

HUMAN ENDOGENOUS RETROVIRUSES
Within the human genome reside thousands of retrovirus-like sequences which comprises nearly 8% of the human genome (Griffiths, 2001). Originating from ancient retroviruses that overcame host defense mechanisms and permanently integrated into the genomes of our early and more recent primate and hominid ancestors, these provirus remnants are referred to as human endogenous retroviruses (HERVs). HERV is a broad heading for numerous families of retroviruses which were able to infect germline cells over the course of human evolution. Subsequent to integration in the genome, transmission of HERVs occurred in a Mendelian fashion, overriding the need to spread by exogenously acquired infection. At this stage, HERV expression within the host may have drastically changed: now all host cells carried integrated provirus, as opposed to the original subset of cells targeted during exogenous HERV infection. The clinical consequences for the affected hosts remain unknown; however, it is clear that epigenetic and antiviral responses were essential in silencing the expression of these integrated proviruses over time. Yet, current research indicates that not all HERVs remain silent passengers, as reactivation of HERVs is associated with several cancers, inflammatory diseases, and neurologic disorders, although they have not been shown to be causative of any human disease.
While this chapter has focused on the putative role of endogenous retroviruses in the inception and exacerbation of neurologic disease, it should be noted that, besides HERVs, multiple copies of Borna virus and human herpesvirus-6-derived sequences have undergone endogenation during human evolution and these viruses have also been implicated in neurologic diseases (Tanaka-Taya et al., 2004; Horie et al., 2010).
HERV lifecycles: the past and the present
Despite the limited number of exogenous retroviruses known to be circulating in modern human populations (human immunodeficiency virus (HIV); human T-cell lymphotropic virus (HTLV); xenotropic murine leukemia virus-related virus (XMRV)), it is clear that numerous exogenous forms of HERVs were propagated during human evolution. The process of retroviral endogenation has resulted in at least 31 independently acquired HERV families in the human genome (Belshaw et al., 2005a; Blikstad et al., 2008). The endogenation process required that HERVs bypass the host’s antiviral defense mechanisms (Douville and Hiscott, 2010), infect germline cells without cytotoxicity, and remain sufficiently non-pathogenic so that the host’s progeny might reproduce, now passing on HERV provirus in a Mendelian fashion. Such vertical transmission of viruses is not unique to HERVs and has been demonstrated in many life forms, including plants (Iskra-Caruana et al., 2010). Successful vertical transmission ensures decreased immune surveillance of these viruses and diminished virulence. The host–pathogen interactions occurring during endogenation profoundly impacted both the host’s and virus’ survival and evolution (Blikstad et al., 2008). The interface between host and virus likely accounted for species-specific acquisition of ERVs, as host susceptibility to retroviral invasion is largely dependent on the expression of cellular proteins acting as viral receptors and the efficacy of the antiviral immune responses (Jia et al., 2009; Nakayama and Shioda, 2010).
Postendogenation events have also shaped HERV proviruses and host cell function. HERV polymorphism occurs within a single cell, an individual, or a host species. Variation in HERV distribution between human populations can occur from insertional polymorphisms (Belshaw et al., 2005a; Jha et al., 2009), resulting in unique HERV placement and copy number depending on genetic background. HERV sequences can affect homologous recombination, giving rise to deletion, duplications, and variable tandem repeats in the host genome (Zhu et al., 1992; Bosch and Jobling, 2003; Belshaw et al., 2007). Finally, single-nucleotide polymorphisms (SNPs) also contribute to the observed variation in HERV sequences between individual humans (Macfarlane and Simmonds, 2004; de Parseval et al., 2005; Mayer et al., 2005; Blikstad et al., 2008). From the host’s perspective, these variations in HERV sequence can divergently impact cellular function (reviewed in Sverdlov, 2005), as HERV long-terminal repeats (LTRs) can act as tissue-specific transcription enhancers (Ting et al., 1992; Dunn et al., 2003), promoters (Sjottem et al., 1996; Medstrand et al., 2001; Landry et al., 2002; Lee et al., 2003a; Ruda et al., 2004) (even bidirectionally: Leupin et al., 2005), and polyadenylation sites (Mager et al., 1999). Moreover, HERVs can contribute to alternative and necessary RNA splicing signals (Feuchter-Murthy et al., 1993; Kowalski et al., 1999). Use of HERVs can even contribute novel components to the human proteome. An example of retroviral domestication is Syncytin-1, an HERV-W envelope protein within chromosome 7q21, which now plays an essential physiologic role in the syncytiotrophoblast structure of the placenta (Bonnaud et al., 2004). Today, as in the past, HERVs contribute to human health and disease – the exaptation of HERV sequences thus results in a dualism between acquisition of novel cellular functions and the intrinsic virulence of these retroviruses.
HERV genomic structures
The first characterization of the HERV sequences occurred in the early 1980s, with the identification of proviral sequences homologous to gammaretroviruses in human DNA (Martin et al., 1981). It is clear that multiple families of HERV reside within the human genome (for a review, see Blikstad et al., 2008). Gammmaretroviruses (HERV-W, HERV-H, HERV-F, HERV-I, HERV-E) represent the majority of sequences, followed by betaretroviruses (HERV-K: HML-1 to 10) and spumaretroviruses (HERV-S, HERV-L) (Blikstad et al., 2008; Christensen, 2010). However, the integrity of these HERV sequences is frequently compromised due to recombination events and mutations: few HERV loci encode proteins and even fewer sites have intact (or almost intact) proviruses (Villesen et al., 2004; Boller et al., 2008). The most recently integrated human-specific ERVs are derived from HML-2 family (human murine mammary tumor virus (MMTV)-like viruses) of HERV-K betaretrovirus. Integrated into the human genome as recently as 1 million years ago, some of the HERV-K (HML-2) proviruses are intact (HERV-K113 (Boller et al., 2008)) or almost intact (HERV-K115), with open reading frames (ORFs) for the major retroviral genes (Turner et al., 2001; Jha et al., 2009). Conversely, no single gammaretroviral sequence contains an intact provirus. Thus, a vast mosaic of HERV structures exists within the human genome, representing diverse families of retrovirus and quasispecies which are dispersed in the genome with multicopy insertions, insertional polymorphisms, and accumulated mutations.
Notwithstanding the diversity of HERVs, a basic genomic organization exists which encodes for the four essential Retroviridae genes, 5′-gag-pro-pol-env-3′ (Fig. 22.1). Gag encodes the matrix (MA), capsid (CA), and nucleocapsid (NC) proteins, pro the viral protease (PR), pol the reverse transcriptase (RT) and integrase (IN), and env encodes a glycoprotein (with surface (SU) and transmembrane (TM) subunits). Of note, the respective size and composition of these viral proteins vary considerably between HERV families. Using the components within this genomic structure, the retrovirus is able to reverse the usual flow of genetic information, starting with an RNA viral genome to make a fixed DNA copy within the host cell genome. Infectious retroviral particles contain a dimer formed of two copies of the single-sense strand RNA genomes.
Fig. 22.1.
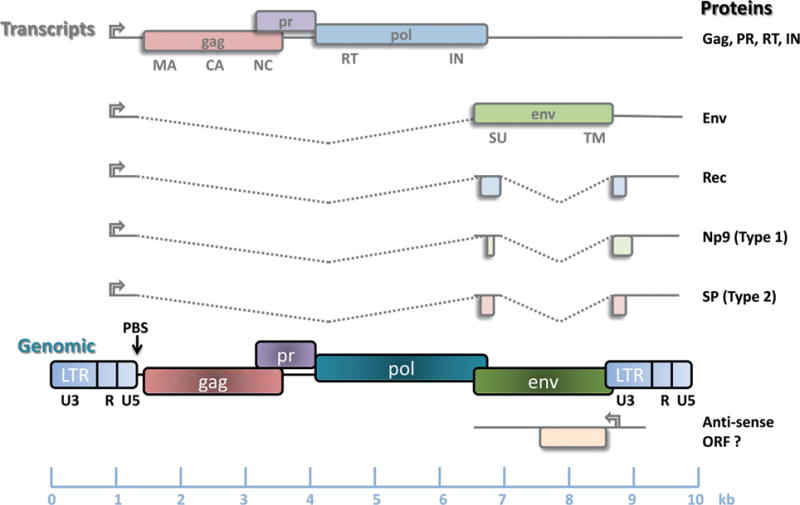
Genomic organization of an integrated human endogenous retrovirus-K (HERV-K) provirus and viral RNA transcripts. Reverse transcription priming differs between HERV families and is used to classify HERVs: because tRNALys is used to initiate reverse transcription at the primer binding site (PBS), the family is named HERV-K (K for lysine). Genomic retroviral RNA packaged in infectious virions is reverse-transcribed in the host cytoplasm to produce a preintegrated provirus. During the integration of retroviral DNA into the host genome, a short duplication of the host DNA occurs (blue arrows), flanking each long terminal repeat (LTR). The LTRs act as viral promoters to initiate transcription of viral RNA. Full-length viral RNA encodes the gag polyprotein which is cleaved by protease (PR) to produce viral matrix (MA), capsid (CA), and nucleocapsid (NC) proteins. A reading frame-shift for the pol gene results in the translation of reverse transcriptase (RT) and integrase (IN). The envelope surface (SU) and transmembrane (TM) units are generated from a sliced transcript. HERV-K type 1 differs from HERV-K type 2 genomes by the absence of a 292 bp sequence at the pol–env boundary. This affects the generation of HERV-K accessory proteins: Np9 is encoded in type 1 HERV-K, whereas SP is found in type 2. Both types also produce transcripts for the analog of HIV Rev, the HERV-K Rec protein. (Courtesy of R. Douville.)
Following internalization, the co-packaged RT enzyme utilizes cellular tRNA as a primer to convert viral RNA into a double-stranded DNA genome. This process results in the formation of preintegrated viral DNA containing LTRs at both ends. Now, the viral integrase enzyme facilitates the entry of the viral DNA genome into the host chromosomal DNA. As a result, the cellular DNA sequence at the integration site is duplicated, with each copy flanking the inserted provirus. Retroviral insertion is random, in the sense that no specific genomic sites are targeted; however, because of epigenetic packaging of DNA, integrations are more prevalent in gene-rich and transcriptionally active sites (Brady et al., 2009). Over time, purifying selection favors the loss of proviruses that alter gene function, thus the majority of older HERVs are found in intergenic DNA or with an antisense orientation within genic regions (Brady et al., 2009).
The integrated proviral genome uses its LTRs as viral promoters which regulate viral RNA transcription (Fig. 22.1). Each LTR contains a U3 (unique 3′), R (repeated), and U5 (unique 5′) region. The transcribed viral genome is a positive single-stranded RNA, capped at the 5′ end and containing a poly(A) tail at its 3′ end. Ribosomal frameshifting during translation of the full-length transcript will produce the gag polyprotein, which is cleaved by PR, as well as the pol-derived proteins. Other provirus-derived transcripts undergo RNA slicing to generate env and accessory proteins. To date, accessory proteins have only been described in the case of HERV-K: these are Rec (Mayer et al., 2004), Np9 (type I HERV-K) (Armbruester et al., 2004), or Sp (type II HERV-K) (Ruggieri et al., 2009) (Fig. 22.1). Finally, interactions with host cellular proteins, such as RNA helicase A, may specifically promote translation of retroviral RNA into viral proteins (Bolinger et al., 2007).
Induction of HERVs in the CNS
The expression of HERVs within the central nervous system (CNS) is likely determined by several factors, such as the insertional and sequence polymorphisms of HERVs, the epigenetic context of HERV proviruses, antiretroviral cellular responses, and environmental triggers. Most HERV sequences are functionally defective, but those retaining potential expression are suppressed by epigenetic modifications of DNA – methylation and supercoiling – which are altered by both age and disease status (Schulz et al., 2006; Jintaridth and Mutirangura, 2010; Stengel et al., 2010). Alternatively, HERV transcription may be actively silenced by host retroviral restriction factors such as TDP-43, TRIM19, or TRIM22 (Ou et al., 1995; Nisole et al., 2005; Douville and Hiscott, 2010), in addition to HERV-expressing cells being targeted by cytotoxic CD8+ T cells (Garrison et al., 2007; Krone and Grange, 2010). As such, HERV proviruses are under selective pressure to be latent and not expressed under normal conditions.
Due to their ancestral ability to infect germline cells, it is understandable that HERV expression is elevated in human reproductive tissues (Prudhomme et al., 2005; Sugimoto and Schust, 2009). Nonetheless, the CNS appears to support elevated expression of HERVs as compared to most other tissue types (Frank et al., 2005; Seifarth et al., 2005; Kim et al., 2008). The reasons for increased HERV expression in CNS tissue remain unclear. Microarray analysis demonstrates that not all HERV families are transcribed in healthy human prefrontal cortex (Frank et al., 2005). Some HERVs are ubiquitously active in brain tissue specimens obtained at autopsy from patients with systemic diseases without any known brain injury (HERV-E, HERV-F, ERV9, HML-2, HML-4, HML-6, HML-9, and HML-10), whereas others are transcriptionally inactive (HERV-I, HERV-Z, ERV3, HML-1) (Frank et al., 2005). Within a given individual, HERV RNA expression is relatively uniform within the brain (Frank et al., 2005), allowing clear evaluation of interindividual differences in HERV expression using techniques such as microarray and digital or quantitative polymerase chain reaction (Q-PCR). Indepth analysis of the PCR product melting temperatures or DNA sequencing can further distinguish sequence differences between amplicons generated by primers targeted toward a specific HERV target (Nellaker et al., 2009; Douville et al., 2011), revealing a more accurate picture of HERV profiles within brain tissue areas in clinical specimens. These unique transcriptional profiles may reflect individual genetic differences (Hughes and Coffin, 2004; Macfarlane and Simmonds, 2004) or similar patterns of gene induction for a given clinical state (Frank et al., 2005; Ahn and Kim, 2009; Krone and Grange, 2010). Alternatively, specific HERV-encoded proteins may serve an unknown biologic function in the CNS, similar to the essential role of syncytin-1 in placental and embryonic tissues (Mi et al., 2000; Bonnaud et al., 2004; Sugimoto and Schust, 2009).
HERVS IN NEUROLOGIC DISEASES
To date, HERV expression is strongly and reproducibly associated with several neurologic diseases, although the etiologic mechanisms to demonstrate that HERVs can cause these diseases have yet to be elucidated (Table 22.1). Without causal evidence, it remains unclear whether HERV expression is indicative of an underlying pathogenesis or a spurious relationship with certain disease states.
Table 22.1.
