

Abstract
Cullin-RING ligases (CRLs), the largest E3 ubiquitin ligase family, promote ubiquitination and degradation of various cellular key regulators involved in a broad array of physiological and pathological processes, including cell cycle progression, signal transduction, transcription, cardiomyopathy, and tumorigenesis. Autophagy, an intracellular catabolic reaction that delivers cytoplasmic components to lysosomes for degradation, is crucial for cellular metabolism and homeostasis. The dysfunction of autophagy has been proved to associate with a variety of human diseases. Recent evidences revealed the emerging roles of CRLs in the regulation of autophagy. In this review, we will focus mainly on recent advances in our understandings of the regulation of autophagy by CRLs and the cross-talk between CRLs and autophagy, two degradation systems. We will also discuss the pathogenesis of human diseases associated with the dysregulation of CRLs and autophagy. Finally, we will discuss current efforts and future perspectives on basic and translational research on CRLs and autophagy.
Keywords: CRL E3 ligase, UPS, Autophagy, mTOR, Ubiquitin, NEDD8, ATG
Background
The long-term health of a cell is closely associated with protein quality control which requires a well-regulated balance between protein synthesis and degradation [1]. It is critical for the maintenance of cellular homeostasis to eliminate unwanted and aberrant intracellular proteins, which is charged by both the ubiquitin–proteasome system (UPS) and the autophagy–lysosome system in a coordinated manner [2]. Thus, the dysregulation of UPS and autophagy disrupts cellular homeostasis and causes many human diseases, such as heart failure, neurodegeneration, and cancer [3, 4].
The UPS, a clearance system, directs target proteins with their lysine residues and the N-terminal methionine residue covalently attached by ubiquitin molecules, to the 26S proteasome for degradation, leading to the elimination of short-lived, misfolded, and damaged proteins [5–8]. Protein ubiquitination is a trio of enzymatic steps mediated by E1 (ubiquitin-activating enzyme), E2 (ubiquitin-conjugating enzyme), and E3 (substrate-specific ubiquitin ligase) [9]. First, ubiquitin is activated in an ATP-dependent reaction catalyzed by E1. Second, the activated ubiquitin is transferred to the active site of an E2. Finally, an E3, which recognizes and recruits the target protein, designated as substrate, mediates the transfer of the activated ubiquitin directly to a lysine residue on the substrate (Fig. 1a). Ubiquitin has seven lysine residues (K6, K11, K27, K29, K33, K48, and K63) and the N-terminal methionine residue, on which the poly-ubiquitin chains can be formed. The distinct fate of ubiquitinated proteins depends on the nature of ubiquitin attachment and the type of isopeptide linkage of the poly-ubiquitin chain. Target proteins marked with K48- or K11-linked poly-ubiquitin chain predominantly are recognized and degraded by the proteasome, whereas mono-ubiquitination and K63-linked polyubiquitination usually alter protein function and subcellular localization [10–12].
Fig. 1.

Ubiquitin system and ubiquitin-like systems. a The ubiquitination of substrates involves a three-step enzymatic reaction. b Neddylation, like ubiquitination, is a trio of enzymatic steps. c Two ubiquitin-like systems, ATG12-conjugation system and LC3-conjugation system, in autophagosome elongation
Autophagy is a highly conserved eukaryotic intracellular catabolic degradation process in which cytoplasmic contents, like misfolded proteins and damaged organelles, are engulfed by double-membrane autophagosomes and degraded in lysosomes fused with autophagosomes [13]. In general, autophagy is thought to be a nonselective degradation system, which is different from UPS by selectively targeting polyubiquitinated proteins for degradation [14]. There are generally three types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) [15, 16]. Macroautophagy, generally termed “autophagy” unless specified, has received the greatest attention and is the best-characterized form of autophagy. Tons of evidences have shown that autophagy plays important roles in diverse biological processes, such as intracellular protein and organelle clearance, starvation adaptation, development, and tumorigenesis [17].
Autophagy, or “self-eating”, plays a vital role in the maintenance of cellular homeostasis. The self-digestion maintains critical physiological functions by providing nutrients during fasting and by eliminating the “garbage” in cells, such as aggregated proteins, damaged organelles, and invading pathogens [4]. Such functions are likely key to autophagy-mediated physiological and pathological processes as diverse as development, aging, immune response, neurodegeneration, heart failure, and cancer [4, 18]. However, the pro-survival functions of autophagy in certain disease settings may be deleterious. A good example is the dual role of autophagy in cancer progression [19]. On one hand, autophagy acts as a tumor suppressive mechanism through the elimination of aggregated proteins and damaged organelles. On the other hand, it is a key cell survival mechanism by which it facilitates the resistance of established tumors to radiation and chemotherapy. Therefore, the recognition of autophagy function might depend on the specific context.
Given the primary role of autophagy in cellular homeostasis, it is not surprising that the whole process is tightly controlled. Including phosphorylation, ubiquitination, and acetylation, multiple types of post-translational modifications have been found in the regulation of autophagy [20]. This review summarizes our current knowledge of the role of ubiquitination mainly mediated by CRLs in the regulation of autophagy. A thorough understanding of the cross-talk between CRLs and autophagy should lead to new insights into the development of novel therapy for associated diseases.
General features of CRLs
Cullin-RING ligases (CRLs), the largest family of E3 ubiquitin ligases, account for ubiquitination of approximately 20 % cellular proteins degraded by UPS [21]. The following part will describe briefly main features of CRLs, including their composition, and dynamic regulation of CRL assembly and activation mainly mediated by neddylation.
The composition of CRLs
Generally, CRLs consist of four elements: cullins, RING-finger proteins, adaptor proteins, and substrate recognition receptors (Fig. 2). The human genome encodes 8 cullins (CUL1, 2, 3, 4A, 4B, 5, 7, and 9, also known as PARC), 2 RING-finger proteins (RBX1 and RBX2, also known as ROC1 and ROC2/SAG, respectively), 4 adaptor proteins (SKP1 for CUL1/7, Elongin B/C for CUL2/5, and DDB1 for CUL4A/B), and more than 400 substrate recognition receptors (69 F-box proteins for CRL1, 80 SOCS proteins for CRL2/5, about 180 BTB proteins for CRL3, and 90 DCAF proteins for CRL4A/B) [22–28]. Thus, at least 400 CRLs can be assembled in human cells and regulate diverse biological processes by targeted ubiquitination and degradation of thousands of substrates (for a recent review, see Ref. [23]).
Fig. 2.
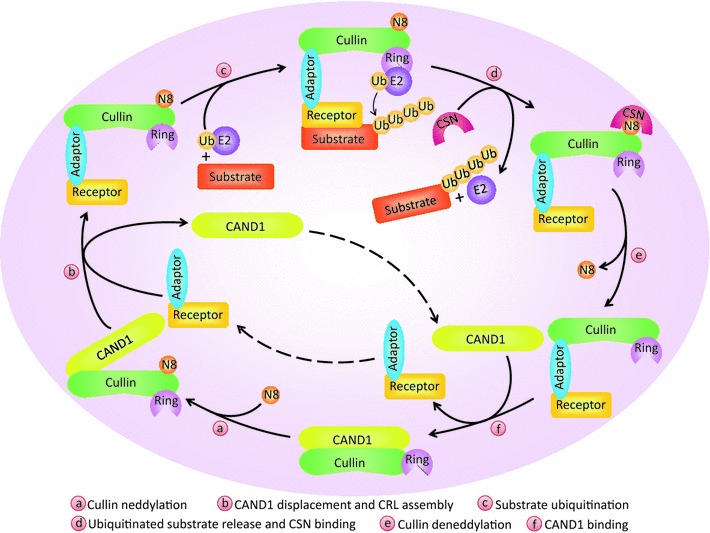
Dynamic regulation of CRLs activity by neddylation and deneddylation. CAND1 binding to unmodified cullin blocks the interaction of cullin with the substrate receptor-adaptor module. Cullin neddylation promotes the dissociation of cullin from CAND1 and restores the CRLs in an active conformation, leading to substrate ubiquitination. After the detachment of ubiquitinated substrate from CRLs, NEDD8 is removed by CSN from cullin for recycling. At last, CAND1 binds to cullin and inactivates CRLs
All CRLs share the similar core architecture with a curved cullin protein acting as a molecular scaffold [22, 29]. Among all CRLs, CRL1, also known as SCF (SKP1-CUL1-F-box), is the most characterized member of CRLs [30]. CUL1 consists of three repeats of a five-helix motif at the N-terminus and a globular domain at the C-terminus. SKP1, the adaptor protein, and RBX1 or RBX2, a RING protein, bind to the N-terminus and the C-terminus of CUL1, respectively. Then, SKP1 binds to an F-box receptor, which specifically recognizes the substrate, whereas the RING protein binds to ubiquitin-charged E2 and effectively catalyzes the transfer of ubiquitin from E2 to the specific substrate [29, 30]. It is well established that the core E3 ligase activity is possessed by the CUL1-RBX1/2 complex in which the RING finger domain of RBX1/2 binds to two zinc atoms via a C3H2C3 motif, and that the substrate specificity of SCF is determined by F-box receptors [24, 31]. Moreover, all cullins contain an evolutionarily conserved lysine residue at its C-terminus for targeted NEDD8 modification, a reaction known as neddylation, which is vital to CRLs activation [32].
The regulation of CRLs activity by neddylation
Neddylation, like ubiquitination, is a process of the attachment of ubiquitin-like molecule NEDD8 to target proteins, involving the successive actions of E1 NEDD8-activating enzyme (a heterodimer of APPBP1/UBA3, also known as NAE), which activates NEDD8; E2 NEDD8-conjugating enzyme (UBE2M, also known as UBC12, or UBE2F), which carries the activated NEDD8; and E3 NEDD8 ligase, which recognizes the substrate and catalyzes the transfer of NEDD8 from E2 to the substrate [33] (Fig. 1b).
