

Abstract
Inadequate dosing and incomplete treatment regimens, coupled with the ability of the tuberculosis bacilli to cause latent infections that are tolerant of currently used drugs, have fueled the rise of multidrug-resistant tuberculosis (MDR-TB). Treatment of MDR-TB infections is a major clinical challenge that has few viable or effective solutions; therefore patients face a poor prognosis and years of treatment. This review focuses on emerging drug classes that have the potential for treating MDR-TB and highlights their particular strengths as leads including their mode of action, in vivo efficacy, and key medicinal chemistry properties. Examples include the newly approved drugs bedaquiline and delaminid, and other agents in clinical and late preclinical development pipeline for the treatment of MDR-TB. Herein, we discuss the challenges to developing drugs to treat tuberculosis and how the field has adapted to these difficulties, with an emphasis on drug discovery approaches that might produce more effective agents and treatment regimens.
Graphical abstract
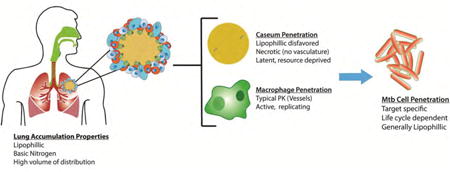
1. Introduction
Mycobacterium tuberculosis (Mtb) has been a major human pathogen since the dawn of modern human existence, with the evolution of modern Mtb sublineages closely correlating with the different waves of human migration out of Africa [1]. As the human race began to form denser population centers, culminating in urbanization, Mtb spread more easily, and it became one of the leading causes of death by the beginning of the twentieth century. The prognosis for patients with tuberculosis (TB) improved dramatically with the discovery and introduction of antitubercular drugs, starting with streptomycin in 1946 and subsequent research that produced today's frontline therapies for drug-sensitive Mtb. It is important to note that despite having an effective treatment regimen, Mtb still caused an estimated 9 million infections and 1.5 million deaths in 2013 [2].
The current standard of care for drug-susceptible Mtb infection is very effective in bacillary clearance, provided full compliance by the patient. A 2-month “intensive” phase of a four-drug cocktail containing rifampicin (RIF), isoniazid (INH), pyrazinamide (PZA) and ethambutol (EMB) (Fig. 1) is followed by a longer “continuation” phase of RIF and INH to eradicate the remaining bacilli that have entered a dormant, slowly-replicating latent phase. A looming global issue hidden in the midst of this epidemic was the emergence of drug-resistant bacteria, a trend that is on the rise, as such strains are easily spread with low fitness costs associated with transmission [3, 4]. The World Health Organization (WHO) reported that globally 3.5% of naïve infections already expressed resistance to the two most efficacious frontline agents used to treat the disease, RIF and INH, thereby classifying the infection as multidrug-resistant tuberculosis (MDR-TB) [2]. Treatment of drug-susceptible Mtb is difficult already, requiring 6 to 9 months of combination therapy in ideal circumstances. Complicating the issue is that Mtb tends to be endemic to the developing world; thus, access to adequate healthcare facilities and drugs can be limited for those patients. This leads to incomplete treatments, a leading driver for conferring drug resistance; 20.5% of patients with relapsed disease have MDR-TB [2].
Fig. 1.

Current frontline agents used to treat drug-susceptible Mtb.
Treatment for MDR-TB can extend upwards of 2 years and relies on more toxic, less efficacious second- or third-line agents, many of which are even more scarce than frontline drugs in affected areas [5]. A very common and deadly complication of Mtb infection is coinfection with human immunodeficiency virus (HIV) [6, 7]. This is particularly troublesome because RIF, a mainstay in Mtb therapy, is a potent inducer of drug-metabolizing enzymes, including cytochrome P450 (CYP) 3A4. This induction dramatically reduces plasma levels of several highly active antiretroviral therapy drugs; thus, patients are often forced to complete their TB treatment before beginning HIV treatment [6-9]. Patients who contract MDR-TB with HIV have a very poor prognosis due to the duration of treatment; these individuals frequently succumb within a few months.
The urgent need to develop new active agents to combat MDR-TB has been compounded by the emergence of extensively drug–resistant tuberculosis (XDR-TB) [10], defined as MDR-TB with additional resistance to fluoroquinolones and one of the injectable second-line agents. Furthermore, cases of totally drug–resistant tuberculosis (TDR-TB) have been noted in China, India, Africa, and Eastern Europe. In TDR-TB, the mycobacterium are resistant to all available therapeutics [11]. Combining the decline in efficacy of known active agents with the dearth of new chemical entities being moved into the clinic setting, we have reached the point where a “postantibiotic” era is a very real possibility [12]. From a clinical perspective, the WHO's approach to reduce the number of TB deaths and infections is to implement directly observed treatment, short course. This regimen implements the following five main elements to increase survival and decrease transmittance for most developing countries: (1) government commitment to monitoring and treating Mtb infections; (2) sputum-smear microscopy for all cases to assess drug susceptibility; (3) standardized treatment regimens with direct observation by a trained healthcare professional; (4) uninterrupted drug supply; and (5) standardized reporting system to maximize data interpretation directly from patients' medical records. This approach has reduced the overall number of TB infections and deaths, but it has not affected the increasing numbers of MDR-TB infections [13]. Therefore, more chemotherapeutic interventions are needed.
New agents would ideally have the following attributes: a novel mechanism of action to attenuate cross resistance; rapid bactericidal activity to reduce duration of therapy; optimized pharmacokinetic/pharmacodynamic (PK/PD) properties for once daily oral administration; low potential for drug–drug interactions to allow combination therapy, especially with other TB drugs and current HIV therapeutics; and excellent safety profile to allow for use in children and pregnant women. These ideal criteria are coupled with other practical goals such as inexpensive manufacturing, high compound stability, narrow spectrum of activity, high tolerability, and a low rate of spontaneous-resistance emergence.
In recent years, we have seen the first regulatory approvals for new Mtb drugs (i.e., bedaquiline and delamanid) in decades (Fig. 2) [14, 15]. Both drugs have been conditionally approved [delamanid in the European Union's European Medicines Agency (EMA)[16]] because adverse events have been noted. The U.S. Food and Drug Agency (FDA) has approved bedaquiline for MDR-TB and delamanid as a compassionate care option for XDR-TB and TDR-TB infections, and the EMA approved both agents for MDR-TB. The fact that these drugs have pronounced issues, including hERG toxicity concerns as well as multiple ADME issues due to their high lipophilicity. A lack of viable alternatives in late-stage clinical development is indicative of the state of drug discovery in this field, and innovation to drive new projects is desperately needed. In this review, we discuss up-and-coming drug classes with demonstrated in vivo efficacy that are on the cusp of entering clinical trials. We focus on the novelty of their mechanisms of action, the advantages they may possess in targeting MDR-TB in replicating and more latent forms, and their prospects moving into the clinic.
Fig. 2.

Two recently approved antitubercular agents developed specifically for MDR-TB infections.
1.1 Resistance in Mtb
Drug resistance in Mtb was noted soon after the introduction of streptomycin, the first drug to treat tuberculosis. Exposure to a drug induces stress responses inside the infected cell that favors both genetic and physiological mechanisms that lead to colony survival. Current antitubercular drugs are commonly associated with specific resistance mutations (Table 1).
Table 1. Clinically relevant antitubercular drugs and their commonly associated resistance mechanisms.
| Drug Name | Mechanism of Action | Common Mechanism of Resistance |
|---|---|---|
| Rifampicin | RNA synthesis inhibition | Mutation of rpoB induces a conformational change at β-subunit of RNA polymerase causing a decrease in binding affinity |
| Isoniazid | Mycolic acid biosynthesis inhibitor and effects on DNA, lipid, carbohydrate, and NAD metabolism | KatG suppression causing decreased pro-drug activation, and a mutation in the promoter region of InhA causing an overexpression of InhA |
| Pyrazinamide | Not fully resolved, may include membrane potential disruption | Mutations in pncA reducing conversion to active acid form |
| Ethambutol | Arabinogalactan biosynthesis inhibition | Mutations in embB at codon embB306 |
| Amikacin/Kanamycin | Protein synthesis inhibition | 16S rRNA target site modulation (1400 and 1401 rrs gene) Increased drug inactivation via overexpression of eis aminoglycoside acetyltransferase |
| Capreomycin | Protein synthesis inhibition | Cross resistance with aminoglycosides plus mutation of tlyA which decreases rRNA methyltransferase activity |
| Streptomycin | Protein synthesis inhibition | Mutations in rpsL and rrs confer binding site modulation |
| Fluoroquinolones | DNA gyrase and Topoisomerase IV inhibitor | Mutations in gyrA and gyrB causing an alteration to DNA Gyrase A/B binding site (later generations not always cross resistant with first generation) and increased ABC-type efflux pump expression |
| Ethionamide | Mycolic acid biosynthesis inhibition | Mutations in ethA and inhA causing decreased pro-drug activation and InhA overexpression (cross resistance with Isoniazid) |
| Cycloserine | Peptidoglycan biosynthesis inhibition | Overexpression of alrA decreasing drug efficiency |
| Para-aminosalicyclic acid | Folic acid and iron metabolism inhibition | Mutations in the thyA causing a decrease in activiated drug concentrations and folC mutations which cause binding site mutations |
| Clofazimine | Release of Reactive Oxygen Species (ROS) and cell membrane disruption | Mutation to Rv0678 causes upregulation of MmpL5, a multisubstrate efflux pump (cross resistance with Bedaquiline) |
| Linezolid | Protein synthesis inhibitor (50S subunit) | T460C mutation in rplC, encoding the 50S ribosomal L3 protein and possible efflux mechanisms |
| ß-lactam/ ß-lactamase inhibitor: Amoxicillin Meropenem Imipenem | Cell wall disruption via peptidoglycan modulation | Overexpression of β-lactamases, (BlaC), point mutations at target site altering deacylation rate and binding affinity, cell permeability (alteration in porins and outer membrane composition) and increased efflux (Rv0194) |
| Thiacetazone | Inhibits methyltransferases in mycolic acid biosynthesis | ethA mutation minimizes pro-drug activation and mutations to hadABC operon affecting dehydratase activity |
| Clarithromycin | Protein synthesis inhibition (50S subunit) | Low cell wall permeability and the expression of emr37, confers 23S rRNA site modulation |
| Bedaquiline | Inhibition of mitochondrial ATP synthase | atpE mutations introduces binding site modulation. Noted efflux via mmpL5 (cross resistance with Clofazimine) |
| Delamanid | Mycolic acid biosynthesis inhibition | Mutation of reductive activating Rv3547 gene |
Most Mtb resistance observed in the clinic can be attributed to independent, spontaneous mutations that interfere with the drug binding to the target protein, reduce prodrug-activating enzymes, or overexpress an essential target [17]. For a more detailed analysis of the molecular basis of resistance to currently used agents, we refer readers to a review by Palomino et al. [18]. In recent years, our ability to study resistance mechanisms has been greatly aided by the availability of whole-genome sequencing. This cutting-edge technology has not only shed light on many direct and indirect routes of resistance but also added insight into the function of some altered genes, thereby providing crucial information about new drug targets and rationale for future drug-design studies. For example, extensive knowledge has been gained in the areas of lipid metabolism, cell wall homeostasis, purine metabolism, and transcriptional regulation [19]. However, many resistant strains that have been sequenced express additional or unique mutations that differ from commonly associated alterations. Resistance mutations often have an associated fitness cost [20], i.e., the physiological penalty for developing said drug resistance is that the Mtb's growth may be limited in vivo. Bacteria can overcome this fitness via compensatory mutations that allow the organism to act more energetically (similar to the wild-type bacteria), while maintaining resistance. An example from MDR-TB clinical isolates in Russia identified rpoA and rpoC as compensatory mutations in RIF-resistant strains [21]. These genes are mutated to alleviate the fitness costs associated with the drug-resistant rpoB mutation that modulates the binding site at the β-subunit of the RNA polymerase. Medicinal chemistry strategies to target drug-resistant TB infections include designing agents that have novel mechanisms of action and are thus not prone to cross-resistance or redesign of inhibitors to target the mutant enzyme form, and identification and development of agents for which the fitness cost of further resistance development is high.
1.2 Mtb life cycle and microenvironments
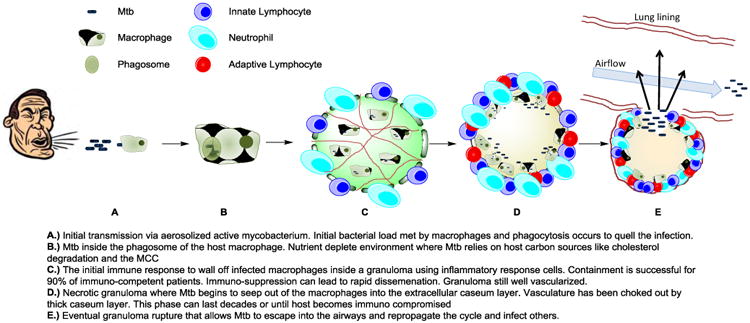
Mtb infection begins upon pulmonary exposure, following inhalation of active bacilli from the surrounding environment. The risk of being infected is greatly influenced by many factors, most of which stem from living and working conditions. Population density, weather, duration and intensity of exposure to the Mtb (cramped living and work environments with poor ventilation), a set of virulence factors unique to the infecting organism, and relative immunocompetency of the would-be host are all variables associated with succumbing to an Mtb infection. Initial infection occurs when a carrier exposes Mtb to the air via coughing. Small droplets are expelled from the lungs that contain a small number of individual organisms (<10) that must be inhaled deep into the lungs. At this point host macrophages in the lung attempt to phagocytize the pathogen (Fig. 3A) and transport it across the alveolar epithelium and into the lung. This triggers a proinflammatory response that will recruit other immune cells to form an encapsulated granuloma, which is a typical immune response to a pathogen. At this point, most of the bacterial burden is contained inside what are now considered foamy macrophages (Fig. 3B), which begin to line the outside of the granuloma. When the granuloma is first formed, it is well vascularized and a lot of immune cells are present (Fig. 3C), which bolsters the ability of drugs to reach the infection and the host's immune system to combat the pathogen. As the granuloma continues to mature against the relentless immune response, the outer wall begins to harden with a thick fibrous capsule, and the inner core is walled-off from the immune cells. Foamy macrophages line the outside of this lesion, cells die and caseum develops at the core, which cuts off the remaining vasculature. At this point, the granuloma is considered necrotic, and the bacilli exist extracellularly in this caseum and can enter their largely dormant state (Fig. 3D). Necrotic granuloma lesions (also known as tubercles) make drug penetration difficult and render mouse infection models that do not recapitulate this complex pathology poor surrogates for the human disease. After years or in cases of immune system compromise, the granulomas will fuse to the airways of the lungs and burst, releasing the pathogen to spread to new tissue and new hosts (Fig. 3E) [22]. Each of these points in the Mtb life cycle have different microenvironments that can affect drug susceptibility. For example, various degrees of vasculature result in lower blood flow, decreased oxygen levels, and pH differences in the intra/extracellular fluids that can affect ionization or activation of drugs, drug targets in Mtb may be turned off, depending on the replication stage (latent vs. actively replicating), and the ability of the drug to penetrate tissues and membranes required to reach the mycobacteria.
Fig. 3. Simplified depiction of the Mtb lifecylce.
2. Drugs under development
The past few years have seen the first approvals of new antitubercular drugs in more than 50 years, in large part due to coordinated efforts of government programs, nongovernmental organizations, and support from pharmaceutical companies. In addition to these new chemical entities available in the clinic, there is a host of small molecules in the development pipeline and at the late-discovery stage. The Stop TB Partnership has been instrumental in organizing information, consolidating clinical and preclinical data, and raising awareness about MDR-TB emergence. The Partnership offers an up-to-date list of agents that are in the pipeline and currently undergoing clinical trials, along with links to relevant publications and research information (Fig. 4; www.newtbdrugs.org). However, the development of some drugs in this pipeline appears slow and even stalled, a phenomena attributed to various flaws the clinical candidates possess that have been discussed at length in other reviews [5]. The paucity of agents in Phase I is also a concern, which is not indicative of a healthy pipeline, as many compounds are needed at this stage to combat usual failure rates as compounds advance through clinical trials.
Fig. 4. Drugs that hold clinical promise that are in development and testing. Pipeline info from www.newtbdrugs.org *conditionally approved agents.