Human endogenous retrovirus (HERV) families associated with neurologic disease
| HERV | Family | Neurologic diseases | Known triggers | Pathogenic contribution |
|---|---|---|---|---|
| HERV-W | Gammaretrovirus | Multiple sclerosis Schizophrenia |
Herpesviruses Toxoplasma Proinflammatory cytokines |
MSRV virions Env superantigen Env alters glial function |
| HERV-H/F | Gammaretrovirus | Multiple sclerosis | Unknown | Virions Env superantigen |
| HERV-K | Betaretrovirus | Schizophrenia ALS HIV infection |
Herpesviruses HIV HTLV-1 Toxoplasma |
Env superantigen Neoepitopes for CTLs |
MSRV, multiple sclerosis-associated retrovirus; ALS, amyotrophic lateral sclerosis; HIV, human immunodeficiency virus; HTLV-1, human T-lymphotropic virus-1; CTLs, cytotoxic T lymphocytes.
HERVs triggered by infectious diseases of the CNS
A potential explanation for the preferential HERV expression seen in pathologic human brain samples comes from evaluating the CNS tropism of environmental triggers such as viruses and bacteria (reviewed in Perron and Lang, 2010). Neuro- or neurovascular-tropic pathogens such as herpesviruses, exogenous retroviruses, Chlamydia pneumoniae, Toxoplasma gondii, and certain strains of influenza virus may cross the blood–brain barrier into the CNS. Also the majority of these neurotropic pathogens lead to abortive infections, but their transient presence in the CNS may be sufficient to trigger HERV expression. For example, multiple sclerosis (MS) is associated with herpesviruses, such as human herpesvirus-6 and Epstein–Barr virus (EBV), and these viruses demonstrate an affinity for infection of white matter (Steiner et al., 2001) or meningeal B lymphocytes (Serafini et al., 2007, 2010). This pattern of tissue tropism mirrors HERV expression in MS patients, as well as the localization of brain areas most affected in MS. Mechanistically, these herpesviruses can induce the expression of HERV during early replication events, such as DNA demethylation (Stengel et al., 2010), expression of transactivating proteins (Hsiao et al., 2006; Nellaker et al., 2006), or aberrant cell signaling (Mameli et al., 2007b) (Fig. 22.2). Thus, both pathogen tissue tropism and pathogen-specific cellular deregulation are essential determinants of the extent and pattern of HERV induction.
Fig. 22.2.
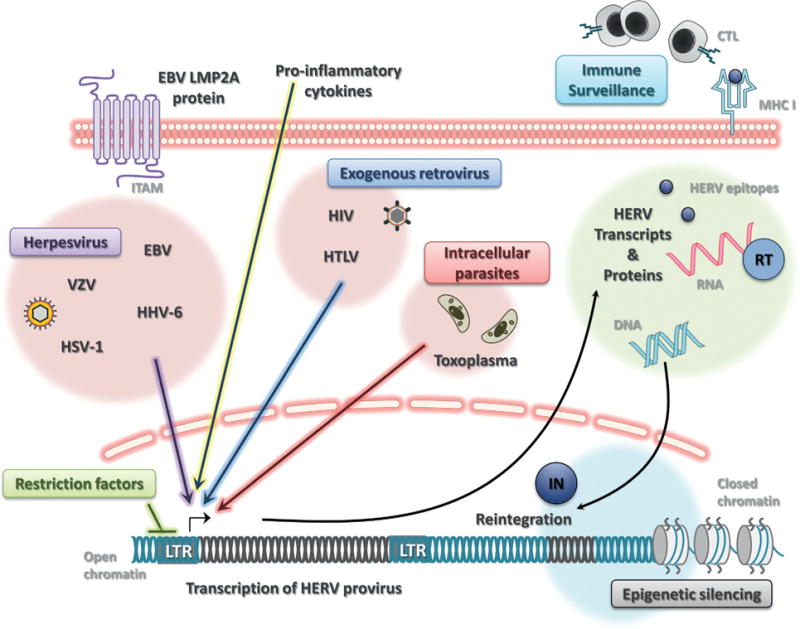
Triggering and repressing human endogenous retrovirus (HERV) expression. Under homeostatic conditions, most HERVs are latent due to epigenetic silencing of proviruses in heterochromatin (closed chromatin) which is inaccessible to transcriptional machinery or actively repressed by DNA-binding retroviral restriction factors. Viral and parasitic infection can elicit the expression of HERVs in cells of the central nervous system. Neurotropic herpesviruses, such as Epstein–Barr virus (EBV), herpes simplex virus type 1 (HSV-1), varicella-zoster virus (VZV), and human herpesvirus type 6 (HHV-6) can trigger HERV expression. The EBV protein LMP2A can also transactivate the HERV-K18 envelope superantigen via its ITAM region. Intracellular parasites, such as Toxoplasma gondii, may also reactivate latent HERVs. Both human immunodeficiency virus (HIV) and human T-cell lymphotropic virus (HTLV) can activate HERVs which further drive adaptive immune responses against exogenous-endogenous retrovirus cross-reactive epitopes. Major histocompatibility complex class I (MHC I) molecules can present these neoepitopes to cytotoxic T lymphocytes (CTL) performing immune surveillance for infected cells. Innate immune sensing of viral RNA and DNA or adaptive immune recognition of viral proteins can elicit a proinflammatory response, which further promotes the transcription of HERV proviruses. Full-length HERV RNA can be converted into DNA by viral reverse transcriptase (RT), allowing de novo insertion into the host DNA by the integrase protein (IN). LTRs, long-terminal repeats. (Courtesy of R. Douville.)
Accumulating evidence suggests that HERV RNA and protein expression may be triggered in the context of other retroviral infections, such as HIV. Activation of HERV-K expression has been repeatedly measured in the serum and peripheral blood mononuclear cells (PBMC) of HIV-infected individuals (Contreras-Galindo et al., 2006), as well as in vitro models of HIV infection (Contreras-Galindo et al., 2007b). As measured by peptide serology or viral RNA, HERV-K102 expression in sera occurs in 70–80% of HIV viremic cases, in contrast to only 2–3% of normal healthy adults (Laderoute et al., 2007). The kinetics of HERV-K expression precede rebounds in HIV titer in patients unresponsive to highly active antiretroviral therapy (HAART), whereas it remains undetectable in patients successfully controlled by HAART (Contreras-Galindo et al., 2007a). The mechanism for HERV-K induction remains unclear, but HERV-K viral load may be useful to predict HIV-1 reactivation. It has been postulated that HERV expression is triggered by HIV’s ability to compromise innate defense mechanisms, thus removing or altering cellular control of HERV activity (Garrison et al., 2007). Our recent findings reveal that, in the prefrontal cortex of HIV-positive patients, HERV-K RT expression is augmented in neurons concomitantly with detection of HIV p24 protein in surrounding tissue (Douville and Nath, unpublished observation). Additionally, HERV-K RT expression was significantly associated with nuclear overexpression and posttranscriptional phosphorylation of TDP-43, a retroviral restriction factor known to silence HIV provirus (Ou et al., 1995), which notably, when deregulated, also plays a role in other neurologic diseases (Buratti and Baralle, 2010; Lagier-Tourenne et al., 2010).
Not only may HERV expression contribute to specific pathogenicity, but it may also augment adaptive immune responses against exogenous retroviruses. Substantial homology in immunogenic viral peptides is shared between HERVs and exogenous retroviruses (Garrison et al., 2007; Thomas et al., 2010). The detection of anti-HERV env antibodies correlated with severe immunosuppression during HIV infection, as defined by at least one opportunistic infection (Stevens et al., 1999). Moreover, HERV expression during HIV infection results in HERV-specific and HIV cross-reactive CD8+ T-cell responses (Garrison et al., 2007). The anti-HERV CD8+ T cells inversely correlated with HIV-1 plasma viral load, as well as demonstrated functional cytotoxic activity against cells presenting their cognate peptide (Garrison et al., 2007). This apparent cross-reactivity between HIV and HERVs may be beneficial for clearing infected cells in the periphery. At the same time, this duality has the potential to enhance lymphocyte recruitment to the CNS and neuroinflammation. Thus, the engagement of protective versus pathologic HERV-specific cytotoxic T-cell and antibody responses has important implications for neuro-AIDS (Petito et al., 2006).
Immunogenic peptides, homologous to HTLV-1 gp21 env (BA-21) and HTLV-1 p24 gag, are believed to drive antibody production in 50% of patients with large granular lymphocytic leukemia (LGLL) (Sokol et al., 2005; Thomas et al., 2010), yet these patients are not HTLV-1-positive. This suggests that LGLL patients are infected with an HTLV-1 variant or exhibit cross-reactivity to homologous non-HTLV-1 antigens. A minority of patients with LGLL are infected with HTLV-2 (7.5%) (Loughran et al., 1994); however, in LGLL patients some degree of HTLV-1 serologic cross-reactivity can be attributed to HERV-K10 (Thomas et al., 2010). HERV-K10 exhibits homologous protein sequences with HTLV-1 p24 and the BA-21 peptide (PLENRVLTG) to which HTLV-1 false-positive LGLL patients seroreact (Thomas et al., 2010). Similarly, a greater frequency of HTLV-1- or HTLV-2-infected patients with myelopathy have anti-HERV-K10 antibodies (31.8%) than patients without myelopathy (16.7%). The example of immunogenic expression of HERV-K10 proteins in LGLL and HTLV-1-infected patients further supports the idea that: (1) exogenous retroviruses can trigger specific HERV expression; (2) anti-HERV immune responses may contribute to the overall pathology of disease; and (3) HERV expression is more frequently associated with neurologic disease.
Herpesviruses may also be a significant trigger of HERV expression in the CNS. Multiple reports have detected EBV, herpes simplex virus type 1 (HSV-1), varicella-zoster virus (VZV), and human herpesvirus type 6 (HHV-6) in MS patient samples (Alvarez-Lafuente et al., 2008; Serafini et al., 2010); all these herpesviruses have also been demonstrated to trigger the expression of HERVs (Christensen, 2005). Induction of HERV expression can be attributed to specific viral proteins. The EBV protein LMP-2A causes the expression of HERV-K18 superantigen (SAg) by mimicking B-cell receptor signaling (Hsiao et al., 2006, 2009), thus promoting SAg-expressing B cells to strongly stimulate antigen-independent TcR vβ13 T-cell activation (Sutkowski et al., 2001). Similarly, the neurotropic strain HHV-6A has also been shown to trigger HERV-K18 SAg during both latency and acute infection (Tai et al., 2009). HSV-1 immediate early proteins can initiate transcription of HERV LTRs: HERV-K is induced by the HSV-1 ICP0 protein via its AP-1 binding site (Kwun et al., 2002), whereas the HSV-1 IE1 protein requires an Oct-1 binding site within the HERV-W LTR (Lee et al., 2003b). Importantly, in vitro infection of neuronal and brain endothelial cells with HSV-1 results in increased levels of HERV-W gag and env proteins (Ruprecht et al., 2006). Conversely, inactivated herpesviruses can also trigger HERV expression when co-cultured with MS patient PBMCs (Brudek et al., 2007), and the combination of antigenic stimuli synergistically enhances interferon-γ (IFN-γ) responses in MS patients (Brudek et al., 2008). Together, these findings point to herpesviruses as playing a dual role in neurodegenerative disease, both as pathologic entities on their own and as inducers of HERVs.
Another potential trigger of HERV expression within the CNS is that of opportunistic infection. For example, Toxoplasma gondii infection can transcriptionally activate HERVs within neuronal cells (Frank et al., 2006). Future studies may benefit from considering additional pathogenic inducers of HERV expression within the CNS, considering the breadth of viral, bacterial, and parasitic infections associated with known HERV-related diseases (Steiner et al., 2001; Mattson, 2004; Brown and Derkits, 2010). This may particularly be worthy of consideration in patients with MS who developed progressive multifocal leukoencephalopathy due to JC virus infection while being treated with natalizumab. Some of these patients developed a persistent JC virus infection in the brain despite normalization of the immune system – a feature not seen in promyelocytic leukemia (PML) associated with other diseases (Ryschkewitsch et al., 2010). The possibility that this may be driven by HERV expression needs to be considered.
HERVs in multiple sclerosis
MS is a demyelinating disease of unknown etiology with clinical symptoms of fatigue and perturbations of sensory, motor, cerebellar, and cognitive function, as well as loss of bowel and bladder control (reviewed in Tremlett et al., 2010). Pathologically, MS is characterized by inflammatory immune cell infiltrates into the CNS, focal CNS inflammation, loss of myelin and cellular apoptosis, leading to lesions and brain atrophy (Hafler et al., 2005; Ramagopalan et al., 2010).
When retrovirus-like particles with RT activity were originally described in primary cells from MS patients (Perron et al., 1989; Haahr et al., 1991), their origin was speculated to be that of HTLV-1 owing to the clinical similarities between HTLV-I-associated myelopathy/tropical spastic paraparesis (HAM/TSP) and MS (Waksman, 1989). Subsequent analysis determined that the pattern of HERV expression in MS includes both HERV-W (MS-associated retrovirus: MSRV) and HERV-H/F sequences (reviewed in Christensen, 2005; Dolei and Perron, 2009; Perron and Lang, 2010). HERV sequences in the cerebrospinal fluid (CSF) of MS patients are biomarkers for disease severity and are strongly associated with disability and the progressive phase of disease (Mameli et al., 2008; Sotgiu et al., 2010), although another group failed to detect HERV-W and HERV-H in CSF of patients with MS following the first clinically evidentde-myelinating event (n = 48) (Alvarez-Lafuente et al., 2008). Ex vivo lymphocytes from MS patients produce transmissible RT-positive virions containing either HERV-H or HERV-W-derived viral RNA (Perron et al., 1992, 1997; Christensen et al., 1999, 2000; Komurian-Pradel et al., 1999). Virion-associated HERV RNA can readily be amplified from serum of MS patients, but rarely in healthy individuals (Christensen et al., 1998; Garson et al., 1998). Perplexingly, no known gammaretroviral HERV contains intact ORFs in all essential retroviral genes, and thus it has been proposed that an exogenous HERV-W virus may exist (Dolei and Perron, 2009). Alternatively, complementation by several sequences with coding potential may be able to produce sufficient viral proteins and genomic RNA for the formation of viable virions. To date the origin of infectious HERV-W particles is unknown. As virion formation requires the envelope protein, several groups have searched for the source of the MSRV envelope protein. The only full-length HERV-W envelope protein is syncytin-1, encoded by the ERVWE1 locus (Mi et al., 2000). Three specific genomic loci on chromosome X may encode the HERV-W env protein found within MSRV (Laufer et al., 2009; Perron et al., 2009; Roebke et al., 2010). Env protein production would be expected to increase in the presence of two X chromosomes in women compared to a single one in men, which may explain the increased incidence of MS in women (Perron et al., 2009).
HERV expression within brain tissue from MS patients has also been reported. Significantly increased amounts of HERV-K and HERV-W RNA are measured in MS brains as compared to controls (Johnston et al., 2001; Mameli et al., 2007a). At the protein level, HERV-W env proteins (MSRV env and syncytin-1) are undetectable in normal white matter of control brains, whereas they are upregulated within acute and chronic MS lesions (Antony et al., 2007; Mameli et al., 2007a). Immunostaining localizes MSRV env expression to glial cells at the periphery of the lesion and astrocytes within the plaque core (Mameli et al., 2007a). Moreover, HERV-W env protein expression correlates with the degree of active demyelination and inflammation (Perron et al., 2005; Mameli et al., 2007a).