An impressive feature of CRLs is that their activity is dynamically regulated by neddylation and deneddylation. The binding of unmodified cullins to CAND1 (cullin-associated and neddylation-dissociated 1) blocks the interaction of the substrate receptor-adaptor complex with the N-terminus of cullins. However, covalent conjugation of cullin with one NEDD8 molecule removes the inhibitory binding to CAND1 and restores the CRLs in an active conformation [34–36]. In addition, neddylation enhances and stabilizes the recruitment of ubiquitin-loaded E2 to CRLs, facilitates the initial ubiquitin transfer and also increases the elongation rate of poly-ubiquitin chain [37–40]. After dissociation of polyubiquitinated substrate from CRLs, NEDD8 is detached by the COP9 signalosome complex (CSN) from cullins for recycling, a reaction known as deneddylation [41] (Fig. 2). The activation cycle of CRLs by dynamic neddylation and deneddylation is essential for the maintenance of cellular homeostasis. Moreover, this cycle assists the recycling of the cullin-RING core that will make it possible for the assembly of other CRLs to allow the ubiquitination of various different substrates as required by the cells in a short time [42]. On the other hand, the inactivation of all CRLs can be achieved by inhibiting cullin neddylation. Indeed, MLN4924, a newly discovered inhibitor of NAE, blocks the entire neddylation pathway, and thus serves as an indirect inhibitor of CRLs [21]. Treatment of MLN4924 causes the accumulation of a number of CRLs substrates and consequently induces cell apoptosis, senescence, and autophagy [43], suggesting that CRLs may regulate autophagy.
Basic concepts of autophagy
Core machinery of autophagy
In mammalian cells, autophagy consists of several sequential steps: initiation, autophagosome formation, cargo recognition and delivery, autophagosome–lysosome fusion, and cargo degradation followed by recycling of the resulting macromolecules via permeases, all of which are coordinated by different sets of ATGs (Fig. 3). Among these steps, autophagosome formation is the key process in autophagy, which is regulated by at least four complexes, known as the core machinery of autophagy, including the ULK1 (a homologue of yeast Atg1) complex, the Beclin-1/Class III PI3K complex, ATG9 and its recycling system, and two ubiquitin-like protein conjugation systems [44–46] (Fig. 3).
Fig. 3.
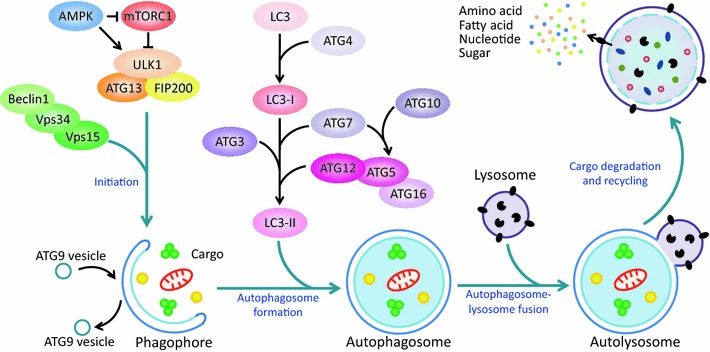
A schematic summary of autophagy process and core machineries in autophagosome formation. Autophagy, a sequential process, consists of initiation, autophagosome formation, autophagosome–lysosome fusion, and cargo degradation, followed by recycling of macromolecules. This process is precisely regulated by different sets of ATGs and involves at least four core machineries, including the ULK1 complex, the Beclin-1/Class III PI3K complex, ATG9 and its recycling system, and two ubiquitin-like protein conjugation systems
The activity of the ULK1 complex (along with ATG13 and FIP200) is required for the autophagy induction. Under physiological conditions, the activated mTORC1 directly binds to the ULK1 complex and inhibits autophagy by phosphorylating ULK1 on Ser757 and ATG13 [47, 48]. Under unfavorable conditions, mTORC1 is inactivated and disconnects from the ULK1 complex. ULK1 is then auto-phosphorylated and then phosphorylates ATG13 and FIP200. As a result, the whole ULK1 complex is activated [49–51], which translocates to ER or other specific places to help with the nucleation of autophagosome formation, followed by the recruitment of downstream effectors including the Beclin-1/Class III PI3K complex and LC3 (a homologue of yeast Atg8) to the site where the nucleation takes place [52].
The Beclin-1/Class III PI3K complex, consisting of Beclin-1, Vps34, and Vps15, is essential for the nucleation of phagophore [53]. It catalyzes the phosphorylation of phosphoinositide to produce phosphatidylinositol-3-phosphate (PI3P), which recruits those effectors containing PX or FYVE domain (such as DFCP1 and WIPI) to mediate the formation of the initial sequestering vesicle (also known as phagophore) that develops into the autophagosome [54, 55]. Accumulating evidences revealed that Beclin-1 serves as an adaptor to recruit multiple proteins, such as ATG14, Ambra1, UVRAG, and Rubicon [56], that modulate the kinase activity of Vps34. Additionally, Beclin-1 is highly regulated in autophagy by post-translational modifications, including phosphorylation, ubiquitination, and cleavage [57].
ATG9, a multipass transmembrane protein, is essential for autophagosome formation. ATG9 is localized to the trans-Golgi network (TGN) and late endosomes. Following the induction of autophagy, ATG9 is rearranged from juxtanuclear to peripheral structures, and is then retrieved from the completed autophagosome [58]. Thus, ATG9 recycling delivers the membrane to the forming autophagosome. Both the ULK1 complex and the Beclin-1/Class III PI3K complex are involved in regulating this process [46, 59].
The elongation and expansion of the phagophore membrane is controlled by two ubiquitin-like conjugation systems. First, the E1-like enzyme ATG7 and the E2-like enzyme ATG10 mediate the covalent conjugation of ATG12 to ATG5 [60]. The resulting ATG5-ATG12 noncovalently interacts with ATG16 to form the ATG12-ATG5-ATG16 complex, which acts as the E3 ligase towards LC3. Next, the E1-like enzyme ATG7 and the E2-like enzyme ATG3 sequentially act to conjugate phosphatidylethanolamine (PE) to a glycine residue of LC3, which constitutes the other conjugation system with ATG12-ATG5-ATG16 complex, the E3-like enzyme [61]. This process achieves the conversion of LC3-I, the soluble form of LC3, to LC3-II (also known as LC3-PE), the autophagic vesicle-associated form (Figs. 1c, 3). In addition, it is worth noting that ATG4, a cysteine protease, plays critical roles in the proteolysis of the full-length isoform of LC3 (pro-LC3), as well as in the deconjugation of the lipidated LC3-PE for recycling [62, 63].
Signals that regulate autophagy
Autophagy is induced by a range of cellular stresses, including nutrient and energy depletion, ER stress, hypoxia, redox stress, and oncogenic activation [64]. It is regulated either negatively or positively by the following two biologically significant molecules.
mTOR is the well-established negative regulator of autophagy. It plays a key role in the coordination of cell growth with autophagy in response to physiological and environmental conditions [65]. mTOR, an evolutionarily conserved serine/threonine protein kinase, forms two structurally and functionally distinct complexes (namely mTORC1 and mTORC2) in mammalian cells. mTORC1 is composed of mTOR, raptor, PRAS40, mLST8, and DEPTOR; mTORC2 also contains mTOR, mLST8, and DEPTOR, but instead of raptor and PRAS40, the proteins rictor, mSin1, and protor exclusively exist in mTORC2 [66]. Moreover, mTORC1, as a negative regulator of the ULK1 complex, inhibits autophagosome formation in response to diverse signals [65], whereas mTORC2 inhibits autophagy through repressing the transcription of some ATGs via AKT-FoxO3 signaling [67, 68] (Figs. 3, 4).
Fig. 4.
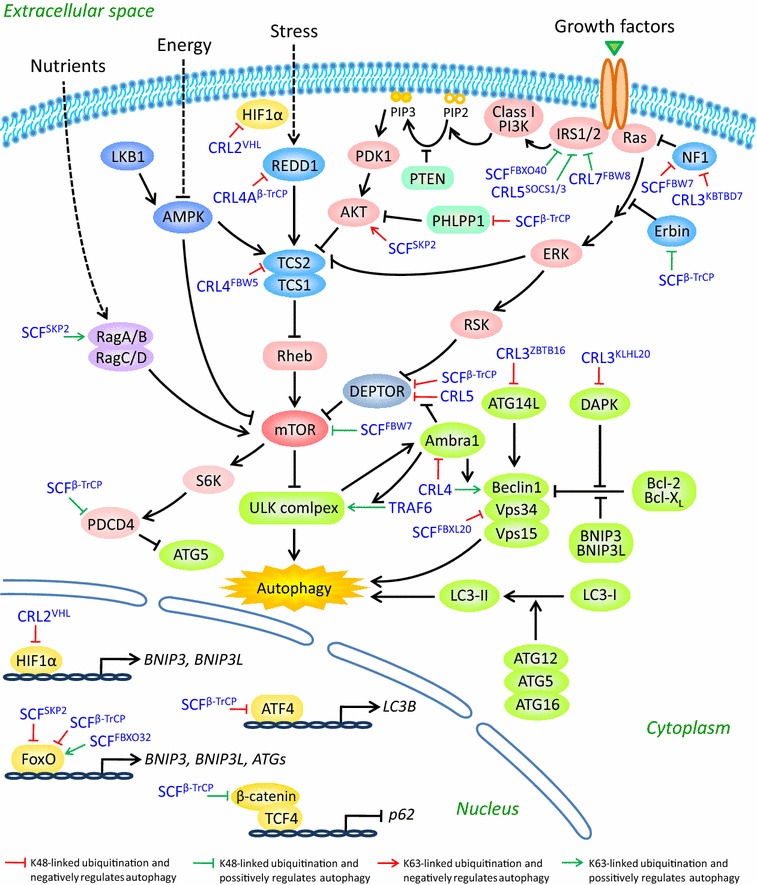
A schematic summary of CRLs substrates in the regulation of autophagy. CRLs control autophagy at multiple levels. First, CRLs mediate the ubiquitination of several components of autophagy machinery. Second, CRLs regulate the activation of mTOR pathway, the central regulator of autophagy. At last, several key transcription factors involved in autophagy are also the substrates of CRLs. See text for details
AMP-activated protein kinase (AMPK), a master regulator of energy metabolism, is a vital positive regulator of autophagy. As a serine/threonine kinase, AMPK is activated when the levels of AMP and ADP in the cells rise owing to various physiological stresses [69]. Upon phosphorylated by activated AMPK, a range of substrates not only acutely affect metabolism and growth, but also are responsible for the long-term metabolic reprogramming. AMPK induces autophagy through phosphorylation of TSC2 and raptor to inhibit mTORC1 [70, 71], and through ULK1 phosphorylation on Ser317 and Ser777 to activate ULK1 [47].
