

Graphical Abstract
Genome sequences of important model organisms are now available, owing to the technological advances in high throughput DNA sequencing. An important next step in understanding biology and disease is to identify protein interactome,1–4 which however, unlike the genome, is highly dynamic and contains weakly associated components that are difficult to detect by affinity-based approaches.5, 6 In order to construct a complete human interactome,7, 8 development of new protein labeling techniques is necessary for mass spectrometric identification of weak and transient protein interactions.9–11 Recently, an enzyme-based labeling technology has been described.12 The next step is to enable spatial and temporal control of enzyme activity for protein labeling. Here we describe the concept and demonstration of a photoactivatable protein labeling technology.
This technology, dubbed SPRAY (singlet oxygen activated protein labeling, coupled to mass spectrometry), uses a genetically encoded singlet oxygen (1O2) photosensitizer (SOG) and 1O2-reactive molecules. The general concept is as follows: SOG is genetically fused to the protein of interest (POI), followed by addition of the labeling molecule that forms a reactive intermediate upon oxidation by 1O2. The intermediate then reacts with specific residues (e.g. cysteine, lysine, histidine, and serine) of closely interacting proteins. After cell lysis, the labeled proteins can be isolated by streptavidin-biotin interaction. Mass spectrometry can be used for identification of the interacting proteins. Specific labeling of closely interacting proteins is likely achieved because of the short diffusion distance of 1O2 in cells (~30 nm within half-life).13 The SPRAY labeling technique can be tested on a variety of small molecules that are known to react with 1O2. These molecules could potentially generate electrophilic species that further react with the side chains of cysteine, lysine, histidine, and serine, resulting in a diverse repertoire of chemical signatures for mass-spectrometric identification.
To realize the concept of SPRAY, first we made use of the genetically encoded small 1O2 photosensitizer miniSOG (106-residue) that requires no exogenous cofactors.14 MiniSOG is green fluorescent and generates 1O2 when illuminated with blue light (450–490 nm). Next, we designed a labeling molecule that is small and likely cell permeable so that it is potentially applicable in intact cells. Most importantly, upon 1O2 activation the molecule should have strong reactivity toward a protein residue. One candidate is a thiol-based molecule which, when oxidized by 1O2, forms a disulfide bond with the side chain of cysteine. In this oxidation process, it is believed that 1O2 is the major reactive oxygen species generated by miniSOG and the contribution from other reactive oxygen species are insignificant as previously shown.15,16 However we cannot completely rule out the partial contribution from other endogenous reactive oxygen species including hydrogen peroxide. Thus, we specifically designed a thiol-containing small molecule with a biotin group attached to it. The spacer arm of the molecule contains a cleavable ester site that may be broken with hydroxylamine to remove the biotin moiety.
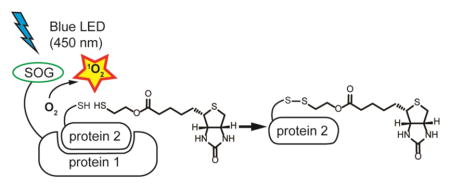
The experimental workflow is as follows: we use blue light to generate 1O2 from miniSOG (Figure 1). 1O2 then oxidizes the thiol group of the labeling molecule, which reacts with cysteine residues on the surface of nearby proteins, forming a disulfide bond.17 The labeled proteins can then be separated from the unlabeled proteins through streptavidin-biotin interaction. The biotin labeled proteins can be eluted by biotin competition, for example, by desthiobiotin. Alternatively, the labeled proteins can be released from the streptavidin by hydroxylamine cleavage (Supplementary Figure 1). The labeled proteins can also be digested with trypsin to enrich only the labeled peptides. Lastly, these labeled peptides can be fragmented and identified by tandem mass spectrometry.
Figure 1. Design of photo-activatable protein labeling via singlet oxygen.

The interacting protein in close proximity to the protein of interest (POI) is labeled. POI is fused to singlet oxygen generator (SOG) to generate the singlet oxygen upon irradiation. Then, the thiol-containing labeling molecule (Y=SH) forms a disulfide bond with a surface cysteine (X=SH) residue of interacting protein. The labeled protein can then be purified by affinity column and released by hydroxylamine followed by trypsin digestion and identified by mass spectrometry.