Cellular response to HERV-W envelope protein stimulates innate inflammatory and Th1-biased cytokine responses from PBMC (Rolland et al., 2005). Antibody responses against HERV antigens are also readily detectable in CSF and serum from MS patients, with evidence of antienvelope reactivity to HERV-W and HERV-H (Jolivet-Reynaud et al., 1999; Christensen et al., 2003, 2007). Flow cytometry analysis reveals that B cells and monocytes from patients with active MS express abundant surface expression of both HERV-H and HERV-W envelope proteins, which directly correlates with the intensity of serologic responses (Brudek et al., 2009). Thus, it is conceivable that anti-HERV reactivity may contribute to the intrathecal oligoclonal bands often observed in MS CSF (Cross and Wu, 2010).
More recently, genomic profiling in MS patients and controls has attempted to determine if there is a genetic risk of disease conferred by specific HERV proviruses. Using a family-based cohort, differential insertional polymorphism of HERV-K113 provirus was tested in over 900 patients with MS and their unaffected parents, yet similar frequencies of HERV-K113 were observed between the affected and control groups (Moyes et al., 2008). In contrast, homozygous carriers of a specific allele of HERV-K18 env (K18.3) have a threefold increased risk of MS (Tai et al., 2008). To date, no study has determined whether HERV-W (MSRV) or HERV-H alleles may modulate the risk or progression of MS; however, it has been shown that treatment with IFN-β decreases the immune responses to HERV-W and HERV-H in patients with MS (Petersen et al., 2009).
HERVs in schizophrenia
Schizophrenia is a complex neuropsychiatric disorder which encompasses a variety of cognitive, emotional, and perceptual disturbances. Pathologically, schizophrenia is characterized by decreased brain volume, loss of myelin, and altered astrocyte function (Archer, 2010). The pattern of known HERV expression in schizophrenia includes both HERV-K and HERV-W sequences.
In 2000, Karlsson et al. first demonstrated an association between HERV expression and recent-onset schizophrenia or schizoaffective disorder (Yolken et al., 2000; Karlsson et al., 2001). Using a panretroviral PCR assay, 28.6% (10/35) of CSF samples from recent-onset schizophrenia and 5% (1/20) of chronic schizophrenia patients were positive for retroviral RNA, as compared to undetectable viral loads in non-inflammatory neurologic disorders and non-neurologic disease controls. Sequencing of the PCR products revealed that the retroviral RNA in schizophrenia patients was mainly derived from HERV-W, and to a lesser extent, ERV9 and FRD C-type retroviruses. Moreover, frontal cortex tissue from schizophrenia patients exhibited 10-fold greater frequency of HERV-W pol RNA transcripts than unaffected individuals. Schizophrenia patients exhibiting retroviral sequences within their CNS compartment were also more likely to be positive for HERV-W in their plasma (Karlsson et al., 2004). Together, this suggests that activation of HERV-W retroviral transcripts occurs at early onset of schizophrenia and may contribute to the initial progression of the illness (Yao et al., 2008). Of note, no differences in the genomic provirus load among the groups studied could account for the observed increased expression of HERVs in schizophrenia. Mechanistically, overexpression of HERV-W env in glial cells induces brain-derived neurotrophic factor, an important schizophrenia-associated gene (Huang et al., 2010).
Subsequently, conflicting reports have suggested that HERV expression is weakly correlated with schizophrenia. A microarray-based analysis of pol genes from representative members of 20 HERV families only showed specific upregulation of HERV-K10 (HML-2 family) in brain tissue from schizophrenia and bipolar disorder patients (Frank et al., 2005). Interestingly, bipolar disorder patients exhibited a loss of HERV expression specifically for HERV-IP (HERV-I family), NMWV7 (HML-7 HERV-K family), HERV-L, and a subgroup of ERV9. Using Q-PCR analysis of RNA used in the micro-array analysis, similar expression of HERV-K10 and HERV-W env gene was detected in schizophrenia versus non-neurologic disease control patients. The disparity between previous reports showing increased HERV-W expression in schizophrenia (Karlsson et al., 2001, 2004) may reflect different experimental approaches (specifically choice of PCR primers) or differential use of anti-psychotic medications by schizophrenia patients, specifically haloperidol and clozapine, which are known to dampen retroviral expression (Jones-Brando et al., 1997; Schifitto et al., 2009). Clinically, long-term treatment of schizophrenia patients with clozapine showed better effectiveness than other typical antipsychotics (Ravanic et al., 2009; Agid et al., 2010), although it is not known if this has any relation to suppression of HERV activity in the CNS. Another group reported the increase in HERV-W in patients with schizophrenia being treated with valproate. However the differences are small and there was no difference between the schizophrenia patients treated with valproate when compared to healthy controls (Diem et al., 2012).
HERVs in motor neuron disease
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by the progressive loss of spinal and cortical motor neurons, the cause of which is unknown. Approximately 20% of familial ALS is associated with mutations in the superoxide dismutase (SOD1) gene (Vucic and Kiernan, 2009), whereas triggers of sporadic ALS have yet to be clearly identified (Kim et al., 2010). Involvement of retroviruses in the pathophysiology of ALS has long been suspected due to the consistent observation of RT in serum of patients with ALS at levels comparable to those of HIV-infected patients (MacGowan et al., 2007; McCormick et al., 2008). Further, rarely HIV-infected patients may develop ALS which responds to treatment with antiretroviral therapy (Alfahad and Nath, 2013).
Recently, we have demonstrated that HERV-K pol transcripts were increased in the brains of patients with ALS compared to those with systemic illness, but could not be detected in Parkinson’s disease or in the accidental death controls (Douville et al., 2011). Sequencing of the transcripts generated using degenerate HERV-K primers revealed several actively transcribed loci in the HML-2 and HML-3 subfamiles of HERV-K. HERV-K sequences from 7q34 (HML-2) are more frequently expressed in patients with ALS as compared to controls. HML-3 is not a ubiquitously active HERV in the human brain (Frank et al., 2005), yet we identified the adjacent region 7q36.1 locus (HML-3) as being uniquely expressed in patients with ALS, and it was not detected in any of the other patient groups. The 7q36.1 locus has a putative ORF for RT, with a unique insertion of two codons that would potentially translate to two amino acids inserted prior to the conserved RT motif LPQG (Berkhout et al., 1999; Sharma et al., 2002), suggesting expression of an RT protein with specific functional properties. Most interestingly, this genomic region has previously been associated with another type of motor neuron disease (Gopinath et al., 2007); however, the contributing genes have not been identified to date. Thus specific HERV-K sequences within this candidate interval (between D7S2511 and D7S798) (Gopinath et al., 2007) may represent a putative disease marker in ALS and other types of motor neuron disease.
We further demonstrated that the HERV-K RT protein was localized to neurons in cortical brain tissue of patients with ALS (Douville et al., 2011). Prefrontal and motor cortex showed the strongest RT protein expression, despite an even distribution of HERV-K transcripts among all brain regions in ALS patients. HERV-K expression was also strongly correlated with TDP-43, a multifunctional protein known to be dysregulated in ALS. Neuronal TDP-43 aggregate formation is believed to play an important pathophysiologic role in motor neuron degeneration. However, TDP-43 aggregation may also promote the overexpression of HERV transcripts in ALS due to impairment of its function as a retroviral restriction factor (Ou et al., 1995; Douville and Hiscott, 2010).
HERVs in Creutzfeldt–Jacob disease
Creutzfeldt–Jacob disease is a rapidly progressive and uniformly fatal spongiform encephalopathy which is thought to be mediated via an abnormal isoform of a cellular prion protein. This abnormal form is protease-resistant (PrPres). Cofactors implicated in the formation of PrPres include HERVs. Most experimental studies have been performed using scrapie, which occurs in sheep, but the infectivity can be transmitted to rodents. Previous studies showed that an exogenous retrovirus, Moloney murine leukemia virus infection, strongly enhances release of scrapie infectivity in cell culture (Leblanc et al., 2006). The expression of endogenous murine leukemia virus is also associated with acceleration of scrapie infection in mice (Lee et al., 2006). In another study where HERV sequences were amplified from the cell pellets from CSF, it was found that several HERVs were activated in patients with Creutzfeldt–Jacob disease compared to controls (Jeong et al., 2010). These observations suggest that the relationship between prion diseases and HERVs need to be further explored.
PATHOLOGIC CONSEQUENCES OF HERV EXPRESSION IN THE NERVOUS SYSTEM
Definitive proof that HERVs are the cause – and not an epiphenomenon – of neurologic disease is still lacking. Arguably, putative pathogenic effects mediated by HERVs have been described both theoretically and experimentally (Fig. 22.3). Either as a genetic element or as viral pathogen, HERVs may alter cellular function in a multitude of ways, including modulation of DNA stability and transcription, altering cell signaling pathways, and stimulating the immune system.
Fig. 22.3.
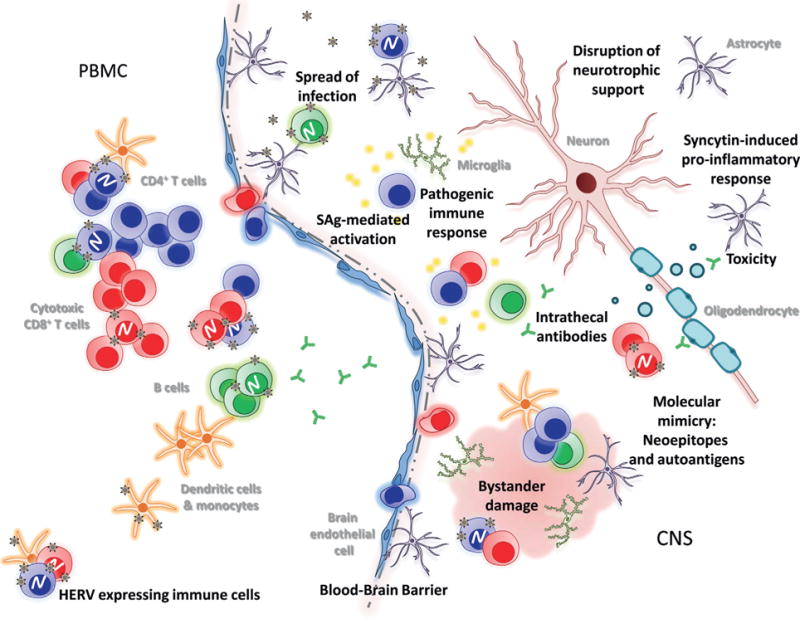
Potential human endogenous retrovirus (HERV)-mediated neurotoxicity in the central nervous system (CNS). HERV activation occurs both in the immune cell compartment of peripheral blood mononuclear cells (PBMC) and in the CNS. PBMC from patients with multiple sclerosis are known to produce infectious HERV-derived retroviral particles. Trafficking of HERV-shedding PBMC across the blood–brain barrier may infect cells of the CNS. Conversely, HERV reactivation may occur in CNS cells and impair their ability to perform neurotrophic support and promote proinflammatory responses. Recruitment of T cells, B cells, and myeloid cells into the CNS may elicit pathologic inflammatory and cytotoxic responses upon recognition of HERV antigens. HERV-encoded envelope superantigens (SAg) may further boost glial cell and T-cell activation, resulting in excessive tissue damage. (Courtesy of R. Douville.)
HERVs, akin to other genetic mobile elements such as retrotransposons, have the ability to undergo transposition, recombination, and reintegration cycles (Belshaw et al., 2005b; Mills et al., 2007). Reintegration requires a subset of viral genes – gag and pol only – as nascent genomic viral RNA is not packaged into virions but instead is reverse-transcribed and reintegrated into the host cell. In fact, some HERV families, such as HERV-K (HML-3), which are found in high copy number within the human genome, are believed to have spread through reintegration of an already present provirus (Belshaw et al., 2005b). De novo accumulation of HERVs, as measured by the integration pattern of a reconstituted consensus HERV-K virus, shows preferential targeting for gene-rich and transcriptionally active sites (Brady et al., 2009). Over time, purifying selection favors the loss of proviruses which disturb gene function within a population. However, within an individual, every new integration event increases the risk that a detrimental insertion will occur, leading to knockout of a cellular gene or DNA damage through recombination events (Bosch and Jobling, 2003). This process may be an important component for HERV-associated cancers, such as primary human glioma, where HERV-K integrations were identified in the tumor suppressor gene BRCA2 and the DNA repair gene XRCC1 (Misra et al., 2001).
Another component related to integrated proviruses is their ability to modulate the expression of adjacent or distal host cell genes. HERV LTRs contain enhancer sequences and transcription factor-responsive elements which participate in host gene expression (Sjottem et al., 1996; Schulte et al., 2000; Landry et al., 2002; Lee et al., 2003a; Ruda et al., 2004; Leupin et al., 2005). HERV proviruses also endow adjacent host sequences with alternative transcription initiation sites, splice acceptor/donor sites, and polyadenylation signals (Gogvadze et al., 2009; Herai and Yamagishi, 2010). Moreover, antisense transcription of HERV-K LTRs can produce regulatory RNA which silences neighboring genes by decreasing their mRNA levels, as is the case for SLC4A8 (sodium bicarbonate cotransporter) and IFT172 (intraflagellar transport protein 172) (Gogvadze et al., 2009). Again, insertional polymorphism, HERV SNPs, and epigenetic profile of cell type will impact the ability of HERVs to exert a regulatory effect on host gene expression.
Finally, transcription and translation of individual retroviral proteins encoded by HERVs may have an impact on a multitude of cellular pathways, in addition to their combinatorial effect during retroviral replication. A remarkable number of intact HERV ORFs exist, yet their potential contribution to neurodegeneration has yet to be fully explored. Conserved retroviral proteins, such as Gag, PR, RT, IN, and Env, as well as accessory proteins may modulate cellular activity. HERV-K is known to encode the accessory protein HIV Rev-like regulatory protein – Rec (Mayer et al., 2004) – and Np9, which is encoded by a splice variant of the rec gene of HERV-K type 1 provirus (Armbruester et al., 2004). Another HERV-K accessory protein in type II provirus is produced by cleavage of the HML-2 Env precursor, forming the nucleolar localized signal peptide (SP) (Ruggieri et al., 2009). The exact functions of these regulatory proteins are beginning to be elucidated. Rec and Np9 may contribute to oncogenesis via inhibition of PML zinc finger protein, thus promoting c-myc expression and subsequent transcription of c-myc target genes (Denne et al., 2007). Rec also binds the testicular zinc-finger protein, which de-represses its complex with the androgen receptor (Kaufmann et al., 2010), another pathway which could promote oncogenesis. Additionally, Np9 has been shown to interact with the RING-type E3 ubiquitin ligase LNX (ligand of Numb protein X), which is involved in the Notch signaling pathway (Armbruester et al., 2004). Similar to other retroviral families such as HTLV and HIV (Ludwig et al., 2006; Halin et al., 2009; Matsuoka and Green, 2009), HERV-K has a large antisense ORF which may encode an accessory protein with pathogenic potential (Fig. 22.1; Douville and Nath, unpublished observation). Thus, as with exogenous retroviruses (Irish et al., 2009), HERV-encoded regulatory proteins may play a key role in neuropathogenesis.