2.1. Diarylquinolones and respiratory chain antagonists
The diarylquinolones are the most advanced series of novel antitubercular drugs. The recently approved bedaquiline falls under this classification. Bedaquiline is highly selective and specifically inhibits essential ATP-synthase activity in replicating and dormant mycobacteria but not in other prokaryotic or eukaryotic cells [23, 24]. Through the growth of drug-resistant mutants, it was discovered that the rotor ring of the organism's F0F1 ATP synthase was the target, specifically binding to the c subunit [25]. Mtb survives in a nonreplicating state using a pool of ATP that is used to maintain an energized membrane produced by the F0F1 ATP synthase [26]. This makes bedaquiline a prime weapon for killing the latent Mtb subpopulation. With a mechanism of action distinct from RIF and INH, bedaquiline is an effective addition to an MDR-TB regimen. Discovered during a phenotypic screen performed by Janssen, bedaquiline shows excellent activity against drug-sensitive and drug-resistant isolates, with a minimum inhibitory concentration (MIC) of 0.03 μg/mL for drug-sensitive Mtb and 0.12 μg/mL for drug-resistant strains, as well as outstanding activity in in vivo murine models. However, bedaquiline's efficacy in mice has not been as pronounced in humans, presumably due to limited distribution within human granulomas. Bedaquiline is a large lipophilic drug that is subject to CYP3A4 metabolism and has been noted to have PK drug–drug interactions with the CYP3A4 inducer RIF, which decreases its activity [27]. Bedaquiline has been associated with QT-prolongation and must be carefully combined with other drugs that share this risk, including fluoroquinolones, macrolides, clofazimine, or drugs that inhibit CYP3A4, which may increase its exposure [15]. Thus, efforts are ongoing to develop second-generation diarylquinolones that will improve the drug's physiochemical properties and remove its cardiac liability. This effort has been recently aided by the co-crystal structure of bedaquiline bound to its active site in the ATP synthase, which should enable structure-guided design of more potent and less toxic analogs[28].
At the time of this writing, Q203 (Fig. 5), an imidazopyridine antitubercular compound that also acts on the respiratory chain, just entered Phase I clinical trials. Q203 targets the cytochrome b subunit (QcrB) of the cytochrome bc1 complex. This complex is an essential component of the respiratory electron transport chain of ATP synthesis. Q203 causes a rapid depletion of intracellular ATP at an IC50 of 1.1 nM and interrupts ATP homeostasis in dormant Mtb at an IC50 of 10 nM. Both of these values are better than bedaquiline's measures, and they explain Q203's excellent killing profile in chronic Mtb-infection models. Sponsored by Qurient Therapeutics (Gyeonggi-do, Korea), Q203 was discovered after medicinal-chemistry optimization and a high-throughput screen (HTS) performed against infected human macrophages [29]. This screen identified 96 compounds that were able to perfuse and accumulate enough in the macrophage to display dose-dependent growth inhibition. The imidazopyridine scaffold was selected due to its activity in both macrophages and free media, and the initial hit was active against clinical MDR-TB isolates [29]. After the initial hits were evaluated, 477 analogs were synthesized, aided by structure-guided design, and the best was Q203, with an MIC50 of 2.7 nM in broth media and 0.28 nM inside the macrophage. Kim et al. reported PK and safety-profile data compatible with once daily oral dosing, with 90% oral bioavailability in mice, and a half-life of almost 24 hours. There was no detection of cytotoxicity in vitro or in vivo, no signs of hERG inhibition (indicative of limited cardiotoxicity), and mice tolerated 1g/kg doses for 2 weeks. Q203 did not inhibit any P450 isoforms and was not a substrate or inhibitor of P-gP efflux, which indicated a low risk of drug–drug interactions. Though it should be noted that like bedaquiline, Q203 is a highly lipophilic drug, with very high serum protein binding. The Phase I clinical trial (clinicaltrials.gov identifier: NCT02530710) enrolling healthy patients is a dose-escalation study starting at 100 mg dosing that will be adjusted based on PK analysis. Further combination trials will be needed to determine whether Q203 can function synergistically in combination with other second-line agents to treat MDR-TB which are no longer susceptible to INH or RIF, in the hopes of shortening the duration of therapy compared to current treatments for MDR-TB.
Fig. 5.

Q203, which is currently in phase I clinical trials, has robust activity against latent Mtb and is a promising option for addition to MDR-TB treatment regimens.
The imidazopyridine QcrB inhibitors [30] were discovered around the same time by several groups applying phenotypic MIC screening approaches of pharmaceutical company screening collections. The imidazopyridine core is widely distributed in pharmaceutical compound screening collections as the core is found in many γ-aminobutyric acid (GABA) receptor agonists, including the sleep aid ambien. Miller et al. used an anagram approach to add structural diversity to adjust the imidazo[1,2-a]pyrimidine core to obtain enhanced Mtb activity and remove human GABA receptor activity. [31]. By simply rearranging functional groups already present on ambien, the investigators increased the mycobacterial activity 2500 fold (ambien: 10 μM → ambien “anagram”: 4nM) and maintaining good safety. The best compounds were cross-resistant with Q203, thereby confirming a similar mechanism of action. The differential display of side groups in this series offers the advantage of providing compounds with alternative PK and ADME properties compared to those of Q203. This is a nice example of scaffold development to provide an additional pool of Mtb drug candidates, though at this time these molecules are best described as backup candidates.
2.2 DprE1 inhibitors
Decaprenylphosphoryl-β-D-ribose 2′-epimerase 1 (DprE1) was first identified as the target of 1,3-benzothiazin-4-ones (benzothiazones), which are potent antimycobacterial agents. DprE1, in combination with DprE2, catalyzes the epimerization of decaprenylphosphoryl-β-D-ribose to the corresponding of decaprenylphosphoryl-β-D-arabinose [32], thereby providing a crucial precursor to the cell wall arabinogalactan polysaccharide via a series of sequential oxidation-reduction reactions [33]. This target is frequently identified as the mechanism of action of hits identified in whole-cell phenotypic MIC screens, most likely due to its key biosynthetic role, high metabolic turnover, presence of nucleophilic residues at its active site, and location on the extracellular face of the cytoplasmic membrane that does not require transport inside the mycobacterium [34]. As an essential aspect of Mtb survival and a novel mechanism of antitubercular activity, optimized DprE1 inhibitors are thought of as powerful new classes for the treatment of MDR-TB, with limited cross-resistance to current therapeutic options [35]. There is a host of promising hits being investigated currently, but in this review, we will focus on those that are furthest advanced.
The benzothiazones have displayed nanomolar Mtb activity in multiple models, including ex vivo and in vivo murine models [36]. PBTZ 169 (Fig. 6) is the lead molecule in this series and is being developed by the Innovative Medicines for Tuberculosis (Lausanne, Switzerland). Both PBTZ 169 and a backup benzothiazone BTZ 043 are being studied preclinically. PBTZ 169 has the advantage of being slightly more potent and not being stereoselective, meaning it is generated via an easier and cheaper synthesis process [37]. The benzothiazones are irreversible inhibitors that require activation of an aromatic nitro group for antitubercular activity. The nitro group is required, as well as a meta electron–withdrawing group, typically either a trifluoro methyl or another nitro (dinitro benzenes). The nitroaromatic group is available for a specific Mtb mediated bioreduction to a corresponding reactive nitrosoarene. The nitroso group then reacts with an active site cysteine residue in the DprE1 enzyme to form a semimercaptal adduct inactivating the enzyme [38]. Importantly, human reductive metabolism does not have the ability to activate this prodrug series providing a suitable safety window and are negative in the Ames test for DNA mutagenesis.
Fig. 6.

PBTZ 169 and BTZ 043 with the covalent inhibition mechanism of DprE1.
A single mutation to the Cys387 residue is the commonly associated resistance mechanism and results in more than a 500-fold reduction in benzothiazone activity. Delamanid is similarly reduced by Mtb to elicit its bactericidal properties but does not result in cross-resistance with the benzothiazones as they are bio-reduced by a different enzyme (F420-deazaflavin–dependent nitroreductase [39]).
An alternative class of DprE1 inhibitors, the 1,4-azaindoles (Fig. 7), were identified via a scaffold-morphing approach by AstraZeneca (Waltham, MA) and now are being advanced by the Global TB alliance (New York, NY). These compounds were derived from an imidazo-pyridine HTS hit that displayed a good Mtb MIC but were not bactericidal. To gain bactericidal activity, multiple scaffold morphs were performed. The resulting 1,4-azaindole scaffold underwent several structure– activity relationship (SAR) optimizations to improve bactericidal activity, shift selectivity away from the human phosphodiesterase 6 (PDE6) and increase metabolic stability [40, 41]. PDE6 is a key off target pharmacology for this class as it is expressed in rod and cone photoreceptor cells of the eye, and extended inhibition can be detrimental to visual acuity. A similar adverse event is noted with prolonged EMB exposure [42]. Lead molecules from these optimizations have a marked decrease in PDE6 inhibition (>100 μM) and a good PK profile, while maintaining or improving Mtb activity. This class of DprE1 inhibitors (at 100 mg/kg) had comparable efficacy to INH in a murine model of acute TB infection and to RIF in a chronic model in vivo [41]. An important distinction for this potential drug class is the lack of cross-resistance with the benzothiazones, despite having the same molecular target. This is due to the noncovalent nature of inhibition the 1,4-azaindoles display. The commonly associated resistance mutation for benzothiazones is the single-point mutation of the nucleophilic Cys387, which does not affect the binding of the 1,4-azaindoles, nor their antitubercular activity [40].
Fig. 7.

The 1,4 azaindoles are a promising class of novel DprE1 inhibitors that are reversible and orally bioavailable.
TCA1 (Fig. 8), a member of a third class of promising DprE1 inhibitors, was discovered using a clever screen aimed at finding inhibitors that prevent mycobacterial biofilm formation. TCA1 has bactericidal activity against wild-type TB and MDR-TB in both active and dormant states and is effective in both acute and chronic in vivo infection models [43]. TCA1 not only has activity under normal growth conditions and biofilm culture conditions but also has the ability to prevent biofilm formation. With a narrow spectrum of activity specific to mycobacterium, TCA1 displayed potent bactericidal activity against XDR-TB, suggesting a novel mechanism and a great potential to be optimized into a new therapeutic option for MDR-TB. Using purified DprE1 enzyme, it was shown that TCA1 prevents the formation of DPA in a dose-dependent manner. Results from a competitive binding assay also showed that TCA1 binds at the same site as the benzothiazone, but there does not appear to be cross-resistance associated with either's resistant mutants [43]. These competition assays were verified upon co-crystallization of TCA1 and DprE1, which confirmed a significant overlap in the binding domains. One key difference to be noted is that the benzothiazones do not display activity in latent Mtb cells, and TCA1 down regulates the expression of genes associated with persistence (i.e., fdxA, a low-redox electron carrier [44]) and drug tolerance. This added benefit of killing latent Mtb is thought to be due to a second target of TCA1, a target that is not essential for growth in optimal conditions but is physiologically relevant when the mycobacterium is under antibiotic stress. This was shown in a series of growth models using DprE1 overexpression and various nutrient-rich or nutrient-depleted media [43]. In a competitive molecular pull-down experiment using a TCA1 analog, it was determined that MoeW was the secondary target. MoeW is a protein involved in molybdenum cofactor biosynthesis, and the gene encoding this protein is conserved only in Mtb and not in any other mycobacterium or bacterial species. The molybdenum cofactor is essential for nitroreductase activity in Mtb, which is important for the hypoxic and nitric oxide–induced stress response.
Fig. 8.

TCA1 is a recently identified DprE1 inhibitor with activity against latent MDR-TB infection in vivo.
2.3 MmpL3 inhibitors
The Mycobacterial membrane protein Large (MmpL) family of export proteins are involved in transportation of metabolites from the cytosol of Mtb. The Mtb genome contains 12 genes that express the MmpL proteins that are considered resistance-nodulation-division proteins, which play an important role in Mtb survival and pathogenesis [45]. MmpL3 is required for the export of mycolic acids in the form of trehalose monomycolates to the periplasmic space or outer membrane. Established as the only protein in this family that is essential for mycobacterial survival, MmpL3 is an attractive drug target. It is an important membrane protein, and a very common target elucidated in hits from whole-cell phenotypic assays, similar to DprE1 and QcrB [34]. Because MmpL3 is an easily druggable target with proven essential roles in Mtb survival, there is immense interest in MmpL3 inhibition as a novel mechanism of action for a new antitubercular agent.
The most advanced MmpL3 inhibitor is SQ109 (Fig. 9), a structural derivative of EMB's diamine moiety [46]. The initial effort in discovering SQ109 was undertaken using a combinatorial chemistry approach to find an EMB analog with improved activity because EMB is the weakest agent of the frontline therapeutics. However, retrospective analysis using SQ109-resistant mutants showed that this inhibitor's mode of action differs from EMB, with action associated with MmpL3 inhibition (Fig. 9). It is now accepted that SQ109 has polypharmacology properties, as it has activity on fungi and bacteria that do not possess mycolic acids [47, 48] and activity against latent cells that do not require active cell wall synthesis [49, 50]. Further investigation of SQ109's mechanism of action divulged additional inhibition of menaquinone synthesis, cellular respiration, and ATP synthesis in part due to dissipation of the proton motif force across the cytoplasmic membrane [50]. These multiple mechanisms of antitubercular activities suggest that SQ109 would be an effective agent to add to MDR-TB therapies and will cause limited instances of resistance if used in such a regimen, though it does have some pharmacological limitations due to its amphipathic structure.
Fig. 9.