Taken together, by integrating both intracellular and extracellular signals, mTOR and AMPK function coordinately in the regulation of autophagy (Figs. 3, 4).
The role of CRLs in the regulation of autophagy
More recently, the study on the roles of post-translational modifications in regulation of autophagic flux by affecting the activity, recruitment, and turnover of autophagic components has become an attractive area due to the implications of dysregulated autophagy in multiple diseases [20]. Ubiquitination, an important cellular post-translational modification, plays a principal role in controlling protein turnover, activation, subcellular localization, and protein–protein interactions. However, current knowledge of the roles of E3 ubiquitin ligases in the regulation of autophagy is fairly limited. It was reported that RNF5, a RING finger E3 ligase, negatively regulates autophagy by controlling the stability of ATG4B [72], and that Parkin, a RING-HECT hybrid E3 ligase, induces mitophagy through ubiquitination of multiple mitochondrial proteins [73, 74]. Furthermore, the role of CRLs, the largest E3 ubiquitin ligase family, in regulating autophagy is rarely mentioned. Here, we discuss the emerging roles of CRLs in the control of autophagy, especially those regulating autophagy machinery and upstream regulators.
CRLs regulate autophagy machinery
Several components of autophagy machinery are subjected to CRLs-mediated regulation. For instance, SCFFBXL20 targets Vps34, the catalytic subunit of the Beclin-1/Class III PI3K complex, for ubiquitination and proteasome degradation, and plays an important role in DNA damage-induced suppression of autophagy [75, 76]. Meanwhile, ATG14L (Atg14 in yeast), a crucial player to initiate autophagosome formation by mediating the production of PI3P, is the substrate of CRL3ZBTB16. Thus, CRL3ZBTB16 controls the initiation of autophagy by regulating the degradation of ATG14L [77]. In addition, Beclin-1, an adaptor protein in the Beclin-1/Class III PI3K complex, can be modified with multiple poly-ubiquitin chains catalyzed by distinct E3 ligases [78–80]. Specifically, CRL4Ambra1 mediates K63-linked polyubiquitination of Beclin-1 and enhances its association with Vps34 to promote the activation of Vps34, which is required for starvation-induced autophagy [78]. Notably, Ambra1 (activating molecule in Beclin-1-regulated autophagy), also identified as DCAF3 (DDB1 and cullin4 associated factor 3), has multiple roles in the regulation of autophagy. First of all, Ambra1 acts as a substrate receptor for Beclin-1 ubiquitination [78]. Second, Ambra1 recruits the E3 ligase TRAF6 to promote K63-linked polyubiquitination of ULK1, resulting in the stabilization and activation of ULK1 [81]. Third, by dynamically interacting with CRL5 and CRL4, Ambra1 temporally controls the onset and the termination of autophagy response to stress [82]. Under unstressed conditions, Ambra1 is maintained at low levels through degradation by cullin4-DDB1, and autophagy is off. In the early starvation, Ambra1 is detached from cullin4-DDB1 and phosphorylated by ULK1. Thus, transiently stabilized Ambra1 binds to cullin5 to inhibit CRL5-mediated DEPTOR degradation (see below for details), leading to the suppression of mTOR activity. As a result, autophagy is on. In the prolonged starvation, cullin4-DDB1 associates with Ambra1 and targets its polyubiquitination and degradation, leading to the termination of autophagy [82, 83]. Altogether, Ambra1, as a bridge between CRLs and autophagy, plays vital roles in the regulation of autophagic flux at different stages in response to stress. Additionally, CRL3KLHL20 regulates IFN-induced autophagic death by the destruction of DAPK, a serine/threonine protein kinase, which is in charge of the dissociation of Beclin-1 from its Bcl-2 inhibitors by phosphorylating Beclin-1 on Thr119 located at a key position within its BH3 domain required for the interaction with Bcl-2 family members [84–86] (Fig. 4).
To date, the studies on the roles of CRLs in the regulation of autophagy machinery mainly focus on the ULK1 complex and the Beclin-1/Class III PI3K complex, both of which function in the early stage of autophagy. The roles of CRLs in other components of autophagy are fairly limited. Given that the whole process of autophagy is precisely coordinated, it will be intriguing and helpful to elucidate the roles of CRLs in regulating other autophagy machineries, such as ATG9 and its recycling system and two ubiquitin-like protein conjugation systems.
CRLs regulate the upstream regulators of autophagy
Since mTOR pathway is a central regulator of autophagy, it is conceivable that CRLs regulate autophagy via modifying a variety of components, both upstream and downstream, of mTOR pathway [87]. First of all, mTOR itself was identified as a substrate of SCFFBW7 that negatively regulates mTOR protein stability [88]. Second, DEPTOR, a direct mTOR inhibitor, was reported to undergo ubiquitin-mediated degradation by SCFβ-TrCP on phosphorylation by the setting S6K1/RSK or mTOR/CK1 [87, 89–91], and was also proved recently to be a substrate of CRL5 [82]. Third, CRL4FBW5 controls TSC2 protein stability and the subsequent turnover of TSC complex, which is the major inhibitor of mTOR pathway [92]. Fourth, HIF1α, a negative regulator of mTORC1 via the REDD1-TSC1/2 axis, is a well-characterized substrate of CRL2VHL [93, 94]. Fifth, REDD1, an inhibitor of mTORC1, was subjected to CRL4Aβ-TrCP-mediated ubiquitination and degradation with GSK-3β as a corresponding kinase for phosphorylation [95]. Sixth, PHLPP1, a protein phosphatase negatively regulating AKT via direct dephosphorylation of activated AKT, was identified as a substrate of SCFβ-TrCP in a manner dependent on CK1 and GSK3β [96]. Seventh, IRS1 and IRS2, as adaptor proteins to mediate insulin/insulin-like growth factor 1 signaling, are the substrates of CRL5SOCS1/3 [97]. IRS1 can also be ubiquitinated and degraded either by CRL7FBW8 upon prerequisite phosphorylation by mTOR and S6K [98] or by SCFFBXO40 [99]. Eighth, NF1, an inhibitor of Ras, was identified as a substrate of SCFFBW7 [100] and CRL3KBTBD7 [101]. Ninth, Erbin, an inhibitor of Ras-Raf signaling, is a newly discovered substrate of SCFβ-TrCP, which targets Erbin for degradation to trigger autophagy by ROS accumulation [102]. At last, PDCD4, a downstream target of mTOR-S6K1 pathway, was reported to be a substrate of SCFβ-TrCP dependent on pre-phosphorylation at Ser67 by S6K1, and negatively regulates autophagy by inhibiting ATG5 protein expression [103, 104] (Fig. 4).
Therefore, given the fact that nearly all these CRLs substrates, except for mTOR and IRS1, are negative regulators of mTOR pathway, the general inhibition of CRLs would likely cause their accumulation to down-regulate mTOR pathway, leading to the induction of autophagy. Indeed, MLN4924, an indirect inhibitor of CRLs, induces autophagy in multiple cancer cell lines resulting from inactivating mTORC1 by the accumulation of DEPTOR and HIF1α [105]. Consistently, silencing of RBX1, one of two RING proteins in CRLs, also triggers autophagy response by the accumulation of DEPTOR [106]. All these findings suggest that modulation of the activity of CRLs regulates autophagy induction, which may provide a novel therapeutic strategy for autophagy-associated human diseases.
In addition, SCFSKP2 promotes K63-linked ubiquitination of RagA, which recruits GATOR1 to hydrolyze RagAGTP and blocks mTORC1 lysosomal localization and activation, leading to autophagy induction [107]. Interestingly, AKT is also a nonproteolytic substrate of SCFSKP2. SCFSKP2 drives K63-linked ubiquitination of AKT, which is vital for ErbB-receptor-mediated AKT membrane recruitment and activation in response to EGF [108]. Notably, it is a paradox that, on one hand, SKP2-mediated RagA ubiquitination suppresses mTORC1 activation; on the other hand, SKP2 promotes ubiquitination of AKT and increases its activity, which further activates mTORC1. Thus, the function of SCFSKP2 in the regulation of autophagy depends on certain cell type and its context (Fig. 4).
Moreover, PHLPP1 not only triggers macroautophagy, but also regulates chaperone-mediated autophagy (CMA) [109, 110]. CMA selectively degrades cytosolic proteins delivered by a cytosolic chaperone in the lysosomes [15, 111]. PHLPP1 induces CMA through its inhibitory effect on AKT [110]. Given that PHLPP1 is a substrate of SCFβ-TrCP, CRLs may regulate CMA by controlling PHLPP1 stability.
CRLs regulate autophagy at the transcriptional level
In addition to regulating autophagy machinery and upstream regulators, CRLs also control autophagy at the transcriptional level through modification of several key transcription factors.
We already discussed that accumulation of HIF1α, as a well-established substrate of CRL2VHL, is partially responsible for MLN4924-induced autophagy [105]. In fact, besides via the HIF1α-REDD1-TSC axis to block mTORC1 activity, resulting in autophagy induction, HIF1α itself, as a transcription factor, could induce autophagy directly by transcriptional regulation of its target genes. In response to hypoxia, HIF1α is activated and promotes the transcription of BNIP3 and BNIP3L (also known as NIX), both of which disrupt the Bcl-2/Beclin-1 complex, leading to the release of Beclin-1 from Bcl-2 and the subsequent induction of autophagy [112, 113]. In addition, NIX/BNIP3, also located at the outer membrane of mitochondria, contains a WXXL/WXXL-like motif that binds to LC3 and its homolog GABARAP, leading to mitophagy induction [114, 115]. Thus, CRLs may also regulate mitophagy by inducing the transcription of NIX/BNIP3 via HIF1α.