Toxicity and immunomodulation by HERV envelope proteins
Sustained activation of an HERV-mediated innate immune response by resident cells within the CNS could promote neural injury and amplify neuroinflammation in concert with the adaptive immune response. Both exogenous and endogenous retrovirus proteins, particularly envelope glycoproteins, show a proclivity for immunomodulation and causing neuropathogenic responses in glial cells (Mangeney et al., 2001; Nath et al., 2002; Medders et al., 2010).
Expression of the HERV-W (MSRV) envelope protein (ENV-W) can cause several immunomodulatory events leading to neurologic damage. The HERV-W provirus located on chromosome 7q21–22 (ERVWE1) encodes an envelope protein, syncytin-1, which is characteristically overexpressed in glia within MS lesions (Antony et al., 2007). Interestingly, another HERV-W provirus located on chromosome Xq22.3 (ERVWE2) encodes a similar but truncated envelope protein of unknown pathogenic potential (Laufer et al., 2009; Roebke et al., 2010). Although it is not clear which genomic loci are used to encode the MSRV envelope protein, there is evidence that ENV-W proteins, and specifically syncytin-1, can mediate neurotoxicity.
ENV-W initiates innate immune response by triggering TLR4 signaling, leading to proinflammatory cytokine secretion (tumor necrosis factor-α (TNF-α), interleukin (IL)-1b and IL-6) in human monocytes (Rolland et al., 2006). In vitro, these proinflammatory cytokines can amplify MSRV release by PBMC of MSRV+individuals, causing a positive-feedback loop of virus production and inflammation (Serra et al., 2003). In concert, inflammatory mediators, such as TNF-α, IFN-γ, IL-6, and IL-1β, activate the ERVWE1 promoter to induce syncytin expression (Mameli et al., 2007b), which could lead to a vicious cycle of uncontrolled inflammation. Here, activation of the ERVWE1 promoter was driven by the transcription factor NF-κB, and supports the notion that triggering innate antiviral pathways – either through recognition of HERV nucleic acids and proteins (O’Neill and Bowie, 2010) or other co-infecting viruses (Christensen, 2005) – could reactivate the expression of latent HERVs. Thus, under specific conditions of neuroinflammation, a vicious HERV-meditated inflammatory loop may ensue and contribute several disease pathologies.
The MSRV envelop protein can also drive aberrant adaptive immune responses. Exposure of monocyte-derived dendritic cells to ENV-W prompts their maturation and promotes Th1-like T-cell differentiation (Rolland et al., 2006). Furthermore, soluble ENV-W or MSRV virions act as a potent superantigen for T cells expressing Vβ16 TcR chains, and non-specifically activates abnormal T-cell-dependent immune responses (Perron et al., 2001). In a humanized mouse model, injection of MSRV virions caused severe neurologic symptoms and death (Firouzi et al., 2003). Neuropathologic examination of these mice revealed multifocal hemorrhages in the brain parenchyma and meninges which were dependent on the presence of T cells – as depletion of T cells resulted in a reduced number of affected mice (Firouzi et al., 2003).
When murine glial cells (astrocytes and oligodendrocytes) are exposed to serum or CSF of MSRV + MS patients, they undergo apoptosis, whereas neurons do not (Menard et al., 1997). The gliotoxic components have yet to be clearly identified, but may relate to neuropathogenic effects of ENV-W or syncytin-1 (Rieger et al., 2000; Antony et al., 2010). Overexpression of ENV-W in human U251 glioma cells upregulates brain-derived neurotrophic factor, neurotrophic tyrosine kinase receptor type 2, and dopamine receptor D3 expression, in addition to increasing the phosphorylation of cyclic adenosine monophosphate response element-binding protein (Huang et al., 2010). Overexpression of syncytin-1 in astrocytes was shown to elicit endoplasmic reticulum stress and a robust inflammatory response mediated by inducible nitric oxide synthase, IL-1β, and redox reactants, which damage neighboring cells (Antony et al., 2004, 2007, 2010). Supernatants from these syncytin-1-expressing astrocytes were toxic to oligodendrocyte cultures in vitro, and in vivo the proinflammatory propensity of syncytin-1 resulted in neuropathologic evidence of demyelination with diminished motor activity and exploratory behavior in a murine model (Antony et al., 2004).
HERV-W ENV also disrupts astrocyte-mediated neurotrophic support by binding to neutral amino acid transporters ASCT-1 and ASCT-2 (reviewed in Antony et al., 2007, 2010). ASCT-1 controls the secretion of L-serine, promoting neuronal survival and differentiation (Furuya and Watanabe, 2003). Reduced detection of ASCT-1 is observed in both MS (astrocytes) and schizophrenia (neurons) and may relate to either HERV-W ENV-inducing receptor internalization or antigen masking during immunohistochemistry (Antony et al., 2007; Weis et al., 2007). However the exact role of HERV-W ENV and ASCT transporters in neurodegeneration merits further examination.
Another example of envelope superantigen is from the HERV-K family. HERV-K18 resides on chromosome 1 in an antisense orientation within the first intron of the CD48 gene (SLAMF2: signaling lymphocyte-activating molecule). The only intact viral gene of this provirus is env, which encodes a superantigen (ENV-K18) that stimulates Vβ7 TcR CD4+ T cells (Meylan et al., 2005). While most ENV-K18 reactive thymocytes are believed to be negatively selected in the thymus, a small portion of Vβ7 thymocytes may escape deletion in certain individuals, leading to increased numbers of circulating Vβ7 TcR CD4+ T cells (Meylan et al., 2005). These ENV-K18 SAg reactive T cells may further be triggered during viral infection, either by type I IFN or directly by herpesvirus proteins. Surprisingly, and unlike other HERV-K proviruses, ENV-K18 expression is inducible by type I IFNs in the non-T-cell fraction of PBMC (Stauffer et al., 2001). Specifically, the IFN-α1 isoform elicits robust ENV-K18 transcription (distinct from CD48), as compared with IFN-α2 or IFN-β (Stauffer et al., 2001). The induction of ENV-K18 can also be synergistically boosted by IFN-γ (Stauffer et al., 2001). Together, this suggests that acute or chronic viral infections which drive IFN responses may trigger ENV-K18 expression and activation of peripheral Vβ7 TcR CD4+ T cells. Alternatively, both the EBV LMP-2A protein and HHV-6 infection can induce ENV-K18 expression (Hsiao et al., 2006, 2009; Tai et al., 2009; Turcanova et al., 2009).
IMMUNE RESPONSE TO HERVS IN THE NERVOUS SYSTEM
Although most HERVs are unable to replicate and produce infectious virions, expression of HERV proteins within the nervous system may have profound effects on disease inception and exacerbation by stimulating both the innate and adaptive immune system. As described above, both resident CNS cells and cells of the adaptive immune system respond to HERV expression. However, considering the complexity by which other retroviruses mediate aberrant host responses and neurovirulent pathologies (Power, 2001; Jacobson, 2002; Kraft-Terry et al., 2010), our current knowledge of immunomodulation by HERVs is very limited.
Can resident CNS cells respond to HERV expression?
Innate and intrinsic immune responses can be mediated by virtually all CNS cell types, although microglia and astrocytes are the main mediators of resident antiviral immunity in the brain (Lewis et al., 2008). These cells can sense retroviral nucleic acids and viral proteins through a variety of pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs), RIG-I-like receptors (RLRs), and NOD-like receptors (Lewis et al., 2008). The activity of PRR signaling results in the induction of both IFN and proinflammatory responses which drive a cellular antiviral state and the recruitment of effector immune cells. Conversely, a plethora of examples exist for retroviral evasion of the innate immune system (Cheng et al., 2009; Fischer et al., 2010; Kirchhoff, 2010; Oliere et al., 2010), but if these mechanisms exist for HERVs, they have yet to be described.
How HERV expression can trigger adaptive immune responses
Once immune cells, such as T cells, are recruited to the CNS they may recognize novel HERV-derived antigens presented by CNS cells or infiltrating antigen-presenting cells. It has been described that, during cellular apoptosis, activated caspases or granzyme B can cleave HERV proteins, potentially resulting in T-cell neoepitopes, similar to the events producing nuclear autoantigens (Beyer et al., 2002). CD4+ T cells as well as cytotoxic CD8+ T cells can amplify ongoing inflammatory responses and directly contribute to neuronal transection, demyelination, and tissue injury.
Genetic polymorphisms in the host may contribute to varied clinical outcomes related to HERV expression
Genetic profiling has now established clear associations between SNPs or a collection of SNPs forming a specific genetic haplotype to the risk of disease inception and morbidity (Heap et al., 2009). This relationship is mediated by the intermediate phenotype of a genetic polymorphism: functional changes in promoter activity, protein structure and activity, expression of miRNAs – all leading to differential outcomes for cellular pathways and host pathophysiology. Thus, individual differences in the way a host will react to the expression of HERVs may dictate the extent of their pathologic contribution towards a particular disease state.
Intrinsic and innate antiviral responses are the first line of defense within retrovirus-infected cells (Douville and Hiscott, 2010). Functional polymorphisms in virus-sensing PRRs and IFN-related genes can have a dramatic impact on innate immune responses to viral nucleic acids and thus retrovirus replication (Diop et al., 2006; Oh et al., 2009). Moreover, few reports show evidence that SNPs in IFN-induced retroviral restriction factors, such as TRIM5α (Speelmon et al., 2006) and Cyclophillin A (Rits et al., 2008), may modulate the extent of retrovirus replication or associates with disease progression. A large portion of cellular miRNAs also target retroviral transcripts (Hakim et al., 2008), but as miRNA expression is variable between individuals (Krol et al., 2010), this may contribute to differential silencing of retrovirus expression. As these antiviral mechanisms are likely acting upon HERVs (Armitage et al., 2008), it follows that the host’s genetic background will play a deterministic role in the type and amplitude of HERV-mediated pathology.
Furthermore, there is now a better appreciation of HERVs as polymorphic genetic elements with precise contributions to cellular function and deregulation. The only full-length proviruses known to be insertionally polymorphic in humans are HERV-K113 and HERV-K115 (Turner et al., 2001). There are significantly higher insertional frequencies in African Americans as compared to Caucasians: HERV-K113 (21% versus 9%) and HERV-K115 (35% versus 6%) respectively. Moreover, within these HERVs, specific haplotypes could be identified, adding to the diversity of these sequences (Jha et al., 2009). Interestingly, HERV-K115 insertion is associated with earlier onset of schizophrenia, conferring not a disease risk per se but an effect on the early manifestations of the disease (Otowa et al., 2006). Moreover, induction of HERVs in neurologic disease may also contribute to the severity of comorbidities. Patients with schizophrenia are at increased risk of type 2 diabetes (odds ratio 9.0) if they carry a unique haplotype defined by two polymorphisms in the envelope region of HERV-K18 (Dickerson et al., 2008). Similarly, HERV-K18 polymorphism may influence the genetic susceptibility to MS (Tai et al., 2008). Together, these examples demonstrate the complexity of HERVs in modulating disease outcomes and the importance of SNP profiling for HERVs as well as human genes.
FUTURE DIRECTIONS IN HERV RESEARCH AND THERAPY FOR HERV-ASSOCIATED NEUROLOGIC DISEASES
Current and novel therapies for HERV-associated neurologic diseases
IFN-β is an essential cytokine which drives innate immune responses subsequent to pathogen recognition by TLR and RLR (Kawai and Akira, 2006; Hiscott, 2007). This initial burst of IFN-β is crucial for the amplification of IFN-stimulated genes, such as retroviral restriction factors (Douville and Hiscott, 2010). It is well known that IFN-β has antiviral effects on retrovirus-infected cells (Oka et al., 1990; Serra et al., 2003; Brule et al., 2007). In clinical use, IFN-β therapy has been used with moderate success for several retroviral-mediated neurologic diseases, such as ALS-like syndrome (HIV) (von Giesen et al., 2002) and HAM/TSP (HTLV-1) (Saito et al., 2004; Oh et al., 2005), although IFN-β alone is not sufficient to control these diseases adequately. However, IFN-β therapy can be effective in HERV-associated diseases such as MS (Vandenbroeck et al., 2010). Recent direct evidence suggests that IFN-β treatment can reduce the expression of HERV-W RNA in MS patients (Mameli et al., 2008), as well as circulating antibodies against HERV-H/F and HERV-W antigens (Petersen et al., 2009).
Interestingly, commonly prescribed schizophrenia medications – lithium, haloperidol, and clozapine metabolites – also have antiretroviral properties (Jones-Brando et al., 1997; Schifitto et al., 2009). The use of lithium in patients with HIV dementia improves measures of neurocognitive function (Letendre et al., 2006). This may be due to lithium’s enhancing effect on canonical Wnt pathway via β-catenin signaling (Sinha et al., 2005; Sutton et al., 2007), which inhibits retroviral replication and the activation of latent provirus (Kumar et al., 2008). This antiviral mechanism may in part be dependent on the β-catenin-dependent pathogen recognition receptor LRRFIP1, which can enhance IFN-β production (Bagashev et al., 2010). This therapeutic strategy may serve as a complementary add-on treatment for HERV-mediated neurologic disorders, enhancing host antiviral pathways, preventing activation of latent provirus, and complementing retroviral protein inhibitors.
One caveat of the therapeutic design for HERV-mediated pathology is the formulation of inhibitors specific to HERV proteins (Kuhelj et al., 2001). Retroviral proteases are essential for viral replication, as well as exerting secondary effects on cellular pathways (Strack et al., 1996; Nie et al., 2008; Solis et al., 2010). Although HERV protease exhibits limited activity on HIV proteins (Towler et al., 1998; Padow et al., 2000), current HIV protease inhibitors, including ritonavir, indinavir, and saquinavir, have no efficacy against HERV-K10 protease (Towler et al., 1998). Despite the inability of HERV-K protease to substitute the functionality of wild-type HIV-1 protease, HERV-encoded protease may in itself contribute to the disruption of cellular pathways or complement for HIV protease when compromised by anti-PR drugs or drug-resistant mutations. Further development of PR, RT, and IN inhibitors for neurodegenerative diseases should consider the family of HERV expressed and empirically test the individual efficacy of known and novel inhibitors (Opii et al., 2007).