MmpL3 inhibitors exhibit structural diversity.
More specific classes of MmpL3 inhibitors were discovered via a whole-cell phenotypic screen performed at the Novartis Institute for Tropical Diseases (NITD; Singapore). Indolcarboxamides (Fig. 9) that have excellent PD properties, including potent bactericidal activity, display both concentration and time-dependent killing [51]. Lead molecules are orally bioavailable and display limited toxicity, including no inhibition of HepG2, hERG, AMES, or CYP450. They possess a narrow spectrum of activity against gram-positive and -negative bacteria, a positive attribute for a potential antitubercular agent. Researchers working with indolcarboxamide-resistant Mtb discovered no cross-resistance associated with any commonly used drugs for Mtb treatment. Cross-resistance was noted with other MmpL3 inhibitors, including SQ109 (16-fold shift in MIC), AU1235 (>1,024-fold), and BM212 (2-fold) [52]. This range of MIC shifts can be explained by different binding sites of the inhibitors in MmpL3. One crucial advantage that the indolcarboxamides display is a dramatic accumulation in the lungs in vivo. Early analogs of the initial HTS hit showed a 5-fold higher Cmax and a 10-fold increase in AUC0-24 h, in respect to lung concentrations vs. plasma concentrations of drug [52]. In general, MmpL3 inhibitors are quite lipophilic, which is not surprising, given the role of MmpL3 in exporting mycolic acids. This lipophilic nature, in turn, causes problems with distribution and propensity for oxidative metabolism of the inhibitors.
2.4 InhA inhibitors
Isoniazid is a staple of frontline tuberculosis therapy. Its molecular target is the mycobacterial enoyl reductase InhA, which is required for the biosynthesis of mycolic acids, the dominant feature of the lipophilic outer mycobacterial cell wall that is essential for growth and virulence. [53, 54]. There are two distinct fatty acid biosynthesis (FAS) routes; mammals rely on the FAS I pathway, and bacteria rely on the FAS II. Mycobacteria contain both pathways, with a distinct subset of enzymes included in FAS II for the biosynthesis of extraordinarily long mycolic acids. These FASII enzymes are where drugs can be designed for mycobacterium specificity, an important consideration for the development of a therapy for MDR-TB. INH is a prodrug that requires activation by the catalase-peroxidase KatG to inhibit InhA, and this bioactivation step is where Mtb primarily develops resistance by inactivating this nonessential enzyme (Fig. 10). Therefore, new prodrugs targeting MDR-TB would run the risk of encountering cross-resistance in cases of Mtb if their bioactivation pathway is similar to that of INH or other prodrugs that are currently used in TB therapy, including EMB and PZA. The resulting INH–NAD adduct inhibits the enzyme and displays remarkable residency time due, in part, to a conformational shift in helix 6 that occurs after the initial binding, a shift that does not occur with rapid reversible inhibitors of InhA [55-57]. Peter Tonge and others have stressed the importance of residency time in drug discovery and lead optimization [58-60]. Just having tight binding affinity is not enough. Typically, it is the dissociation rate of the inhibitor from the enzyme that is more indicative of the biological activity in vivo [61]. FabI, the S. aureus ortholog of InhA, has been the subject of much antibacterial structure–based drug design, with at least two candidates advancing into clinical trials (NCT02426918) [62, 63]. This information provides a useful knowledge base for ongoing efforts to discover direct-acting inhibitors of InhA that are not susceptible to KatG-associated cross-resistance.
Fig. 10.

INH bioactivation of the NAD adduct that is responsible for InhA inhibition.
Researchers at GSK (Middlesex, UK) and subsequently AstraZeneca have produced lead molecules with a methyl thiazole core (Fig. 11) that are nanomolar InhA inhibitors. Both have excellent MIC values against Mtb [64, 65] and display a favorable preclinical toxicity profile, with limited CYP450 activity and favorable preliminary PK properties. The GSK lead has a whole-cell MIC of 1 μM and an intracellular MIC of 0.48 μM. However, the series falls short of INH in vivo potency and requires more optimization to be clinically relevant. The NITD recently published a thorough study of another chemical class to directly inhibit InhA, the 4-hydroxy-2-pyridones (Fig. 11) [66]. This class of orally active compounds, the lead of which is NITD-916, displays potent bactericidal activity against multiple INH-resistant strains of Mtb and in vivo efficacy in acute- and chronic-infection models. The primary concerns with this class of inhibitors are similar to those associated with the MmpL3 series (i.e., activity against dormant bacteria and high lipophilicity) and poor resultant physiochemical properties of inhibitors that engage mycolic acid–binding sites.
Fig. 11.

Direct InhA inhibitors that are under development by GSK and the NITD.
2.5 Novel β-lactam combinations
The β-lactams are the most populous class of antibiotics and are the mainstay of most antibacterial drug treatment regimens; however, their efficacy against Mtb has always been limited. With the pressing situation of the advance of MDR-TB and a greater knowledge of the mechanisms that render Mtb insensitive to β-lactams, efforts have been renewed to determine whether β-lactam agents can be developed to treat MDR-TB [67]. Case reports of amoxicillin plus clavulanate have shown some limited success treating MDR-TB [68] [69]. However, carbapenems are the most suitable β-lactams for treating Mtb as they potently target the high-molecular-weight penicillin-binding proteins, and also inactivate the unusual ι,D-transpeptidases that form the 3→3 crosslinks found in the unique Mtb cell wall [70]. Additionally, Carbapenems are less favorable substrates for the Mtb β-lactamase BlaC, which is expressed at high levels and inactivates most other β-lactams. Meropenem and imipenem have been tested for MDR-TB, in combination with a β-lactamase inhibitor, such as clavulanate, which inactives BlaC [69, 71-73]. Clinical efficacy has been demonstrated in XDR-TB by using meropenem–clavulanate combinations, which requires administration three times daily via infusion [74, 75]. One advantage the β-lactams offer as a series over other second-line alternatives is that they have a well-defined safety profile [76]. However, complications with the use of these powerful broad-spectrum antibiotics in a long-term course of therapy can originate with disturbances of the patients' microbiome and could lead to regiment compliance issues.
Faropenem (Fig. 12) a penem, is structurally similar to the carbapenems. The penem ring system is slightly less strained and consequently has improved chemical stability, and has been modified to a prodrug ester (faropenem medoxomil) that allows for oral administration, both desirable advantages for treating MDR-TB [77, 78]. Already approved for treatment of respiratory infections in humans, faropenem has displayed promising bactericidal activity in both active and non-growing, metabolically active Mtb cells [79], which is comparable to that seen with meropenem. Such results combined with renewed efforts to design a new generation of Mtb-targeted β-lactams, especially in conjunction with recent advances in β-lactamase inhibitor design, hold promise for clinical development as MDR-TB therapies. The primary limitations of this class of inhibitors relate to issues of obtaining sufficient drug exposure, for instance meropenem's dosing regimen requires 1g infusions, three times daily.
Fig. 12.

Faropenem, a β-lactam antibiotic used in pulmonary infections that may soon be used in MDR-TB regimen.
2.6 Oxazolidinones and other protein-synthesis inhibitors
Linezolid, the first-in-class oxazolidinone antibacterial agent, has demonstrated excellent clinical efficacy in treating drug-resistant gram-positive pulmonary infections. Oxazolidinones are protein synthesis inhibitors that bind to the 50s ribosomal subunit of the 23S rRNA. Linezolid possesses robust efficacy in treating MDR-TB infections, despite having modest activity in vitro and in mouse models of acute TB infection [80]. These results have encouraged further examination of the oxazolidinones class of antibiotics for the treatment of MDR-TB [80-84]. Only two agents are currently clinically approved, linezolid and tedizolid, but expansive efforts have been made to develop next-generation oxazolidinones that have greater antibacterial potency and less adverse effects. For other indications, linezolid use is generally restricted to less than 2 weeks, because the agent can cause myelotoxicity (as a result of mitochondrial protein synthesis inhibition), cytopenia, neuropathies, lactic acidosis, and rhabdomyolysis. This is particularly troublesome when undergoing prolonged therapy that is required for MDR-TB. Avoiding these mechanistic toxicities is a top priority in the design of future analogs. Over the past decade, many oxazolidinone analogs have been created in antibacterial-discovery programs at major pharmaceutical companies, some of which are pharmacologically validated leads that are well suited for repurposing for MDR-TB applications with larger safety indices. Sutezolid (Fig. 13) has emerged as a promising example and has completed a Phase II clinical trial (NCT01225640). In addition AstraZeneca is pursuing a second-generation oxazolidinone AZD5847 that also displays promising efficacy in animal models in a Phase II clinical trials [85] (NCT01516203).
Fig. 13.

Second-generation oxalidinones designed to combat MDR-TB pulmonary infections with fewer adverse events than their parent compound linezolid.
Sanofi (Paris, France) has recently begun to reinvestigate macrolide antibiotics from their compound archive for MDR-TB application because of the efficacy of other protein synthesis inhibitors and the reported use of the macrolide clarithromycin. From the natural product sequanamycin A (SEQ-503), chemical optimization has obtained SEQ-9 (Fig. 14). SEQ-9 has better mycobacterial potency and much better PK properties, including increased plasma stability, a better metabolic profile (high stability and no CYP inhibition), and it is efficacious in acute in vivo murine models. SEQ-9 does not appear to be bactericidal yet, it has notable activity against replicating Mtb, latent Mtb, and intramacrophage Mtb displaying submicromolar MIC80 activity in all three models. An exciting aspect of SEQ-9 is that there is no MIC shift in multiple lines of human clinical Mtb isolates and does not appear to be hampered by inducible macrolide mediated resistance that restricts the use of other macrolides [86]. When Sanofi researchers added SEQ-9 to combination studies, they noted a dramatic increase in the total bactericidal activity of those combinations. The greatest increase was observed in combination with bedaquiline and PA-824, with nearly complete elimination of chronic-TB infection in mice by 8 weeks. There is still plenty of optimization to be performed on this macrolide, but this study adds more evidence that adding a protein-synthesis inhibitor to an antitubercular regimen could dramatically reduce treatment duration. Other macrolides are being investigated and should produce additional options to MDR-TB treatment.
Fig. 14.

SEQ-9, a macrolide antitubercular agent derived from a natural extract from Allokutzaneria albata.
Leucyl-tRNA synthase (LeuRS) is an attractive antibiotic target that is required for protein synthesis. A unique class of boron-containing inhibitors has been identified that act on the editing site of LeuRS and disrupt its proofreading role in leucyl-tRNA synthesis. These inhibitors trap tRNA of the LeuRS away from the active site to a separate editing site, approximately 30 Å away [87]. Locking this terminal ribonucleotide via covalent attachment of the ribosyl diols to the boron atom (Fig. 15) blocking the tRNA from being properly aminoacylated and preventing protein synthesis. Recently, this inhibitor class has produced an approved agent for fungal infections [87] and late stage clinical candidates for the treatment of gram-negative infections [88, 89]. Anacor Pharmacueticals (Palo Alto, CA) in collaboration with GSK recently opened new investigations to identify oxaboroles that are active against Mtb identifying a promising Mtb selective subclass. These low molecular weight and hydrophilic inhibitors have notably good oral bioavailability and PK properties. The LeuRS editing site is structurally unique across species, which enables scientists to develop species-specific inhibitors [88]. Anacor Pharmaceuticals is now optimizing this series of Mtb LeuRS–specific protein synthesis inhibitors. This class of agents has been described to the StopTB Partnership as having potent, Mtb-specific activity in both acute- and chronic-infection murine models in vivo at very low doses (<10 mg/kg per day).
Fig. 15.

Mechanism of action for a representative oxaborole LueRS inhibitor.
2.7 Rekindling old antibiotics with natural product chemistry
The first effective treatment for TB followed the isolation of streptomycin in 1943 and its clinical introduction in 1946. Streptomycin was first isolated by Albert Schatz, a graduate student in Selman Waksman's laboratory at Rutgers University (New Brunswick, NJ). The antibiotic was isolated from the bacterial species from which its name is derived, Streptomyces griseus. This discovery was the genesis of studying natural products produced by bacteria rather than those produced by plants or fungi. This discovery method of screening soil-derived microbes for antibiotic production helped launch the “Golden Age” of antibiotic drug discovery (1943–1965), when one half of clinically relevant antibiotics were discovered [90]. The most important application of this approach in the area of TB therapy was the discovery of rifamycin antibiotics by Piero Sensi at Lepetit Pharmaceuticals research laboratory in Milan, Italy. Semisynthetic modification of rifamycin produced rifampicin, which remains one of the most effective antitubercular agents ever developed. In the course of soil-microbe screening efforts, many potent series were left behind due to chemical instability, rapid metabolism, toxicity, or an undesirable spectrum of activity. However, with the rise of antibiotic resistance, there has been an increased interest in reinvestigating some of the scaffolds that have displayed a unique mechanism of action or where new structural or mechanistic target information can be exploited. As the last group of TB drugs under development to be discussed in this review, we will focus on some synthetic adaptations made to underexploited natural-product entities that have the potential to be adapted to clinically relevant drug classes for the treatment of MDR-TB.
Spectinamides are semisynthetic derivatives of the antibiotic spectinomycin produced by Streptomyces spectabilis. Spectinomycin works by inhibiting ribosomal translocation and consequent protein synthesis by binding to a unique binding site on the 16S ribosomal subunit [91, 92]. A second-line agent used in the treatment of gonorrhea infections, spectinomycin has a documented excellent safety profile, with no associated nephrotoxicity or ototoxicity, which are common adverse effects of aminoglycosides, consistent with its distinct mechanism of action. However, spectinomycin only has modest antitubercular activity. Using structure-guided design, workers in the Lee laboratory generated a novel series of spectinamide analogs, including lead 1599 (Fig. 16), that have greatly increased antitubercular activity (spectinomycin Mtb MIC = 50 μg/mL → 1599, 0.8 μg/mL). Structural modifications in the amide side chain enable the spectinamides to escape an intrinsic efflux mechanism through the Rv1258c pump that limits the activity of spectinomycin against Mtb [93]. Spectinamides are highly selective for Mtb, display a marked postantibiotic effect, and lack cross-resistance with any other protein synthesis inhibitors, and retained activity against MDR-TB and XDR-TB. 1599 is active in mouse models of acute and chronic TB infection and demonstrates synergy in vivo when combined with RIF and PZA and additivity when combined with secondline agents used for treating MDR-TB suggesting they are good potential partners agents for novel treatment regimens. The hydrophilic nature, low protein binding, lack of hepatic metabolism and ability to avoid efflux also makes them favorable combination partners, though this series is limited by their lack of oral bioavailability.
Fig. 16.

Lee 1599, an optimized spectinomycin analog that possesses high selectivity for bacterial RNA and escapes efflux pump Rv1258c.
Griselmycins are Streptomyces-derived antibiotic class that have been recently re-explored to generate new agents to treat MDR-TB. Griselimycin (Fig. 17) was discovered in the 1960s at which time its anti-tubercular potency was noted by Sanofi but as it possessed poor PK properties it was eventually dropped in favor of other programs due to their rapid elimination following oral administration [94, 95]. Sanofi recently reinvestigated the griselmycin scaffold, generating several analogs of the cyclic peptide by total synthesis, which appears to have improved therapeutic potential. Most interestingly, the investigators identified the molecular target as the DNA-polymerase sliding clamp DnaN, validating it as a novel target for future drug discovery [96]. In this study, analogs of griselimycin were synthesized by structure-guided design, with the hopes of increasing metabolic stability and maintaining in vitro and in vivo efficacy. A cyclohexyl analog proved to be efficacious and blocked the primary point of metabolism by sterically limiting access to cleavage of the Proline-8 amide bond. Of note, this preclinical class showed a low emergence of spontaneous resistance mutations, even those that arise at a drastic fitness cost to the mycobacteria. The primary resistance mechanism identified is the amplification of the dnaN gene, but any mutants with such a mutation grew markedly slower [96]. This highly disfavored, rarely occurring resistance risk, coupled with a novel mechanism of action and promising oral PK properties, have propelled this class of naturally derived antitubercular compounds into the upper echelon of preclinical leads for MDR-TB.
Fig. 17.

