

Abstract
Background
Worldwide, over 350 million people are chronically infected with the hepatitis B virus (HBV) and are at increased risk of developing progressive liver diseases. The confinement of HBV replication to the liver, which also acts as the central hub for metabolic and nutritional regulation, emphasizes the interlinked nature of host metabolism and the disease. Still, the metabolic processes operational during the distinct clinical phases of a chronic HBV infection—immune tolerant, immune active, inactive carrier, and HBeAg-negative hepatitis phases—remains unexplored.
Methods
To investigate this, we conducted a targeted metabolomics approach on serum to determine the metabolic progression over the clinical phases of chronic HBV infection, using patient samples grouped based on their HBV DNA, alanine aminotransferase, and HBeAg serum levels.
Results
Our data illustrate the strength of metabolomics to provide insight into the metabolic dysregulation experienced during chronic HBV. The immune tolerant phase is characterized by the speculated viral hijacking of the glycerol-3-phosphate–NADH shuttle, explaining the reduced glycerophospholipid and increased plasmalogen species, indicating a strong link to HBV replication. The persisting impairment of the choline glycerophospholipids, even during the inactive carrier phase with minimal HBV activity, alludes to possible metabolic imprinting effects. The progression of chronic HBV is associated with increased concentrations of very long chain triglycerides together with citrulline and ornithine, reflective of a dysregulated urea cycle peaking in the HBV envelope antigen-negative phase.
Conclusions
The work presented here will aid in future studies to (i) validate and understand the implication of these metabolic changes using a thorough systems biology approach, (ii) monitor and predict disease severity, as well as (iii) determine the therapeutic value of the glycerol-3-phosphate–NADH shuttle.
Electronic supplementary material
The online version of this article (doi:10.1186/s13073-016-0318-8) contains supplementary material, which is available to authorized users.
Keywords: Metabolomics, Chronic hepatitis B, Liver, Viral hijacking, Clinical phases
Background
Worldwide, over 350 million people are chronically infected with the hepatitis B virus (HBV) and are at increased risk of developing progressive liver diseases, including fibrosis, liver failure, or hepatocellular carcinoma (HCC), over the course of several decades [1, 2]. Chronic HBV infection can be divided into four progressively distinct clinical phases based upon serum levels of HBV DNA, alanine aminotransferase (ALT), and HBV envelope antigen (HBeAg). These four phases are the immune tolerant (IT), immune active (IA), inactive carrier (IC), and HBeAg-negative (ENEG) phases. The increased ALT levels in the IA and ENEG phases reflect hepatic injury due to viral activity and immune activity. Even though these phases are used in clinical practice for deciding on therapeutic interventions [3], not much is known about the underlying metabolic mechanisms associated with each and/or the progressive nature of the disease. HBV replicates in the liver, which also acts as the central hub for metabolic and nutritional regulation, emphasizing the interlinked nature of the metabolism and the disease. Hence, a clear metabolic understanding of the progressive driving forces and diverse clinical outcomes is important in the management of chronic hepatitis B—for instance, to provide prediction markers for disease progression and candidate targets for therapeutic intervention. In vitro systems are not reliable models for chronic HBV infection since they do not represent the persistence, complexity, and progressive nature of the disease. Animal models do not appropriately mimic the pathogenic and immunological responses experienced during human HBV infection, hindering effective translational studies [4, 5]. Thus, the use of material obtained from patients is crucial to study and understand chronic HBV infection.
Metabolomics is an established, continuously improving tool used to study the metabolome through targeted or untargeted approaches, providing static observational and/or longitudinal readouts from a dynamic system [6–9]. Targeted metabolomics, which focuses on a predefined biology-driven subset of metabolite classes, is ideal for low concentration metabolites, whereas untargeted metabolomics is more suitable for measuring high concentration metabolites [10]. Metabolomics data aid in identifying and understanding the metabolic perturbations leading to dysregulated homeostasis and stress, e.g., during viral infection [11–14]. Subsequently, the contribution of these metabolic perturbations to the pathogenicity of the virus can be determined. Several metabolomics studies have been done to assess the metabolic host–pathogen interactions during chronic HBV infection. Targeted metabolomics approaches have identified the phospholipid [15, 16], triglyceride [15, 17, 18], sphingomyelin [15, 19], and free fatty acid [16, 20] metabolic pathways as affected during chronic HBV infection. Untargeted approaches have also been used to investigate metabolic changes experienced during chronic HBV infection [21–23] and have primarily focused on HBV-related cirrhosis and HCC [15, 24–26].
Here we present the first discovery metabolomics study of chronic HBV infection, shedding light on the progressive metabolic alterations over the different clinical phases. We used targeted metabolomics platforms to illuminate the phospholipid, triglyceride, sphingomyelin, amino acid, acyl-carnitine, and signaling lipid profiles to characterize the progressive nature of chronic HBV infection. We identified the most profound changes to occur in choline glycerophospholipids, plasmalogens, very long chain triglyceride species, and urea cycle intermediates across the progression of the four clinical phases of chronic HBV infection. Our data support the viral hijacking and persistent impairment of the glycerol-3-phosphate–NADH shuttle, modulating the host lipid profile. These events are central to understanding the metabolic perturbations reflective of the natural progression of chronic HBV infection.
Methods
Study population
Sixty-nine chronic HBV patients and 19 healthy controls (HCs) were included in this discovery study. Blood was prospectively collected in SST tubes at the Erasmus MC and centrifuged to separate the serum and stored at −80 °C until analysis. Patients were treatment naïve and excluded if they had other chronic infections (HIV, hepatitis C virus) or a body mass index of 31 or more. This study was conducted in accordance with the guidelines of the Declaration of Helsinki and the principles of Good Clinical Practice. The ethical review board of the Erasmus MC approved the study and informed consent was obtained from all patients who were asked to donate blood.
Definition of chronic HBV clinical phases
Serum ALT was measured on an automated analyzer, qualitative serum HBV surface antigen (HBsAg), HBeAg, and anti-HBeAg antibodies were measured on an Architect Abbott analyzer, and serum HBV DNA levels were measured using the COBAS AmpliPrep-COBAS Taq-Man HBVv2test (CAP-CTM; Roche Molecular Systems, Indianapolis, IN, USA). Based on serum HBV DNA, ALT levels, and HBeAg presence at the time of sampling, patients were categorized into four clinical HBV phases according to international guidelines [27]. IT patients (n = 18) had detectable serum HBeAg and repetitive normal ALT values (<40 U/L). IA patients (n = 12) and ENEG patients (n = 19) had repetitive or intermittent abnormal serum ALT (>40 U/L) values and HBV DNA levels >2000 IU/mL. IC patients (n = 20) were HBeAg-negative and had both repetitive normal ALT values (<40 IU/L) and HBV DNA levels below 20,000 IU/mL.
Targeted liquid chromatography–mass spectrometry metabolomics
Targeted metabolomics analyses were done using standard operating procedures from previously published methods. Detailed procedures and target lists are provided in the Additional file 1: Methods and a brief overview of the five platforms used is given in Table 1.
Table 1.