Meanwhile, the transcription factor FoxO3 regulates autophagy in skeletal muscle by transactivating NIX/BNIP3 [116]. In addition, FoxO factors (such as FoxO1 and FoxO3) induce autophagy by promoting the expression of multiple ATG genes, including ATG4B, ATG8, ATG12, Vps34, and Beclin-1, during muscle atrophy [116–118]. And cytosolic FoxO1 is also required for autophagy induction in a transcription-independent manner via the interaction of acetylated FoxO1 with ATG7 [119]. Given the critical role of FoxO factors in regulating autophagy, the specific CRLs in charge of their stability were identified. Both FoxO1 and FoxO3 were ubiquitinated and degraded by SCFSKP2 [120, 121]. Additionally, FoxO3 is also the substrate of SCFβ-TrCP in an IKKβ-dependent manner [122]. Moreover, Atrogin-1 (also known as MAFbx or FBXO32), as a muscle-specific F-box protein that forms a complex with SKP1-CUL1-RBX1, mediates K63-linked polyubiquitination and consequent transactivation of FoxO1/FoxO3 and is a central node in the regulation of autophagy during muscle atrophy [117, 123].
ATF4, a transcription factor induced by severe hypoxia and involved in the unfolded protein response (UPR), up-regulates LC3B by directly binding to its promoter to facilitate autophagy [124, 125]. ATF4, a short-lived protein with a half-life time of about 30 min, is degraded rapidly by proteasome, following SCFβ-TrCP-mediated polyubiquitination [126]. More importantly, bortezomib, a potent inhibitor of the 26S proteasome, activates autophagy by proteasomal stabilization of ATF4 and ATF4-induced up-regulation of LC3B [124]. β-catenin, another well-known substrate of SCFβ-TrCP, inhibits autophagosome formation by suppressing p62 (also known as SQSTM1, an autophagy adaptor protein) expression via TCF4 [127]. Thus, SCFβ-TrCP paradoxically regulates autophagy through repressing LC3B or inducing p62, two key proteins in the process of autophagy (Fig. 4).
In conclusion, all these studies highlight the importance and complexity of CRLs in the regulation of autophagy. Given that 1) these findings are mostly associated with mTOR pathway, the ULK1 complex, and the Beclin-1/Class III PI3K complex; 2) one specific CRL can target various substrates; 3) the specific substrate is subjected to the regulation of multiple CRLs; 4) autophagy can be regulated at the transcriptional, translational, and post-translational levels, further studies should be directed to elucidate the functional network of CRLs in the whole process of autophagy.
The effects of autophagy on CRLs and UPS
Accumulating evidences indicate that the active cross-talk exists between UPS and autophagy, two major intracellular clearance systems [2, 128, 129]. Inhibition of UPS enhances autophagic activity possibly as a compensatory mechanism [129, 130]. In contrast, long-term inhibition of autophagy has been shown to compromise the degradation of proteasomal substrates, which leads to the accumulation of short-lived regulatory proteins, particularly some oncoproteins, with predicted deleterious consequences [131]. For example, p62, a selective autophagy receptor for the ubiquitinated protein aggregates, is degraded by autophagy. Twist1, an oncogenic transcription factor, is polyubiquitinated by SCFFBXL14 and subsequently degraded by the proteasome [132]. However, accumulated p62 caused by autophagy deficiency binds to polyubiquitinated Twist1 and inhibits its proteasomal destruction, in consequence, promoting tumor cell growth and metastasis [133]. p62 abrogates the clearance of ubiquitinated short-lived proteins destined for proteasomal degradation through two possible manners: (1) p62 disrupts the binding of ubiquitinated proteins with their partners that escort them to the proteasome [131, 133]; (2) p62 together with proteasomal substrate forms oligomer, which would be too bulky to be degraded by the proteasome in its narrow catalytic pore [128]. Paradoxically, p62 was also reported to interact with ubiquitinated proteins and deliver them (such as Tau) to the proteasome for degradation [134, 135]. This discrepancy may be caused by diverse protein substrates, specific cellular context, and different cell types. Notably, p62 also can impair CRLs-mediated ubiquitination. Specifically, p62 was accumulated in autophagy-defective cells and interacts with Keap1 on the NRF2-binding site to disrupt the ubiquitination of NRF2 mediated by CRL3Keap1, resulting in the hyperactivation of NRF2, which may contribute to hepatoma development [136–138]. Moreover, ATG16L1, an essential component of the autophagosome, is necessary for the neddylation of CUL3 with unknown mechanism, which is required for the ligase activity of CRL3 [139]. Taken together, autophagy can adjust UPS via multiple mechanisms. Future studies to explore precise molecular mechanisms should facilitate the development of novel therapeutic strategies for autophagy-defective human diseases.
CRLs and autophagy in diseases
Given the facts that UPS and autophagy are two cornerstones in the maintenance of cellular homeostasis, and CRLs are the largest E3 ligase family, it is conceivable that the dysfunction of CRLs and autophagy contributes to the pathogenesis of various human diseases. In this part, we will mainly discuss the diseases associated with the dysfunction of both CRLs and autophagy.
3-M syndrome
Genetic studies have demonstrated a crucial role of CUL7 E3 ligase in controlling growth. CUL7 germline mutations, resulting in loss of its functional cullin domain, are responsible for 3-M syndrome, characterized by prenatal and postnatal growth retardation [140]. The cause of these growth defects with CUL7 germline mutations may owe to the accumulation of CRL7 substrates. Indeed, IRS1, one of CRL7 substrates, was stabilized in Cul7−/− MEFs with senescence phenotype. The increased IRS1 activates its downstream AKT and MEK/ERK pathways, both of which were shown to induce senescence [98]. This kind of senescence, also known as oncogene-induced senescence, is closely associated with development and tumorigenesis [141, 142]. Accumulating evidences revealed that autophagy facilitates oncogene-induced senescence [102, 143, 144]. Thus, the accumulation of IRS1 or other unknown substrate(s) of CRL7 may contribute to the senescence through affecting autophagic flux. Recently, CRL5 and CRL4 were found to control the onset and the termination of autophagy, respectively, by dynamically interacting with Ambra1 [82]. In fact, the data also showed that Ambra1 could bind to CUL7. However, the underlying physiological functions are not further explored [82]. These may offer one potential hint that CUL7-mediated autophagy by interacting with Ambra1 may also contribute to senescence.
Neural disease
FBXL20 (also known as SCRAPPER), a synapse-localized F-box protein, was proved to regulate neuronal synaptic tuning via the destruction of RIM1, which is required for synaptic vesicle release [145]. Scrapper knock-out mice displayed abnormal electrophysiological synaptic activity resulting from upregulation of RIM1. Moreover, FBXL20 is responsible for the ubiquitination and proteasomal degradation of Vps34, which controls intracellular vesicular processes, such as autophagy and endocytosis [75]. In light of the roles of endocytosis [146] and autophagy [147] in regulating synaptic development and plasticity, the control of Vps34 levels by SCFFBXL20 may provide an important regulatory mechanism for synaptic transmission and plasticity. Since many neural diseases are caused by excessive neurotransmitter release, future studies on FBXL20 might help elucidate their molecular pathogenesis.
Although a causal pathogenetic linkage between CRLs and neural disorders and diseases has not been established, the ubiquitin conjugates and/or inclusion bodies associated with ubiquitin have been discovered to be accumulated in a wide array of chronic neurodegenerative diseases [3]. In addition, NEDD8, one of the ubiquitin-like proteins, is also accumulated in ubiquitin-positive inclusions in various neurodegenerative disorders [148, 149]. Given that cullins are the best-characterized substrates of neddylation, the possible involvement of CRLs in the pathogenesis of neurodegeneration should not be neglected. In the mouse model of Huntington’s disease, inhibition of GPCR signaling by AMD3100, a selective GPCR antagonist, can induce autophagy by suppressing CRL3ZBTB16-mediated ATG14L degradation, leading to the expanded polyQ degradation and the preservation of neuronal functions [77]. Meanwhile, activated IRS2, a substrate of CRL5 [97], can induce autophagy in modified neural cell lines, used as models for Huntington’s disease, to enhance the clearance of polyQ proteins [150]. Thus, CRLs would play important roles in the pathogenesis of neurodegenerative diseases through their key substrates associated with autophagy.
Cardiac disease
Atrogin-1, also known as FBXO32, is a skeletal and cardiac muscle-specific F-box protein [151]. Atrogin-1 was demonstrated as a critical player in skeletal muscle atrophy programs, and it is tightly regulated at the transcriptional level by FoxO factors [152]. Meanwhile, Atrogin-1 also induces the transcriptional activity of FoxO1/FoxO3 dependent on their K63-linked polyubiquitination mediated by SCFAtrogin-1. These findings were confirmed in Atrogin-1 transgenic and knock-out mouse models, strongly indicating its crucial role in the inhibition of cardiac hypertrophy [123]. Moreover, accumulating data have proved that the autophagic activity governed by FoxO factors at multiple steps significantly contributes to cardiac homeostasis and disease [153]. All these studies suggest CRLs affect cardiac remodeling through regulating autophagic flux, which was further demonstrated in vivo. In Atrogin-1 knock-out mice, Atrogin-1 depletion causes cardiomyopathy and premature death resulting from impaired autophagy [154]. Mechanistically, SCFAtrogin-1 promotes the ubiquitination and degradation of CHMP2B, which is part of an endosomal sorting complex required for autophagy [155]. Atrogin-1 deficiency failed to destroy CHMP2B, resulting in sequential serious consequences: autophagy impairment, protein aggregation, activation of unfolded protein response (UPR) signaling pathways, and ultimately, cardiomyocyte apoptosis [154]. In addition, cardiomyocyte-restricted Csn8 knock-out (CR-Csn8KO) mouse model also proved the important role of Atrogin-1 in cardiomyocyte necrosis and dilated cardiomyopathy via autophagy impairment caused by down-regulation of Rab7, which is indispensable for autophagosome-lysosome fusion [156]. The underlying molecular mechanisms could be that (1) Atrogin-1 is down-regulated in Csn8-null heart, which is consistent with the theory that CSN-mediated deneddylation stabilizes F-box proteins [157, 158]; (2) Atrogin-1 enhances the transactivation of FoxO1/FoxO3 by promoting their ubiquitination; (3) Rab7 is a target gene of FoxO factors [159]. Taken together, Atrogin-1 plays a vital role in maintaining the homeostasis of cardiac myocytes through regulating autophagic flux.