Griselimycin scaffold. R position is hydrogen on the natural product and cyclohexyl on the orally available analog.
Capuramycin is naturally occurring antibiotic secreted by S. griseus around which much antibiotic chemistry has been performed. Capuramycin inhibits peptidoglycan biosynthesis by targeting translocase I, an essential enzyme in this pathway and a distinct target from others discussed in this review [97]. Many capuramycin derivatives have been synthetically generated and subclasses identified that show impressive anti-mycobacterial potency and selectivity, but also commonly suffer from poor aqueous solubility. However, they exhibit rapid bactericidal activity and a lengthy post-antibiotic effect, making promising potential additions to Mtb therapy. Daiichi-Sankyo (Parsippany, NJ) created a library of more than 7,000 capuramycin analogs, including many that have potent antimycobacterial activity; the lead from this series is SQ641 (Fig. 18) [98, 99]. Sequella (Rockville, MD) has licensed the right to develop this compound though little progress has recently been reported. As nucleoside-peptide antibiotics, capuramycins have limited oral bioavailability and pharmaceutical properties, which limit their development. To get around these limitations Lilly (Indianapolis, IN) has explored aerosol delivery of their capuramycin lead (CPZEN-45; Fig. 18) with positive results [100]. This study shows that CPZEN-45 is efficiently absorbed by lung tissue through inhalation, and can reach therapeutically relevant concentrations at the primary site of Mtb infection opening the door to clinical trials.
Fig. 18.

Two lead capuramycin analogs, SQ641 and CPZEN-45, that are currently undergoing preclinical testing and optimizations.
Pyridomycin was first isolated in 1953 from Streptomyces pyridomyceticus (Fig 19). Recent studies by the Cole group demonstrated that Pyridomycin is a direct inhibitor of InhA [101]. However, in contrast to INH and ETH that also target InhA, pyridomycins do not require bioactivation by KatG or EthA mitigating the primary potential source of cross-resistance to these existing drugs. Structural studies demonstrated that pyridomycin spans the active site of InhA, making key hydrogen bonds at both the substrate and NADH binding sites [102]. Another interesting aspect of pyridomycin is its ability for its aromatic rings to intramolecularly π-stack, a feature that might aid in cellular entry across the Mtb cell envelope. The drug is able to penetrate host macrophages, displaying bacteriocidal activity at 10 μg/mL but is inactive against slowly-replicating bacteria consistent with its mode of action against cell wall biosynthesis. The structural and the SAR knowledge from other inhibitor programs available on InhA combined with pyridomycins chemically tractable scaffold leave this natural product ripe for synthetic derivatization. However, much work remains to advance this molecule class into preclinical development.
Fig. 19. Pyridomycin is a naturally occurring InhA inhibitor. Possessing the same mechanism as known drugs lends credence to the importance of InhA as a drug target.