Metabolomics platforms: volumes, sample preparation, and analytical instruments
| Targeted metabolomics platform | Volume of serum used (μL) | Sample prep method | Analytical platform | Metabolite class coverage | Platform targets (n) | ||
|---|---|---|---|---|---|---|---|
| Total | Quality control passed | Percentage of missing data | |||||
| Biogenic amine [51] | 5 | Protein precipitation and AccQTag derivatization | UPLC-Xevo-TQMS | Amino acids, catecholamines and polyamines | 100 | 38 | 0.03 |
| Positive lipid [52] | 10 | Isopropyl alcohol extraction | UPLC-QToF | Phospholipids, cholesterol esters, di/triglycerides and sphingomyelins | 250 | 140 | 0.11 |
| Negative lipid [52] | 20 | Methanol extraction | UPLC-QToF | Free fatty acids and phospholipids | 150 | 59 | 0.17 |
| Oxylipins [53] | 250 | Oasis HLB solid-phase extraction | HPLC–QqQ-MS | Hydroxylated fatty acids, prostaglandins, and thromboxanes | 120 | 35 | 0.07 |
| Acyl-carnitines | 10 | Protein precipitation | UPLC- Xevo-TQMS | Acylcarnitines, TMAO, choline, betaine | 50 | 26 | 0 |
QqQ-MS triple quadrupole mass spectrometry, QToF quadrupole time-of-flight, TQMS triple quadrupole mass spectrometry, TMAO trimethylamine-N-oxide, UPLC ultra performance liquid chromatography
Quality control (QC) samples consisted of equally pooled volumes of all study samples. A set of QC samples was then included during the analyses of the experimental groups on the individual metabolomic platforms and evenly distributed through the randomized samples prior to liquid chromatography–mass spectrometry (LC–MS) analyses. In addition, independent duplicate samples (10–15 %) were randomly selected where sample volume allowed.
After LC–MS analyses, peak integration was done using the instrument’s software and the relative ratios between metabolites and their corresponding internal standards were determined. Using the QC samples and duplicate samples, a double QC approach was applied to include metabolites that were reliably measured by the individual metabolomics platforms by reporting and using only those metabolites for which both duplicate samples and QC samples showed a relative standard deviation <30 %. After QC, a data set comprising 88 cases and 314 metabolites was constructed, with all missing data being replaced by half the minimal intensity value of the corresponding metabolite.
Statistical data analyses
SPSS 21.0 (SPSS Inc., Chicago, IL, USA) was used for Fisher’s exact tests on the patient cohort characteristics presented as frequencies and ANOVA on the continuous values. A combination of univariate and multivariate bioinformatics approaches was performed using the R script-based online tool Metaboanalyst 3.0, a comprehensive tool suitable for analyzing metabolomics data [28, 29]. The metabolomics data sets were log transformed and auto-scaled prior to bioinformatics analyses. For the analyses between HCs and IT patients, significant metabolites were identified per metabolomics platform based on the following criteria: (i) a p value <0.05 using the independent student t-test; and (ii) a fold change ≥1.20 or ≤0.80, indicating a 20 % increase or decrease. The false discovery rate’s q values are reported for every reported p value. Multivariate principal component analysis was also done to visualize the natural distribution of the data.
The multivariate ANOVA test was used to identify the significant changes (p < 0.05) across the four clinical phases of chronic HBV infection in conjunction with K-means clustering. The K-means clustering was performed in MATLAB using kmeans and “correlation” as the distance measure on the metabolites in order to examine changes in their levels during the course of chronic HBV infection, using all 314 metabolites and 88 patients. Its algorithm partitions the metabolites into K mutually exclusive clusters (k is the number of desired clusters), taking into account the measurement of each metabolite under multiple conditions, in this case HCs plus all four clinical phases. Within each cluster, metabolite measurement patterns are as close to each other as possible, while as far away from those in other clusters as possible. The partition was repeated with metabolites reassigned among clusters at each iteration until the sum of point-to-centroid distance reached a minimum [30]. The pattern analysis was performed sequentially with k assigned with 12, 16, 20, and 24 each time.
Results
Baseline characteristics of the study population
For the metabolomics characterization of chronic HBV infection, a cohort of treatment-naïve patients was chosen and patients with other comorbidities and/or advanced liver fibrosis were excluded from the study. Due to the progressive nature of chronic HBV infection over time, IT and IA patients are younger than IC and ENEG patients (p = 0.03; Table 2). The asymptomatic IT and IC groups have a larger female representation due to routine HBsAg testing during pregnancy and subsequent referrals. This gender ratio is reversed in the IA and ENEG groups (Table 2). The ratios of HBV genotypes are evenly spread in the cohort, except for the IT and ENEG groups where genotypes B/C and D are dominant, respectively (Table 2). Owing to the stringent definition criteria, differences in ALT (Fig. 1a) and HBV DNA (Fig. 1b) levels are observed between the clinical phases.
Table 2.
Baseline characteristics of the patient cohort
| Characteristics | Total cohort | Healthy control (HC) | Immune tolerant (IT) | HBeAg-positive active hepatitis (IA) | Inactive carrier (IC) | HBeAg-negative active hepatitis (ENEG) |
|---|---|---|---|---|---|---|
| Number of patients | 88 | 19 | 18 | 12 | 20 | 19 |
| Demography | ||||||
| Age, years (SE) | 35.26 (11.58) | - | 30.89 (1.77) | 31.75 (2.60) | 39.85 (3.02) | 37.68 (2.75) |
| Gender, n** | ||||||
| Female | 41 | 7 (17.1 %) | 13 (31.7 %) | 5 (12.2 %) | 14 (34.2 %) | 2 (4.9 %) |
| Male | 41 | 6 (14.6 %) | 5 (12.2 %) | 7 (17.1 %) | 6 (14.6 %) | 17 (41.5 %) |
| 7 missing | ||||||
| BMI, kg/m2 (SE) | 24.64 (6.40) | - | 25.93 (3.27) | 23.08 (1.12) | 24.41 (0.70) | 24.66 (0.79) |
| Race, n* | ||||||
| Asian | 44 (63.77 %) | - | 16 (36.4 %) | 9 (22.7 %) | 8 (18.21 %) | 10 (22.7 %) |
| Caucasian | 9 (13.04 %) | - | 0 (0.0 %) | 3 (33.3 %) | 2 (22.2 %) | 4 (44.4 %) |
| African | 8 (11.59 %) | - | 0 (0.0 %) | 0 (0.0 %) | 5 (62.5 %) | 3 (37.5 %) |
| Other | 8 (11.59 %) | - | 2 (25.0 %) | 0 (0.0 %) | 4 (50 %) | 2 (25.0 %) |
| Virology | ||||||
| Log HBV DNA, IU/ml (SE)*** | 5.55 (2.82) | - | 8.84 (0.07) | 7.40 (0.57) | 2.43 (0.15) | 4.39 (0.30) |
| HBV genotype A/B/C/D** | 13/16/20/20 | - | 0/8/8/2 | 3/2/6/1 | 6/5/2/6 | 4/1/3/11 |
| Chemistry/hematology | ||||||
| ALT, IU/l (SE)*** | 48.17 (43.56) | - | 24.50 (1.72) | 88.83 (16.93) | 23.90 (1.43) | 69.53 (10.91) |
BMI body mass index, SE standard error
Fisher’s exact p value: *p < 0.05, **p < 0.01
***ANOVA p value < 0.01
Fig. 1.