Cancer
Given that CRLs play a fundamental role in regulating a wide range of biological processes, including signal transduction, gene transcription, DNA replication, cell cycle progression, and apoptosis among others, it is anticipated that deregulation of CRLs is related to uncontrolled cell proliferation, ultimately leading to cancer [23]. It is widely accepted that autophagy plays an important role in tumorigenesis, hence autophagy regulated by CRLs more or less contributes to cancer development. For instance, (1) RBX1 knock-down triggers protective autophagy. Blockage of autophagy pathway significantly enhances the inhibition of tumor cell growth induced by RBX1 knock-down [106]. Similarly, (2) MLN4924, a general inhibitor of CRLs, also triggers a protective autophagy in many human cancer cell lines through mTORC1 inhibition resulting from the accumulation of DEPTOR and HIF1α, two well-known substrates of CRLs. Thus, autophagy inhibitors remarkably increase the apoptosis induced by MLN4924 [105]. Nevertheless, (3) Rbx2 deletion in the skin inhibits autophagy and oncogene-activated senescence induced by KrasG12D, and consequently, promotes skin papillomagenesis. Thus Rbx2 acts as a skin-specific tumor suppressor by promoting autophagy via targeting its substrates: Erbin and Nrf2 [102].
Conclusions and future perspectives
In summary, autophagy and UPS are crucial in the maintenance of cellular homeostasis, hence both of them need to be precisely orchestrated. CRLs, the largest E3 ubiquitin ligase family, mediate the degradation or activity alteration of many components and regulators in the autophagy pathway to control autophagic activity. Meanwhile, autophagy also conversely affects the activity of CRLs and UPS. The cross-talk between CRLs and autophagy deserves further intensive investigation to elucidate how the dysfunction of CRLs and autophagy contributes to the development of various human disorders, such as neural diseases, cardiac diseases, and cancer, which should provide new insights into drug discovery targeting CRLs and autophagy. In consideration of the facts that (1) CRLs are being validated as promising anti-cancer targets; (2) MLN4924, a small molecule indirect inhibitor of CRLs, which is currently in several Phase I clinical trials against a number of human malignancies, induces protective autophagy; (3) the inhibitors of autophagy significantly sensitize cancer cells, particularly resistant cancer cells, to MLN4924 treatment, future studies focused on CRLs and autophagy would eventually benefit human anti-cancer therapy.
Although some associations of CRLs and autophagy have been clarified, many fundamental questions still remain to be addressed: (1) what are other components of autophagy machinery and regulators associated with autophagy as the substrates of CRLs? (2) How does autophagy regulate the activity of CRLs and UPS? (3) Are the findings in cell culture settings consistent to those in physiological settings (knock-out/knock-in mouse models and patient samples)? (4) What is the function of deubiquitination in autophagy? Some recent studies have shown that deubiquitinases, enzymes catalyzing a reverse process for protein ubiquitination, also play a regulatory role in autophagy, such as USP36 and A20 [79, 160]. The answers to these fundamental questions would certainly uncover the precise roles of CRLs in the regulation of autophagy and autophagy-associated diseases, and provide molecular basis for rational drug design by targeting CRLs and autophagy.
Authors’ contributions
DC, XX, YZ drafted and revised the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We sincerely apologize that we were not able to cite all related original articles due to space limitation. We would like to thank Dr. Yi Sun for critical reading of the manuscript.
Competing interests
The authors declare that they have no competing interests.
Funding
Yongchao Zhao is funded by the National Natural Science Foundation of China (Grant No. 31470753) and the Natural Science Foundation of Zhejiang Province (Grant No. LR16C050001). Xiufang Xiong is funded by the National Natural Science Foundation of China (Grant No. 81572708).
Abbreviations
- Ambra1
activating molecule in Beclin-1-regulated autophagy
- AMPK
AMP-activated protein kinase
- ATF4
activating transcription factor 4
- ATG
autophagy related gene
- Bcl-2
B-cell lymphoma 2
- BTB
bric-a-brac, tramtrack, broad-complex domain
- β-TrCP
β-transducin repeat-containing protein
- CAND1
cullin-associated and neddylation-dissociated 1
- CHMP2B
charged multivesicular body protein 2B
- CMA
chaperone-mediated autophagy
- CRLs
cullin-RING ligases
- CSN
COP9 signalosome complex
- DAPK
death associated protein kinase
- DCAF
DDB1-CUL4 associated factor
- DDB1
DNA damage-binding protein 1
- DEPTOR
DEP domain containing mTOR-interacting protein
- DFCP1
double FYVE-containing protein 1
- FBW
F-box and WD-40 domain protein
- FBXL
F-box and leucine-rich repeat protein
- FBXO
F-box only protein
- HECT
homologous to E6-AP C-terminus
- HIF1
hypoxia-inducible factor 1
- IKK
inhibitor of κB kinase
- IRS1
insulin receptor substrate 1
- Keap1
kelch-like ECH-associated protein 1
- KLHL20
kelch-like family member 20
- LC3
microtubule-associated protein light chain 3
- mTOR
mammalian target of rapamycin
- NAE
NEDD8-activating enzyme
- NEDD8
neural precursor cell expressed, developmentally down-regulated 8
- NF1
neurofibromatosis type 1
- NRF2
NF-E2 related factor 2
- PDCD4
programmed cell death 4
- PHLPP1
PH domain and leucine rich repeat protein phosphatase 1
- PI3K
phosphatidylinositol-3-kinase
- polyQ
polyglutamine
- RBX1/2
RING box protein 1/2
- REDD1
regulated in development and DNA damage responses 1
- RIM1
Rab3-interacting molecule 1
- RING
really interesting new gene
- RNF5
RING finger protein 5
- ROC1/2
regulator of cullins 1/2
- Rubicon
RUN domain protein as Beclin-1 interacting and cysteine-rich containing
- S6K1
ribosomal S6 kinase 1
- SAG
sensitive to apoptosis gene
- SCF
SKP1, cullin and F-box protein
- SKP1/2
S-phase kinase-associated protein 1/2
- SOCS
suppressors of cytokine signaling
- SQSTM1
sequestosome 1
- TRAF6
TNF-receptor-associated factor 6
- TSC1/2
tuberous sclerosis 1/2
- UPR
unfolded protein response
- UPS
ubiquitin-proteasomal system
- USP
ubiquitin specific protease
- UVRAG
UV radiation resistance associated gene
- VHL
Von Hippel–Lindau
- WIPI
WD-repeat domain protein interacting with phosphoinositides
- ZBTB16
zinc finger and BTB domain containing 16
Contributor Information
Danrui Cui, Email: cuidanrui@zju.edu.cn.
Xiufang Xiong, Email: xiufang@zju.edu.cn.
Yongchao Zhao, Phone: +86-571-86971963, Email: yongchao@zju.edu.cn.