3. Notable recent advances in drug discovery technologies for Mtb
As discovery and development of novel chemical entities to treat Mtb has increased significantly in recent years [50, 103-105] so have the technologies used to discover new agents. New technologies have been developed to complement our increased understanding in the biology of the Mtb life cycle and the success and failures of prior drug development strategies. While there is not space to adequately discuss all these advances in this review we do briefly want to discuss the impact of three key technologies – genomics, screening under defined growth conditions that recapitulate various microenviroments of the Mtb life cycle, and MALDI-MS imaging of drug distribution within the infected lung.
As discussed previously, the accessibility of whole genome sequencing and genetic technologies that can be used to modulate gene expression levels has revolutionized our understanding of Mtb. The ability to search the genome for suitable therapeutic targets, to look for target conservation across multiple strains and lineages of Mtb and the ability to compare tractability with similar targets in other bacteria now provides a rich pool of information at the start of any discovery program [21, 106-109]. It is now typically required that any new target based drug discovery programs are first validated by gene knockout and/or gene knockdown experiments [110, 111] to help ensure the druggability of the target. These experiments give key insight into the sensitivity of the Mtb cell to inhibition of a biochemical target, as it is clear that not all therapeutic targets are created equal and inhibition of some are more lethal than others.
The ability to screen Mtb under defined growth conditions that better represent the microenviroments of the Mtb life cycle is also an advancing technology in this field displaying great promise [112]. A good example is the high throughput screening of macrophage-resident Mtb that identified inhibitors active against cholesterol catabolism in Mtb [113]. Mtb is an intracellular pathogen that infects and multiplies in host macrophages, which is typically a very resource-deprived environment. In this case, the pathogen must scavenge carbon sources from the host, so Mtb has evolved specific catabolic enzymes to break down substrates like cholesterol and particle remnants of low-density lipids that are also scavenged by macrophages. Once inhibitors were identified in the macrophage based screen, the researchers tested for inhibition of cholesterol catabolic pathway (or methyl citrate cycle) inhibition by limiting the carbon source of in vitro grown cells in the presence of the inhibitor. These studies led to novel inhibitors of HsaAB, a two-component, flavin-dependent hydroxylase that is crucial to the degradation of the “A” ring of cholesterol [113]. While these inhibitors are still under development, they are best considered as chemical probes with which to study the role of cholesterol catabolism in Mtb infection. The Russell group's work is a landmark study because it has identified and validated a new area of Mtb metabolism for chemotherapeutic intervention and drug design.
Technology that enables us to detect and image drug distribution allows researchers to gain a better understanding of how TB drugs distribute within the lungs of patients, and how this exposure compares to rodent models of Mtb infection, is another significant recent advance. It is quite easy to look at a single parameter (e.g., plasma concentration) and be deceived about how a drug will act in a patient. However, plasma concentration may be a poor indicator of tissue and granuloma perfusion, especially perfusion into necrotic granulomas' caseum-filled cores [114]. This phenomenon could help explain the failure of the Rapid Evaluation of Moxifloxacin in Tuberculosis (REMoxTB) clinical trial, one of the largest Phase III TB trials ever conducted. The trial, which included more than 1,900 patients, was proposed to shorten the duration of Mtb therapy from 6 to 4 months by substituting moxifloxacin for either INH or EMB. The trial was supported by results from extensive combination testing in mice [115], however it was unable to confirm the treatment shortening potential of moxifloxacin seen in mice. Subsequent data has shown that moxifloxacin is not able to fully penetrate human caseous lesions that have a low degree of celluarity [114], despite showing high concentrations inside activated macrophages [116, 117].
These results have further spurred on efforts to study location-dependent drug concentrations in the Mtb infected lung, taking advantage of new MALDI-MS imaging technologies [116, 118-121]. In a small study of drug distribution in granulomas taken directly from the lungs of MDR-TB patients who were treated with drug combinations at varying time points prior to lung resection, moxifloxacin exhibited several-fold lower concentrations in caseous regions of pulmonary lesions compared to RIF, concentrating around the periphery of the lesion. RIF exhibited a slow accumulation until high steady-state concentrations were achieved deep within lesions, and PZA was seen to rapidly perfuse throughout the lesion. This inability to efficiently drug the entire lesion probably explains why shortening the course of REMoxTB trail was not successful. These findings support a previous in vivo study in rabbits that used similar matrix-assisted laser-desorption/ionization mass spectroscopy imaging [122, 123], effect. These results suggest that penetration into caseous lesions is critical for future efforts towards treatment shortening regimens (Fig. 20).
Fig. 20.
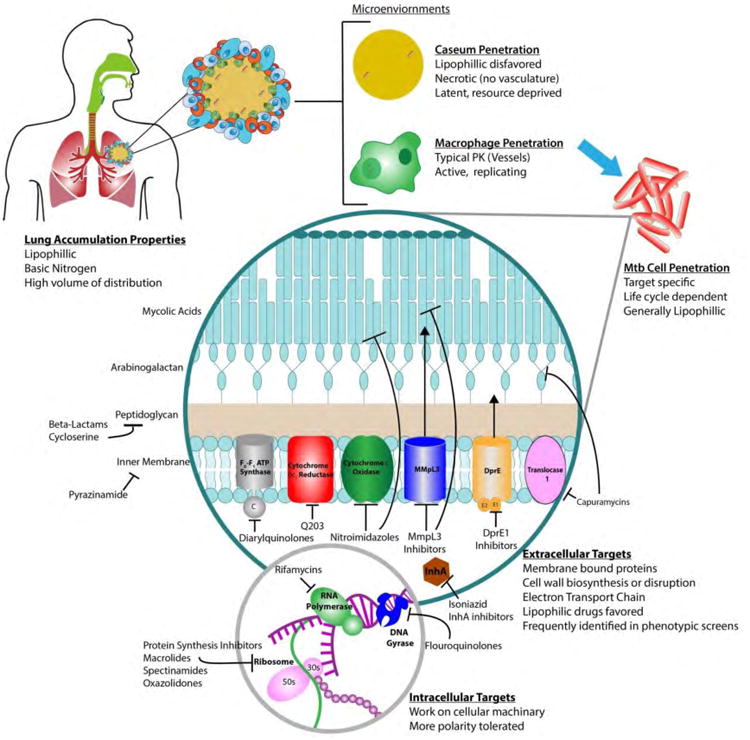
The various environments a drug must thrive in to be an effective antitubercular agent.
4. Discussion
There is an obvious need to continue the push to discover novel chemotherapeutic options to treat MDR-TB infections. Resistance continues to spread across the world, and an ever-expanding global population and global strife will only serve to aid in this expansion. This review has highlighted some of the more promising classes of agents developed by various research groups, but this global effort must be maintained to ensure adequate weapons are available in this ever-evolving fight. Advances in drug discovery approaches, technological tools, a better understanding of the mycobacterium biology and disease states, and some hard learned lessons from failures in clinical trials have made this paradigm appear more promising as of late. Thus, there is reason to hope for improved outcomes for this serious disease are in the pipeline. In the past decade four primary strategies have been applied to discover the new agents discussed previously in this review: target based screening; phenotypic based screening; semisynthetic natural product chemistry driven approach; and repurposing of antibacterial agents. Each of these approaches has its own advantages and disadvantages.
When the whole Mtb genome sequence became available in 1998 [124], it greatly aided the identification of essential genes and facilitated the recognition of many novel Mtb drug targets. This increased target knowledge was thought to hold great potential and ushered in an era of target-based drug discovery primarily through the use of high throughput screening (HTS). However, the HTS target-based approach has to date only produced a few advanced leads with the current most advanced compounds being the InhA inhibitor class discussed earlier [125]. This mirrors the experience of the broader antibacterial drug discovery community, though lessons in this regard were slower to be accepted in the Mtb field. The primary limitations of this approach relate to target selection, screening library composition, and the concomitant hurdle of generating compounds that can penetrate and accumulate in the Mtb bacilli and remain stable to human metabolism. To address these limitations there needs to be a better understanding of which targets are suitable for this approach, chemical libraries need to be improved upon and new approaches like fragment based drug design should be implemented, enabling the building out of high affinity inhibitors with concomitant drug like and Mtb penetrant properties [126, 127].
Once the difficulties of target based approaches to TB drug discovery became apparent, whole-cell MIC driven HTS approaches dominated this discovery space, efforts that were fueled by the discovery of bedaquiline. This is in essence a reversion to an old approach that identified INH and EMB, but on a much larger screening scale, taking advantage of vast pharmaceutical screening libraries that have been put together in the intervening decades. The primary advantages of this approach are: is not limited to inherent target bias, thus allowing all potential chemically tractable targets to be explored simultaneously; common efflux and penetration issues are also addressed along with target-sensitivity validation. After an active compound is identified, resistant mutants can be generated and sequenced to identify genetic variations and thus assist in defining the mechanism of action. The most common targets identified in Mtb via typical phenotypic whole-cell screening were MmpL3, DprE1, and QcrB [30, 105, 128] all of which have produced preclinical leads discussed earlier. Another advantage of this method includes the relatively low costs associated with this approach. Many companies have legacy compound libraries can be quickly repurposed for TB drug discovery, which can be compared to published hit data sets for comparative purposes to identify novel leads. Disadvantages include: an often difficult in vitro to in vivo active transition; mitigating off target pharmacology as the hits are often derived from compound classes generated for other human disease indications; repeated discovery of compounds targeting the same enzymes (MmpL3, DprE1, and QcrB); frequent discovery of prodrug activated electrophiles, for which care must be taken to ensure these agents are not activated by central human metabolism, though it must be noted as evidenced by the nitroimidazoles, delaminid and pretomanid this is achievable. Finally, despite genomic evaluation of resistant mutants, elucidating the molecular targets of drugs identified phenotypically can be more complex, time consuming, expensive, and is generally considered a bottleneck in this process.
These challenges are often compounded in lead optimization process, as it is common to increase lipophilicity from the initial HTS hit [129], and drug discovery in TB is no different. The NITD recommends monitoring lipophilic efficiency to guide phenotypic-hit selection and development, an opinion we share. Development of such compounds will be a delicate balance of maintaining potency and optimizing exposure via ADME (absorption, distribution, metabolism, and excretion) properties. As shown in Fig. 21, there has been a trend in developing new oral agents from phenotypic screening to better adhere to Lipinski's rule of five, such as BTZ043, Azaindoles and TCA1 [130, 131]. However it should be noted that most of the newer agents are still on the high end of lipophilicity range (cLogP > 3). This not only makes formulation difficult and increases the risk of drug-drug interactions, but may also play a role in limiting drug distribution into the caesum microenviornment. The success of bedaquiline and delaminid are examples that high lipophillicity (cLogP values of 7.3 and 5.6, respectively) can be overcome with advances in formulation to achieve proper oral-exposure levels and advanced medicinal chemistry to block sites of oxidative metabolism. However, they have had limited success in reducing treatment duration, as in vitro and in vivo mouse efficacy testing may have suggested. The results of Q203 in clinical trials could add more evidence to the theory that these large lipophillic molecules have trouble clearing mycobacterium from necrotic granulomas. The feasibility of combining multiple high-liphophilic drugs in MDR-TB combination therapy will likely need to be addressed. This is due to additive liabilities and consequent drug-drug interactions causing decreased bioavailability and additive toxicities. These complications, combined with the more favorable distribution across multiple microenviornments, favors the identification and prioritization of phenotypic MIC screening hits with more hydrophilic properties.
Fig. 21.
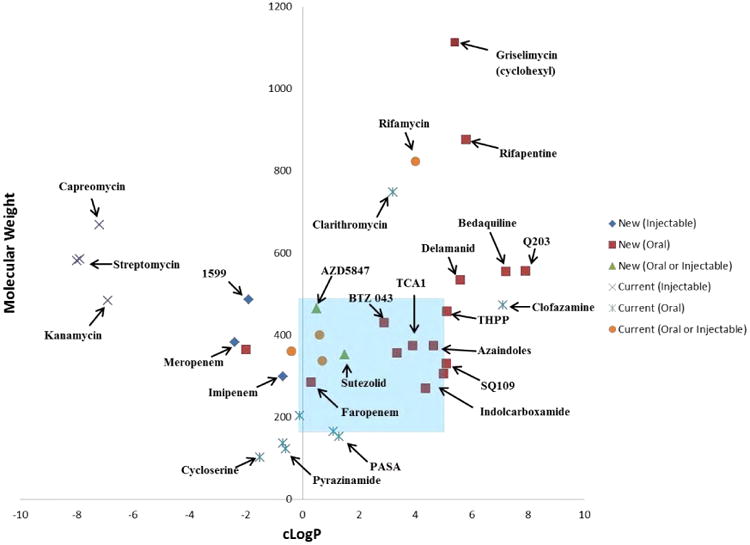
Physical chemical properties of currently used therapeutics vs newer agents in the discovery pipeline with their route of administration. The blue-shaded areas indicate the stereotypical Lipinski's rules for oral bioavailability. cLogP values taken from PubChem database.
During the golden age of antibiotic discovery, many unique natural product scaffolds were discarded in favor of compounds with more favorable characteristics, such as ease of production or lack of antibacterial spectrum of activity. Recent efforts to optimize some of these previously identified natural products for MDR-TB therapy, using a semisynthetic chemical modification approach has also proven highly productive, yielding the Macrolide SEQ-9, Spectinamides, Griselimycins, and Capuramycins, all of which are suitable to treat MDR-TB. The advantages of these molecules include: in vitro to in vivo efficacy conversion is higher than with purely synthetic compounds; they hit targets complementary to those identified in phenotypic screens; they represent novel chemical classes with different physicochemical properties from those found in synthetic screening libraries. The primary disadvantages include: these are often resource intensive and difficult programs to initiate; the natural product scaffolds are often of high molecular weight, contain metabolic hotspots and have poor consequent oral bioavailability requiring parenteral administration; this approach often employs difficult and sometimes costly chemistry to generate these analogs.
Repurposing existing antibiotics such as β-lactams (faropenem, meropenem), oxazolidiones (linezolid), and fluoroquinolones (moxifloxacin) is another productive strategy to identify new MDR-TB treatment options [5, 132]. Linezolid is the prototypical example in terms of MDR-TB treatment [80]. Its proven efficacy in the treatment of MDR-TB has generated further studies on sutezolid and AZD5847, second generation oxazolidiones with greater mycobacterium selectivity and lower side effect potential. This strategy has the advantage of very rapid advancement into clinical trials, as ample human safety and PK data often exists. One factor that limits this approach is the number of new antibacterial agents that are available for repurposing, as the number of new antibacterial agents approved has dropped significantly in recent years. Currently the best untapped opportunity for this approach probably lies in the new wave of β-lactamase inhibitors being developed to tackle multidrug resistant Gram-negative bacterial infections. It remains to be seen if they have sufficient in vivo potency against Mtb BlaC enzyme [133,134] to enable future β-lactam β-lactamase inhibitor combinations to move forward for MDR-TB. However, the failure of the recent REMoxTB Phase III clinical trial designed to reduce treatment time is a cautionary note for this approach [115].
At the time of this writing, 10 Mtb drug clinical trials are registered in the United States (Table 2), including six compounds classified as “novel chemical entities” by the Stop TB Partnership. There are also 3 Phase III trials for optimized combination studies ongoing with bedaquiline and delamindib and pretomanid (formerly PA-824, another nitroimidazole) being added to optimized background regimens for patients with MDR-TB infections. These studies are seeking to enroll more than 1,000 patients suffering from MDR-TB, including some who are co-infected with HIV (NCT00910871, NCT01600963, and NCT01424670). This progress is much improved over past decades, but the need for new drugs that are safer and more efficacious with the ability for treatment shortening is still apparent. A strong discovery pipeline requires many more agents in early stage trials, as attrition rates increase the further drugs progress down the pipeline. Thus we believe more new entities are urgently required, as some of these agents will inevitably fail. It is our opinion that agents that have good pharmacokinetic properties including caseum penetration and anti-tubercular activity in this environment are most needed.