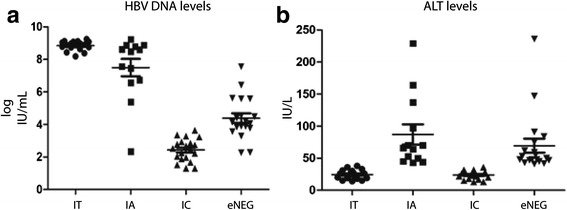
Clinical parameters. Baseline serum characteristics of patients with chronic HBV infection divided into four clinical phases based on ALT and HBV DNA levels. Serum samples of 69 chronic HBV patients were measured for their levels of a HBV DNA and b ALT levels
Metabolic characterization of the IT phase of chronic HBV infection versus the HCs
To investigate and understand the metabolic alterations during the progressive clinical phases of a chronic HBV infection, we first characterized HCs versus the IT phase, representing the onset of chronic HBV. Multivariate and univariate data analyses were performed to identify metabolic pathways altered in the IT phase. The principal component analysis showed natural clustering with partial overlap observed between the HC and IT sample groups (Additional file 2: Figure S1). For univariate analysis, fold change in combination with t-tests was used and the identified significant metabolites visualized in a volcano plot (Fig. 2; Additional file 2: Table S1). The volcano plot highlighted 92 significant metabolites belonging to the phospholipid, plasmalogen, triglyceride, amino acid, sphingomyelin, free fatty acid, and acyl-carnitine metabolic pathways listed in Table 3. Under closer inspection, the univariate/supervised analysis reveals primarily uniform class responses, indicating definite metabolic rearrangement. The down-regulated metabolites are dominated by triglycerides, phospholipids, lysophospholipids, and sphingomyelins in the IT phase compared with HCs. In contrast, plasmalogens (specialised vinyl ether-linked phospholipids), free fatty acids, and acyl-carnitines had elevated levels in the IT phase compared with HCs. Clinically, the IT phase is characterized by high levels of HBV replication and negligible hepatic injury; hence, the dysregulated lipid profile observed is most likely due to metabolic hijacking occurring during the HBV life cycle.
Fig. 2.
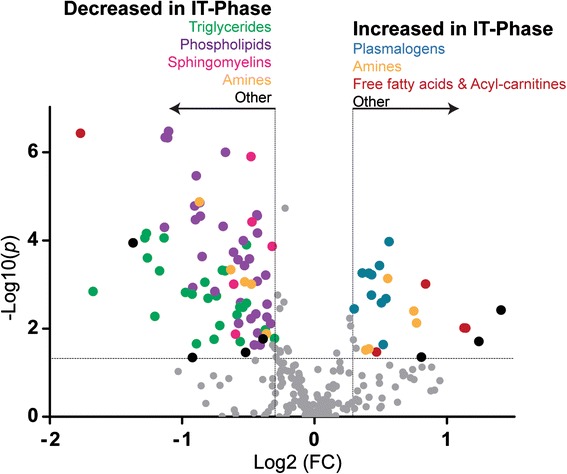
Volcano plot of the significant serum metabolites between the HCs and the IT phase. Metabolites with a fold change (FC; x-axis) threshold of ≥1.20 or ≤0.80 and t-tests (y-axis) threshold of 0.05 are identified as significant and represented by colored circles ordered by the metabolite class. The further its position away from (0,0), the more significant the metabolite. Metabolite classes are as follows: triglycerides, green; phospholipids, purple; sphingomyelins, pink; amines, orange; plasmalogens, blue; free fatty acids and acyl carnitines, maroon; others, black
Table 3.
Significant serum metabolites identified between HCs and the IT phase
| Targeted metabolomics platform | Class | Metabolite species identified as significant between the HCs and IT phase | Trend in the IT phase |
|---|---|---|---|
| Phospholipids | |||
| Positive lipids | Phosphatidylcholines (PC) | C32:1, C32:2, C34:1, C34:3, C34:4, C36:3, C36:4, C36:6, C38:3 | Decreased |
| Phosphatidylethanolamines (PE) | C38:4 | Decreased | |
| Plasmalogens | |||
| Positive lipids | Plasmalogen phosphatidylcholines (pPC) | C34:2, C36:2, C36:3, C38:6 | Increased |
| Plasmalogen phosphatidylethanolamines (PE) | C38:5, C38:7# | Increased | |
| Lysophospholipids | |||
| Negative lipids | Lysophosphatidylcholines (LPC) | sn1: C14:0, C15:0, C16:1, C18:1, C18:2, C18:3 (ω3ω6), C20:3 (ω3ω6), C20:3 (ω9), C20:4, C22:4, C22:5 (ω6) | Decreased |
| sn2: C14:0, C16:1, C18:1, C18:2, C18:3 (ω3ω6), C20:3 (ω3ω6), C20:4 | |||
| Lysophosphatidylethanolamines (LPE) | C18:0, C18:1, C18:2#, C20:3 (ω3ω6), C20:4, C22:5# (ω3) | Decreased | |
| Plasmalogen lysophospholipids | |||
| Negative lipids | Plasmalogen lysophosphatidylcholines (pLPC) | C16:0, C16:1, C18:1, C18:2 | Increased |
| Triglycerides | |||
| Positive lipids | Triglycerides | C42:1, C44:2, C46:1, C46:2, C46:3, C48:1, C48:2, C48:3, C48:4#, C50:0, C50:1, C50:2, C50:3, C50:4, C50:5, C51:1, C51:2, C51:3, C52:1, C52:2, C54:1, C54:2, C55:2, C56:5, C58:5 | Decreased |
| Amines | |||
| Biogenic amines | Amino acids | Tryptophan# | Decreased |
| Glutathione | Glutathione | Decreased | |
| Dipeptides | γ-Glutamylglutamine | Decreased | |
| Amino acids metabolites | Kynurenine, saccharopine | Decreased | |
| Hepatic injury | 4-Hydroxyproline, pipecolic acid | Increased | |
| Glycine metabolism | Betaine#, sarcosine# | Increased | |
| Other | s-Methylcysteine | Increased | |
| Sphingomyelins | |||
| Positive lipids | Sphingomyelins (SM) | C18:1/14:0, C18:1/21:0, C18:1/23:0, C18:1/25:0, C18:1/25:1 | Decreased |
| Free fatty acids and Acyl-carnitines | |||
| Negative lipids | Free fatty acids (FFA) | C18:2, C20:2 | Increased |
| Acyl-carnitines | Acyl-carnitines | Oleylcarnitine (C18:1)#, Linoleylcarnitine (C18:2) | Increased |
| Nonanoylcarnitine (C9:0) | Decreased | ||
| Others | |||
| Positive lipids | Cholestrol esters (CE) | C18:3 | Decreased |
| Oxylipins | Oxylipins | 11-HDoHE#, 14-HDoHE#, 10-HDoHE# | Increased |
| 5-HETrE, 8,9-DiHETre,#, 12,13-DiHOME,# | Decreased | ||
The metabolites identified as significant based on t-test and fold change are divided into their different classes together with their trend. All metabolites had a q value <0.05 unless otherwise indicated
# q value >0.05
Metabolomics characterization of the natural course of a chronic HBV infection
Given the interdependent nature of metabolic pathways, functionally related metabolites might have similar response patterns. To examine this, a K-means pattern analysis was performed on the total metabolomics data set comprising 88 cases and 314 metabolites. A pattern of 24 clusters was selected as the most distinct pattern describing the metabolomics data (Additional file 2: Figure S2; Additional file 3: Table S2). Subsequently, 6 of the 24 clusters were chosen for further study based on the profile and the number of metabolites (n > 5) clustering within each and the correlation between profiles and fluctuation of ALT and HBV DNA levels (Fig. 3). Pattern analysis identifies similarly responding metabolites but is not a measure of significance and, therefore, an ANOVA approach was used between the four clinical phases to identify significantly changing metabolites (Table 4; Additional file 2: Table S3).
Fig. 3.