References
- 1.Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008;22(11):1427–1438. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wong E, Cuervo AM. Integration of clearance mechanisms: the proteasome and autophagy. Cold Spring Harb Perspect Biol. 2010;2(12):a006734. doi: 10.1101/cshperspect.a006734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ciechanover A. The ubiquitin proteolytic system and pathogenesis of human diseases: a novel platform for mechanism-based drug targeting. Biochem Soc Trans. 2003;31(2):474–481. doi: 10.1042/bst0310474. [DOI] [PubMed] [Google Scholar]
- 4.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ciechanover A. The ubiquitin–proteasome pathway: on protein death and cell life. EMBO J. 1998;17(24):7151–7160. doi: 10.1093/emboj/17.24.7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 7.Glickman MH, Ciechanover A. The ubiquitin–proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82(2):373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- 8.Ciechanover A, Ben-Saadon R. N-terminal ubiquitination: more protein substrates join in. Trends Cell Biol. 2004;14(3):103–106. doi: 10.1016/j.tcb.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 9.Hershko A. The ubiquitin system for protein degradation and some of its roles in the control of the cell-division cycle (Nobel lecture) Angew Chem Int Ed Engl. 2005;44(37):5932–5943. doi: 10.1002/anie.200501724. [DOI] [PubMed] [Google Scholar]
- 10.Peng J, Schwartz D, Elias JE, Thoreen CC, Cheng D, Marsischky G, et al. A proteomics approach to understanding protein ubiquitination. Nat Biotechnol. 2003;21(8):921–926. doi: 10.1038/nbt849. [DOI] [PubMed] [Google Scholar]
- 11.Pickart CM. Ubiquitin in chains. Trends Biochem Sci. 2000;25(11):544–548. doi: 10.1016/S0968-0004(00)01681-9. [DOI] [PubMed] [Google Scholar]
- 12.Xu P, Duong DM, Seyfried NT, Cheng D, Xie Y, Robert J, et al. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell. 2009;137(1):133–145. doi: 10.1016/j.cell.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ. 2005;12(Suppl 2):1542–1552. doi: 10.1038/sj.cdd.4401765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21(22):2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 15.Kaushik S, Bandyopadhyay U, Sridhar S, Kiffin R, Martinez-Vicente M, Kon M, et al. Chaperone-mediated autophagy at a glance. J Cell Sci. 2011;124(4):495–499. doi: 10.1242/jcs.073874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12(9):814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mizushima N. The pleiotropic role of autophagy: from protein metabolism to bactericide. Cell Death Differ. 2005;12(Suppl 2):1535–1541. doi: 10.1038/sj.cdd.4401728. [DOI] [PubMed] [Google Scholar]
- 18.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368(7):651–662. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 19.Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015;34(7):856–880. doi: 10.15252/embj.201490784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McEwan DG, Dikic I. The three musketeers of autophagy: phosphorylation, ubiquitylation and acetylation. Trends Cell Biol. 2011;21(4):195–201. doi: 10.1016/j.tcb.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458(7239):732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 22.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6(1):9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 23.Zhao YC, Sun Y. Cullin-RING ligases as attractive anti-cancer targets. Curr Pharm Design. 2013;19(18):3215–3225. doi: 10.2174/13816128113199990300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jin JP, Cardozo T, Lovering RC, Elledge SJ, Pagano M, Harper JW. Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev. 2004;18(21):2573–2580. doi: 10.1101/gad.1255304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Linossi EM, Nicholson SE. The SOCS box-adapting proteins for ubiquitination and proteasomal degradation. IUBMB Life. 2012;64(4):316–323. doi: 10.1002/iub.1011. [DOI] [PubMed] [Google Scholar]
- 26.Stogios PJ, Downs GS, Jauhal JJ, Nandra SK, Prive GG. Sequence and structural analysis of BTB domain proteins. Genome Biol. 2005;6(10):R82. doi: 10.1186/gb-2005-6-10-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He YJ, McCall CM, Hu J, Zeng YX, Xiong Y. DDB1 functions as a linker to recruit receptor WD40 proteins to CUL4-ROC1 ubiquitin ligases. Genes Dev. 2006;20(21):2949–2954. doi: 10.1101/gad.1483206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Z, Liu P, Inuzuka H, Wei W. Roles of F-box proteins in cancer. Nat Rev Cancer. 2014;14(4):233–247. doi: 10.1038/nrc3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng N, Schulman BA, Song LZ, Miller JJ, Jeffrey PD, Wang P, et al. Structure of the Cul1–Rbx1–Skp1–F box(Skp2) SCF ubiquitin ligase complex. Nature. 2002;416(6882):703–709. doi: 10.1038/416703a. [DOI] [PubMed] [Google Scholar]
- 30.Cardozo T, Pagano M. The SCF ubiquitin ligase: insights into a molecular machine. Nat Rev Mol Cell Biol. 2004;5(9):739–751. doi: 10.1038/nrm1471. [DOI] [PubMed] [Google Scholar]
- 31.Wu K, Fuchs SY, Chen A, Tan PL, Gomez C, Ronai Z, et al. The SCFHOS/β-TRCP-ROC1 E3 ubiquitin ligase utilizes two distinct domains within CUL1 for substrate targeting and ubiquitin ligation. Mol Cell Biol. 2000;20(4):1382–1393. doi: 10.1128/MCB.20.4.1382-1393.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pan ZQ, Kentsis A, Dias DC, Yamoah K, Wu K. Nedd8 on cullin: building an expressway to protein destruction. Oncogene. 2004;23(11):1985–1997. doi: 10.1038/sj.onc.1207414. [DOI] [PubMed] [Google Scholar]
- 33.Xirodimas DP. Novel substrates and functions for the ubiquitin-like molecule NEDD8. Biochem Soc Trans. 2008;36(5):802–806. doi: 10.1042/BST0360802. [DOI] [PubMed] [Google Scholar]
- 34.Liu J, Furukawa M, Matsumoto T, Xiong Y. NEDD8 modification of CUL1 dissociates p120(CAND1), an inhibitor of CUL1-SKP1 binding and SCF ligases. Mol Cell. 2002;10(6):1511–1518. doi: 10.1016/S1097-2765(02)00783-9. [DOI] [PubMed] [Google Scholar]
- 35.Zheng J, Yang X, Harrell JM, Ryzhikov S, Shim EH, Lykke-Andersen K, et al. CAND1 binds to unneddylated CUL1 and regulates the formation of SCF ubiquitin E3 ligase complex. Mol Cell. 2002;10(6):1519–1526. doi: 10.1016/S1097-2765(02)00784-0. [DOI] [PubMed] [Google Scholar]
- 36.Goldenberg SJ, Cascio TC, Shumway SD, Garbutt KC, Liu J, Xiong Y, et al. Structure of the Cand1–Cul1–Roc1 complex reveals regulatory mechanisms for the assembly of the multisubunit cullin-dependent ubiquitin ligases. Cell. 2004;119(4):517–528. doi: 10.1016/j.cell.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 37.Kawakami T, Chiba T, Suzuki T, Iwai K, Yamanaka K, Minato N, et al. NEDD8 recruits E2-ubiquitin to SCF E3 ligase. EMBO J. 2001;20(15):4003–4012. doi: 10.1093/emboj/20.15.4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saha A, Deshaies RJ. Multimodal activation of the ubiquitin ligase SCF by Nedd8 conjugation. Mol Cell. 2008;32(1):21–31. doi: 10.1016/j.molcel.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu K, Chen A, Pan ZQ. Conjugation of Nedd8 to CUL1 enhances the ability of the ROC1–CUL1 complex to promote ubiquitin polymerization. J Biol Chem. 2000;275(41):32317–32324. doi: 10.1074/jbc.M004847200. [DOI] [PubMed] [Google Scholar]
- 40.Sakata E, Yamaguchi Y, Miyauchi Y, Iwai K, Chiba T, Saeki Y, et al. Direct interactions between NEDD8 and ubiquitin E2 conjugating enzymes upregulate cullin-based E3 ligase activity. Nat Struct Mol Biol. 2007;14(2):167–168. doi: 10.1038/nsmb1191. [DOI] [PubMed] [Google Scholar]
- 41.Lyapina S, Cope G, Shevchenko A, Serino G, Tsuge T, Zhou CS, et al. Promotion of NEDD8–CUL1 conjugate cleavage by COP9 signalosome. Science. 2001;292(5520):1382–1385. doi: 10.1126/science.1059780. [DOI] [PubMed] [Google Scholar]
- 42.Lo SC, Hannink M. CAND1-mediated substrate adaptor recycling is required for efficient repression of Nrf2 by Keap1. Mol Cell Biol. 2006;26(4):1235–1244. doi: 10.1128/MCB.26.4.1235-1244.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao Y, Morgan MA, Sun Y. Targeting neddylation pathways to inactivate Cullin-RING ligases for anticancer therapy. Antioxid Redox Signal. 2014;21(17):2383–2400. doi: 10.1089/ars.2013.5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9(10):1102–1109. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- 45.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 46.Kraft C, Martens S. Mechanisms and regulation of autophagosome formation. Curr Opin Cell Biol. 2012;24(4):496–501. doi: 10.1016/j.ceb.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 47.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient-dependent mTORC1 association with the ULK1–Atg13–FIP200 complex required for autophagy. Mol Biol Cell. 2009;20(7):1981–1991. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abada A, Elazar Z. Getting ready for building: signaling and autophagosome biogenesis. EMBO Rep. 2014;15(8):839–852. doi: 10.15252/embr.201439076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jung CH, Jun CB, Ro SH, Kim Y-M, Otto NM, Cao J, et al. ULK–Atg13–FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20(7):1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ganley IG, Lam DH, Wang J, Ding X, Chen S, Jiang X. ULK1·ATG13·FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284(18):12297–12305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. 2010;6(6):764–776. doi: 10.4161/auto.6.6.12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.He C, Levine B. The beclin 1 interactome. Curr Opin Cell Biol. 2010;22(2):140–149. doi: 10.1016/j.ceb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182(4):685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Polson HE, de Lartigue J, Rigden DJ, Reedijk M, Urbe S, Clague MJ, et al. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy. 2010;6(4):506–522. doi: 10.4161/auto.6.4.11863. [DOI] [PubMed] [Google Scholar]
- 56.Simonsen A, Tooze SA. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J Cell Biol. 2009;186(6):773–782. doi: 10.1083/jcb.200907014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McKnight NC, Zhenyu Y. Beclin 1, an essential component and master regulator of PI3K-III in health and disease. Curr Pathobiol Rep. 2013;1(4):231–238. doi: 10.1007/s40139-013-0028-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Young AR, Chan EY, Hu XW, Kochl R, Crawshaw SG, High S, et al. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci. 2006;119(18):3888–3900. doi: 10.1242/jcs.03172. [DOI] [PubMed] [Google Scholar]
- 59.Webber JL, Tooze SA. New insights into the function of Atg9. FEBS Lett. 2010;584(7):1319–1326. doi: 10.1016/j.febslet.2010.01.020. [DOI] [PubMed] [Google Scholar]