State-of-the-art in HERV research: HERV polymorphisms, transcriptome, and proteome
Together, these findings reveal the importance of monitoring not only HERV sequence polymorphism but also insertional polymorphism when analyzing genome-wide disease association and intermediate molecular phenotypes. With the advent and increased availability of whole genome sequencing, future studies will be better equipped to predict disease risk based on more complete HERV genomic profiles (Villesen et al., 2004; Mayer et al., 2005; Moyes et al., 2007). RNA and microRNA deep sequencing in CNS specimens versus other tissues from clinically characterized HERV-associated diseases will also contribute to a clearer appreciation of the diversity of HERV transcriptomes (Seifarth et al., 2005; Flockerzi et al., 2008). Finally, with the development of HERV consensus plasmids (Dewannieux et al., 2006; Brady et al., 2009), virion-producing cell lines (Ruprecht et al., 2008), and anti-HERV antibodies, this emerging field of virology will continue to generate knowledge relevant for the diagnosis and treatment of HERV-associated neurologic diseases.
CAPITULATION
With the continued discovery of new retroviruses, such as XMRV (which to date has a disputed association with chronic fatigue syndrome) (Lombardi et al., 2009; Switzer et al., 2010), there is the potential for an under-appreciated number of neurologic disorders to be caused by unidentified exogenous or endogenous retroviruses. In species other than human, endogenous retroviruses are known pathogens with clear pathologic outcomes (reviewed in Weiss, 2006). Recent findings highlight an association between precise patterns of HERV expression and diseases previously associated with specific pathogens or of a sporadic nature, such as ALS with HERV-K (Douville et al., 2011) and Creutzfeldt–Jakob prion disease with HERV-W (Jeong et al., 2010). Thus, the expansion of HERV research into sporadic, idiopathic, rare or orphan neurologic disorders is likely a worthwhile endeavor which will illuminate novel pathologic mechanisms underpinning various neurodegenerative disorders.
References
- Agid O, Foussias G, Singh S, et al. Where to position clozapine: re-examining the evidence. Can J Psychiatry. 2010;55:677–684. doi: 10.1177/070674371005501007. [DOI] [PubMed] [Google Scholar]
- Ahn K, Kim HS. Structural and quantitative expression analyses of HERV gene family in human tissues. Mol Cells. 2009;28:99–103. doi: 10.1007/s10059-009-0107-y. [DOI] [PubMed] [Google Scholar]
- Alfahad T, Nath A. Retroviruses and amyotropic lateral sclerosis. Antiviral Res. 2013;99:180–187. doi: 10.1016/j.antiviral.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Lafuente R, Garcia-Montojo M, De Las Heras V, et al. Herpesviruses and human endogenous retroviral sequences in the cerebrospinal fluid of multiple sclerosis patients. Mult Scler. 2008;14:595–601. doi: 10.1177/1352458507086425. [DOI] [PubMed] [Google Scholar]
- Antony JM, Van Marle G, Opii W, et al. Human endogenous retrovirus glycoprotein-mediated induction of redox reactants causes oligodendrocyte death and demyelination. Nat Neurosci. 2004;7:1088–1095. doi: 10.1038/nn1319. [DOI] [PubMed] [Google Scholar]
- Antony JM, Ellestad KK, Hammond R, et al. The human endogenous retrovirus envelope glycoprotein, syncytin-1, regulates neuroinflammation and its receptor expression in multiple sclerosis: a role for endoplasmic reticulum chaperones in astrocytes. J Immunol. 2007;179:1210–1224. doi: 10.4049/jimmunol.179.2.1210. [DOI] [PubMed] [Google Scholar]
- Antony JM, Deslauriers AM, Bhat RK, et al. Human endogenous retroviruses and multiple sclerosis: innocent bystanders or disease determinants? Biochim Biophys Acta. 2010;1812:162–176. doi: 10.1016/j.bbadis.2010.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer T. Neurodegeneration in schizophrenia. Expert Rev Neurother. 2010;10:1131–1141. doi: 10.1586/ern.09.152. [DOI] [PubMed] [Google Scholar]
- Armbruester V, Sauter M, Roemer K, et al. Np9 protein of human endogenous retrovirus K interacts with ligand of numb protein X. J Virol. 2004;78:10310–10319. doi: 10.1128/JVI.78.19.10310-10319.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armitage AE, Katzourakis A, De Oliveira T, et al. Conserved footprints of APOBEC3G on hypermutated human immunodeficiency virus type 1 and human endogenous retrovirus HERV-K(HML2) sequences. J Virol. 2008;82:8743–8761. doi: 10.1128/JVI.00584-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagashev A, Fitzgerald MC, Larosa DF, et al. Leucine-rich repeat (in flightless I) interacting protein-1 regulates a rapid type I interferon response. J Interferon Cytokine Res. 2010;30:843–852. doi: 10.1089/jir.2010.0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belshaw R, Dawson AL, Woolven-Allen J, et al. Genomewide screening reveals high levels of insertional polymorphism in the human endogenous retrovirus family HERV-K(HML2): implications for present-day activity. J Virol. 2005a;79:12507–12514. doi: 10.1128/JVI.79.19.12507-12514.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belshaw R, Katzourakis A, Paces J, et al. High copy number in human endogenous retrovirus families is associated with copying mechanisms in addition to reinfection. Mol Biol Evol. 2005b;22:814–817. doi: 10.1093/molbev/msi088. [DOI] [PubMed] [Google Scholar]
- Belshaw R, Watson J, Katzourakis A, et al. Rate of recombinational deletion among human endogenous retroviruses. J Virol. 2007;81:9437–9442. doi: 10.1128/JVI.02216-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkhout B, Jebbink M, Zsiros J. Identification of an active reverse transcriptase enzyme encoded by a human endogenous HERV-K retrovirus. J Virol. 1999;73:2365–2375. doi: 10.1128/jvi.73.3.2365-2375.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer TD, Kolowos W, Dumitriu IE, et al. Apoptosis of the teratocarcinoma cell line Tera-1 leads to the cleavage of HERV-K10gag proteins by caspases and/or granzyme B. Scand J Immunol. 2002;56:303–309. doi: 10.1046/j.1365-3083.2002.01139.x. [DOI] [PubMed] [Google Scholar]
- Blikstad V, Benachenhou F, Sperber GO, et al. Evolution of human endogenous retroviral sequences: a conceptual account. Cell Mol Life Sci. 2008;65:3348–3365. doi: 10.1007/s00018-008-8495-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolinger C, Yilmaz A, Hartman TR, et al. RNA helicase A interacts with divergent lymphotropic retroviruses and promotes translation of human T-cell leukemia virus type 1. Nucleic Acids Res. 2007;35:2629–2642. doi: 10.1093/nar/gkm124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boller K, Schonfeld K, Lischer S, et al. Human endogenous retrovirus HERV-K113 is capable of producing intact viral particles. J Gen Virol. 2008;89:567–572. doi: 10.1099/vir.0.83534-0. [DOI] [PubMed] [Google Scholar]
- Bonnaud B, Bouton O, Oriol G, et al. Evidence of selection on the domesticated ERVWE1 env retroviral element involved in placentation. Mol Biol Evol. 2004;21:1895–1901. doi: 10.1093/molbev/msh206. [DOI] [PubMed] [Google Scholar]
- Bosch E, Jobling MA. Duplications of the AZFa region of the human Y chromosome are mediated by homologous recombination between HERVs and are compatible with male fertility. Hum Mol Genet. 2003;12:341–347. doi: 10.1093/hmg/ddg031. [DOI] [PubMed] [Google Scholar]
- Brady T, Lee YN, Ronen K, et al. Integration target site selection by a resurrected human endogenous retrovirus. Genes Dev. 2009;23:633–642. doi: 10.1101/gad.1762309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AS, Derkits EJ. Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am J Psychiatry. 2010;167:261–280. doi: 10.1176/appi.ajp.2009.09030361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brudek T, Luhdorf P, Christensen T, et al. Activation of endogenous retrovirus reverse transcriptase in multiple sclerosis patient lymphocytes by inactivated HSV-1, HHV-6 and VZV. J Neuroimmunol. 2007;187:147–155. doi: 10.1016/j.jneuroim.2007.04.003. [DOI] [PubMed] [Google Scholar]
- Brudek T, Christensen T, Hansen HJ, et al. Synergistic immune responses induced by endogenous retrovirus and herpesvirus antigens result in increased production of inflammatory cytokines in multiple sclerosis patients. Scand J Immunol. 2008;67:295–303. doi: 10.1111/j.1365-3083.2007.02067.x. [DOI] [PubMed] [Google Scholar]
- Brudek T, Christensen T, Aagaard L, et al. B cells and monocytes from patients with active multiple sclerosis exhibit increased surface expression of both HERV-H Env and HERV-W Env, accompanied by increased seroreactivity. Retrovirology. 2009;6:104. doi: 10.1186/1742-4690-6-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brule F, Khatissian E, Benani A, et al. Inhibition of HIV replication: a powerful antiviral strategy by IFN-beta gene delivery in CD4+ cells. Biochem Pharmacol. 2007;74:898–910. doi: 10.1016/j.bcp.2007.06.036. [DOI] [PubMed] [Google Scholar]
- Buratti E, Baralle FE. The multiple roles of TDP-43 in pre-mRNA processing and gene expression regulation. RNA Biol. 2010;7 doi: 10.4161/rna.7.4.12205. [DOI] [PubMed] [Google Scholar]
- Cheng SM, LI JC, Lin SS, et al. HIV-1 transactivator protein induction of suppressor of cytokine signaling-2 contributes to dysregulation of IFNγ signaling. Blood. 2009;113:5192–5201. doi: 10.1182/blood-2008-10-183525. [DOI] [PubMed] [Google Scholar]
- Christensen T. Association of human endogenous retroviruses with multiple sclerosis and possible interactions with herpes viruses. Rev Med Virol. 2005;15:179–211. doi: 10.1002/rmv.465. [DOI] [PubMed] [Google Scholar]
- Christensen T. HERVs in Neuropathogenesis. J Neuroimmune Pharmacol. 2010;5:326–335. doi: 10.1007/s11481-010-9214-y. [DOI] [PubMed] [Google Scholar]
- Christensen T, Dissing Sorensen P, Riemann H, et al. Expression of sequence variants of endogenous retrovirus RGH in particle form in multiple sclerosis. Lancet. 1998;352:1033. doi: 10.1016/s0140-6736(05)60075-x. [DOI] [PubMed] [Google Scholar]
- Christensen T, Tonjes RR, Zur Megede J, et al. Reverse transcriptase activity and particle production in B lymphoblastoid cell lines established from lymphocytes of patients with multiple sclerosis. AIDS Res Hum Retroviruses. 1999;15:285–291. doi: 10.1089/088922299311466. [DOI] [PubMed] [Google Scholar]
- Christensen T, Dissing Sorensen P, Riemann H, et al. Molecular characterization of HERV-H variants associated with multiple sclerosis. Acta Neurol Scand. 2000;101:229–238. doi: 10.1034/j.1600-0404.2000.101004229.x. [DOI] [PubMed] [Google Scholar]
- Christensen T, Sorensen PD, Hansen HJ, et al. Antibodies against a human endogenous retrovirus and the preponderance of env splice variants in multiple sclerosis patients. Mult Scler. 2003;9:6–15. doi: 10.1191/1352458503ms867oa. [DOI] [PubMed] [Google Scholar]
- Christensen T, Petersen T, Thiel S, et al. Gene– environment interactions in multiple sclerosis: innate and adaptive immune responses to human endogenous retrovirus and herpesvirus antigens and the lectin complement activation pathway. J Neuroimmunol. 2007;183:175–188. doi: 10.1016/j.jneuroim.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Contreras-Galindo R, Kaplan MH, Markovitz DM, et al. Detection of HERV-K(HML-2) viral RNA in plasma of HIV type 1-infected individuals. AIDS Res Hum Retroviruses. 2006;22:979–984. doi: 10.1089/aid.2006.22.979. [DOI] [PubMed] [Google Scholar]
- Contreras-Galindo R, Almodovar-Camacho S, Gonzalez-Ramirez S, et al. Comparative longitudinal studies of HERV-K and HIV-1 RNA titers in HIV-1-infected patients receiving successful versus unsuccessful highly active antiretroviral therapy. AIDS Res Hum Retroviruses. 2007a;23:1083–1086. doi: 10.1089/aid.2007.0054. [DOI] [PubMed] [Google Scholar]
- Contreras-Galindo R, Lopez P, Velez R, et al. HIV-1 infection increases the expression of human endogenous retroviruses type K (HERV-K) in vitro. AIDS Res Hum Retroviruses. 2007b;23:116–122. doi: 10.1089/aid.2006.0117. [DOI] [PubMed] [Google Scholar]
- Cross AH, Wu GF. Multiple sclerosis: oligoclonal bands still yield clues about multiple sclerosis. Nat Rev Neurol. 2010;6:588–589. doi: 10.1038/nrneurol.2010.142. [DOI] [PubMed] [Google Scholar]
- De Parseval N, Diop G, Blaise S, et al. Comprehensive search for intra- and inter-specific sequence polymorphisms among coding envelope genes of retroviral origin found in the human genome: genes and pseudogenes. BMC Genomics. 2005;6:117. doi: 10.1186/1471-2164-6-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denne M, Sauter M, Armbruester V, et al. Physical and functional interactions of human endogenous retrovirus proteins Np9 and rec with the promyelocytic leukemia zinc finger protein. J Virol. 2007;81:5607–5616. doi: 10.1128/JVI.02771-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewannieux M, Harper F, Richaud A, et al. Identification of an infectious progenitor for the multiple-copy HERV-K human endogenous retroelements. Genome Res. 2006;16:1548–1556. doi: 10.1101/gr.5565706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson F, Rubalcaba E, Viscidi R, et al. Polymorphisms in human endogenous retrovirus K-18 and risk of type 2 diabetes in individuals with schizophrenia. Schizophr Res. 2008;104:121–126. doi: 10.1016/j.schres.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Diem O, Schaffner M, Seifarth W, et al. Influence of antipsychotic drugs on human endogenous retrovirus (HERV) transcription in brain cells. PLoS One. 2012;7:e30054. doi: 10.1371/journal.pone.0030054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diop G, Hirtzig T, Do H, et al. Exhaustive genotyping of the interferon alpha receptor 1 (IFNAR1) gene and association of an IFNAR1 protein variant with AIDS progression or susceptibility to HIV-1 infection in a French AIDS cohort. Biomed Pharmacother. 2006;60:569–577. doi: 10.1016/j.biopha.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Dolei A, Perron H. The multiple sclerosis-associated retrovirus and its HERV-W endogenous family: a biological interface between virology, genetics, and immunology in human physiology and disease. J Neurovirol. 2009;15:4–13. doi: 10.1080/13550280802448451. [DOI] [PubMed] [Google Scholar]