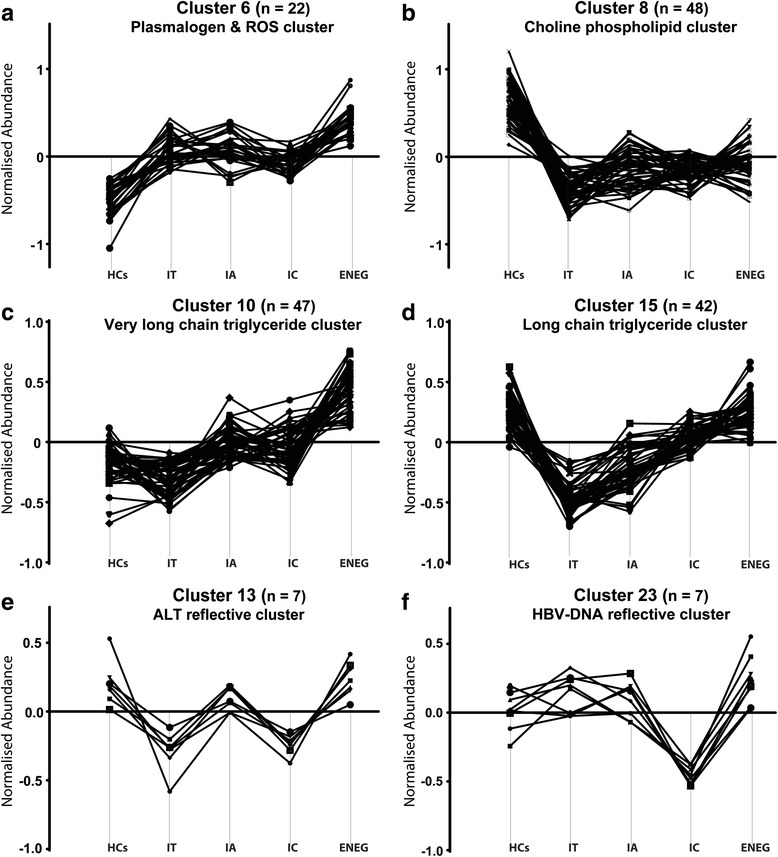
Serum metabolite pattern analyses. a Cluster 6, the plasmalogen and reactive oxygen species (ROS) cluster, showed a “stable” elevated trend against the HCs during the progression of the four clinical phases. b Cluster 8, the choline phospholipid cluster, showed a “stable” reduced trend over the four clinical phases against the HCs. c Cluster 10, the very long chain triglyceride cluster, represents metabolites with subtle changes at the start of chronic HBV infection but increased significantly over the progression of the four clinical phases. d Cluster 15, the long chain triglyceride cluster, grouped metabolites with a significant reduction between the HCs and the IT phase, after which their levels increased again as the disease progressed. e Cluster 13, the ALT reflective cluster, grouped metabolites following the same trend as ALT with increased levels in the IA and ENEG phases. f Cluster 23, the HBV DNA cluster, represents metabolites reflective of HBV DNA levels, which are lowest during the IC phase. The y-axis is the abundance of metabolites normalized to the overall mean of the metabolites across all samples
Table 4.
Serum metabolites identified as changing significantly over the progression of chronic HBV infection
| Targeted metabolomics platform | Metabolite class | Metabolite species identified as changing significantly during the progression of chronic HBV |
|---|---|---|
| Cluster 6: stable increased | ||
| Biogenic amines | Amines | Carnitine, ornithine |
| Cluster 8: stable decreased | ||
| Acyl-carnitines | Acylcarnitines | Nonaylcarnitine |
| Negative lipids | Lysophosphatidylcholine (LPC) | sn1, C20:3 (ω3ω6), sn2, C20:3 (ω3ω6) |
| Cluster 10: increasing over the four phases | ||
| Acyl-carnitines | Acyl-carnitines | Isovalerylcarnitine, 2-methylbutyroylcarnitine, stearoylcarnitine |
| Biogenic amines | Amino acids | Phenylalanine, glutamic acid, methionine |
| Amines | Choline | |
| Positive lipids | Diacylglycerol (DG) | C36:2 |
| Triglycerides (TG) | C54:2, C54:3, C54:4, C55:3, C56:3, C56:4, C56:5, C56:6, C58:5 | |
| Negative lipids | Free fatty acids | C20:4 (ω6) |
| Cluster 13: reflective of ALT levels | ||
| Biogenic amines | Amino acid metabolite | Kynurenine |
| Cluster 15: increasing over the four phases | ||
| Positive lipids | Phosphatidylcholine (PC) | C38:4 |
| Sphingomeylins (SM) | C18:1/25:0 | |
| Triglycerides (TG) | C42:1, C52:2, C55:2 | |
| Negative lipids | Lysophosphatidylethanolamine (LPE) | C18:1, C20:3 (ω3ω6), C20:4 |
| Cluster 23: reflective of HBV DNA levels | ||
| Acyl-carnitines | Acylcarnitines | Decanoylcarnitine |
| Negative lipids | Free fatty acids | C20:3 (ω3ω6), C22:4 |
The significant metabolites are divided into their clusters indicative of their trend and all had an ANOVA p value <0.05
Metabolite clusters showing a “stable” increased or decreased trend over the four clinical phases of chronic HBV
Clusters 6 (Fig. 3a) and 8 (Fig. 3b) represent metabolites significantly altered in the IT phase compared with HCs and whose alteration persisted throughout the progression of chronic HBV infection with stable increased or decreased trends. Cluster 6 contained seven plasmalogen phospholipid and lysophospholipid species, of which three were significantly up-regulated in the IT phase compared with the HCs, having sustained increased levels across the four clinical phases. We named this cluster the “ROS-plasmalogen cluster”. Four non-enzymatic route oxylipins (10-, 11-, 13-, and 14-HDoHEs) grouped within cluster 6 indicate maintained oxidative stress experienced during the progression of chronic HBV infection. Interestingly, the choline catabolic metabolites betaine and sarcosine were also in the ROS-plasmalogen cluster, which may suggest some degree of choline metabolism perturbation. The amine metabolites carnitine and ornithine were identified as significantly changing over the clinical phases (Table 4). Carnitine is the free fatty acid transporter metabolite and indicates significant dysregulation during chronic HBV infection (p < 0.001). The IT and IC phases had lower carnitine levels compared with the IA and ENEG phases, reflecting increased energy requirements during these phases. Ornithine is a urea cycle intermediate together with citrulline (cluster 9, p = 0.006), indicating a dysregulated urea cycle, worsening with the progression of chronic HBV infection as both had their highest levels in the ENEG phase.
Cluster 8 represents the opposite trend, showing a stable decline of metabolite levels across the four clinical phases compared with HCs. Cluster 8 was labeled the “choline phospholipid cluster” as it comprises 14 phosphatidylcholines and 21 lysophosphatidylcholines. The strong initial reduction of choline phospholipids, together with the subsequent maintenance of the reduced levels over the four clinical phases, hints towards an imprinted metabolic alteration. Only the sn1 and sn2 lysophosphatidylcholine C20:3 species showed significant fluctuations during the progression of the clinical phases (Table 4). Three amines, including saccharopine, glutathione and γ-glutamyl glutamine, had stable decreased levels compared with HCs. The glutathione levels reflect diminished antioxidant capacity during chronic HBV, supporting the increased levels of lipid peroxidation products observed within cluster 6.
Metabolite clusters showing a trend for increasing over the clinical phases of chronic HBV
From the 24 clusters, clusters 10 (Fig. 3c) and 15 (Fig. 3d) were the most dense, containing 47 and 42 metabolites, respectively. Both clusters showed an increasing trend across the four clinical phases of chronic HBV infection, with their defining difference observed when taking the HCs into account.
Cluster 10 shows subtle changes between the HCs and the IT phase but significantly increased during the progression of chronic HBV infection to levels well above HC levels in the ENEG phase. Cluster 10 is dominated by 13 amino acids and 17 triglyceride species, composed of long and very long acyl species with a combined acyl chain length of between C54 and C60. Thus, this cluster is called the “very long chain triglyceride cluster”. During the progression over the four clinical phases, 9 of the 17 triglycerides were identified as significantly increasing, revealing a perturbed triglyceride metabolism (Table 4). The essential amino acids methionine and phenylalanine were identified as significantly changing over the clinical phases and have both been identified as markers of hepatic injury and had their highest levels in the ENEG phase [31–33]. Choline, identified as significant and in cluster 10, is an integral part of glycerophospholipid and sphingolipid metabolism, acting as a functional lipid moiety. The increasing choline levels oppose that of its elevated catabolic metabolites betaine and sarcosine (cluster 6).