Table 2. List of on-going and completed clinical trials for new drugs to treat TB and MDR-TB. Information compiled from StopTB Partnership (www.newtbdrugs.org) and ClinicalTrials.gov as of April 2016.
| Drugs | Trial # | Phase | Sponsor | Start Date | Target Patient Enrollment | Status |
|---|---|---|---|---|---|---|
| TBA-354 | NCT02288481 | 1 | TB Alliance | Jan-15 | 48 | Closed |
| Q203 | NCT02530710 | 1 | Qurient Co., Ltd. | Aug-15 | 56 | Open (Recruiting) |
| AZD5847 | NCT01516203 | 2 | NIAID | Dec-12 | 75 | Closed |
| Sutezolid | NCT00871949 | 1 | Pfizer | May-09 | 19 | Closed |
| NCT00990990 | 1 | Pfizer | Nov-09 | 59 | Closed | |
| NCT01225640 | 2 | Pfizer | Aug-11 | 59 | Closed | |
| SQ109 | NCT01218217 | 2 | University of Munich/Sequella | Dec-10 | 90 | Closed |
| NCT01585636 | 1 | Sequella, Inc. | Nov-10 | 62 | Closed | |
| NCT01358162 | 1 | NIH | Nov-10 | 10 | Closed | |
| NCT01785186 | 2 | Sequella, Inc., PanACEA, EDCTP | April-13 | 365 | Closed | |
| Clofazimine/Moxifloxacin, | NCT01691534 | 2 | TB Alliance | Oct-12 | 105 | Unknown |
| Bedaquiline, Pretomanid, Pyrazinamide | NCT02193776 | 2 | TB Alliance | Nov-15 | 240 | Open (Not recruiting) |
| Levofloxacin | NCT01918397 | 2 | Boston University | Jan-15 | 100 | Open (Recruiting) |
| Bedaquiline with OBR | NCT00449644 | 2 | Tibotec BVBA | May-07 | 208 | Closed |
| NCT00910871 | 2 | Tibotec BVBA | Jul-09 | 225 | Closed | |
| NCT01803373 | 1 | Janssen | Mar-13 | 36 | Closed | |
| NCT01464762 | 3 | Janssen | Oct-11 | N/A | Open | |
| Delaminid with OBR | NCT00685360 | 2 | Otsuka Pharmaceutical | Apr-08 | 481 | Closed |
| NCT01424670 | 3 | Otsuka Pharmaceutical | Sep-11 | 390 | Closed | |
| NCT01856634 | 1 | Otsuka Pharmaceutical | May-13 | 12 | Open (Recruiting Pediatrics) | |
| Pretomanid, Moxifloxacin, Pyrazinamide | NCT01498419 | 2 | TB Alliance | Mar-12 | 230 | Closed |
| NCT01215851 | 2 | TB Alliance | Oct-10 | 85 | Closed | |
| NCT02342886 | 3 | TB Alliance | Jan-15 | 1500 | On hold |
In conclusion, as outlined in this review, it is evident that the discovery and development of new agents to treat Mtb is a very active drug discovery field with many agents in preclinical development. This area is however not without major challenges. Today we are nowhere close to achieving the often-stated primary goal of developing a shorter, more effective therapeutic regimen for drug susceptible TB. There are worryingly few drugs currently in early stage clinical trials with only 2 compounds in phase I. There are also major pharmacological redundancies with many of the new agents in the pipeline that share a similar mechanism of action, cross-resistance and / or side effect profiles and thus cannot be used in combination together. The discovery and development of new antibiotics in general is difficult due to inherent safety concerns relating to high and frequent compound dosing of agents required in comparison to other classes of human medicines. These problems are further compounded in the therapy of tuberculosis by the long treatment times and mandatory combination therapy such that toxicities may not become apparent until late stage clinical trials with large patient enrollments. The field also faces financial and logistical challenges compounded by the long development times for compounds and slow rate of discovery requiring a very long term commitment, which many sponsoring agencies and pharmaceutical companies struggle to maintain, as these timelines are much slower than their usual business cycles. Examples of this problem include the entrance and exit of pharmaceutical companies such as Novartis, Vertex and AstraZeneca, and the recent reduction in support for TB drug discovery efforts in the European Union. Fortunately some agencies such as the NIAID, WHO and Bill and Melinda Gates Foundation are committed to the long term, playing critical roles in both raising awareness and securing desperately needed funding to continue fueling growth and development in this field. To tackle some of the drug discovery issues large consortia of investigators comprising of academics and industrial members have been formed, such as the TB drug accelerator, New Medicines for Tuberculosis (NM4TB) in addition to the work of the TB alliance and the Critical Path Institute. These consortia offer venues to share complementary expertise and tackle complex problems. These units have made notable progress as evidenced by discovery and development of the benzothiazone class developed by NM4TB. However there is a propensity for large conglomerate organizations to become slowed by the competing priorities within the groupings, such that the overall return on the sum of investment in terms of delivery of new preclinical and clinical candidates has been less than had been hoped for at the time of their formation.
In conclusion, despite the challenges outlined we firmly believe the field of tuberculosis drug discovery is much better placed to achieve significant therapeutic advances in the coming decade. This results from a much-improved understanding of Mtb lifecycle, the pharmacological requirements for successful Mtb drugs, and the new anti-tubercular chemical matter derived in recent years. The importance of recent improvements in animal infection models that better mimic the pathology of the human tuberculosis lung, coupled with new imaging technologies provide us with much better predictive preclinical models to produce drug combinations efficacious against hard to treat slow growing sub-populations, should not be understated. Finally, we believe there is much strength in the expertise, experience and diversity of investigators currently working in this area, which needs to be nurtured to continue the important fight against MDR-TB.
Acknowledgments
This study was supported by U.S. National Institutes of Health (NIH) grants AI090810 and the American Lebanese Syrian Associated Charities, St. Jude Children's Research Hospital. We thank Dr. Angela McArthur for assistance in editing this article and Timothy Hammond for assistance in preparation of the color figures.
Abbreviations
- Mtb
Mycobacterium tuberculosis
- TB
Tuberculosis
- MDR-TB
Multidrug-resistant tuberculosis
- RIF
Rifampicin
- INH
Isoniazid
- PZA
Pyrazinamide
- ETH
Ethambutol
- WHO
World Health Organization
- CYP
Cytochrome P450
- ADME
Absorption, Distribution, Metabolism, and Excretion
- HIV
Human Immunodeficiency Virus
- XDR-TB
Extensively drug resistant tuberculosis
- TDR-TB
Totally drug resistant tuberculosis
- PK
Pharmacokinetic
- PD
Pharmacodynamic
- EMA
European Medicines Agency
- FDA
Food and Drug Administration
- MIC
Minimum inhibitory concentration
- OBR
Optimized Background Regimen
Footnotes
Chemical compounds studied in this article: Bedaquiline, CID: 5388906
Delamanid, CID: 6480466
Q203, CID: 68234908
PBTZ 169, CID: 57331386
TCA 1, CID: 4557640
SQ109, CID: 5274428
Faropenem, CID: 65894
Sutezolid, CID: 465951
Lee1599, CID: 60173108
SQ641, CID: 3013049
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Cited literature
- 1.Comas I, Coscolla M, Luo T, Borrell S, Holt KE, Kato-Maeda M, Parkhill J, Malla B, Berg S, Thwaites G, Yeboah-Manu D, Bothamley G, Mei J, Wei L, Bentley S, Harris SR, Niemann S, Diel R, Aseffa A, Gao Q, Young D, Gagneux S. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat Genet. 2013;45:1176–1182. doi: 10.1038/ng.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.WHO. WHO Global Tuberculosis Report 2014. 2014 [Google Scholar]
- 3.Luciani F, Sisson SA, Jiang H, Francis AR, Tanaka MM. The epidemiological fitness cost of drug resistance in Mycobacterium tuberculosis. Proceedings of the National Academy of Sciences. 2009;106:14711–14715. doi: 10.1073/pnas.0902437106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Udwadia ZF, Amale RA, Ajbani KK, Rodrigues C. Totally Drug-Resistant Tuberculosis in India. Clinical Infectious Diseases. 2012;54:579–581. doi: 10.1093/cid/cir889. [DOI] [PubMed] [Google Scholar]
- 5.Zumla A, Nahid P, Cole ST. Advances in the development of new tuberculosis drugs and treatment regimens. Nature reviews Drug discovery. 2013;12:388–404. doi: 10.1038/nrd4001. [DOI] [PubMed] [Google Scholar]
- 6.Drobniewski F. Is Death Inevitable with Multiresistant TB plus HIV Infection. Lancet. 1997;349:71–73. doi: 10.1016/S0140-6736(05)60878-1. [DOI] [PubMed] [Google Scholar]
- 7.Breen RAM. Tuberculosis and HIV Co-Infection: A Practical Therapeutic Approach. Drugs. 2006;66:2299–2308. doi: 10.2165/00003495-200666180-00003. [DOI] [PubMed] [Google Scholar]
- 8.Burger DM. Pharmacokinetic interaction between rifampin and zidovudine. Antimicrobial Agents and Chemotherapy. 1993;37:1426–1432. doi: 10.1128/aac.37.7.1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niemi M. Pharmacokinetic Interactions with Rifampicin: Clinical Relevance. Clinical Pharmacokinetics. 2003;42:819–851. doi: 10.2165/00003088-200342090-00003. [DOI] [PubMed] [Google Scholar]
- 10.Esposito S, D'Ambrosio L, Tadolini M, Schaaf HS, Caminero Luna J, Marais B, Centis R, Dara M, Matteelli A, Blasi F, Migliori GB. ERS/WHO Tuberculosis Consilium assistance with extensively drug-resistant tuberculosis management in a child: case study of compassionate delamanid use. European Respiratory Journal. 2014;44:811–815. doi: 10.1183/09031936.00060414. [DOI] [PubMed] [Google Scholar]
- 11.Marisa K, Robin Mark W, Cindy H, Nicolaas Claudius Gey van P, Elizabeth Maria S, Borna M, Frederick Adriaan S, Mamisa CN, Ebrahim H, Gerrit C, Paul David van H, Thomas Calldo V, André Phillip T. Emergence and Spread of Extensively and Totally Drug-Resistant Tuberculosis, South Africa. Emerging Infectious Disease journal. 2013;19:449. doi: 10.3201//EID1903.120246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raviglione M. XDR-TB: entering the post-antibiotic era? The international journal of tuberculosis and lung disease : the official journal of the International Union against Tuberculosis and Lung Disease. 2006;10:1185–1187. [PubMed] [Google Scholar]
- 13.Kumar K, Abubakar I. Clinical implications of the global multidrug-resistant tuberculosis epidemic. Clinical Medicine. 2015;15:s37–s42. doi: 10.7861/clinmedicine.15-6-s37. [DOI] [PubMed] [Google Scholar]
- 14.Gler MT, Skripconoka V, Sanchez-Garavito E, Xiao H, Cabrera-Rivero JL, Vargas-Vasquez DE, Gao M, Awad M, Park SK, Shim TS, Suh GY, Danilovits M, Ogata H, Kurve A, Chang J, Suzuki K, Tupasi T, Koh WJ, Seaworth B, Geiter LJ, Wells CD. Delamanid for multidrug-resistant pulmonary tuberculosis. N Engl J Med. 2012;366:2151–2160. doi: 10.1056/NEJMoa1112433. [DOI] [PubMed] [Google Scholar]
- 15.Cohen J. Approval of Novel TB Drug Celebrated—With Restraint. Science. 2013;339:130. doi: 10.1126/science.339.6116.130. [DOI] [PubMed] [Google Scholar]
- 16.Ryan N, Lo J. Delamanid: First Global Approval. Drugs. 2014;74:1041–1045. doi: 10.1007/s40265-014-0241-5. [DOI] [PubMed] [Google Scholar]
- 17.Cohen KA, Bishai WR, Pym AS. Molecular Basis of Drug Resistance in Mycobacterium tuberculosis. Microbiology Spectrum. 2014;2 doi: 10.1128/microbiolspec.MGM2-0036-2013. [DOI] [PubMed] [Google Scholar]
- 18.Palomino J, Martin A. Drug Resistance Mechanisms in Mycobacterium tuberculosis. Antibiotics. 2014;3:317. doi: 10.3390/antibiotics3030317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fonseca JD, Knight GM, McHugh TD. The complex evolution of antibiotic resistance in Mycobacterium tuberculosis. International Journal of Infectious Diseases. 2015;32:94–100. doi: 10.1016/j.ijid.2015.01.014. [DOI] [PubMed] [Google Scholar]
- 20.Gagneux S, Long CD, Small PM, Van T, Schoolnik GK, Bohannan BJ. The competitive cost of antibiotic resistance in Mycobacterium tuberculosis. Science. 2006;312:1944–1946. doi: 10.1126/science.1124410. [DOI] [PubMed] [Google Scholar]
- 21.Casali N, Nikolayevskyy V, Balabanova Y, Harris SR, Ignatyeva O, Kontsevaya I, Corander J, Bryant J, Parkhill J, Nejentsev S, Horstmann RD, Brown T, Drobniewski F. Evolution and transmission of drug-resistant tuberculosis in a Russian population. Nat Genet. 2014;46:279–286. doi: 10.1038/ng.2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dartois V. The path of anti-tuberculosis drugs: from blood to lesions to mycobacterial cells. Nat Rev Micro. 2014;12:159–167. doi: 10.1038/nrmicro3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koul A, Dendouga N, Vergauwen K, Molenberghs B, Vranckx L, Willebrords R, Ristic Z, Lill H, Dorange I, Guillemont J, Bald D, Andries K. Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat Chem Biol. 2007;3:323–324. doi: 10.1038/nchembio884. [DOI] [PubMed] [Google Scholar]
- 24.Diacon AH, Pym A, Grobusch M, Patientia R, Rustomjee R, Page-Shipp L, Pistorius C, Krause R, Bogoshi M, Churchyard G, Venter A, Allen J, Palomino JC, De Marez T, van Heeswijk RP, Lounis N, Meyvisch P, Verbeeck J, Parys W, de Beule K, Andries K, Mc Neeley DF. The diarylquinoline TMC207 for multidrug-resistant tuberculosis. N Engl J Med. 2009;360:2397–2405. doi: 10.1056/NEJMoa0808427. [DOI] [PubMed] [Google Scholar]
- 25.Andries K, Verhasselt P, Guillemont J, Göhlmann HWH, Neefs JM, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V. A Diarylquinoline Drug Active on the ATP Synthase of Mycobacterium tuberculosis. Science. 2005;307:223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 26.Watanabe S, Zimmermann M, Goodwin MB, Sauer U, Barry CE, 3rd, Boshoff HI. Fumarate Reductase Activity Maintains an Energized Membrane in Anaerobic Mycobacterium tuberculosis. PLoS pathogens. 2011;7:e1002287. doi: 10.1371/journal.ppat.1002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lounis N, Gevers T, Van Den Berg J, Andries K. Impact of the Interaction of R207910 with Rifampin on the Treatment of Tuberculosis Studied in the Mouse Model. Antimicrobial Agents and Chemotherapy. 2008;52:3568–3572. doi: 10.1128/AAC.00566-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Preiss L, Langer JD, Yildiz Ö, Eckhardt-Strelau L, Guillemont JEG, Koul A, Meier T. Structure of the mycobacterial ATP synthase Fo rotor ring in complex with the anti-TB drug bedaquiline. Science Advances. 2015;1 doi: 10.1126/sciadv.1500106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pethe K, Bifani P, Jang J, Kang S, Park S, Ahn S, Jiricek J, Jung J, Jeon HK, Cechetto J, Christophe T, Lee H, Kempf M, Jackson M, Lenaerts AJ, Pham H, Jones V, Seo MJ, Kim YM, Seo M, Seo JJ, Park D, Ko Y, Choi I, Kim R, Kim SY, Lim S, Yim SA, Nam J, Kang H, Kwon H, Oh CT, Cho Y, Jang Y, Kim J, Chua A, Tan BH, Nanjundappa MB, Rao SP, Barnes WS, Wintjens R, Walker JR, Alonso S, Lee S, Kim J, Oh S, Oh T, Nehrbass U, Han SJ, No Z, Lee J, Brodin P, Cho SN, Nam K, Kim J. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat Med. 2013;19:1157–1160. doi: 10.1038/nm.3262. [DOI] [PubMed] [Google Scholar]
- 30.Abrahams KA, Cox JAG, Spivey VL, Loman NJ, Pallen MJ, Constantinidou C, Fernández R, Alemparte C, Remuiñán MJ, Barros D, Ballell L, Besra GS. Identification of Novel Imidazo[1,2-a]pyridine Inhibitors Targeting M. tuberculosis QcrB. PLoS ONE. 2012;7:e52951. doi: 10.1371/journal.pone.0052951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moraski GC, Miller PA, Bailey MA, Ollinger J, Parish T, Boshoff HI, Cho S, Anderson JR, Mulugeta S, Franzblau SG, Miller MJ. Putting Tuberculosis (TB) To Rest: Transformation of the Sleep Aid, Ambien, and “Anagrams” Generated Potent Antituberculosis Agents. ACS Infectious Diseases. 2015;1:85–90. doi: 10.1021/id500008t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee RE, Mikusova K, Brennan PJ, Besra GS. Synthesis of the Arabinose Donor .beta.-D-Arabinofuranosyl-1-monophosphoryldecaprenol, Development of a Basic Arabinosyl-Transferase Assay, and Identification of Ethambutol as an Arabinosyl Transferase Inhibitor. Journal of the American Chemical Society. 1995;117:11829–11832. [Google Scholar]