As a proof of concept, we directly fused miniSOG to a phycobiliprotein, allophycocyanin (APC) that contains three cysteine residues, and tested the labeling method. We also synthesized a thiol molecule conjugated to biotin by ester formation and reduction (Scheme 1). This compound showed a low to moderate membrane permeability in PAMPA assay (Pe = 1.7×10−6 cm/s, PBS buffer (pH=7.4)).
Scheme 1.

Reagents and Conditions: (a) 2-hydroxyethyl disulfide, EDC, HOBt, Et3N, DMF,12 h, rt; (b) TCEP, CH3CN-H2O (2:1), 2h, rt (53% over 2 steps)
We then mixed 1 μM freshly purified miniSOG-APC protein with various concentrations of the biotin-thiol molecule, and performed labeling experiments with different illumination times. The reaction mixture was illuminated with a customized 450 ± 20 nm blue LED (Innovations in Optics, Woburn MA) at an intensity of ~ 200 mW/cm2. After illumination, we first removed the free labeling molecules by centrifugal filtration. Then we purified the labeled proteins by streptavidin, and assayed the labeled proteins by LDS-PAGE (Figure 2c, Supplementary Figure 2). The data indicated that the amount of labeled proteins depends on both the illumination time and the concentration of labeling molecules. For example, with 10 μM biotin-thiol, the longer the illumination time was, the higher the labeling efficiency. Efficient labeling was achieved with 100 μM biotin-thiol and 30-minute illumination time. Under this condition, about 67% of the fusion protein was recovered (Supplementary Figure 3). On the other hand, when the illumination time was reduced to 5 minutes, negligible amount of proteins were labeled with 100 μM biotin-thiol.
Figure 2. Demonstration of the photo-activatable protein labeling.

[A] Primary sequence of allophycocyanin (APC) (cysteines are shown in Magenta and Red and the underlined residue represented the digested peptide fragment [B] Modeled structure of APC (Swiss-Model) The conserved Cys52 is likely linked to phycocyanobilin (in blue) [C] LDS-PAGE of a mixture of purified miniSOG-APC fusion protein (1 μM) and free APC (1 μM) [D] Ion trap CID of the identified tryptic peptide [E] Fragment ions detected are labeled according to the nomenclature.20 The N-terminal Cys was identified as bearing the biotin-label
To demonstrate the dependence of labeling on protein proximity, we compared the extent of labeling of miniSOG-APC fusion with that of free APC in the same mixture (Figure 2c). The center-to-center distance between miniSOG and APC in the fusion is estimated to be around 5 nm, which is short compared to the average intermolecular distance of >50 nm between miniSOG-APC and APC in an equimolar mixture of 1 μM each. The LDS-PAGE data showed that the miniSOG-APC fusion featured a significantly stronger labeling efficiency (> 5 fold) than free APC under all conditions tested, demonstrating that SPRAY preferentially labels proximal proteins.
To confirm the specific chemical modification, we digested the purified miniSOG-APC with trypsin and subjected the sample to LC/MS/MS analysis using an LTQ-Orbitrap XL mass spectrometer. While miniSOG does not contain cysteine, APC contains three cysteine residues (Figure 2a): Cys52, Cys66 and Cys103. Furthermore, based on the modeled structure (Swiss-Model)18, Cys103 (in red) resides on the protein surface (Figure 2b), whereas Cys52 and Cys66 (in magenta) are buried inside the protein, with Cys52 linked to the cofactor phycocyanobilin (in purple). Therefore, we expected that the exposed cysteine Cys103 would be labeled by the biotin-thiol molecule. Indeed, the Cys103-containing tryptic peptide 103CLKEASLTLLDEEDAKK119 was identified from its ion trap CID (collision induced dissociation) spectrum and Cys103 was assigned as the amino acid carrying the biotin-thiol modification (Figure 2e). On the other hand, the mass spectrometry results also suggested that the buried cysteines Cys52 and Cys66 were not labeled, presumably because they were not accessible to the labeling molecules.