Cluster 15 includes metabolites that were significantly reduced in the IT phase compared with the HCs, after which metabolite levels started to increase back to HC levels. More than half of the 42 metabolites in cluster 15 are triglycerides, with a combined acyl chain length ranging from C42 to C55, effectively labeling this the “long chain triglyceride cluster”. These results show a biphasic triglyceride response during chronic HBV infection. Three of the five clustered lysophosphatidylethanolamines species, including C18:1, C20:3, and C20:4, were subsequently identified as significantly increasing over the four clinical phases, with levels almost resembling the HCs in the ENEG phase. These results also show that lysophosphatidylethanolamines (cluster 15) respond differently during chronic HBV infection to lysophosphatidylcholines (cluster 8).
Metabolite clusters correlating to ALT and HBV DNA levels
During chronic HBV infection, ALT levels (Fig. 1a) were highest during the IA and ENEG phases, whereas HBV DNA levels (Fig. 1b) were high in the IT, IA, and ENEG phases and lowest in the IC phase. Clusters 13 (Fig. 3e) and 23 (Fig. 3f) represented metabolites correlating to ALT and HBV DNA levels, respectively.
Kynurenine identified in cluster 13 correlated to ALT levels and had significantly higher levels in the IA and ENEG phases compared with the IT and IC phases (Table 4). Kynurenine and its precursor amino acid tryptophan (cluster 15) were both also identified as significantly down-regulated during the IT phase compared with the HCs. Due to these fluctuating levels during the progression of chronic HBV, we inspected the tryptophan/kynurenine ratio relating to indoleamine 2,3-dioxygenase (IDO) activity. The tryptophan/kynurenine ratio had a p value of 0.038 across the five groups with a reduced IDO ratio in the IT, IA, and IC phases of chronic HBV infection compared with an ENEG phase ratio comparable to that of the HCs, implying altered IDO regulation during the first three phases of chronic HBV infection.
Cluster 23, reflecting HBV DNA levels during chronic HBV infection, consisted solely of five acyl-carnitines and four free fatty acids. The long chain free fatty acids C20:3 (ω3/ω6) and C22:4, together with decanoylcarnitine, had significantly lower levels in the IC phase compared with the other clinical phases, possibly correlating to HBV-induced secreted phospholipases [34, 35].
Discussion
The present study is the first targeted and biology driven metabolomics profiling of chronic HBV infection, characterizing the natural progression through its distinct clinical phases. With the liver being the central organ in nutritional regulation and metabolism, it is not surprising that chronic HBV infections have been shown to induce multiple metabolic alterations in lipid metabolism of the host [15–21]. However, these studies have not addressed and positioned metabolic changes in relation to the progression of chronic HBV disease.
The IT phase is clinically characterized by high levels of HBV replication and minimal hepatic injury. We now show that this phase exhibits major lipid alterations, with increased free fatty acids, acyl-carnitines, and plasmalogens concurrent with decreased triglyceride, phospholipid (ester-linked), and sphingomyelin levels (Fig. 4). The glycolysis intermediate dihydroxyacetone phosphate (DHAP) is the precursor metabolite via glycerol-3-phosphate (G3P) for the de novo glycerophospholipids and triglyceride synthesis pathways. Alternatively, DHAP can also be transported to the peroxisomes where it is the precursor for the synthesis of vinyl ether-linked plasmalogen phospholipids. The DHAP to G3P reaction is catalyzed by glycerol-3-phosphate dehydrogenase (GPDH) and is known as the G3P–NADH shuttle, simultaneously converting NADH to NAD+ necessary during the glycolysis cycle. Our results suggest that HBV hijacks this G3P–NADH shuttle, resulting in reduced levels of glycerophospholipids, lysoglycerophospholipids, triglycerides, and sphingomyelins. The secretion of glycerophospholipids from the liver is the main contributor to their serum levels, substantiating the hepatic metabolic fingerprint and the link to HBV activity [36]. Plasmalogen phosphatidylcholine has been identified as the preferred lipid species in the viral envelope and surface antigen particles of HBV, accounting for approximately 60 % of total lipid content [37]. Additionally, Li et al. [16] reported the upregulation of mRNA transcripts in the phosphatidylcholine biosynthesis pathway during HBV infection in HepG2 cell lines and its necessity for HBV replication. Collectively, the observed increase in choline plasmalogen levels reveals the metabolic engineering capacity of HBV and supports our hypothesis of HBV hijacking of the G3P–NADH shuttle. Importantly, this altered lipid profile of down-regulated phospholipids and increased plasmalogens persists during the progression of chronic HBV infection (Fig 3a, b), even during the IC phase where virtually no HBV replication is taking place, implying a HBV-induced metabolic imprinting effect. Although other reasons could be used to explain metabolic observations (such as an altered metabolic flux or consumption rate of metabolites and even diet), the use of pattern analyses and the progressive nature of our clinical phase-defined chronic HBV samples provide concrete evidence to illustrate an altered metabolic state.
Fig. 4.
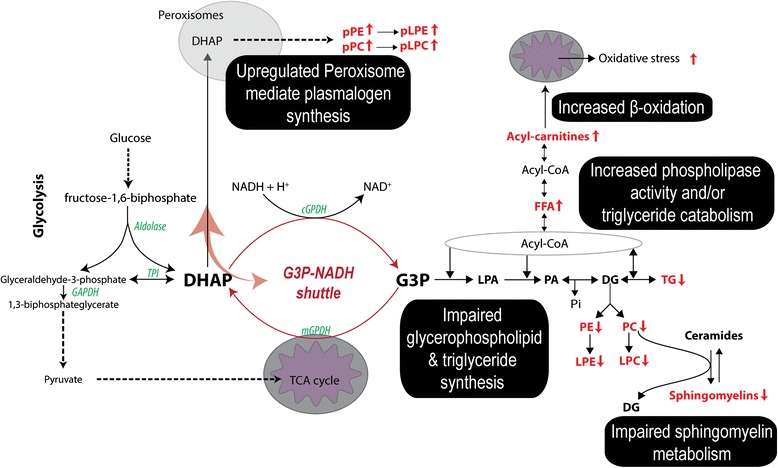
Metabolic alterations identified in the IT phase of chronic HBV. Increased levels of DHAP-derived plasmalogen phospholipid species, free fatty acids, and acyl carnitines were found. A significant decrease in glycerophospholipids, triglycerides, and sphingomyelins was found, suggesting the HBV hijacking of the cytosolic glycerol-3-phosphate dehydrogenase (GPDH) enzyme, favoring the synthesis of plasmalogen lipid species. Detected metabolic species are highlighted in red with the arrow indicating its trend. DHAP dihydroxyacetone phosphate, G3P glycerol-3-phosphate, LPA lysophosphatidic acid, PA phosphatidic acid, DG diacylglycerol, PE phosphatidylethanolamine, PC phosphatidylcholine, LPE lysophosphatidylethanolamine, LPC lysophosphatidylcholine, TG triglyceride, FFA free fatty acids, pPE plasmalogen phosphatidylethanolamine, pPC plasmalogen phosphatidylcholine, pLPE plasmalogen lysophosphatidylethanolamine, pLPC plasmalogen lysophosphatidylcholine, GAPDH glyceraldehyde-3-phosphate dehydrogenase, TPI triosephosphate isomerase, cGPDH cytosolic glycerol-3-phosphate dehydrogenase, mGPDH mitochondrial glycerol-3-phosphate dehydrogenase
During the natural progression of chronic HBV, we measured increased choline, methionine, and very long acyl chain triglyceride levels together with reduced phosphatidylcholine and lysophosphatidylcholine levels, indicating a perturbed choline metabolism. Dietary choline and methionine depravation is strongly linked to the development of steatosis, non-alcoholic fatty liver disease, cirrhosis, and HCC [38–41]. During conditions of choline restriction, the reduced levels of phosphatidylcholine, a critical component of the very low density lipoprotein particle, impair hepatic lipoprotein synthesis and result in the accumulation of free triglycerides within hepatocytes [42, 43]. Decreased phosphatidylcholine species in the presence of high choline levels during the IA, IC, and ENEG phases support the permanent G3P–NADH shuttle hijacking hypothesis, impairing lipoprotein synthesis during chronic HBV, while also explaining the accumulation of triglycerides. Even with decreased levels of long chain free fatty acids in the IC phase, no attenuation of long chain triglyceride levels was observed in this phase. The stable elevated levels of betaine, sarcosine, and methionine indicate enhanced choline catabolism, while increased levels of methionine are also reflective of hepatic injury [32, 33]. Previous studies demonstrated that host factors were responsible for the development of steatosis rather than viral factors [44, 45]. Our metabolomics data suggest that initiation of steatosis may be a consequence of HBV hijacking of the host’s glycerophopholipid metabolism, as liver fat content closely correlates with serum triglyceride levels [46, 47].