- Douville RN, Hiscott J. The interface between the innate interferon response and expression of host retroviral restriction factors. Cytokine. 2010;52:108–115. doi: 10.1016/j.cyto.2010.04.010. [DOI] [PubMed] [Google Scholar]
- Douville RN, Liu J, Rothstein J, et al. Identification of active loci of a human endogenous retrovirus in neurons of patients with amyotrophic lateral sclerosis. Ann Neurol. 2011;69:141–151. doi: 10.1002/ana.22149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn CA, Medstrand P, Mager DL. An endogenous retroviral long terminal repeat is the dominant promoter for human beta1,3-galactosyltransferase 5 in the colon. Proc Natl Acad Sci U S A. 2003;100:12841–12846. doi: 10.1073/pnas.2134464100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuchter-Murthy AE, Freeman JD, Mager DL. Splicing of a human endogenous retrovirus to a novel phospholipase A2 related gene. Nucleic Acids Res. 1993;21:135–143. doi: 10.1093/nar/21.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firouzi R, Rolland A, Michel M, et al. Multiple sclerosis-associated retrovirus particles cause T lymphocyte-dependent death with brain hemorrhage in humanized SCID mice model. J Neurovirol. 2003;9:79–93. doi: 10.1080/13550280390173328. [DOI] [PubMed] [Google Scholar]
- Fischer W, Ganusov VV, Giorgi EE, et al. Transmission of single HIV-1 genomes and dynamics of early immune escape revealed by ultra-deep sequencing. PLoS One. 2010;5:e12303. doi: 10.1371/journal.pone.0012303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flockerzi A, Ruggieri A, Frank O, et al. Expression patterns of transcribed human endogenous retrovirus HERV-K(HML-2) loci in human tissues and the need for a HERV Transcriptome Project. BMC Genomics. 2008;9:354. doi: 10.1186/1471-2164-9-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank O, Giehl M, Zheng C, et al. Human endogenous retrovirus expression profiles in samples from brains of patients with schizophrenia and bipolar disorders. J Virol. 2005;79:10890–10901. doi: 10.1128/JVI.79.17.10890-10901.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank O, Jones-Brando L, Leib-Mosch C, et al. Altered transcriptional activity of human endogenous retroviruses in neuroepithelial cells after infection with Toxoplasma gondii. J Infect Dis. 2006;194:1447–1449. doi: 10.1086/508496. [DOI] [PubMed] [Google Scholar]
- Furuya S, Watanabe M. Novel neuroglial and glioglial relationships mediated by L-serine metabolism. Arch Histol Cytol. 2003;66:109–121. doi: 10.1679/aohc.66.109. [DOI] [PubMed] [Google Scholar]
- Garrison KE, Jones RB, Meiklejohn DA, et al. T cell responses to human endogenous retroviruses in HIV-1 infection. PLoS Pathog. 2007;3:e165. doi: 10.1371/journal.ppat.0030165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garson JA, Tuke PW, Giraud P, et al. Detection of virion-associated MSRV-RNA in serum of patients with multiple sclerosis. Lancet. 1998;351:33. doi: 10.1016/s0140-6736(98)24001-3. [DOI] [PubMed] [Google Scholar]
- Gogvadze E, Stukacheva E, Buzdin A, et al. Human-specific modulation of transcriptional activity provided by endogenous retroviral insertions. J Virol. 2009;83:6098–6105. doi: 10.1128/JVI.00123-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopinath S, Blair IP, Kennerson ML, et al. A novel locus for distal motor neuron degeneration maps to chromosome 7q34–q36. Hum Genet. 2007;121:559–564. doi: 10.1007/s00439-007-0348-9. [DOI] [PubMed] [Google Scholar]
- Griffiths DJ. Endogenous retroviruses in the human genome sequence. Genome Biol. 2001;2 doi: 10.1186/gb-2001-2-6-reviews1017. reviews 1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haahr S, Sommerlund M, Moller-Larsen A, et al. Just another dubious virus in cells from a patient with multiple sclerosis? Lancet. 1991;337:863–864. doi: 10.1016/0140-6736(91)92580-u. [DOI] [PubMed] [Google Scholar]
- Hafler DA, Slavik JM, Anderson DE, et al. Multiple sclerosis. Immunol Rev. 2005;204:208–231. doi: 10.1111/j.0105-2896.2005.00240.x. [DOI] [PubMed] [Google Scholar]
- Hakim ST, Alsayari M, Mclean DC, et al. A large number of the human microRNAs target lentiviruses, retroviruses, and endogenous retroviruses. Biochem Biophys Res Commun. 2008;369:357–362. doi: 10.1016/j.bbrc.2008.02.025. [DOI] [PubMed] [Google Scholar]
- Halin M, Douceron E, Clerc I, et al. Human T-cell leukemia virus type 2 produces a spliced antisense transcript encoding a protein that lacks a classic bZIP domain but still inhibits Tax2-mediated transcription. Blood. 2009;114:2427–2438. doi: 10.1182/blood-2008-09-179879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heap GA, Trynka G, Jansen RC, et al. Complex nature of SNP genotype effects on gene expression in primary human leucocytes. BMC Med Genomics. 2009;2:1. doi: 10.1186/1755-8794-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herai RH, Yamagishi ME. Detection of human inter-chromosomal trans-splicing in sequence databanks. Brief Bioinform. 2010;11:198–209. doi: 10.1093/bib/bbp041. [DOI] [PubMed] [Google Scholar]
- Hiscott J. Triggering the innate antiviral response through IRF-3 activation. J Biol Chem. 2007;282:15325–15329. doi: 10.1074/jbc.R700002200. [DOI] [PubMed] [Google Scholar]
- Horie M, Honda T, Suzuki Y, et al. Endogenous non-retroviral RNA virus elements in mammalian genomes. Nature. 2010;463:84–87. doi: 10.1038/nature08695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao FC, Lin M, Tai A, et al. Cutting edge: Epstein–Barr virus transactivates the HERV-K18 superantigen by docking to the human complement receptor 2 (CD21) on primary B cells. J Immunol. 2006;177:2056–2060. doi: 10.4049/jimmunol.177.4.2056. [DOI] [PubMed] [Google Scholar]
- Hsiao FC, Tai AK, Deglon A, et al. EBV LMP-2A employs a novel mechanism to transactivate the HERV-K18 superantigen through its ITAM. Virology. 2009;385:261–266. doi: 10.1016/j.virol.2008.11.025. [DOI] [PubMed] [Google Scholar]
- Huang W, Li S, Hu Y, et al. Implication of the env gene of the human endogenous retrovirus W family in the expression of BDNF and DRD3 and development of recent-onset schizophrenia. Schizophr Bull. 2010;37:988–1000. doi: 10.1093/schbul/sbp166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JF, Coffin JM. Human endogenous retrovirus K solo-LTR formation and insertional polymorphisms: implications for human and viral evolution. Proc Natl Acad Sci U S A. 2004;101:1668–1672. doi: 10.1073/pnas.0307885100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irish BP, Khan ZK, Jain P, et al. Molecular mechanisms of neurodegenerative diseases induced by human retroviruses: a review. Am J Infect Dis. 2009;5:231–258. doi: 10.3844/ajidsp.2009.231.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iskra-Caruana ML, Baurens FC, Gayral P, et al. A four-partner plant-virus interaction: enemies can also come from within. Mol Plant Microbe Interact. 2010;23:1394–1402. doi: 10.1094/MPMI-05-10-0107. [DOI] [PubMed] [Google Scholar]
- Jacobson S. Immunopathogenesis of human T cell lymphotropic virus type I-associated neurologic disease. J Infect Dis. 2002;186(Suppl 2):S187–S192. doi: 10.1086/344269. [DOI] [PubMed] [Google Scholar]
- Jeong BH, Lee YJ, Carp RI, et al. The prevalence of human endogenous retroviruses in cerebrospinal fluids from patients with sporadic Creutzfeldt–Jakob disease. J Clin Virol. 2010;47:136–142. doi: 10.1016/j.jcv.2009.11.016. [DOI] [PubMed] [Google Scholar]
- Jha AR, Pillai SK, York VA, et al. Cross-sectional dating of novel haplotypes of HERV-K 113 and HERV-K 115 indicate these proviruses originated in Africa before Homo sapiens. Mol Biol Evol. 2009;26:2617–2626. doi: 10.1093/molbev/msp180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia B, Serra-Moreno R, Neidermyer W, et al. Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by tetherin/BST2. PLoS Pathog. 2009;5:e1000429. doi: 10.1371/journal.ppat.1000429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jintaridth P, Mutirangura A. Distinctive patterns of age-dependent hypomethylation in interspersed repetitive sequences. Physiol Genomics. 2010;41:194–200. doi: 10.1152/physiolgenomics.00146.2009. [DOI] [PubMed] [Google Scholar]
- Johnston JB, Silva C, Holden J, et al. Monocyte activation and differentiation augment human endogenous retrovirus expression: implications for inflammatory brain diseases. Ann Neurol. 2001;50:434–442. doi: 10.1002/ana.1131. [DOI] [PubMed] [Google Scholar]
- Jolivet-Reynaud C, Perron H, Ferrante P, et al. Specificities of multiple sclerosis cerebrospinal fluid and serum antibodies against mimotopes. Clin Immunol. 1999;93:283–293. doi: 10.1006/clim.1999.4789. [DOI] [PubMed] [Google Scholar]
- Jones-Brando LV, Buthod JL, Holland LE, et al. Metabolites of the antipsychotic agent clozapine inhibit the replication of human immunodeficiency virus type 1. Schizophr Res. 1997;25:63–70. doi: 10.1016/s0920-9964(97)00007-8. [DOI] [PubMed] [Google Scholar]
- Karlsson H, Bachmann S, Schroder J, et al. Retroviral RNA identified in the cerebrospinal fluids and brains of individuals with schizophrenia. Proc Natl Acad Sci U S A. 2001;98:4634–4639. doi: 10.1073/pnas.061021998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson H, Schroder J, Bachmann S, et al. HERV-W-related RNA detected in plasma from individuals with recent-onset schizophrenia or schizoaffective disorder. Mol Psychiatry. 2004;9:12–13. doi: 10.1038/sj.mp.4001439. [DOI] [PubMed] [Google Scholar]
- Kaufmann S, Sauter M, Schmitt M, et al. Human endogenous retrovirus protein Rec interacts with the testicular zinc-finger protein and androgen receptor. J Gen Virol. 2010;91:1494–1502. doi: 10.1099/vir.0.014241-0. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol. 2006;7:131–137. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- Kim HS, Ahn K, Kim DS. Quantitative expression of the HERV-W env gene in human tissues. Arch Virol. 2008;153:1587–1591. doi: 10.1007/s00705-008-0159-x. [DOI] [PubMed] [Google Scholar]
- Kim YJ, Fan Y, Laurie P, et al. No evidence of HIV pol gene in spinal cord tissues in sporadic ALS by real-time RT-PCR. Amyotroph Lateral Scler. 2010;11:91–96. doi: 10.3109/17482960902835988. [DOI] [PubMed] [Google Scholar]
- Kirchhoff F. Immune evasion and counteraction of restriction factors by HIV-1 and other primate lentiviruses. Cell Host Microbe. 2010;8:55–67. doi: 10.1016/j.chom.2010.06.004. [DOI] [PubMed] [Google Scholar]
- Komurian-Pradel F, Paranhos-Baccala G, Bedin F, et al. Molecular cloning and characterization of MSRV-related sequences associated with retrovirus-like particles. Virology. 1999;260:1–9. doi: 10.1006/viro.1999.9792. [DOI] [PubMed] [Google Scholar]
- Kowalski PE, Freeman JD, Mager DL. Intergenic splicing between a HERV-H endogenous retrovirus and two adjacent human genes. Genomics. 1999;57:371–379. doi: 10.1006/geno.1999.5787. [DOI] [PubMed] [Google Scholar]
- Kraft-Terry SD, Stothert AR, Buch S, et al. HIV-1 neuroimmunity in the era of antiretroviral therapy. Neurobiol Dis. 2010;37:542–548. doi: 10.1016/j.nbd.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11:597–610. doi: 10.1038/nrg2843. [DOI] [PubMed] [Google Scholar]
- Krone B, Grange JM. Melanoma, Darwinian medicine and the inner world. J Cancer Res Clin Oncol. 2010;136:1787–1794. doi: 10.1007/s00432-010-0949-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhelj R, Rizzo CJ, Chang CH, et al. Inhibition of human endogenous retrovirus-K10 protease in cell-free and cell-based assays. J Biol Chem. 2001;276:16674–16682. doi: 10.1074/jbc.M008763200. [DOI] [PubMed] [Google Scholar]
- Kumar A, Zloza A, Moon RT, et al. Active beta-catenin signaling is an inhibitory pathway for human immunodeficiency virus replication in peripheral blood mononuclear cells. J Virol. 2008;82:2813–2820. doi: 10.1128/JVI.02498-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwun HJ, Han HJ, Lee WJ, et al. Transactivation of the human endogenous retrovirus K long terminal repeat by herpes simplex virus type 1 immediate early protein 0. Virus Res. 2002;86:93–100. doi: 10.1016/s0168-1702(02)00058-8. [DOI] [PubMed] [Google Scholar]
- Laderoute MP, Giulivi A, Larocque L, et al. The replicative activity of human endogenous retrovirus K102 (HERV-K102) with HIV viremia. AIDS. 2007;21:2417–2424. doi: 10.1097/QAD.0b013e3282f14d64. [DOI] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Polymenidou M, Cleveland DW. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet. 2010;19:R46–R64. doi: 10.1093/hmg/ddq137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry JR, Rouhi A, Medstrand P, et al. The Opitz syndrome gene Mid1 is transcribed from a human endogenous retroviral promoter. Mol Biol Evol. 2002;19:1934–1942. doi: 10.1093/oxfordjournals.molbev.a004017. [DOI] [PubMed] [Google Scholar]
- Laufer G, Mayer J, Mueller BF, et al. Analysis of transcribed human endogenous retrovirus W env loci clarifies the origin of multiple sclerosis-associated retrovirus env sequences. Retrovirology. 2009;6:37. doi: 10.1186/1742-4690-6-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblanc P, Alais S, Porto-Carreiro I, et al. Retrovirus infection strongly enhances scrapie infectivity release in cell culture. EMBO J. 2006;25:2674–2685. doi: 10.1038/sj.emboj.7601162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WJ, Kwun HJ, Jang KL. Analysis of transcriptional regulatory sequences in the human endogenous retrovirus W long terminal repeat. J Gen Virol. 2003a;84:2229–2235. doi: 10.1099/vir.0.19076-0. [DOI] [PubMed] [Google Scholar]