To demonstrate application of the SPRAY method in protein labeling in living cells, we fused miniSOG to Skp2, an F-box protein of SCF (Skp1-cullin-F-Box) ubiquitin ligase. SCF is a multi-protein complex, composed of four subunits: Skp1 (S-phase kinase-associated protein 1), cullin1, a RING finger protein (RBX1/HRT1/ROC1) and a variable F-box protein. We expressed the Skp2-miniSOG fusion protein in cultured human cells (HeLa). The green fluorescence of miniSOG in the nucleus suggests correct localization of the fusion protein (Figure 3a). Through the SPRAY labeling process (20 min irradiation on live cells and 10 min irradiation after lysis to minimize the possible reduction of disulfide bond), we identified one of the known interacting proteins, Skp1 (Figure 3b). The Skp1 contains three cysteine residues (Figure 3c): Cys62, Cys120 and Cys160. Based on the crystal structure of Skp1 and Skp2 complex (PDB: 1FS1), Cys160 (in red) resides on the protein surface (Figure 3d), whereas Cys62 (in magenta) is buried inside the protein, and Cys120 (in magenta) is buried within the interacting interface with Skp2. We used hydroxylamine to remove the biotin during elution and identified the labeled peptide containing surface cysteine Cys160 with the ‘leftover’ beta-mercapto-ethanol modification (Cys-S)-S-CH2-CH2-OH using mass spectrometry (Figure 3e). Thus the SPRAY method is feasible to label interacting proteins in living cells.
Figure 3. Photo-activatable protein labeling in mammalian cells.

[A] Fluorescence images of miniSOG tagged Skp2, an F-box protein of SCF (Skp1-cullin-F-Box) complex. [B] Cartoon showing Skp2 and Skp1 (human S-phase kinase-associated protein 1) complex with Skp2 tagged by miniSOG. [C] Primary sequence of Skp1. Cysteines are shown in Magenta and Red and the underlined residues represented the digested peptide fragment [D] Modeled structure of miniSOG fused Skp1-Skp2 complex based on the crystal structure of Skp1-Skp2 complex (PDB ID 2ASS) using Swissmodel. [E] CID spectrum of m/z 635.2659(2+) representing tryptic peptide 155KENQWC(DeStreak)EEK163 of Skp1. ‘DeStreak’ is the Unimod identification for cysteine mercaptoethanol [www.unimod.org]. Detected fragment ions are labeled according to the nomenclature20.
However, we noted that Skp2 interacting partners other than Skp1 was not detected, suggesting that the current method requires further improvement. For instance, we may increase the number of cells used in the labeling step. In addition, development of highly cell-permeable probe molecule could increase the detection of the protein-protein interaction and this is the subject of future work. With further improvement, the SPRAY method might overcome the limitations of affinity-based approaches in identifying weak and transient protein-protein interactions.
It also needs to be pointed out that proteins are likely to be oxidized by singlet oxygen generated during the labeling procedure. For example, we detected oxidation of methionine and tyrosine (Supplementary Figure 4). To minimize protein oxidation, in the future the labeling molecules should be more reactive toward singlet oxygen than the singlet oxygen-sensitive residues such as methionine so that the interacting proteins are labeled with low concentrations of singlet oxygen.
In summary, we have developed a singlet oxygen-mediated, photo-activatable protein labeling technology SPRAY. It uses a genetically encoded photosensitizer miniSOG to photogenerate 1O2, which reacts with the biotin-thiol molecule. The oxidized/activated biotin-thiol then forms a disulfide bond with exposed cysteine residues of nearby proteins. This labeling produces a unique chemical signature that can be identified by mass spectrometry.
The light-inducible nature of this technology also allows spatial and temporal control of protein labeling. Future directions for technology development include application of this technology to identify interacting proteins in cellular context; design of new labeling molecules that label other amino acid residues such as lysine; and design of a new class of labeling molecules that label other biological molecules such as nucleic acids or glucose. For example, although each protein on average contains ~3 exposed cysteines,19 some of them may be post-translationally modified. Thus, in the future a lysine residue may be a better target using a similar approach with an appropriate labeling molecule.