Another metabolic pathway with increased activity reflective of the natural progression of chronic HBV infection is composed of urea cycle intermediates: enhanced levels of citrulline and ornithine were detected in the IC and ENEG phases, respectively. The urea cycle is predominantly active in hepatocytes and responsible for detoxifying ammonia. Moreover, it has an intimate relationship with the aspartate–malate NADH shuttle functioning across the mitochondrial membrane. One could speculate that the HBV hijacking of the G3P–NADH shuttle, as explained above, will affect the redox status (NADH/NAD+) of the cell and cause an up-regulation/stress of the malate–aspartate NADH/NAD+ shuttle to rectify this imbalance (Fig. 5). The mitochondrial transporter citrin (AGC), encoded by the gene SLC25A13, is responsible for the transport of aspartate and glutamate during aspartate–malate NADH shuttling. Cytosolic aspartate binds to citrulline to form the urea cycle intermediate argininosuccinate, which can be converted to arginine, which in turn is converted to ornithine with the release of urea. We detected increased levels of citrulline, ornithine and glutamate, all implicating impaired aspartate transport, and may reflect reduced integrity and functioning of the mitochondrial citrin transporter. A dysregulated urea cycle precedes the histological manifestations of irreversible liver damage [48], and thus might be a prediction marker for chronic HBV progression and severity.
Fig. 5.
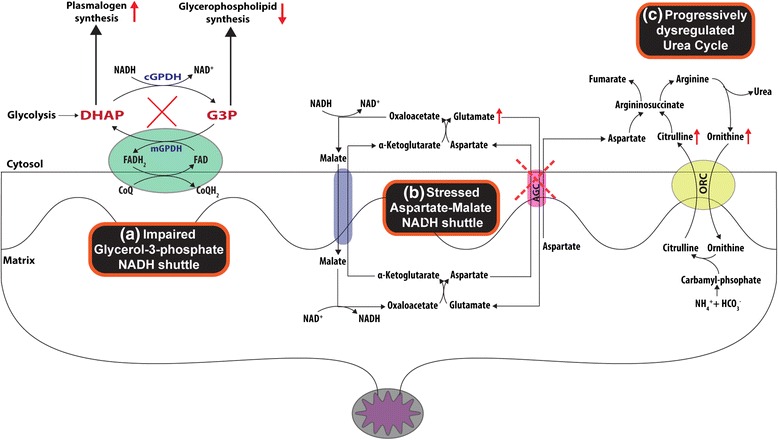
Cellular NADH shuttles and the urea cycle. The interplay between a the reduced G3P–NADH shuttle and b a stressed aspartate–malate NADH shuttle and its influence on c the urea cycle. The aspartate transporter AGC (aka citrin) facilitates the transport of aspartate across the mitochondrial membrane to the cytosol where it binds to citrulline in the urea cycle to help detoxify ammonium. Ineffective aspartate transport will lead to the accumulation of glutamate, citrulline, and ornithine identified in the IC and ENEG clinical phases of HBV infection
Collectively, the data presented here comprise the first metabolic study on the natural progression of chronic HBV infection using patient samples. We found impaired choline glycerophospholipid metabolism across the four chronic HBV clinical phases, together with increasing triglyceride and urea cycle intermediate levels as a liver metabolic fingerprint of the progression of chronic HBV infection. This metabolic fingerprint relates to HBV’s hijacking of the G3P–NADH shuttle, a key player in the plasmalogen, choline, and glycerophospholipid metabolic pathways, and its potential as a therapeutic target deserves further investigation. Elegant work done by Zeissig et al. [34] demonstrated the role of lysophospholipids as endogenous antigenic lipid species able to illicit protective immunological responses, which enhanced HBV clearance during acute infection. This may imply that, in addition to redirecting host lipid metabolism to produce the plasmalogens, hijacking of glycerophospholipid metabolism during acute HBV infection could act as a switch to determine HBV clearance or persistence. Furthermore, the diabetes drug metformin, which inhibits mitochondrial GPDH [49], part of the G3P–NADH shuttle, was found to inhibit HBV protein production and replication [50]. These findings substantiate the therapeutic value of the G3P–NADH shuttle in chronic HBV infection.
Conclusions
The present study provides many insights and leads to design follow-up studies and, at the same time, highlights the need for a systems biology approach to better understand chronic HBV infection. We identified liver-related metabolic and injury perturbations, which reflect the natural progression of the disease. The altered glycerophospholipid metabolism in the IT phase attributed to the HBV hijacking of the G3P–NADH shuttle has an intimate relationship with the persistent lipid dysregulation observed in the IA, IC, and ENEG clinical phases. Increased levels of the very long chain triglycerides in the IA phase and urea cycle intermediates in the IC phase highlight the risk for developing secondary liver complications during chronic HBV infection. These metabolites might prove useful as markers of disease progression and severity.
Abbreviations
ALT, alanine aminotransferase; DHAP, dihydroxyacetone phosphate; ENEG, HBeAg-negative; G3P, glycerol-3-phosphate; GPDH, glycerol-3-phosphate dehydrogenase; HBeAg, HBV envelope antigen; HBsAg, HBV surface antigen; HBV, hepatitis B virus; HC, healthy control; HCC, hepatocellular carcinoma; IA, immune active; IC, inactive carrier; IDO, indoleamine 2,3-dioxygenase; IT, immune tolerant; LC–MS, liquid chromatography–mass spectrometry; QC, quality control.
Acknowledgements
This study was supported by the NWO-ZonMW (grant number 435002027) and the Virgo consortium, funded by the Dutch government project number FES0908. We would also like to thank the technical staff, including L. Lamont, B. Gonzalez Amoros, and H. Elfrink at the Netherlands Metabolomics Centre, Leiden University for performing the metabolomics sample analyses.
Availability of data and materials
The metabolomics datasets supporting the conclusions of this article are available in the online MetaboLights data repository, under accession numbers MTBLS253, MTBLS279, and MTBLS280.
Authors’ contributions
AB and TH served as principal investigators and conceived and designed the study; JCS, JH, and ACH performed the experiments; JH and AB assisted in acquisition of the samples and clinical data; JCS, JH, RB, RJV, TH, and AB contributed to the interpretation of the data; JCS wrote the first draft of the manuscript with input from all authors; and all authors approved the final manuscript revisions.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
All authors give consent for the publication of the manuscript in Genome Medicine.
