biol Chem. 1995;270(2):770–774. doi: 10.1074/jbc.270.2.770. [DOI] [PubMed] [Google Scholar]
- 19.Toomre D, Keller P, White J, Olivo JC, Simons K. Dual-color visualization of trans-Golgi network to plasma membrane traffic along microtubules in living cells. J Cell Sci. 1999;112(Pt 1):21–33. doi: 10.1242/jcs.112.1.21. [DOI] [PubMed] [Google Scholar]
- 20.Gillingham AK, Munro S. Finding the Golgi: golgin coiled-coil proteins show the way. Trends Cell Biol. 2016;26(6):399–408. doi: 10.1016/j.tcb.2016.02.005. [DOI] [PubMed] [Google Scholar]
- 21.Witkos TM, Lowe M. The Golgin Family of Coiled-Coil Tethering Proteins. Front Cell Dev Biol. 2016;11(3):86. doi: 10.3389/fcell.2015.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adamkin DH. Neonatal hypoglycemia. Curr Opin Pediatr. 2016;28(2):150–155. doi: 10.1097/MOP.0000000000000319. [DOI] [PubMed] [Google Scholar]
- 23.Arya VB, et al. Activating AKT2 mutation: hypoinsulinemic hypoketotic hypoglycemia. J Clin Endocrinol Metab. 2014;99(2):391–394. doi: 10.1210/jc.2013-3228. [DOI] [PubMed] [Google Scholar]
- 24.Hussain K, et al. An activating mutation of AKT2 and human hypoglycemia. Science. 2011;334(6055):474. doi: 10.1126/science.1210878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dutta P, et al. Non-islet cell tumor-induced hypoglycemia: a report of five cases and brief review of the literature. Endocrinol Diabetes Metab Case Rep. 2013;2013:130046. doi: 10.1530/EDM-13-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zambrowicz BP, et al. Wnk1 kinase deficiency lowers blood pressure in mice: a gene-trap screen to identify potential targets for therapeutic intervention. Proc Natl Acad Sci USA. 2003;100(24):14109–14114. doi: 10.1073/pnas.2336103100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.(MGP) MGIatWTSIMGP (2011) Obtaining and loading phenotype annotations from the Wellcome Trust Sanger Institute (WTSI) Mouse Resources Portal. Database Release.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable,because we do not have any readily reproducible materials in the manuscript, no new software, no databases and relevant raw data are already included in the text and figures for the reviewers.