- Lee WJ, Kwun HJ, Kim HS, et al. Activation of the human endogenous retrovirus W long terminal repeat by herpes simplex virus type 1 immediate early protein 1. Mol Cells. 2003b;15:75–80. [PubMed] [Google Scholar]
- Lee KH, Jeong BH, Jin JK, et al. Scrapie infection activates the replication of ecotropic, xenotropic, and polytropic murine leukemia virus (MuLV) in brains and spinal cords of senescence-accelerated mice: implication of MuLV in progression of scrapie pathogenesis. Biochem Biophys Res Commun. 2006;349:122–130. doi: 10.1016/j.bbrc.2006.08.016. [DOI] [PubMed] [Google Scholar]
- Letendre SL, Woods SP, Ellis RJ, et al. Lithium improves HIV-associated neurocognitive impairment. AIDS. 2006;20:1885–1888. doi: 10.1097/01.aids.0000244208.49123.1b. [DOI] [PubMed] [Google Scholar]
- Leupin O, Attanasio C, Marguerat S, et al. Transcriptional activation by bidirectional RNA polymerase II elongation over a silent promoter. EMBO Rep. 2005;6:956–960. doi: 10.1038/sj.embor.7400502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis SD, Butchi NB, Khaleduzzaman M, et al. Toll-like receptor 7 is not necessary for retroviral neuropathogenesis but does contribute to virus-induced neuroinflammation. J Neurovirol. 2008;14:492–502. doi: 10.1080/13550280802345723. [DOI] [PubMed] [Google Scholar]
- Lombardi VC, Ruscetti FW, Das Gupta J, et al. Detection of an infectious retrovirus, XMRV, in blood cells of patients with chronic fatigue syndrome. Science. 2009;326:585–589. doi: 10.1126/science.1179052. [DOI] [PubMed] [Google Scholar]
- Loughran TP, Jr, Sherman MP, Ruscetti FW, et al. Prototypical HTLV-I/II infection is rare in LGL leukemia. Leuk Res. 1994;18:423–429. doi: 10.1016/0145-2126(94)90078-7. [DOI] [PubMed] [Google Scholar]
- Ludwig LB, Ambrus JL, Jr, Krawczyk KA, et al. Human immunodeficiency virus-type 1 LTR DNA contains an intrinsic gene producing antisense RNA and protein products. Retrovirology. 2006;3:80. doi: 10.1186/1742-4690-3-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlane C, Simmonds P. Allelic variation of HERV-K(HML-2) endogenous retroviral elements in human populations. J Mol Evol. 2004;59:642–656. doi: 10.1007/s00239-004-2656-1. [DOI] [PubMed] [Google Scholar]
- Macgowan DJ, Scelsa SN, Imperato TE, et al. A controlled study of reverse transcriptase in serum and CSF of HIV-negative patients with ALS. Neurology. 2007;68:1944–1946. doi: 10.1212/01.wnl.0000263188.77797.99. [DOI] [PubMed] [Google Scholar]
- Mager DL, Hunter DG, Schertzer M, et al. Endogenous retroviruses provide the primary polyadenylation signal for two new human genes (HHLA2 and HHLA3) Genomics. 1999;59:255–263. doi: 10.1006/geno.1999.5877. [DOI] [PubMed] [Google Scholar]
- Mameli G, Astone V, Arru G, et al. Brains and peripheral blood mononuclear cells of multiple sclerosis (MS) patients hyperexpress MS-associated retrovirus/HERV-W endogenous retrovirus, but not Human herpesvirus 6. J Gen Virol. 2007a;88:264–274. doi: 10.1099/vir.0.81890-0. [DOI] [PubMed] [Google Scholar]
- Mameli G, Astone V, Khalili K, et al. Regulation of the syncytin-1 promoter in human astrocytes by multiple sclerosis-related cytokines. Virology. 2007b;362:120–130. doi: 10.1016/j.virol.2006.12.019. [DOI] [PubMed] [Google Scholar]
- Mameli G, Serra C, Astone V, et al. Inhibition of multiple-sclerosis-associated retrovirus as biomarker of interferon therapy. J Neurovirol. 2008;14:73–77. doi: 10.1080/13550280701801107. [DOI] [PubMed] [Google Scholar]
- Mangeney M, De Parseval N, Thomas G, et al. The full-length envelope of an HERV-H human endogenous retrovirus has immunosuppressive properties. J Gen Virol. 2001;82:2515–2518. doi: 10.1099/0022-1317-82-10-2515. [DOI] [PubMed] [Google Scholar]
- Martin MA, Bryan T, Rasheed S, et al. Identification and cloning of endogenous retroviral sequences present in human DNA. Proc Natl Acad Sci U S A. 1981;78:4892–4896. doi: 10.1073/pnas.78.8.4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka M, Green PL. The HBZ gene, a key player in HTLV-1 pathogenesis. Retrovirology. 2009;6:71. doi: 10.1186/1742-4690-6-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Infectious agents and age-related neurodegenerative disorders. Ageing Res Rev. 2004;3:105–120. doi: 10.1016/j.arr.2003.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer J, Ehlhardt S, Seifert M, et al. Human endogenous retrovirus HERV-K(HML-2) proviruses with Rec protein coding capacity and transcriptional activity. Virology. 2004;322:190–198. doi: 10.1016/j.virol.2004.01.023. [DOI] [PubMed] [Google Scholar]
- Mayer J, Stuhr T, Reus K, et al. Haplotype analysis of the human endogenous retrovirus locus HERV-K(HML-2. HOM) and its evolutionary implications. J Mol Evol. 2005;61:706–715. doi: 10.1007/s00239-005-0066-7. [DOI] [PubMed] [Google Scholar]
- Mccormick AL, Brown RH, Jr, Cudkowicz ME, et al. Quantification of reverse transcriptase in ALS and elimination of a novel retroviral candidate. Neurology. 2008;70:278–283. doi: 10.1212/01.wnl.0000297552.13219.b4. [DOI] [PubMed] [Google Scholar]
- Medders KE, Sejbuk NE, Maung R, et al. Activation of p38 MAPK is required in monocytic and neuronal cells for HIV glycoprotein 120-induced neurotoxicity. J Immunol. 2010;185:4883–4895. doi: 10.4049/jimmunol.0902535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medstrand P, Landry JR, Mager DL. Long terminal repeats are used as alternative promoters for the endothelin B receptor and apolipoprotein C-I genes in humans. J Biol Chem. 2001;276:1896–1903. doi: 10.1074/jbc.M006557200. [DOI] [PubMed] [Google Scholar]
- Menard A, Paranhos-Baccala G, Pelletier J, et al. A cytotoxic factor for glial cells: a new avenue of research for multiple sclerosis? Cell Mol Biol (Noisy-le-grand) 1997;43:889–901. [PubMed] [Google Scholar]
- Meylan F, De Smedt M, Leclercq G, et al. Negative thymocyte selection to HERV-K18 superantigens in humans. Blood. 2005;105:4377–4382. doi: 10.1182/blood-2004-07-2596. [DOI] [PubMed] [Google Scholar]
- Mi S, Lee X, Li X, et al. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature. 2000;403:785–789. doi: 10.1038/35001608. [DOI] [PubMed] [Google Scholar]
- Mills RE, Bennett EA, Iskow RC, et al. Which transposable elements are active in the human genome? Trends Genet. 2007;23:183–191. doi: 10.1016/j.tig.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Misra A, Chosdol K, Sarkar C, et al. Alteration of a sequence with homology to human endogenous retrovirus (HERV-K) in primary human glioma: implications for viral repeat mediated rearrangement. Mutat Res. 2001;484:53–59. doi: 10.1016/s0027-5107(01)00240-8. [DOI] [PubMed] [Google Scholar]
- Moyes D, Griffiths DJ, Venables PJ. Insertional polymorphisms: a new lease of life for endogenous retroviruses in human disease. Trends Genet. 2007;23:326–333. doi: 10.1016/j.tig.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Moyes DL, Goris A, Ban M, et al. HERV-K113 is not associated with multiple sclerosis in a large family-based study. AIDS Res Hum Retroviruses. 2008;24:363–365. doi: 10.1089/aid.2007.0196. [DOI] [PubMed] [Google Scholar]
- Nakayama EE, Shioda T. Anti-retroviral activity of TRIM5 alpha. Rev Med Virol. 2010;20:77–92. doi: 10.1002/rmv.637. [DOI] [PubMed] [Google Scholar]
- Nath A, Hauser KF, Wojna V, et al. Molecular basis for interactions of HIV and drugs of abuse. J Acquir Immune Defic Syndr. 2002;31(Suppl 2):S62–S69. doi: 10.1097/00126334-200210012-00006. [DOI] [PubMed] [Google Scholar]
- Nellaker C, Yao Y, Jones-Brando L, et al. Transactivation of elements in the human endogenous retrovirus W family by viral infection. Retrovirology. 2006;3:44. doi: 10.1186/1742-4690-3-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nellaker C, Li F, Uhrzander F, et al. Expression profiling of repetitive elements by melting temperature analysis: variation in HERV-W gag expression across human individuals and tissues. BMC Genomics. 2009;10:532. doi: 10.1186/1471-2164-10-532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Z, Bren GD, Rizza SA, et al. HIV protease cleavage of procaspase 8 is necessary for death of HIV-infected cells. Open Virol J. 2008;2:1–7. doi: 10.2174/1874357900802010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisole S, Stoye JP, Saib A. TRIM family proteins: retroviral restriction and antiviral defence. Nat Rev Microbiol. 2005;3:799–808. doi: 10.1038/nrmicro1248. [DOI] [PubMed] [Google Scholar]
- Oh U, Yamano Y, Mora CA, et al. Interferon-beta1a therapy in human T-lymphotropic virus type I-associated neurologic disease. Ann Neurol. 2005;57:526–534. doi: 10.1002/ana.20429. [DOI] [PubMed] [Google Scholar]
- Oh DY, Baumann K, Hamouda O, et al. A frequent functional Toll-like receptor 7 polymorphism is associated with accelerated HIV-1 disease progression. AIDS. 2009;23:297–307. doi: 10.1097/QAD.0b013e32831fb540. [DOI] [PubMed] [Google Scholar]
- Oka T, Ohtsuki Y, Sonobe H, et al. Suppressive effects of interferons on the production and release of human T-lymphotropic virus type-I (HTLV-I) Arch Virol. 1990;115:63–73. doi: 10.1007/BF01310623. [DOI] [PubMed] [Google Scholar]
- Oliere S, Hernandez E, Lezin A, et al. HTLV-1 evades type I interferon antiviral signaling by inducing the suppressor of cytokine signaling 1 (SOCS1) PLoS Pathog. 2010;6:e1001177. doi: 10.1371/journal.ppat.1001177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’neill LA, Bowie AG. Sensing and signaling in antiviral innate immunity. Curr Biol. 2010;20:R328–R333. doi: 10.1016/j.cub.2010.01.044. [DOI] [PubMed] [Google Scholar]
- Opii WO, Sultana R, Abdul HM, et al. Oxidative stress and toxicity induced by the nucleoside reverse transcriptase inhibitor (NRTI)–2′,3′-dideoxycytidine (ddC): relevance to HIV-dementia. Exp Neurol. 2007;204:29–38. doi: 10.1016/j.expneurol.2006.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otowa T, Tochigi M, Rogers M, et al. Insertional polymorphism of endogenous retrovirus HERV-K115 in schizophrenia. Neurosci Lett. 2006;408:226–229. doi: 10.1016/j.neulet.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Ou SH, Wu F, Harrich D, et al. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol. 1995;69:3584–3596. doi: 10.1128/jvi.69.6.3584-3596.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padow M, Lai L, Fisher RJ, et al. Analysis of human immunodeficiency virus type 1 containing HERV-K protease. AIDS Res Hum Retroviruses. 2000;16:1973–1980. doi: 10.1089/088922200750054701. [DOI] [PubMed] [Google Scholar]
- Perron H, Lang A. The human endogenous retrovirus link between genes and environment in multiple sclerosis and in multifactorial diseases associating neuroinflammation. Clin Rev Allergy Immunol. 2010;39:51–61. doi: 10.1007/s12016-009-8170-x. [DOI] [PubMed] [Google Scholar]
- Perron H, Geny C, Laurent A, et al. Leptomeningeal cell line from multiple sclerosis with reverse transcriptase activity and viral particles. Res Virol. 1989;140:551–561. doi: 10.1016/s0923-2516(89)80141-4. [DOI] [PubMed] [Google Scholar]
- Perron H, Gratacap B, Lalande B, et al. In vitro transmission and antigenicity of a retrovirus isolated from a multiple sclerosis patient. Res Virol. 1992;143:337–350. doi: 10.1016/s0923-2516(06)80122-6. [DOI] [PubMed] [Google Scholar]
- Perron H, Garson JA, Bedin F, et al. Molecular identification of a novel retrovirus repeatedly isolated from patients with multiple sclerosis. The Collaborative Research Group on Multiple Sclerosis. Proc Natl Acad Sci U S A. 1997;94:7583–7588. doi: 10.1073/pnas.94.14.7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perron H, Jouvin-Marche E, Michel M, et al. Multiple sclerosis retrovirus particles and recombinant envelope trigger an abnormal immune response in vitro, by inducing polyclonal Vbeta16 T-lymphocyte activation. Virology. 2001;287:321–332. doi: 10.1006/viro.2001.1045. [DOI] [PubMed] [Google Scholar]
- Perron H, Lazarini F, Ruprecht K, et al. Human endogenous retrovirus (HERV)-W ENV and GAG proteins: physiological expression in human brain and pathophysiological modulation in multiple sclerosis lesions. J Neurovirol. 2005;11:23–33. doi: 10.1080/13550280590901741. [DOI] [PubMed] [Google Scholar]
- Perron H, Bernard C, Bertrand JB, et al. Endogenous retroviral genes, Herpesviruses and gender in Multiple Sclerosis. J Neurol Sci. 2009;286:65–72. doi: 10.1016/j.jns.2009.04.034. [DOI] [PubMed] [Google Scholar]
- Petersen T, Moller-Larsen A, Thiel S, et al. Effects of interferon-beta therapy on innate and adaptive immune responses to the human endogenous retroviruses HERV-H and HERV-W, cytokine production, and the lectin complement activation pathway in multiple sclerosis. J Neuroimmunol. 2009;215:108–116. doi: 10.1016/j.jneuroim.2009.08.015. [DOI] [PubMed] [Google Scholar]
- Petito CK, Torres-Munoz JE, Zielger F, et al. Brain CD8+ and cytotoxic T lymphocytes are associated with, and may be specific for, human immunodeficiency virus type 1 encephalitis in patients with acquired immunodeficiency syndrome. J Neurovirol. 2006;12:272–283. doi: 10.1080/13550280600879204. [DOI] [PubMed] [Google Scholar]
- Power C. Retroviral diseases of the nervous system: pathogenic host response or viral gene-mediated neurovirulence? Trends Neurosci. 2001;24:162–169. doi: 10.1016/s0166-2236(00)01737-9. [DOI] [PubMed] [Google Scholar]
- Prudhomme S, Bonnaud B, Mallet F. Endogenous retroviruses and animal reproduction. Cytogenet Genome Res. 2005;110:353–364. doi: 10.1159/000084967. [DOI] [PubMed] [Google Scholar]
- Ramagopalan SV, Dobson R, Meier UC, et al. Multiple sclerosis: risk factors, prodromes, and potential causal pathways. Lancet Neurol. 2010;9:727–739. doi: 10.1016/S1474-4422(10)70094-6. [DOI] [PubMed] [Google Scholar]