Supplementary Material
Acknowledgments
We thank David Maltby and Kathleen Molnar for assistance in mass spectrometry. This work was supported by a NIH Director’s New Innovator Award [1DP2GM105446 (to X.S.)], NIH [GM54616 (to W.F.D)], [NIGMS 8P41GM103481 (to the Bio-Organic Biomedical Mass Spectrometry Resource at UCSF, Director: A.L.B)]. T.-L.T was partly supported by NIH T32 training grant.
Footnotes
Supplementary data (methods and supplementary figures) associated with this article can be found, in the online version, at doi: xx.xxxxx
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Seebacher J, Gavin AC. Cell. 2011;144:1000. doi: 10.1016/j.cell.2011.02.025. [DOI] [PubMed] [Google Scholar]
- 2.Bonetta L. Nature. 2010;468:851. doi: 10.1038/468851a. [DOI] [PubMed] [Google Scholar]
- 3.Perkel JM. Science. 2010;329:463. [Google Scholar]
- 4.Alberts B. Cell. 1998;92:291. doi: 10.1016/s0092-8674(00)80922-8. [DOI] [PubMed] [Google Scholar]
- 5.Bensimon A, Heck AJR, Aebersold R. Annu Rev Biochem. 2012;81:379. doi: 10.1146/annurev-biochem-072909-100424. [DOI] [PubMed] [Google Scholar]
- 6.Gingras AC, Gstaiger M, Raught B, Aebersold R. Nat Rev Mol Cell Bio. 2007;8:645. doi: 10.1038/nrm2208. [DOI] [PubMed] [Google Scholar]
- 7.Stumpf MPH, Thorne T, de Silva E, Stewart R, An HJ, Lappe M, Wiuf C. Proc Natl Acad Sci USA. 2008;105:6959. doi: 10.1073/pnas.0708078105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N, Klitgord N, Simon C, Boxem M, Milstein S, Rosenberg J, Goldberg DS, Zhang LV, Wong SL, Franklin G, Li SM, Albala JS, Lim JH, Fraughton C, Llamosas E, Cevik S, Bex C, Lamesch P, Sikorski RS, Vandenhaute J, Zoghbi HY, Smolyar A, Bosak S, Sequerra R, Doucette-Stamm L, Cusick ME, Hill DE, Roth FP, Vidal M. Nature. 2005;437:1173. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- 9.Acuner Ozbabacan SE, Engin HB, Gursoy A, Keskin O. Protein Eng Des Sel. 2011;24:635. doi: 10.1093/protein/gzr025. [DOI] [PubMed] [Google Scholar]
- 10.Perkins JR, Diboun I, Dessailly BH, Lees JG, Orengo C. Structure. 2010;18:1233. doi: 10.1016/j.str.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 11.Russell RB, Aloy P. Nat Chem Biol. 2008;4:666. doi: 10.1038/nchembio.119. [DOI] [PubMed] [Google Scholar]
- 12.Rhee HW, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA, Ting AY. Science. 2013;339:1328. doi: 10.1126/science.1230593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jacobson K, Rajfur Z, Vitriol E, Hahn K. Trends Cell Biol. 2008;18:443. doi: 10.1016/j.tcb.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shu X, Lev-Ram V, Deerinck TJ, Qi Y, Ramko EB, Davidson MW, Jin Y, Ellisman MH, Tsien RY. PLoS Biol. 2011;9:e1001041. doi: 10.1371/journal.pbio.1001041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.To T-L, Fadul MJ, Shu X. Nat Comm. 2011;5:4072. doi: 10.1038/ncomms5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruben RG, Cortajarena AL, Mejias SH, Agut M, Nonell S, Flors C. J Am Chem Soc. 2013;135:9564. doi: 10.1021/ja4020524. [DOI] [PubMed] [Google Scholar]
- 17.Buettner GR, Hall RD. Biochim Biophys Acta. 1987;923:501. doi: 10.1016/0304-4165(87)90060-2. [DOI] [PubMed] [Google Scholar]
- 18.Schwede T, Kopp J, Guex N, Peitsch MC. Nucleic Acids Res. 2003;31:3381. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Requejo R, Hurd TR, Costa NJ, Murphy MP. FEBS J. 2010;277:1465. doi: 10.1111/j.1742-4658.2010.07576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Biemann K. Methods Enzymol. 1990;193:886. doi: 10.1016/0076-6879(90)93460-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.