- 33.Wolucka BA. Biosynthesis of D-arabinose in mycobacteria – a novel bacterial pathway with implications for antimycobacterial therapy. FEBS Journal. 2008;275:2691–2711. doi: 10.1111/j.1742-4658.2008.06395.x. [DOI] [PubMed] [Google Scholar]
- 34.Goldman RC. Why are membrane targets discovered by phenotypic screens and genome sequencing in Mycobacterium tuberculosis? Tuberculosis (Edinburgh, Scotland) 2013;93:569–588. doi: 10.1016/j.tube.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 35.Riccardi G, Pasca M, Chiarelli L, Manina G, Mattevi A, Binda C. The DprE1 enzyme, one of the most vulnerable targets of Mycobacterium tuberculosis. Appl Microbiol Biotechnol. 2013;97:8841–8848. doi: 10.1007/s00253-013-5218-x. [DOI] [PubMed] [Google Scholar]
- 36.Makarov V, Manina G, Mikusova K, Möllmann U, Ryabova O, Saint-Joanis B, Dhar N, Pasca MR, Buroni S, Lucarelli AP, Milano A, De Rossi E, Belanova M, Bobovska A, Dianiskova P, Kordulakova J, Sala C, Fullam E, Schneider P, McKinney JD, Brodin P, Christophe T, Waddell S, Butcher P, Albrethsen J, Rosenkrands I, Brosch R, Nandi V, Bharath S, Gaonkar S, Shandil RK, Balasubramanian V, Balganesh T, Tyagi S, Grosset J, Riccardi G, Cole ST. Benzothiazinones Kill Mycobacterium tuberculosis by Blocking Arabinan Synthesis. Science. 2009;324:801–804. doi: 10.1126/science.1171583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Makarov V, Lechartier B, Zhang M, Neres J, van der Sar AM, Raadsen SA, Hartkoorn RC, Ryabova OB, Vocat A, Decosterd LA, Widmer N, Buclin T, Bitter W, Andries K, Pojer F, Dyson PJ, Cole ST. Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO molecular medicine. 2014;6:372–383. doi: 10.1002/emmm.201303575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trefzer C, Rengifo-Gonzalez M, Hinner MJ, Schneider P, Makarov V, Cole ST, Johnsson K. Benzothiazinones: Prodrugs That Covalently Modify the Decaprenylphosphoryl-β-d-ribose 2′ -epimerase DprE1 of Mycobacterium tuberculosis. Journal of the American Chemical Society. 2010;132:13663–13665. doi: 10.1021/ja106357w. [DOI] [PubMed] [Google Scholar]
- 39.Matsumoto M, Hashizume H, Tomishige T, Kawasaki M, Tsubouchi H, Sasaki H, Shimokawa Y, Komatsu M. OPC-67683, a Nitro-Dihydro-Imidazooxazole Derivative with Promising Action against Tuberculosis In Vitro and In Mice. PLoS Med. 2006;3:e466. doi: 10.1371/journal.pmed.0030466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shirude PS, Shandil R, Sadler C, Naik M, Hosagrahara V, Hameed S, Shinde V, Bathula C, Humnabadkar V, Kumar N, Reddy J, Panduga V, Sharma S, Ambady A, Hegde N, Whiteaker J, McLaughlin RE, Gardner H, Madhavapeddi P, Ramachandran V, Kaur P, Narayan A, Guptha S, Awasthy D, Narayan C, Mahadevaswamy J, Vishwas KG, Ahuja V, Srivastava A, Prabhakar KR, Bharath S, Kale R, Ramaiah M, Choudhury NR, Sambandamurthy VK, Solapure S, Iyer PS, Narayanan S, Chatterji M. Azaindoles: Noncovalent DprE1 Inhibitors from Scaffold Morphing Efforts, Kill Mycobacterium tuberculosis and Are Efficacious in Vivo. Journal of Medicinal Chemistry. 2013;56:9701–9708. doi: 10.1021/jm401382v. [DOI] [PubMed] [Google Scholar]
- 41.Shirude PS, Shandil RK, Manjunatha MR, Sadler C, Panda M, Panduga V, Reddy J, Saralaya R, Nanduri R, Ambady A, Ravishankar S, Sambandamurthy VK, Humnabadkar V, Jena LK, Suresh RS, Srivastava A, Prabhakar KR, Whiteaker J, McLaughlin RE, Sharma S, Cooper CB, Mdluli K, Butler S, Iyer PS, Narayanan S, Chatterji M. Lead Optimization of 1,4-Azaindoles as Antimycobacterial Agents. Journal of Medicinal Chemistry. 2014;57:5728–5737. doi: 10.1021/jm500571f. [DOI] [PubMed] [Google Scholar]
- 42.Donald PR, Schaaf HS. Old and new drugs for the treatment of tuberculosis in children. Paediatric respiratory reviews. 2007;8:134–141. doi: 10.1016/j.prrv.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 43.Wang F, Sambandan D, Halder R, Wang J, Batt SM, Weinrick B, Ahmad I, Yang P, Zhang Y, Kim J, Hassani M, Huszar S, Trefzer C, Ma Z, Kaneko T, Mdluli KE, Franzblau S, Chatterjee AK, Johnsson K, Mikusova K, Besra GS, Fütterer K, Robbins SH, Barnes SW, Walker JR, Jacobs WR, Schultz PG. Identification of a small molecule with activity against drug-resistant and persistent tuberculosis. Proceedings of the National Academy of Sciences. 2013;110:E2510–E2517. doi: 10.1073/pnas.1309171110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muttucumaru DGN, Roberts G, Hinds J, Stabler RA, Parish T. Gene expression profile of Mycobacterium tuberculosis in a non-replicating state. Tuberculosis. 2004;84:239–246. doi: 10.1016/j.tube.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 45.Domenech P, Reed MB, Barry CE. Contribution of the Mycobacterium tuberculosis MmpL Protein Family to Virulence and Drug Resistance. Infection and Immunity. 2005;73:3492–3501. doi: 10.1128/IAI.73.6.3492-3501.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee RE, Protopopova M, Crooks E, Slayden RA, Terrot M, Barry CE. Combinatorial Lead Optimization of [1,2]-Diamines Based on Ethambutol as Potential Antituberculosis Preclinical Candidates. Journal of Combinatorial Chemistry. 2003;5:172–187. doi: 10.1021/cc020071p. [DOI] [PubMed] [Google Scholar]
- 47.Biava M, Porretta GC, Manetti F. New derivatives of BM212: A class of antimycobacterial compounds based on the pyrrole ring as a scaffold. Mini Rev Med Chem. 2007;7:65–78. doi: 10.2174/138955707779317786. [DOI] [PubMed] [Google Scholar]
- 48.Li W, Upadhyay A, Fontes FL, North EJ, Wang Y, Crans DC, Grzegorzewicz AE, Jones V, Franzblau SG, Lee RE, Crick DC, Jackson M. Novel Insights into the Mechanism of Inhibition of MmpL3, a Target of Multiple Pharmacophores in Mycobacterium tuberculosis. Antimicrobial Agents and Chemotherapy. 2014;58:6413–6423. doi: 10.1128/AAC.03229-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Poce G, Bates RH, Alfonso S, Cocozza M, Porretta GC, Ballell L, Rullas J, Ortega F, De Logu A, Agus E, La Rosa V, Pasca MR, De Rossi E, Wae B, Franzblau SG, Manetti F, Botta M, Biava M. Improved BM212 MmpL3 inhibitor analogue shows efficacy in acute murine model of tuberculosis infection. PLoS One. 2013;8:e56980. doi: 10.1371/journal.pone.0056980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li K, Schurig-Briccio LA, Feng X, Upadhyay A, Pujari V, Lechartier B, Fontes FL, Yang H, Rao G, Zhu W, Gulati A, No JH, Cintra G, Bogue S, Liu YL, Molohon K, Orlean P, Mitchell DA, Freitas-Junior L, Ren F, Sun H, Jiang T, Li Y, Guo RT, Cole ST, Gennis RB, Crick DC, Oldfield E. Multitarget Drug Discovery for Tuberculosis and Other Infectious Diseases. Journal of Medicinal Chemistry. 2014;57:3126–3139. doi: 10.1021/jm500131s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rao SP, Lakshminarayana SB, Kondreddi RR, Herve M, Camacho LR, Bifani P, Kalapala SK, Jiricek J, Ma NL, Tan BH, Ng SH, Nanjundappa M, Ravindran S, Seah PG, Thayalan P, Lim SH, Lee BH, Goh A, Barnes WS, Chen Z, Gagaring K, Chatterjee AK, Pethe K, Kuhen K, Walker J, Feng G, Babu S, Zhang L, Blasco F, Beer D, Weaver M, Dartois V, Glynne R, Dick T, Smith PW, Diagana TT, Manjunatha UH. Indolcarboxamide is a preclinical candidate for treating multidrug-resistant tuberculosis. Science translational medicine. 2013;5:214ra168. doi: 10.1126/scitranslmed.3007355. [DOI] [PubMed] [Google Scholar]
- 52.Lun S, Guo H, Onajole OK, Pieroni M, Gunosewoyo H, Chen G, Tipparaju SK, Ammerman NC, Kozikowski AP, Bishai WR. Indoleamides are active against drug-resistant Mycobacterium tuberculosis. Nat Commun. 2013;4 doi: 10.1038/ncomms3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wright HT, Reynolds KA. Antibacterial Targets in Fatty Acid Biosynthesis. Current opinion in microbiology. 2007;10:447–453. doi: 10.1016/j.mib.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang YM, White SW, Rock CO. Inhibiting Bacterial Fatty Acid Synthesis. Journal of Biological Chemistry. 2006;281:17541–17544. doi: 10.1074/jbc.R600004200. [DOI] [PubMed] [Google Scholar]
- 55.Li HJ, Lai CT, Pan P, Yu W, Liu N, Bommineni GR, Garcia-Diaz M, Simmerling C, Tonge PJ. A Structural and Energetic Model for the Slow-Onset Inhibition of the Mycobacterium tuberculosis Enoyl-ACP Reductase InhA. ACS Chemical Biology. 2014;9:986–993. doi: 10.1021/cb400896g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rawat R, Whitty A, Tonge PJ. The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: Adduct affinity and drug resistance. Proceedings of the National Academy of Sciences. 2003;100:13881–13886. doi: 10.1073/pnas.2235848100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lai CT, Li HJ, Yu W, Shah S, Bommineni GR, Perrone V, Garcia-Diaz M, Tonge PJ, Simmerling C. Rational Modulation of the Induced-Fit Conformational Change for Slow-Onset Inhibition in Mycobacterium tuberculosis InhA. Biochemistry. 2015;54:4683–4691. doi: 10.1021/acs.biochem.5b00284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu H, Tonge PJ. Drug–target residence time: critical information for lead optimization. Current Opinion in Chemical Biology. 2010;14:467–474. doi: 10.1016/j.cbpa.2010.06.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tummino PJ, Copeland RA. Residence Time of Receptor-Ligand Complexes and Its Effect on Biological Function. Biochemistry. 2008;47:5481–5492. doi: 10.1021/bi8002023. [DOI] [PubMed] [Google Scholar]
- 60.Copeland RA, Pompliano DL, Meek TD. Drug-target residence time and its implications for lead optimization. Nature reviews Drug discovery. 2006;5:730–739. doi: 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- 61.Lu H, England K, am Ende C, Truglio JJ, Luckner S, Reddy BG, Marlenee NL, Knudson SE, Knudson DL, Bowen RA, Kisker C, Slayden RA, Tonge PJ. Slow-Onset Inhibition of the FabI Enoyl Reductase from Francisella tularensis: Residence Time and in Vivo Activity. ACS Chemical Biology. 2009;4:221–231. doi: 10.1021/cb800306y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kaplan N, Albert M, Awrey D, Bardouniotis E, Berman J, Clarke T, Dorsey M, Hafkin B, Ramnauth J, Romanov V, Schmid MB, Thalakada R, Yethon J, Pauls HW. Mode of Action, In Vitro Activity, and In Vivo Efficacy of AFN-1252, a Selective Antistaphylococcal FabI Inhibitor. Antimicrobial Agents and Chemotherapy. 2012;56:5865–5874. doi: 10.1128/AAC.01411-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Escaich S, Prouvensier L, Saccomani M, Durant L, Oxoby M, Gerusz V, Moreau F, Vongsouthi V, Maher K, Morrissey I, Soulama-Mouze C. The MUT056399 Inhibitor of FabI Is a New Antistaphylococcal Compound. Antimicrobial Agents and Chemotherapy. 2011;55:4692–4697. doi: 10.1128/AAC.01248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Šink R, Sosič I, Živec M, Fernandez-Menendez R, Turk S, Pajk S, Alvarez-Gomez D, Lopez-Roman EM, Gonzales-Cortez C, Rullas-Triconado J, Angulo-Barturen I, Barros D, Ballell-Pages L, Young RJ, Encinas L, Gobec S. Design, Synthesis, and Evaluation of New Thiadiazole-Based Direct Inhibitors of Enoyl Acyl Carrier Protein Reductase (InhA) for the Treatment of Tuberculosis. Journal of Medicinal Chemistry. 2015;58:613–624. doi: 10.1021/jm501029r. [DOI] [PubMed] [Google Scholar]
- 65.Shirude PS, Madhavapeddi P, Naik M, Murugan K, Shinde V, Nandishaiah R, Bhat J, Kumar A, Hameed S, Holdgate G, Davies G, McMiken H, Hegde N, Ambady A, Venkatraman J, Panda M, Bandodkar B, Sambandamurthy VK, Read JA. Methyl-Thiazoles: A Novel Mode of Inhibition with the Potential to Develop Novel Inhibitors Targeting InhA in Mycobacterium tuberculosis. Journal of Medicinal Chemistry. 2013;56:8533–8542. doi: 10.1021/jm4012033. [DOI] [PubMed] [Google Scholar]
- 66.Manjunatha UH, Rao SPS, Kondreddi RR, Noble CG, Camacho LR, Tan BH, Ng SH, Ng PS, Ma NL, Lakshminarayana SB, Herve M, Barnes SW, Yu W, Kuhen K, Blasco F, Beer D, Walker JR, Tonge PJ, Glynne R, Smith PW, Diagana TT. Direct inhibitors of InhA are active against Mycobacterium tuberculosis. Science translational medicine. 2015;7:269ra263. doi: 10.1126/scitranslmed.3010597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Keener AB. Oldie but goodie: Repurposing penicillin for tuberculosis. Nat Med. 2014;20:976–978. doi: 10.1038/nm0914-976. [DOI] [PubMed] [Google Scholar]
- 68.Nadler JP, Berger J, Nord JA, Cofsky R, Saxena M. AMoxicillin-clavulanic acid for treating drug-resistant mycobacterium tuberculosis. Chest. 1991;99:1025–1026. doi: 10.1378/chest.99.4.1025. [DOI] [PubMed] [Google Scholar]
- 69.Dooley KE, Obuku EA, Durakovic N, Belitsky V, Mitnick C, Nuermberger EL R.-T. on behalf of the Efficacy Subgroup. World Health Organization Group 5 Drugs for the Treatment of Drug-Resistant Tuberculosis: Unclear Efficacy or Untapped Potential? Journal of Infectious Diseases. 2013;207:1352–1358. doi: 10.1093/infdis/jis460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dubée V, Triboulet S, Mainardi JL, Ethève-Quelquejeu M, Gutmann L, Marie A, Dubost L, Hugonnet JE, Arthur M. Inactivation of Mycobacterium tuberculosis l,d-Transpeptidase LdtMt1 by Carbapenems and Cephalosporins. Antimicrobial Agents and Chemotherapy. 2012;56:4189–4195. doi: 10.1128/AAC.00665-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wivagg CN, Bhattacharyya RP, Hung DT. Mechanisms of [beta]-lactam killing and resistance in the context of Mycobacterium tuberculosis. J Antibiot. 2014;67:645–654. doi: 10.1038/ja.2014.94. [DOI] [PubMed] [Google Scholar]
- 72.De Lorenzo S, Alffenaar JW, Sotgiu G, Centis R, D'Ambrosio L, Tiberi S, Bolhuis MS, van Altena R, Viggiani P, Piana A, Spanevello A, Migliori GB. Efficacy and safety of meropenem–clavulanate added to linezolid-containing regimens in the treatment of MDR-/XDR-TB. European Respiratory Journal. 2013;41:1386–1392. doi: 10.1183/09031936.00124312. [DOI] [PubMed] [Google Scholar]
- 73.Chambers HF, Turner J, Schecter GF, Kawamura M, Hopewell PC. Imipenem for Treatment of Tuberculosis in Mice and Humans. Antimicrobial Agents and Chemotherapy. 2005;49:2816–2821. doi: 10.1128/AAC.49.7.2816-2821.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hugonnet JE, Tremblay LW, Boshoff HI, Barry CE, Blanchard JS. Meropenem-Clavulanate Is Effective Against Extensively Drug-Resistant Mycobacterium tuberculosis. Science. 2009;323:1215–1218. doi: 10.1126/science.1167498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Payen MC, De Wit S, Martin C, Sergysels R, Muylle I, Van Laethem Y, Clumeck N. Clinical use of the meropenem-clavulanate combination for extensively drug-resistant tuberculosis. The international journal of tuberculosis and lung disease : the official journal of the International Union against Tuberculosis and Lung Disease. 2012;16:558–560. doi: 10.5588/ijtld.11.0414. [DOI] [PubMed] [Google Scholar]
- 76.Hoagland D, Zhao Y, Lee R. Advances in Drug Discovery and Development for Pediatric Tuberculosis. Mini Rev Med Chem. 2015 doi: 10.2174/1389557515666150722101723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Milazzo I, Blandino G, Caccamo F, Musumeci R, Nicoletti G, Speciale A. Faropenem, a new oral penem: antibacterial activity against selected anaerobic and fastidious periodontal isolates. Journal of Antimicrobial Chemotherapy. 2003;51:721–725. doi: 10.1093/jac/dkg120. [DOI] [PubMed] [Google Scholar]
- 78.Schurek KN, Wiebe R, Karlowsky JA, Rubinstein E, Hoban DJ, Zhanel GG. Faropenem: review of a new oral penem. Expert Rev Anti Infect Ther. 2007;5:185–198. doi: 10.1586/14787210.5.2.185. [DOI] [PubMed] [Google Scholar]
- 79.Dhar N, Dubée V, Ballell L, Cuinet G, Hugonnet JE, Signorino-Gelo F, Barros D, Arthur M, McKinney JD. Rapid Cytolysis of Mycobacterium tuberculosis by Faropenem, an Orally Bioavailable β-Lactam Antibiotic. Antimicrobial Agents and Chemotherapy. 2015;59:1308–1319. doi: 10.1128/AAC.03461-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee M, Lee J, Carroll MW, Choi H, Min S, Song T, Via LE, Goldfeder LC, Kang E, Jin B, Park H, Kwak H, Kim H, Jeon HS, Jeong I, Joh JS, Chen RY, Olivier KN, Shaw PA, Follmann D, Song SD, Lee JK, Lee D, Kim CT, Dartois V, Park SK, Cho SN, Barry CE., 3rd Linezolid for treatment of chronic extensively drug-resistant tuberculosis. N Engl J Med. 2012;367:1508–1518. doi: 10.1056/NEJMoa1201964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ashtekar DR, Costa-Periera R, Shrinivasan T, Iyyer R, Vishvanathan N, Rittel W. Oxazolidinones, a new class of synthetic antituberculosis agent. In vitro and in vivo activities of DuP-721 against Mycobacterium tuberculosis. Diagnostic microbiology and infectious disease. 1991;14:465–471. doi: 10.1016/0732-8893(91)90002-w. [DOI] [PubMed] [Google Scholar]