- Ravanic DB, Dejanovic SM, Janjic V, et al. Effectiveness of clozapine, haloperidol and chlorpromazine in schizophrenia during a five-year period. Arq Neuropsiquiatr. 2009;67:195–202. doi: 10.1590/s0004-282x2009000200005. [DOI] [PubMed] [Google Scholar]
- Rieger F, Pierig R, Cifuentes-Diaz C, et al. New perspectives in multiple sclerosis: retroviral involvement and glial cell death. Pathol Biol (Paris) 2000;48:15–24. [PubMed] [Google Scholar]
- Rits MA, Van Dort KA, Kootstra NA. Polymorphisms in the regulatory region of the Cyclophilin A gene influence the susceptibility for HIV-1 infection. PLoS One. 2008;3:e3975. doi: 10.1371/journal.pone.0003975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roebke C, Wahl S, Laufer G, et al. An N-terminally truncated envelope protein encoded by a human endogenous retrovirus W locus on chromosome Xq22.3. Retrovirology. 2010;7:69. doi: 10.1186/1742-4690-7-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolland A, Jouvin-Marche E, Saresella M, et al. Correlation between disease severity and in vitro cytokine production mediated by MSRV (multiple sclerosis associated retroviral element) envelope protein in patients with multiple sclerosis. J Neuroimmunol. 2005;160:195–203. doi: 10.1016/j.jneuroim.2004.10.019. [DOI] [PubMed] [Google Scholar]
- Rolland A, Jouvin-Marche E, Viret C, et al. The envelope protein of a human endogenous retrovirus-W family activates innate immunity through CD14/TLR4 and promotes Th1-like responses. J Immunol. 2006;176:7636–7644. doi: 10.4049/jimmunol.176.12.7636. [DOI] [PubMed] [Google Scholar]
- Ruda VM, Akopov SB, Trubetskoy DO, et al. Tissue specificity of enhancer and promoter activities of a HERV-K(HML-2) LTR. Virus Res. 2004;104:11–16. doi: 10.1016/j.virusres.2004.02.036. [DOI] [PubMed] [Google Scholar]
- Ruggieri A, Maldener E, Sauter M, et al. Human endogenous retrovirus HERV-K(HML-2) encodes a stable signal peptide with biological properties distinct from Rec. Retrovirology. 2009;6:17. doi: 10.1186/1742-4690-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruprecht K, Obojes K, Wengel V, et al. Regulation of human endogenous retrovirus W protein expression by herpes simplex virus type 1: implications for multiple sclerosis. J Neurovirol. 2006;12:65–71. doi: 10.1080/13550280600614973. [DOI] [PubMed] [Google Scholar]
- Ruprecht K, Ferreira H, Flockerzi A, et al. Human endogenous retrovirus family HERV-K(HML-2) RNA transcripts are selectively packaged into retroviral particles produced by the human germ cell tumor line Tera-1 and originate mainly from a provirus on chromosome 22q11.21. J Virol. 2008;82:10008–100016. doi: 10.1128/JVI.01016-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryschkewitsch CF, Jensen PN, Monaco MC, et al. JC virus persistence following progressive multifocal leukoencephalopathy in multiple sclerosis patients treated with natalizumab. Ann Neurol. 2010;68:384–391. doi: 10.1002/ana.22137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M, Nakagawa M, Kaseda S, et al. Decreased human T lymphotropic virus type I (HTLV-I) provirus load and alteration in T cell phenotype after interferon-alpha therapy for HTLV-I-associated myelopathy/tropical spastic paraparesis. J Infect Dis. 2004;189:29–40. doi: 10.1086/380101. [DOI] [PubMed] [Google Scholar]
- Schifitto G, Zhong J, Gill D, et al. Lithium therapy for human immunodeficiency virus type 1-associated neurocognitive impairment. J Neurovirol. 2009;15:176–186. doi: 10.1080/13550280902758973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte AM, Malerczyk C, Cabal-Manzano R, et al. Influence of the human endogenous retrovirus-like element HERV-E.PTN on the expression of growth factor pleiotrophin: a critical role of a retroviral Sp1-binding site. Oncogene. 2000;19:3988–3998. doi: 10.1038/sj.onc.1203742. [DOI] [PubMed] [Google Scholar]
- Schulz WA, Steinhoff C, Florl AR. Methylation of endogenous human retroelements in health and disease. Curr Top Microbiol Immunol. 2006;310:211–250. doi: 10.1007/3-540-31181-5_11. [DOI] [PubMed] [Google Scholar]
- Seifarth W, Frank O, Zeilfelder U, et al. Comprehensive analysis of human endogenous retrovirus transcriptional activity in human tissues with a retrovirus-specific microarray. J Virol. 2005;79:341–352. doi: 10.1128/JVI.79.1.341-352.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini B, Rosicarelli B, Franciotta D, et al. Dysregulated Epstein–Barr virus infection in the multiple sclerosis brain. J Exp Med. 2007;204:2899–2912. doi: 10.1084/jem.20071030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini B, Severa M, Columba-Cabezas S, et al. Epstein–Barr virus latent infection and BAFF expression in B cells in the multiple sclerosis brain: implications for viral persistence and intrathecal B-cell activation. J Neuropathol Exp Neurol. 2010;69:677–693. doi: 10.1097/NEN.0b013e3181e332ec. [DOI] [PubMed] [Google Scholar]
- Serra C, Mameli G, Arru G, et al. In vitro modulation of the multiple sclerosis (MS)-associated retrovirus by cytokines: implications for MS pathogenesis. J Neurovirol. 2003;9:637–643. doi: 10.1080/13550280390246462. [DOI] [PubMed] [Google Scholar]
- Sharma B, Kaushik N, Singh K, et al. Substitution of conserved hydrophobic residues in motifs B and C of HIV-1 RT alters the geometry of its catalytic pocket. Biochemistry. 2002;41:15685–15697. doi: 10.1021/bi026311z. [DOI] [PubMed] [Google Scholar]
- Sinha D, Wang Z, Ruchalski KL, et al. Lithium activates the Wnt and phosphatidylinositol 3-kinase Akt signaling pathways to promote cell survival in the absence of soluble survival factors. Am J Physiol Renal Physiol. 2005;288:F703–F713. doi: 10.1152/ajprenal.00189.2004. [DOI] [PubMed] [Google Scholar]
- Sjottem E, Anderssen S, Johansen T. The promoter activity of long terminal repeats of the HERV-H family of human retrovirus-like elements is critically dependent on Sp1 family proteins interacting with a GC/GT box located immediately 3′ to the TATA box. J Virol. 1996;70:188–198. doi: 10.1128/jvi.70.1.188-198.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol L, Agrawal D, Loughran TP., Jr Characterization of HTLV envelope seroreactivity in large granular lymphocyte leukemia. Leuk Res. 2005;29:381–387. doi: 10.1016/j.leukres.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Solis M, Nakhaei P, Jalalirad M, et al. In revision. RIG-I mediated antiviral signaling is inhibited in HIV-1 infection by a protease-mediated sequestration of RIG-I. J Virol. 2010;85:1224–1236. doi: 10.1128/JVI.01635-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotgiu S, Mameli G, Serra C, et al. Multiple sclerosis-associated retrovirus and progressive disability of multiple sclerosis. Mult Scler. 2010;16:1248–1251. doi: 10.1177/1352458510376956. [DOI] [PubMed] [Google Scholar]
- Speelmon EC, Livingston-Rosanoff D, Li SS, et al. Genetic association of the antiviral restriction factor TRIM5alpha with human immunodeficiency virus type 1 infection. J Virol. 2006;80:2463–2471. doi: 10.1128/JVI.80.5.2463-2471.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauffer Y, Marguerat S, Meylan F, et al. Interferon-alpha-induced endogenous superantigen. a model linking environment and autoimmunity. Immunity. 2001;15:591–601. doi: 10.1016/s1074-7613(01)00212-6. [DOI] [PubMed] [Google Scholar]
- Steiner I, Nisipianu P, Wirguin I. Infection and the etiology and pathogenesis of multiple sclerosis. Curr Neurol Neurosci Rep. 2001;1:271–276. doi: 10.1007/s11910-001-0030-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stengel S, Fiebig U, Kurth R, et al. Regulation of human endogenous retrovirus-K expression in melanomas by CpG methylation. Genes Chromosomes Cancer. 2010;49:401–411. doi: 10.1002/gcc.20751. [DOI] [PubMed] [Google Scholar]
- Stevens RW, Baltch AL, Smith RP, et al. Antibody to human endogenous retrovirus peptide in urine of human immunodeficiency virus type 1-positive patients. Clin Diagn Lab Immunol. 1999;6:783–786. doi: 10.1128/cdli.6.6.783-786.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack PR, Frey MW, Rizzo CJ, et al. Apoptosis mediated by HIV protease is preceded by cleavage of Bcl-2. Proc Natl Acad Sci U S A. 1996;93:9571–9576. doi: 10.1073/pnas.93.18.9571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto J, Schust DJ. Review: human endogenous retroviruses and the placenta. Reprod Sci. 2009;16:1023–1033. doi: 10.1177/1933719109336620. [DOI] [PubMed] [Google Scholar]
- Sutkowski N, Conrad B, Thorley-Lawson DA, et al. Epstein–Barr virus transactivates the human endogenous retrovirus HERV-K18 that encodes a superantigen. Immunity. 2001;15:579–589. doi: 10.1016/s1074-7613(01)00210-2. [DOI] [PubMed] [Google Scholar]
- Sutton LP, Honardoust D, Mouyal J, et al. Activation of the canonical Wnt pathway by the antipsychotics haloperidol and clozapine involves dishevelled-3. J Neurochem. 2007;102:153–169. doi: 10.1111/j.1471-4159.2007.04527.x. [DOI] [PubMed] [Google Scholar]
- Sverdlov ED. Retroviruses and primate genome evolution. Landes Bioscience; Georgetown, Tex: 2005. [Google Scholar]
- Switzer WM, Jia H, Hohn O, et al. Absence of evidence of xenotropic murine leukemia virus-related virus infection in persons with chronic fatigue syndrome and healthy controls in the United States. Retrovirology. 2010;7:57. doi: 10.1186/1742-4690-7-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai AK, O’Reilly EJ, Alroy KA, et al. Human endogenous retrovirus-K18 Env as a risk factor in multiple sclerosis. Mult Scler. 2008;14:1175–1180. doi: 10.1177/1352458508094641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai AK, Luka J, Ablashi D, et al. HHV-6A infection induces expression of HERV-K18-encoded superantigen. J Clin Virol. 2009;46:47–48. doi: 10.1016/j.jcv.2009.05.019. [DOI] [PubMed] [Google Scholar]
- Tanaka-Taya K, Sashihara J, Kurahashi H, et al. Human herpesvirus 6 (HHV-6) is transmitted from parent to child in an integrated form and characterization of cases with chromosomally integrated HHV-6 DNA. J Med Virol. 2004;73:465–473. doi: 10.1002/jmv.20113. [DOI] [PubMed] [Google Scholar]
- Thomas A, Perzova R, Abbott L, et al. LGL leukemia and HTLV. AIDS Res Hum Retroviruses. 2010;26:33–40. doi: 10.1089/aid.2009.0124. [DOI] [PubMed] [Google Scholar]
- Ting CN, Rosenberg MP, Snow CM, et al. Endogenous retroviral sequences are required for tissue-specific expression of a human salivary amylase gene. Genes Dev. 1992;6:1457–1465. doi: 10.1101/gad.6.8.1457. [DOI] [PubMed] [Google Scholar]
- Towler EM, Gulnik SV, Bhat TN, et al. Functional characterization of the protease of human endogenous retrovirus, K10: can it complement HIV-1 protease? Biochemistry. 1998;37:17137–17144. doi: 10.1021/bi9818927. [DOI] [PubMed] [Google Scholar]
- Tremlett H, Zhao Y, Rieckmann P, et al. New perspectives in the natural history of multiple sclerosis. Neurology. 2010;74:2004–2015. doi: 10.1212/WNL.0b013e3181e3973f. [DOI] [PubMed] [Google Scholar]
- Turcanova VL, Bundgaard B, Hollsberg P. Human herpesvirus-6B induces expression of the human endogenous retrovirus K18-encoded superantigen. J Clin Virol. 2009;46:15–19. doi: 10.1016/j.jcv.2009.05.015. [DOI] [PubMed] [Google Scholar]
- Turner G, Barbulescu M, Su M, et al. Insertional polymorphisms of full-length endogenous retroviruses in humans. Curr Biol. 2001;11:1531–1535. doi: 10.1016/s0960-9822(01)00455-9. [DOI] [PubMed] [Google Scholar]
- Vandenbroeck K, Urcelay E, Comabella M. IFN-beta pharmacogenomics in multiple sclerosis. Pharmacogenomics. 2010;11:1137–1148. doi: 10.2217/pgs.10.108. [DOI] [PubMed] [Google Scholar]
- Villesen P, Aagaard L, Wiuf C, et al. Identification of endogenous retroviral reading frames in the human genome. Retrovirology. 2004;1:32. doi: 10.1186/1742-4690-1-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Giesen HJ, Kaiser R, Koller H, et al. Reversible ALS-like disorder in HIV infection. An ALS-like syndrome with new HIV infection and complete response to antiretroviral therapy. Neurology. 2002;59:474. doi: 10.1212/wnl.59.3.474. author reply 474–475. [DOI] [PubMed] [Google Scholar]
- Vucic S, Kiernan MC. Pathophysiology of neurodegeneration in familial amyotrophic lateral sclerosis. Curr Mol Med. 2009;9:255–272. doi: 10.2174/156652409787847173. [DOI] [PubMed] [Google Scholar]
- Waksman BH. Multiple sclerosis. Relationship to a retrovirus? Nature. 1989;337:599. doi: 10.1038/337599a0. [DOI] [PubMed] [Google Scholar]
- Weis S, Llenos IC, Dulay JR, et al. Changes in region-and cell type-specific expression patterns of neutral amino acid transporter 1 (ASCT-1) in the anterior cingulate cortex and hippocampus in schizophrenia, bipolar disorder and major depression. J Neural Transm. 2007;114:261–271. doi: 10.1007/s00702-006-0544-0. [DOI] [PubMed] [Google Scholar]
- Weiss RA. The discovery of endogenous retroviruses. Retrovirology. 2006;3:67. doi: 10.1186/1742-4690-3-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Schroder J, Nellaker C, et al. Elevated levels of human endogenous retrovirus-W transcripts in blood cells from patients with first episode schizophrenia. Genes Brain Behav. 2008;7:103–112. doi: 10.1111/j.1601-183X.2007.00334.x. [DOI] [PubMed] [Google Scholar]
- Yolken RH, Karlsson H, Yee F, et al. Endogenous retroviruses and schizophrenia. Brain Res Brain Res Rev. 2000;31:193–199. doi: 10.1016/s0165-0173(99)00037-5. [DOI] [PubMed] [Google Scholar]
- Zhu ZB, Hsieh SL, Bentley DR, et al. A variable number of tandem repeats locus within the human complement C2 gene is associated with a retroposon derived from a human endogenous retrovirus. J Exp Med. 1992;175:1783–1787. doi: 10.1084/jem.175.6.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]