Ethics approval and consent to participate
This study was conducted in accordance with the guidelines of the Declaration of Helsinki and the principles of Good Clinical Practice. The ethical review board of the Erasmus MC approved the study and informed consent was obtained from all patients who were asked to donate blood.
Additional files
Supplementary methods. (DOCX 33 kb)
Supplementary Figures S1 and S2 and Tables S1 and S3. (DOCX 397 kb)
Supplementary Table S2. (XLSX 38 kb)
References
- 1.Locarnini S, Hatzakis A, Chen D-S, Lok A. Strategies to control hepatitis B: Public policy, epidemiology, vaccine and drugs. J Hepatol. 2015;62(1 Suppl):S76–86. doi: 10.1016/j.jhep.2015.01.018. [DOI] [PubMed] [Google Scholar]
- 2.Fattovich G, Bortolotti F, Donato F. Natural history of chronic hepatitis B: special emphasis on disease progression and prognostic factors. J Hepatol. 2008;48(2):335–52. doi: 10.1016/j.jhep.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 3.Kwon H, Lok AS. Hepatitis B therapy. Nat Rev Gastroenterol Hepatol. 2011;8(5):275–84. doi: 10.1038/nrgastro.2011.33. [DOI] [PubMed] [Google Scholar]
- 4.Inuzuka T, Takahashi K, Chiba T, Marusawa H. Mouse models of hepatitis B virus infection comprising host-virus immunologic interactions. Pathogens. 2014;3(2):377–89. doi: 10.3390/pathogens3020377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ishida Y, Yamasaki C, Yanagi A, Yoshizane Y, Fujikawa K, Watashi K, et al. Novel robust in vitro hepatitis B virus infection model using fresh human hepatocytes isolated from humanized mice. Am J Pathol. 2015;185(5):1275–85. doi: 10.1016/j.ajpath.2015.01.028. [DOI] [PubMed] [Google Scholar]
- 6.Putri SP, Nakayama Y, Matsuda F, Uchikata T, Kobayashi S, Matsubara A, et al. Current metabolomics: practical applications. J Biosci Bioeng. 2013;115(6):579–89. doi: 10.1016/j.jbiosc.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 7.Shulaev V. Metabolomics technology and bioinformatics. Brief Bioinform. 2006;7(2):128–39. doi: 10.1093/bib/bbl012. [DOI] [PubMed] [Google Scholar]
- 8.Kell DB. Metabolomics and systems biology: making sense of the soup. Curr Opin Microbiol. 2004;7(3):296–307. doi: 10.1016/j.mib.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 9.Xiao JF, Zhou B, Ressom HW. Metabolite identification and quantitation in LC–MS/MS-based metabolomics. Trends Anal Chem. 2012;32:1–14. doi: 10.1016/j.trac.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramautar R, Berger R, van der Greef J, Hankemeier T. Human metabolomics: strategies to understand biology. Curr Opin Chem Biol. 2013;17(5):841–6. doi: 10.1016/j.cbpa.2013.06.015. [DOI] [PubMed] [Google Scholar]
- 11.Pacchiarotta T, Deelder AM, Mayboroda OA. Metabolomic investigations of human infections. Bioanalysis. 2012;4(8):919–25. doi: 10.4155/bio.12.61. [DOI] [PubMed] [Google Scholar]
- 12.Vinayavekhin N, Homan EA, Saghatelian A. Exploring disease through metabolomics. ACS Chem Biol. 2010;5(1):91–103. doi: 10.1021/cb900271r. [DOI] [PubMed] [Google Scholar]
- 13.Munger J, Bajad SU, Coller HA, Shenk T, Rabinowitz JD. Dynamics of the cellular metabolome during human cytomegalovirus infection. PLoS Pathog. 2006;2(12):e132. doi: 10.1371/journal.ppat.0020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vastag L, Koyuncu E, Grady SL, Shenk TE, Rabinowitz JD. Divergent effects of human cytomegalovirus and herpes simplex virus-1 on cellular metabolism. PLoS Pathog. 2011;7(7):e1002124. doi: 10.1371/journal.ppat.1002124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen S, Yin P, Zhao X, Xing W, Hu C, Zhou L, et al. Serum lipid profiling of patients with chronic hepatitis B, cirrhosis, and hepatocellular carcinoma by ultra fast LC/IT-TOF MS. Electrophoresis. 2013;34(19):2848–56. [PubMed] [Google Scholar]
- 16.Li H, Zhu W, Zhang L, Lei H, Wu X, Guo L, et al. The metabolic responses to hepatitis B virus infection shed new light on pathogenesis and targets for treatment. Sci Rep. 2015;5:8421. doi: 10.1038/srep08421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Teng C-F, Hsieh W-C, Yang C-W, Su H-M, Tsai T-F, Sung W-C, et al. A biphasic response pattern of lipid metabolomics in the stage progression of hepatitis B virus X tumorigenesis. Mol Carcinog. 2016;55(1):105–14. doi: 10.1002/mc.22266. [DOI] [PubMed] [Google Scholar]
- 18.Kwarteng J, Owusu L, Afihene M. Lowered serum triglyceride levels among chronic hepatitis B-infected patients in Ghana. J Sci Technol. 2012;32(3):1–10. [Google Scholar]
- 19.Zheng S-J, Qu F, Li J-F, Zhao J, Zhang J-LJ-Y, Liu M, et al. Serum sphingomyelin has potential to reflect hepatic injury in chronic hepatitis B virus infection. Int J Infect Dis. 2015;33:149–55. doi: 10.1016/j.ijid.2015.01.020. [DOI] [PubMed] [Google Scholar]
- 20.Zhou L, Wang Q, Yin P, Xing W, Wu Z, Chen S, et al. Serum metabolomics reveals the deregulation of fatty acids metabolism in hepatocellular carcinoma and chronic liver diseases. Anal Bioanal Chem. 2012;403(1):203–13. doi: 10.1007/s00216-012-5782-4. [DOI] [PubMed] [Google Scholar]
- 21.Zhang A, Sun H, Han Y, Yan G, Wang X. Urinary metabolic biomarker and pathway study of hepatitis B virus infected patients based on UPLC–MS system. PLoS One. 2013;8(5):1–8. doi: 10.1371/journal.pone.0064381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun S, Dai J, Fang J, Gou X, Cao H, Zheng N, et al. Differences of excess and deficiency in patients with chronic hepatitis B by urinary metabonomics. Evid Based Complement Altern Med. 2013;2013:1–10. doi: 10.1155/2013/738245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soga T, Sugimoto M, Honma M, Mori M, Igarashi K, Kashikura K, et al. Serum metabolomics reveals γ-glutamyl dipeptides as biomarkers for discrimination among different forms of liver disease. J Hepatol. 2011;55(4):896–905. doi: 10.1016/j.jhep.2011.01.031. [DOI] [PubMed] [Google Scholar]
- 24.Xue R, Dong L, Wu H, Liu T, Wang J, Shen X. Gas chromatography/mass spectrometry screening of serum metabolomic biomarkers in hepatitis B virus infected cirrhosis patients. Clin Chem Lab Med. 2009;47(3):305–10. doi: 10.1515/CCLM.2009.083. [DOI] [PubMed] [Google Scholar]
- 25.Beyoǧlu D, Imbeaud S, Maurhofer O, Bioulac-Sage P, Zucman-Rossi J, Dufour JF, et al. Tissue metabolomics of hepatocellular carcinoma: Tumor energy metabolism and the role of transcriptomic classification. Hepatology. 2013;58(1):229–38. doi: 10.1002/hep.26350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yin P, Wan D, Zhao C, Chen J, Zhao X, Wang W, et al. A metabonomic study of hepatitis B-induced liver cirrhosis and hepatocellular carcinoma by using RP-LC and HILIC coupled with mass spectrometry. Mol Biosyst. 2009;5(8):868–76. doi: 10.1039/b820224a. [DOI] [PubMed] [Google Scholar]