- 60.Romanov J, Walczak M, Ibiricu I, Schuchner S, Ogris E, Kraft C, et al. Mechanism and functions of membrane binding by the Atg5–Atg12/Atg16 complex during autophagosome formation. EMBO J. 2012;31(22):4304–4317. doi: 10.1038/emboj.2012.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Klionsky DJ, Schulman BA. Dynamic regulation of macroautophagy by distinctive ubiquitin-like proteins. Nat Struct Mol Biol. 2014;21(4):336–345. doi: 10.1038/nsmb.2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nair U, Yen WL, Mari M, Cao Y, Xie Z, Baba M, et al. A role for Atg8-PE deconjugation in autophagosome biogenesis. Autophagy. 2012;8(5):780–793. doi: 10.4161/auto.19385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nakatogawa H, Ishii J, Asai E, Ohsumi Y. Atg4 recycles inappropriately lipidated Atg8 to promote autophagosome biogenesis. Autophagy. 2012;8(2):177–186. doi: 10.4161/auto.8.2.18373. [DOI] [PubMed] [Google Scholar]
- 64.Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584(7):1287–1295. doi: 10.1016/j.febslet.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Efeyan A, Sabatini DM. mTOR and cancer: many loops in one pathway. Curr Opin Cell Biol. 2010;22(2):169–176. doi: 10.1016/j.ceb.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, et al. Ablation in mice of the mTORC components raptor, rictor, or mlST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCα but not S6K1. Dev Cell. 2006;11(6):859–871. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 68.Zhao JH, Brault JJ, Schild A, Goldberg AL. Coordinate activation of autophagy and the proteasome pathway by FoxO transcription factor. Autophagy. 2008;4(3):378–380. doi: 10.4161/auto.5633. [DOI] [PubMed] [Google Scholar]
- 69.Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13(9):1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Inoki K, Zhu TQ, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115(5):577–590. doi: 10.1016/S0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 71.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30(2):214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kuang E, Okumura CY, Sheffy-Levin S, Varsano T, Shu VC, Qi J, et al. Regulation of ATG4B stability by RNF5 limits basal levels of autophagy and influences susceptibility to bacterial infection. PLoS Genet. 2012;8(10):e1003007. doi: 10.1371/journal.pgen.1003007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12(2):119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 74.Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, et al. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496(7445):372–376. doi: 10.1038/nature12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xiao J, Zhang T, Xu DC, Wang HB, Cai Y, Jin TJ, et al. FBXL20-mediated Vps34 ubiquitination as a p53 controlled checkpoint in regulating autophagy and receptor degradation. Genes Dev. 2015;29(2):184–196. doi: 10.1101/gad.252528.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cheng G, Kong D, Hou X, Liang B, He M, Liang N, et al. The tumor suppressor, p53, contributes to radiosensitivity of lung cancer cells by regulating autophagy and apoptosis. Cancer Biother Radiopharm. 2013;28(2):153–159. doi: 10.1089/cbr.2012.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang T, Dong K, Liang W, Xu D, Xia H, Geng J, et al. G-protein-coupled receptors regulate autophagy by ZBTB16-mediated ubiquitination and proteasomal degradation of Atg14L. eLife. 2015;4:e06734. doi: 10.7554/eLife.06734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xia PY, Wang S, Du Y, Zhao ZN, Shi L, Sun L, et al. WASH inhibits autophagy through suppression of Beclin 1 ubiquitination. EMBO J. 2013;32(20):2685–2696. doi: 10.1038/emboj.2013.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shi CS, Kehrl JH. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal. 2010;3(123):ra42. doi: 10.1126/scisignal.2000751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Platta Harald W, Abrahamsen H, Thoresen Sigrid B, Stenmark H. Nedd4-dependent lysine-11-linked polyubiquitination of the tumour suppressor Beclin 1. Biochem J. 2012;441(1):399–406. doi: 10.1042/BJ20111424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol. 2013;15(4):406–416. doi: 10.1038/ncb2708. [DOI] [PubMed] [Google Scholar]
- 82.Antonioli M, Albiero F, Nazio F, Vescovo T, Perdomo AB, Corazzari M, et al. AMBRA1 interplay with cullin E3 ubiquitin ligases regulates autophagy dynamics. Dev Cell. 2014;31(6):734–746. doi: 10.1016/j.devcel.2014.11.013. [DOI] [PubMed] [Google Scholar]
- 83.McEwan David G, Dikic I. Cullins keep autophagy under control. Dev Cell. 2014;31(6):675–676. doi: 10.1016/j.devcel.2014.12.010. [DOI] [PubMed] [Google Scholar]
- 84.Pattingre S, Tassa A, Qu XP, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 85.Lee YR, Yuan WC, Ho HC, Chen CH, Shih HM, Chen RH. The Cullin 3 substrate adaptor KLHL20 mediates DAPK ubiquitination to control interferon responses. EMBO J. 2010;29(10):1748–1761. doi: 10.1038/emboj.2010.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M, et al. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009;10(3):285–292. doi: 10.1038/embor.2008.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhao Y, Sun Y. Targeting the mTOR-DEPTOR pathway by CRL E3 ubiquitin ligases: therapeutic application. Neoplasia. 2012;14(5):360–367. doi: 10.1593/neo.12532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mao JH, Kim IJ, Wu D, Climent J, Kang HC, DelRosario R, et al. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science. 2008;321(5895):1499–1502. doi: 10.1126/science.1162981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhao YC, Xiong XF, Sun Y. DEPTOR, an mTOR inhibitor, is a physiological substrate of SCF βTrCP E3 ubiquitin ligase and regulates survival and autophagy. Mol Cell. 2011;44(2):304–316. doi: 10.1016/j.molcel.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gao D, Inuzuka H, Tan MKM, Fukushima H, Locasale Jason W, Liu P, et al. mTOR drives its own activation via SCF βTrCP-dependent degradation of the mTOR inhibitor DEPTOR. Mol Cell. 2011;44(2):290–303. doi: 10.1016/j.molcel.2011.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Duan S, Skaar Jeffrey R, Kuchay S, Toschi A, Kanarek N, Ben-Neriah Y, et al. mTOR generates an auto-amplification loop by triggering the βTrCP-and CK1α-dependent degradation of DEPTOR. Mol Cell. 2011;44(2):317–324. doi: 10.1016/j.molcel.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hu J, Zacharek S, He YJ, Lee H, Shumway S, Duronio RJ, et al. WD40 protein FBW5 promotes ubiquitination of tumor suppressor TSC2 by DDB1–CUL4–ROC1 ligase. Genes Dev. 2008;22(7):866–871. doi: 10.1101/gad.1624008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-α to the von Hippel–Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292(5516):468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 94.Ivan M, Kondo K, Yang HF, Kim W, Valiando J, Ohh M, et al. HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292(5516):464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 95.Katiyar S, Liu E, Knutzen CA, Lang ES, Lombardo CR, Sankar S, et al. REDD1, an inhibitor of mTOR signalling, is regulated by the CUL4A-DDB1 ubiquitin ligase. EMBO Rep. 2009;10(8):866–872. doi: 10.1038/embor.2009.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li X, Liu J, Gao T. β-TrCP-mediated ubiquitination and degradation of PHLPP1 are negatively regulated by Akt. Mol Cell Biol. 2009;29(23):6192–6205. doi: 10.1128/MCB.00681-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rui L, Yuan M, Frantz D, Shoelson S, White MF. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem. 2002;277(44):42394–42398. doi: 10.1074/jbc.C200444200. [DOI] [PubMed] [Google Scholar]
- 98.Xu X, Sarikas A, Dias-Santagata DC, Dolios G, Lafontant PJ, Tsai SC, et al. The CUL7 E3 ubiquitin ligase targets insulin receptor substrate 1 for ubiquitin-dependent degradation. Mol Cell. 2008;30(4):403–414. doi: 10.1016/j.molcel.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shi J, Luo L, Eash J, Ibebunjo C, Glass DJ. The SCF-Fbxo40 complex induces IRS1 ubiquitination in skeletal muscle, limiting IGF1 signaling. Dev Cell. 2011;21(5):835–847. doi: 10.1016/j.devcel.2011.09.011. [DOI] [PubMed] [Google Scholar]
- 100.Tan M, Zhao Y, Kim S-J, Liu M, Jia L, Saunders Thomas L, et al. SAG/RBX2/ROC2 E3 ubiquitin ligase is essential for vascular and neural development by targeting NF1 for degradation. Dev Cell. 2011;21(6):1062–1076. doi: 10.1016/j.devcel.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hollstein PE, Cichowski K. Identifying the ubiquitin ligase complex that regulates the NF1 tumor suppressor and Ras. Cancer Discov. 2013;3(8):880–893. doi: 10.1158/2159-8290.CD-13-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Xie CM, Wei DP, Zhao LL, Marchetto S, Mei L, Borg JP, et al. Erbin is a novel substrate of the Sag-βTrCP E3 ligase that regulates Kras(G12D)-induced skin tumorigenesis. J Cell Biol. 2015;209(5):721–737. doi: 10.1083/jcb.201411104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dorrello NV, Peschiaroli A, Guardavaccaro D, Colburn NH, Sherman NE, Pagano M. S6K1-and βTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science. 2006;314(5798):467–471. doi: 10.1126/science.1130276. [DOI] [PubMed] [Google Scholar]
- 104.Song X, Zhang X, Wang X, Zhu F, Guo C, Wang Q, et al. Tumor suppressor gene PDCD4 negatively regulates autophagy by inhibiting the expression of autophagy-related gene ATG5. Autophagy. 2013;9(5):743–755. doi: 10.4161/auto.24069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhao Y, Xiong X, Jia L, Sun Y. Targeting Cullin-RING ligases by MLN4924 induces autophagy via modulating the HIF1–REDD1–TSC1–mTORC1–DEPTOR axis. Cell Death Dis. 2012;3:e386. doi: 10.1038/cddis.2012.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yang D, Li L, Liu H, Wu L, Luo Z, Li H, et al. Induction of autophagy and senescence by knockdown of ROC1 E3 ubiquitin ligase to suppress the growth of liver cancer cells. Cell Death Differ. 2013;20(2):235–247. doi: 10.1038/cdd.2012.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jin G, Lee SW, Zhang X, Cai Z, Gao Y, Chou PC, et al. Skp2-mediated RagA ubiquitination elicits a negative feedback to prevent amino-acid-dependent mTORC1 hyperactivation by recruiting GATOR1. Mol Cell. 2015;58(6):989–1000. doi: 10.1016/j.molcel.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chan CH, Li CF, Yang WL, Gao Y, Lee SW, Feng Z, et al. The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell. 2012;149(5):1098–1111. doi: 10.1016/j.cell.2012.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nitsche C, Edderkaoui M, Moore RM, Eibl G, Kasahara N, Treger J, et al. The phosphatase PHLPP1 regulates Akt2, promotes pancreatic cancer cell death, and inhibits tumor formation. Gastroenterology. 2012;142(2):377–387. doi: 10.1053/j.gastro.2011.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Arias E, Koga H, Diaz A, Mocholi E, Patel B, Cuervo AM. Lysosomal mTORC2/PHLPP1/Akt regulate chaperone-mediated autophagy. Mol Cell. 2015;59(2):270–284. doi: 10.1016/j.molcel.2015.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. 2012;22(8):407–417. doi: 10.1016/j.tcb.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouyssegur J, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29(10):2570–2581. doi: 10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mazure NM, Pouysségur J. Hypoxia-induced autophagy: cell death or cell survival? Curr Opin Cell Biol. 2010;22(2):177–180. doi: 10.1016/j.ceb.2009.11.015. [DOI] [PubMed] [Google Scholar]