- 82.Rose PC, Hallbauer UM, Seddon JA, Hesseling AC, Schaaf HS. Linezolid-containing regimens for the treatment of drug-resistant tuberculosis in South African children. The international journal of tuberculosis and lung disease : the official journal of the International Union against Tuberculosis and Lung Disease. 2012;16:1588–1593. doi: 10.5588/ijtld.12.0322. [DOI] [PubMed] [Google Scholar]
- 83.Anger HA, Dworkin F, Sharma S, Munsiff SS, Nilsen DM, Ahuja SD. Linezolid use for treatment of multidrug-resistant and extensively drug-resistant tuberculosis, New York City, 2000-06. The Journal of antimicrobial chemotherapy. 2010;65:775–783. doi: 10.1093/jac/dkq017. [DOI] [PubMed] [Google Scholar]
- 84.Garcia-Prats AJ, Rose PC, Hesseling AC, Schaaf HS. Linezolid for the treatment of drug-resistant tuberculosis in children: a review and recommendations. Tuberculosis (Edinburgh, Scotland) 2014;94:93–104. doi: 10.1016/j.tube.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 85.Balasubramanian V, Solapure S, Iyer H, Ghosh A, Sharma S, Kaur P, Deepthi R, Subbulakshmi V, Ramya V, Ramachandran V, Balganesh M, Wright L, Melnick D, Butler SL, Sambandamurthy VK. Bactericidal Activity and Mechanism of Action of AZD5847, a Novel Oxazolidinone for Treatment of Tuberculosis. Antimicrobial Agents and Chemotherapy. 2014;58:495–502. doi: 10.1128/AAC.01903-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bruhn DF, Scherman MS, Liu J, Scherbakov D, Meibohm B, Böttger EC, Lenaerts AJ, Lee RE. In vitro and in vivo Evaluation of Synergism between Anti-Tubercular Spectinamides and Non-Classical Tuberculosis Antibiotics. Scientific Reports. 2015;5:13985. doi: 10.1038/srep13985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rock FL, Mao W, Yaremchuk A, Tukalo M, Crépin T, Zhou H, Zhang YK, Hernandez V, Akama T, Baker SJ, Plattner JJ, Shapiro L, Martinis SA, Benkovic SJ, Cusack S, Alley MRK. An Antifungal Agent Inhibits an Aminoacyl-tRNA Synthetase by Trapping tRNA in the Editing Site. Science. 2007;316:1759–1761. doi: 10.1126/science.1142189. [DOI] [PubMed] [Google Scholar]
- 88.Hu QH, Liu RJ, Fang ZP, Zhang J, Ding YY, Tan M, Wang M, Pan W, Zhou HC, Wang ED. Discovery of a potent benzoxaborole-based anti-pneumococcal agent targeting leucyl-tRNA synthetase. Sci Rep. 2013;3 doi: 10.1038/srep02475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hernandez V, Crépin T, Palencia A, Cusack S, Akama T, Baker SJ, Bu W, Feng L, Freund YR, Liu L, Meewan M, Mohan M, Mao W, Rock FL, Sexton H, Sheoran A, Zhang Y, Zhang YK, Zhou Y, Nieman JA, Anugula MR, Keramane EM, Savariraj K, Reddy DS, Sharma R, Subedi R, Singh R, O'Leary A, Simon NL, De Marsh PL, Mushtaq S, Warner M, Livermore DM, Alley MRK, Plattner JJ. Discovery of a Novel Class of Boron-Based Antibacterials with Activity against Gram-Negative Bacteria. Antimicrobial Agents and Chemotherapy. 2013;57:1394–1403. doi: 10.1128/AAC.02058-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Davies J. Where have All the Antibiotics Gone? The Canadian Journal of Infectious Diseases & Medical Microbiology. 2006;17:287–290. doi: 10.1155/2006/707296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Borovinskaya MA, Shoji S, Holton JM, Fredrick K, Cate JHD. A Steric Block in Translation Caused by the Antibiotic Spectinomycin. ACS Chemical Biology. 2007;2:545–552. doi: 10.1021/cb700100n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kehrenberg C, Schwarz S. Mutations in 16S rRNA and Ribosomal Protein S5 Associated with High-Level Spectinomycin Resistance in Pasteurella multocida. Antimicrobial Agents and Chemotherapy. 2007;51:2244–2246. doi: 10.1128/AAC.00229-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee RE, Hurdle JG, Liu J, Bruhn DF, Matt T, Scherman MS, Vaddady PK, Zheng Z, Qi J, Akbergenov R, Das S, Madhura DB, Rathi C, Trivedi A, Villellas C, Lee RB, Rakesh, Waidyarachchi SL, Sun D, Mc Neil MR, Ainsa JA, Boshoff HI, Gonzalez-Juarrero M, Meibohm B, Bottger EC, Lenaerts AJ. Spectinamides: a new class of semisynthetic antituberculosis agents that overcome native drug efflux. Nat Med. 2014;20:152–158. doi: 10.1038/nm.3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Noufflard-Guy-Loe H, Berteaux S. Experimental antituberculous action of a new antibiotic: RP 11,072. Revue de tuberculose et de pneumologie. 1965;29:301–326. [PubMed] [Google Scholar]
- 95.Terlain B, Thomas JP. Structure of griselimycin, polypeptide antibiotic extracted from streptomyces cultures. II. Structure of griselimycin. Bulletin de la Societe chimique de France. 1971;6:2357–2362. [PubMed] [Google Scholar]
- 96.Kling A, Lukat P, Almeida DV, Bauer A, Fontaine E, Sordello S, Zaburannyi N, Herrmann J, Wenzel SC, König C, Ammerman NC, Barrio MB, Borchers K, Bordon-Pallier F, Brönstrup M, Courtemanche G, Gerlitz M, Geslin M, Hammann P, Heinz DW, Hoffmann H, Klieber S, Kohlmann M, Kurz M, Lair C, Matter H, Nuermberger E, Tyagi S, Fraisse L, Grosset JH, Lagrange S, Müller R. Targeting DnaN for tuberculosis therapy using novel griselimycins. Science. 2015;348:1106–1112. doi: 10.1126/science.aaa4690. [DOI] [PubMed] [Google Scholar]
- 97.Reddy VM, Einck L, Nacy CA. In Vitro Antimycobacterial Activities of Capuramycin Analogues. Antimicrobial agents and chemotherapy. 2008;52:719–721. doi: 10.1128/AAC.01469-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nikonenko BV, Reddy VM, Protopopova M, Bogatcheva E, Einck L, Nacy CA. Activity of SQ641, a Capuramycin Analog, in a Murine Model of Tuberculosis. Antimicrobial Agents and Chemotherapy. 2009;53:3138–3139. doi: 10.1128/AAC.00366-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nikonenko B, Reddy VM, Bogatcheva E, Protopopova M, Einck L, Nacy CA. Therapeutic Efficacy of SQ641-NE against Mycobacterium tuberculosis. Antimicrobial Agents and Chemotherapy. 2014;58:587–589. doi: 10.1128/AAC.01254-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Salomon JJ, Galeron P, Schulte N, Morow PR, Severynse-Stevens D, Huwer H, Daum N, Lehr CM, Hickey AJ, Ehrhardt C. Biopharmaceutical in vitro characterization of CPZEN-45, a drug candidate for inhalation therapy of tuberculosis. Therapeutic Delivery. 2013;4:915–923. doi: 10.4155/tde.13.62. [DOI] [PubMed] [Google Scholar]
- 101.Hartkoorn RC, Sala C, Neres J, Pojer F, Magnet S, Mukherjee R, Uplekar S, Boy-Röttger S, Altmann KH, Cole ST. Towards a new tuberculosis drug: pyridomycin – nature's isoniazid. EMBO molecular medicine. 2012;4:1032–1042. doi: 10.1002/emmm.201201689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hartkoorn RC, Pojer F, Read JA, Gingell H, Neres J, Horlacher OP, Altmann KH, Cole ST. Pyridomycin bridges the NADH- and substrate-binding pockets of the enoyl reductase InhA. Nat Chem Biol. 2014;10:96–98. doi: 10.1038/nchembio.1405. [DOI] [PubMed] [Google Scholar]
- 103.Zumla AI, Gillespie SH, Hoelscher M, Philips PPJ, Cole ST, Abubakar I, McHugh TD, Schito M, Maeurer M, Nunn AJ. New antituberculosis drugs, regimens, and adjunct therapies: needs, advances, and future prospects. The Lancet Infectious Diseases. 2014;14:327–340. doi: 10.1016/S1473-3099(13)70328-1. [DOI] [PubMed] [Google Scholar]
- 104.Riccardi G, Pasca MR. Trends in discovery of new drugs for tuberculosis therapy. J Antibiot. 2014;67:655–659. doi: 10.1038/ja.2014.109. [DOI] [PubMed] [Google Scholar]
- 105.Mdluli K, Kaneko T, Upton A. The Tuberculosis Drug Discovery and Development Pipeline and Emerging Drug Targets. Cold Spring Harbor Perspectives in Medicine. 2015 doi: 10.1101/cshperspect.a021154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ioerger TR, O'Malley T, Liao R, Guinn KM, Hickey MJ, Mohaideen N, Murphy KC, Boshoff HIM, Mizrahi V, Rubin EJ, Sassetti CM, Barry CE, III, Sherman DR, Parish T, Sacchettini JC. Identification of New Drug Targets and Resistance Mechanisms in Mycobacterium tuberculosis. PLoS ONE. 2013;8:e75245. doi: 10.1371/journal.pone.0075245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schubert Olga T, Mouritsen J, Ludwig C, Röst Hannes L, Rosenberger G, Arthur Patrick K, Claassen M, Campbell David S, Sun Z, Farrah T, Gengenbacher M, Maiolica A, Kaufmann Stefan HE, Moritz Robert L, Aebersold R. The Mtb Proteome Library: A Resource of Assays to Quantify the Complete Proteome of Mycobacterium tuberculosis. Cell Host & Microbe. 2013;13:602–612. doi: 10.1016/j.chom.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cohen KA, Abeel T, McGuire AM, Desjardins CA, Munsamy V, Shea TP, Walker BJ, Bantubani N, Almeida DV, Alvarado L, Chapman S, Mvelase NR, Duffy EY, Fitzgerald MG, Govender P, Gujja S, Hamilton S, Howarth C, Larimer JD, Maharaj K, Pearson MD, Priest ME, Zeng Q, Padayatchi N, Grosset J, Young SK, Wortman J, Mlisana KP, O'Donnell MR, Birren BW, Bishai WR, Pym AS, Earl AM. Evolution of extensively drug-resistant tuberculosis over four decades revealed by whole genome sequencing of Mycobacterium tuberculosis from KwaZulu-Natal, South Africa. International Journal of Mycobacteriology. 2015;4(1):24–25. [Google Scholar]
- 109.Kuan CS, Chan CL, Yew SM, Toh YF, Khoo JS, Chong J, Lee KW, Tan YC, Yee WY, Ngeow YF, Ng KP. Genome Analysis of the First Extensively Drug-Resistant (XDR) Mycobacterium tuberculosis in Malaysia Provides Insights into the Genetic Basis of Its Biology and Drug Resistance. PLoS ONE. 2015;10:e0131694. doi: 10.1371/journal.pone.0131694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schnappinger D, O'Brien K, Ehrt S. Construction of Conditional Knockdown Mutants in Mycobacteria. In: Parish T, Roberts DM, editors. Mycobacteria Protocols. Springer; New York: 2015. pp. 151–175. [DOI] [PubMed] [Google Scholar]
- 111.Woong Park S, Klotzsche M, Wilson DJ, Boshoff HI, Eoh H, Manjunatha U, Blumenthal A, Rhee K, Barry CE, III, Aldrich CC, Ehrt S, Schnappinger D. Evaluating the Sensitivity of Mycobacterium tuberculosis to Biotin Deprivation Using Regulated Gene Expression. PLoS pathogens. 2011;7:e1002264. doi: 10.1371/journal.ppat.1002264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gold B, Warrier T, Nathan C. A Multi-stress Model for High Throughput Screening Against Non-replicating Mycobacterium tuberculosis. In: Parish T, Roberts DM, editors. Mycobacteria Protocols. Springer; New York: 2015. pp. 293–315. [DOI] [PubMed] [Google Scholar]
- 113.VanderVen BC, Fahey RJ, Lee W, Liu Y, Abramovitch RB, Memmott C, Crowe AM, Eltis LD, Perola E, Deininger DD, Wang T, Locher CP, Russell DG. Novel Inhibitors of Cholesterol Degradation in Mycobacterium tuberculosis Reveal How the Bacterium's Metabolism Is Constrained by the Intracellular Environment. PLoS pathogens. 2015;11:e1004679. doi: 10.1371/journal.ppat.1004679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Prideaux B, Via LE, Zimmerman MD, Eum S, Sarathy J, O'Brien P, Chen C, Kaya F, Weiner DM, Chen PY, Song T, Lee M, Shim TS, Cho JS, Kim W, Cho SN, Olivier KN, Barry CE, Iii, Dartois V. The association between sterilizing activity and drug distribution into tuberculosis lesions. Nat Med. 2015 doi: 10.1038/nm.3937. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gillespie SH, Crook AM, McHugh TD, Mendel CM, Meredith SK, Murray SR, Pappas F, Phillips PPJ, Nunn AJ. Four-Month Moxifloxacin-Based Regimens for Drug-Sensitive Tuberculosis. New England Journal of Medicine. 2014;371:1577–1587. doi: 10.1056/NEJMoa1407426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kjellsson MC, Via LE, Goh A, Weiner D, Low KM, Kern S, Pillai G, Barry CE, Dartois V. Pharmacokinetic Evaluation of the Penetration of Antituberculosis Agents in Rabbit Pulmonary Lesions. Antimicrobial Agents and Chemotherapy. 2012;56:446–457. doi: 10.1128/AAC.05208-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Michot JM, Seral C, Van Bambeke F, Mingeot-Leclercq MP, Tulkens PM. Influence of Efflux Transporters on the Accumulation and Efflux of Four Quinolones (Ciprofloxacin, Levofloxacin, Garenoxacin, and Moxifloxacin) in J774 Macrophages. Antimicrobial Agents and Chemotherapy. 2005;49:2429–2437. doi: 10.1128/AAC.49.6.2429-2437.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Barclay WR, Ebert RH, Le Roy GV, Manthei RW, Roth LJ. Distribution and excretion of radioactive isoniazid in tuberculous patients. J Am Med Assoc. 1953;151:1384–1388. [PubMed] [Google Scholar]
- 119.Canetti G, Parrot R, Porven G, Le Lirzin M. Rifomycin levels in the lung and tuberculous lesions in man. Acta tuberculosea et pneumologica Belgica. 1969;60:315–322. [PubMed] [Google Scholar]
- 120.Ellard GA, Humphries MJ, Allen BW. Cerebrospinal fluid drug concentrations and the treatment of tuberculous meningitis. The American review of respiratory disease. 1993;148:650–655. doi: 10.1164/ajrccm/148.3.650. [DOI] [PubMed] [Google Scholar]
- 121.Ellard GA, Humphries MJ, Gabriel M, Teoh R. Penetration of pyrazinamide into the cerebrospinal fluid in tuberculous meningitis. British medical journal (Clinical research ed) 1987;294:284–285. doi: 10.1136/bmj.294.6567.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Prideaux B, Dartois V, Staab D, Weiner DM, Goh A, Via LE, Barry CE, 3rd, Stoeckli M. High-sensitivity MALDI-MRM-MS imaging of moxifloxacin distribution in tuberculosis-infected rabbit lungs and granulomatous lesions. Anal Chem. 2011;83:2112–2118. doi: 10.1021/ac1029049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Prideaux B, Stoeckli M. Mass spectrometry imaging for drug distribution studies. Journal of proteomics. 2012;75:4999–5013. doi: 10.1016/j.jprot.2012.07.028. [DOI] [PubMed] [Google Scholar]
- 124.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 125.Brown ED, Wright GD. New targets and screening approaches in antimicrobial drug discovery. Chemical reviews. 2005;105:759–774. doi: 10.1021/cr030116o. [DOI] [PubMed] [Google Scholar]
- 126.Wunberg T, Hendrix M, Hillisch A, Lobell M, Meier H, Schmeck C, Wild H, Hinzen B. Improving the hit-to-lead process: data-driven assessment of drug-like and lead-like screening hits. Drug Discovery Today. 2006;11:175–180. doi: 10.1016/S1359-6446(05)03700-1. [DOI] [PubMed] [Google Scholar]
- 127.Elinder M, Geitmann M, Gossas T, Källblad P, Winquist J, Nordström H, Hämäläinen M, Danielson UH. Experimental Validation of a Fragment Library for Lead Discovery Using SPR Biosensor Technology. Journal of Biomolecular Screening. 2011;16:15–25. doi: 10.1177/1087057110389038. [DOI] [PubMed] [Google Scholar]
- 128.Brown JR, North EJ, Hurdle JG, Morisseau C, Scarborough JS, Sun D, Kordulakova J, Scherman MS, Jones V, Grzegorzewicz A, Crew RM, Jackson M, McNeil MR, Lee RE. The structure-activity relationship of urea derivatives as anti-tuberculosis agents. Bioorg Med Chem. 2011;19:5585–5595. doi: 10.1016/j.bmc.2011.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Keseru GM, Makara GM. The influence of lead discovery strategies on the properties of drug candidates. Nature reviews Drug discovery. 2009;8:203–212. doi: 10.1038/nrd2796. [DOI] [PubMed] [Google Scholar]
- 130.Lipinski C. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 131.Lipinski C. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Tox Meth. 2000;44:235–249. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 132.Maitra A, Bates S, Kolvekar T, Devarajan PV, Guzman JD, Bhakta S. Repurposing—a ray of hope in tackling extensively drug resistance in tuberculosis. International Journal of Infectious Diseases. 2015;32:50–55. doi: 10.1016/j.ijid.2014.12.031. [DOI] [PubMed] [Google Scholar]
- 133.Zhang D, Wang Y, Lu J, Pang Y. In vitro activity of β-lactams in combination with β-lactamase inhibitors against multidrug-resistant Mycobacterium tuberculosis isolates. Antimicrobial Agents and Chemotherapy. 2015 doi: 10.1128/AAC.01035-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hazra S, Xu H, Blanchard JS. Tebipenem, a New Carbapenem Antibiotic, Is a Slow Substrate That Inhibits the β-Lactamase from Mycobacterium tuberculosis. Biochemistry. 2014;53:3671–3678. doi: 10.1021/bi500339j. [DOI] [PMC free article] [PubMed] [Google Scholar]