- 27.Association E. EASL clinical practice guidelines: management of chronic hepatitis B virus infection. J Hepatol. 2012;57(1):167–85. doi: 10.1016/j.jhep.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 28.Xia J, Sinelnikov IV, Han B, Wishart DS. MetaboAnalyst 3.0—making metabolomics more meaningful. Nucleic Acids Res. 2015;43(W1):W251–7. doi: 10.1093/nar/gkv380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xia J, Wishart DS. Web-based inference of biological patterns, functions and pathways from metabolomic data using MetaboAnalyst. Nat Protoc. 2011;6(6):743–60. doi: 10.1038/nprot.2011.319. [DOI] [PubMed] [Google Scholar]
- 30.Arthur D, Vassilvitskii S. k-means++: The advantages of careful seeding. Proc Eighteenth Annu ACM-SIAM Symp Discret Algorithms. 2007;8:1027–35. [Google Scholar]
- 31.Dicpinigaitis PV, Bhat R, Rhoton WA, Tibb AS, Negassa A. Effect of viral upper respiratory tract infection on the urge-to-cough sensation. Respir Med. 2011;105(4):615–8. doi: 10.1016/j.rmed.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 32.Morgan MY, Marshall AW, Milsom JP, Sherlock S. Plasma amino-acid patterns in liver disease. Gut. 1982;23(5):362–70. doi: 10.1136/gut.23.5.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lieber CS. S-Adenosyl-L-methionine: its role in the treatment of liver disorders. Am J Clin Nutr. 2002;76(suppl):1183–7S. doi: 10.1093/ajcn/76/5.1183S. [DOI] [PubMed] [Google Scholar]
- 34.Zeissig S, Murata K, Sweet L, Publicover J, Hu Z, Kaser A, et al. Hepatitis B virus–induced lipid alterations contribute to natural killer T cell–dependent protective immunity. Nat Med. 2012;18(7):1060–8. doi: 10.1038/nm.2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ito M, Ishikawa Y, Kiguchi H, Komiyama K, Murakami M, Kudo I, et al. Distribution of type V secretory phospholipase A2 expression in human hepatocytes damaged by liver disease. J Gastroenterol Hepatol. 2004;19(10):1140–9. doi: 10.1111/j.1440-1746.2004.03435.x. [DOI] [PubMed] [Google Scholar]
- 36.Quehenberger O, Dennis EA. The human plasma lipidome. N Engl J Med. 2011;365(19):1812–23. doi: 10.1056/NEJMra1104901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Satoh O, Umeda M, Imai H, Tunoo H, Inoue K. Lipid composition of hepatitis B virus surface antigen particles and the particle-producing human hepatoma cell lines. J Lipid Res. 1990;31(7):1293–300. [PubMed] [Google Scholar]
- 38.Corbin KD, Zeisel SH. Choline metabolism provides novel insights into nonalcoholic fatty liver disease and its progression. Curr Opin Gastroenterol. 2012;2:159–65. doi: 10.1097/MOG.0b013e32834e7b4b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zeisel SH, Blusztajn JK. Choline and human nutrition. Annu Rev Nutr. 1994;14:269–96. doi: 10.1146/annurev.nu.14.070194.001413. [DOI] [PubMed] [Google Scholar]
- 40.Ghoshal AK, Farber E. The induction of liver cancer by dietary deficiency of choline and methionine without added carcinogens. Carcinogenesis. 1984;5(10):1367–70. doi: 10.1093/carcin/5.10.1367. [DOI] [PubMed] [Google Scholar]
- 41.Ghoshal AK, Ahluwalia M, Farber E. The rapid induction of liver cell death in rats fed a choline-deficient methionine-low diet. Am J Pathol. 1983;113(3):309–14. [PMC free article] [PubMed] [Google Scholar]
- 42.Gibbons GF. Assembly and secretion of hepatic very-low-density lipoprotein. Biochem J. 1990;286(1):1–13. doi: 10.1042/bj2680001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mason TM. The role of factors that regulate the synthesis and secretion of very-low-density lipoprotein by hepatocytes. Crit Rev Clin Lab Sci. 1998;35(6):461–87. doi: 10.1080/10408369891234246. [DOI] [PubMed] [Google Scholar]
- 44.Peng D, Han Y, Ding H, Wei L. Hepatic steatosis in chronic hepatitis B patients is associated with metabolic factors more than viral factors. J Gastroenterol Hepatol. 2008;23(7 Pt 1):1082–8. doi: 10.1111/j.1440-1746.2008.05478.x. [DOI] [PubMed] [Google Scholar]
- 45.Machado MV, Oliveira AG, Cortez-Pinto H. Hepatic steatosis in hepatitis B virus infected patients: meta-analysis of risk factors and comparison with hepatitis C infected patients. J Gastroenterol Hepatol. 2011;26(9):1361–7. doi: 10.1111/j.1440-1746.2011.06801.x. [DOI] [PubMed] [Google Scholar]
- 46.Adiels M, Taskinen MR, Packard C, Caslake MJ, Soro-Paavonen A, Westerbacka J, et al. Overproduction of large VLDL particles is driven by increased liver fat content in man. Diabetologia. 2006;49(4):755–65. doi: 10.1007/s00125-005-0125-z. [DOI] [PubMed] [Google Scholar]
- 47.Marchesini G, Brizi M, Bianchi G, Tomassetti S, Bugianesi E, Lenzi M, et al. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes. 2001;50(8):1844–50. doi: 10.2337/diabetes.50.8.1844. [DOI] [PubMed] [Google Scholar]
- 48.Maier KP, Talke H, Gerok W. Activities of urea-cycle enzymes in chronic liver disease. Klin Wochenschr. 1979;57(13):661–5. doi: 10.1007/BF01477666. [DOI] [PubMed] [Google Scholar]
- 49.Madiraju AK, Erion DM, Rahimi Y, Zhang X-M, Braddock DT, Albright RA, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510(7506):542–6. doi: 10.1038/nature13270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xun Y-H, Zhang Y-J, Pan Q-C, Mao R-C, Qin Y-L, Liu H-Y, et al. Metformin inhibits hepatitis B virus protein production and replication in human hepatoma cells. J Viral Hepat. 2014;21(8):597–603. doi: 10.1111/jvh.12187. [DOI] [PubMed] [Google Scholar]
- 51.Noga MJ, Dane A, Shi S, Attali A, van Aken H, Suidgeest E, et al. Metabolomics of cerebrospinal fluid reveals changes in the central nervous system metabolism in a rat model of multiple sclerosis. Metabolomics. 2012;8(2):253–63. doi: 10.1007/s11306-011-0306-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hu C, van Dommelen J, van der Heijden R, Spijksma G, Reijmers TH, Wang M, et al. RPLC-ion-trap-FTMS method for lipid profiling of plasma: method validation and application to p53 mutant mouse model. J Proteome Res. 2008;7(11):4982–91. doi: 10.1021/pr800373m. [DOI] [PubMed] [Google Scholar]
- 53.Strassburg K, Huijbrechts AML, Kortekaas KA, Lindeman JH, Pedersen TL, Dane A, et al. Quantitative profiling of oxylipins through comprehensive LC–MS/MS analysis: application in cardiac surgery. Anal Bioanal Chem. 2012;404(5):1413–26. doi: 10.1007/s00216-012-6226-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The metabolomics datasets supporting the conclusions of this article are available in the online MetaboLights data repository, under accession numbers MTBLS253, MTBLS279, and MTBLS280.