- 114.Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009;16(7):939–946. doi: 10.1038/cdd.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12(1):9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6(6):458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 117.Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, et al. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007;6(6):472–483. doi: 10.1016/j.cmet.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 118.Sengupta A, Molkentin JD, Yutzey KE. FoxO transcription factors promote autophagy in cardiomyocytes. J Biol Chem. 2009;284(41):28319–28331. doi: 10.1074/jbc.M109.024406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhao Y, Yang J, Liao WJ, Liu XY, Zhang H, Wang S, et al. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat Cell Biol. 2010;12(7):665–675. doi: 10.1038/ncb2069. [DOI] [PubMed] [Google Scholar]
- 120.Huang H, Regan KM, Wang F, Wang D, Smith DI, van Deursen JM, et al. Skp2 inhibits FOXO1 in tumor suppression through ubiquitin-mediated degradation. Proc Natl Acad Sci USA. 2005;102(5):1649–1654. doi: 10.1073/pnas.0406789102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang F, Chan CH, Chen K, Guan X, Lin HK, Tong Q. Deacetylation of FOXO3 by SIRT1 or SIRT2 leads to Skp2-mediated FOXO3 ubiquitination and degradation. Oncogene. 2012;31(12):1546–1557. doi: 10.1038/onc.2011.347. [DOI] [PubMed] [Google Scholar]
- 122.Tsai WB, Chung YM, Zou Y, Park SH, Xu Z, Nakayama K, et al. Inhibition of FOXO3 tumor suppressor function by βTrCP1 through ubiquitin-mediated degradation in a tumor mouse model. PLoS One. 2010;5(7):e11171. doi: 10.1371/journal.pone.0011171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Li HH, Willis MS, Lockyer P, Miller N, McDonough H, Glass DJ, et al. Atrogin-1 inhibits Akt-dependent cardiac hypertrophy in mice via ubiquitin-dependent coactivation of forkhead proteins. J Clin Invest. 2007;117(11):3211–3223. doi: 10.1172/JCI31757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Milani M, Rzymski T, Mellor HR, Pike L, Bottini A, Generali D, et al. The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with bortezomib. Cancer Res. 2009;69(10):4415–4423. doi: 10.1158/0008-5472.CAN-08-2839. [DOI] [PubMed] [Google Scholar]
- 125.Rzymski T, Milani M, Pike L, Buffa F, Mellor HR, Winchester L, et al. Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene. 2010;29(31):4424–4435. doi: 10.1038/onc.2010.191. [DOI] [PubMed] [Google Scholar]
- 126.Lassot I, Segeral E, Berlioz-Torrent C, Durand H, Groussin L, Hai T, et al. ATF4 degradation relies on a phosphorylation-dependent interaction with the SCF(βTrCP) ubiquitin ligase. Mol Cell Biol. 2001;21(6):2192–2202. doi: 10.1128/MCB.21.6.2192-2202.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Petherick KJ, Williams AC, Lane JD, Ordonez-Moran P, Huelsken J, Collard TJ, et al. Autolysosomal β-catenin degradation regulates Wnt-autophagy-p62 crosstalk. EMBO J. 2013;32(13):1903–1916. doi: 10.1038/emboj.2013.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Korolchuk VI, Menzies FM, Rubinsztein DC. Mechanisms of cross-talk between the ubiquitin–proteasome and autophagy–lysosome systems. FEBS Lett. 2010;584(7):1393–1398. doi: 10.1016/j.febslet.2009.12.047. [DOI] [PubMed] [Google Scholar]
- 129.Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol. 2010;12(9):836–841. doi: 10.1038/ncb0910-836. [DOI] [PubMed] [Google Scholar]
- 130.Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447(7146):859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 131.Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin–proteasome pathway substrates. Mol Cell. 2009;33(4):517–527. doi: 10.1016/j.molcel.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lander R, Nordin K, LaBonne C. The F-box protein Ppa is a common regulator of core EMT factors Twist, Snail, Slug, and Sip1. J Cell Biol. 2011;194(1):17–25. doi: 10.1083/jcb.201012085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Qiang L, Zhao BZ, Ming M, Wang N, He TC, Hwang S, et al. Regulation of cell proliferation and migration by p62 through stabilization of Twist1. Proc Natl Acad Sci USA. 2014;111(25):9241–9246. doi: 10.1073/pnas.1322913111. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 134.Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol. 2004;24(18):8055–8068. doi: 10.1128/MCB.24.18.8055-8068.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Babu JR, Geetha T, Wooten MW. Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J Neurochem. 2005;94(1):192–203. doi: 10.1111/j.1471-4159.2005.03181.x. [DOI] [PubMed] [Google Scholar]
- 136.Jaramillo MC, Zhang DD. The emerging role of the Nrf2–Keap1 signaling pathway in cancer. Genes Dev. 2013;27(20):2179–2191. doi: 10.1101/gad.225680.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12(3):213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 138.Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O, et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol. 2011;193(2):275–284. doi: 10.1083/jcb.201102031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lee J, Kim HR, Quinley C, Kim J, Gonzalez-Navajas J, Xavier R, et al. Autophagy suppresses interleukin-1β (IL-1β) signaling by activation of p62 degradation via lysosomal and proteasomal pathways. J Biol Chem. 2012;287(6):4033–4040. doi: 10.1074/jbc.M111.280065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Huber C, Dias-Santagata D, Glaser A, O’Sullivan J, Brauner R, Wu K, et al. Identification of mutations in CUL7 in 3-M syndrome. Nat Genet. 2005;37(10):1119–1124. doi: 10.1038/ng1628. [DOI] [PubMed] [Google Scholar]
- 141.Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AHFM, Schlegelberger B, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436(7051):660–665. doi: 10.1038/nature03841. [DOI] [PubMed] [Google Scholar]
- 142.Mooi WJ, Peeper DS. Oncogene-induced cell senescence-halting on the road to cancer. New Engl J Med. 2006;355(10):1037–1046. doi: 10.1056/NEJMra062285. [DOI] [PubMed] [Google Scholar]
- 143.Young ARJ, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JFJ, et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23(7):798–803. doi: 10.1101/gad.519709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Narita M, Young ARJ, Narita M. Autophagy facilitates oncogene-induced senescence. Autophagy. 2009;5(7):1046–1047. doi: 10.4161/auto.5.7.9444. [DOI] [PubMed] [Google Scholar]
- 145.Yao I, Takagi H, Ageta H, Kahyo T, Sato S, Hatanaka K, et al. SCRAPPER-dependent ubiquitination of active zone protein RIM1 regulates synaptic vesicle release. Cell. 2007;130(5):943–957. doi: 10.1016/j.cell.2007.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wu LG, Hamid E, Shin W, Chiang HC. Exocytosis and endocytosis: modes, functions, and coupling mechanisms. Annu Rev Physiol. 2014;76:301–331. doi: 10.1146/annurev-physiol-021113-170305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Shen W, Ganetzky B. Autophagy promotes synapse development in Drosophila. J Cell Biol. 2009;187(1):71–79. doi: 10.1083/jcb.200907109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Mori F, Nishie M, Piao YS, Kito K, Kamitani T, Takahashi H, et al. Accumulation of NEDD8 in neuronal and glial inclusions of neurodegenerative disorders. Neuropathol Appl Neurobiol. 2005;31(1):53–61. doi: 10.1111/j.1365-2990.2004.00603.x. [DOI] [PubMed] [Google Scholar]
- 149.Kuazi AD, Kito K, Abe Y, Shin RW, Kamitani T, Ueda N. NEDD8 protein is involved in ubiquitinated inclusion bodies. J Pathol. 2003;199(2):259–266. doi: 10.1002/path.1283. [DOI] [PubMed] [Google Scholar]
- 150.Yamamoto A, Cremona ML, Rothman JE. Autophagy-mediated clearance of huntingtin aggregates triggered by the insulin-signaling pathway. J Cell Biol. 2006;172(5):719–731. doi: 10.1083/jcb.200510065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bodine SC, Latres E, Baumhueter S, Lai VKM, Nunez L, Clarke BA, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294(5547):1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 152.Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117(3):399–412. doi: 10.1016/S0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ferdous A, Battiprolu PK, Ni YG, Rothermel BA, Hill JA. FoxO, autophagy, and cardiac remodeling. J Cardiovasc Transl. 2010;3(4):355–364. doi: 10.1007/s12265-010-9200-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zaglia T, Milan G, Ruhs A, Franzoso M, Bertaggia E, Pianca N, et al. Atrogin-1 deficiency promotes cardiomyopathy and premature death via impaired autophagy. J Clin Invest. 2014;124(6):2410–2424. doi: 10.1172/JCI66339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lee JA, Beigneux A, Ahmad ST, Young SG, Gao FB. ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr Biol. 2007;17(18):1561–1567. doi: 10.1016/j.cub.2007.07.029. [DOI] [PubMed] [Google Scholar]
- 156.Su HB, Li FQ, Ranek MJ, Wei N, Wang XJ. COP9 signalosome regulates autophagosome maturation. Circulation. 2011;124(19):2117–2128. doi: 10.1161/CIRCULATIONAHA.111.048934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Su HB, Li J, Menon S, Liu JB, Kumarapeli AR, Wei N, et al. Perturbation of cullin deneddylation via conditional Csn8 ablation impairs the ubiquitin–proteasome system and causes cardiomyocyte necrosis and dilated cardiomyopathy in mice. Circ Res. 2011;108(1):40–50. doi: 10.1161/CIRCRESAHA.110.230607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wei N, Serino G, Deng XW. The COP9 signalosome: more than a protease. Trends Biochem Sci. 2008;33(12):592–600. doi: 10.1016/j.tibs.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 159.Hariharan N, Maejima Y, Nakae J, Paik J, DePinho RA, Sadoshima J. Deacetylation of FoxO by SIRT1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ Res. 2010;107(12):1470–1482. doi: 10.1161/CIRCRESAHA.110.227371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Taillebourg E, Gregoire I, Viargues P, Jacomin AC, Thevenon D, Faure M, et al. The deubiquitinating enzyme USP36 controls selective autophagy activation by ubiquitinated proteins. Autophagy. 2012;8(5):767–779. doi: 10.4161/auto.19381. [DOI] [PubMed] [Google Scholar]