

Abstract
Type 1 diabetes (T1D) is an organ-specific autoimmune disease characterized by T cell-mediated destruction of the insulin-producing pancreatic β cells. A combination of genetic and environmental factors eventually leads to the loss of functional β cells mass and hyperglycemia. Both innate and adaptive immunity are involved in the development of T1D. In this review, we have highlighted the most recent findings on the role of innate immunity, especially the pattern recognition receptors (PRRs), in disease development. In murine models and human studies, different PRRs, such as toll-like receptors (TLRs) and nucleotide-binding domain, leucine-rich repeat-containing (or NOD-like) receptors (NLRs), have different roles in the pathogenesis of T1D. These PRRs play a critical role in defending against infection by sensing specific ligands derived from exogenous microorganisms to induce innate immune responses and shape adaptive immunity. Animal studies have shown that TLR7, TLR9, MyD88 and NLPR3 play a disease-predisposing role in T1D, while controversial results have been found with other PRRs, such as TLR2, TLR3, TLR4, TLR5 and others. Human studies also shown that TLR2, TLR3 and TLR4 are expressed in either islet β cells or infiltrated immune cells, indicating the innate immunity plays a role in β cell autoimmunity. Furthermore, some human genetic studies showed a possible association of TLR3, TLR7, TLR8 or NLRP3 genes, at single nucleotide polymorphism (SNP) level, with human T1D. Increasing evidence suggest that the innate immunity modulates β cell autoimmunity. Thus, targeting pathways of innate immunity may provide novel therapeutic strategies to fight this disease.
Keywords: Type 1 diabetes, innate immunity, pattern recognition receptors, β cells, adaptive immunity
1. INTRODUCTION
Type 1 diabetes (T1D) is an organ-specific autoimmune disease characterized by T cell-mediated destruction of the insulin-producing pancreatic β cells. A combination of genetic and environmental factors eventually leads to the loss of functional β cell mass and hyperglycemia [1]. While the precise immunological mechanisms to control disease initiation and progression remain to be fully elucidated, there are three critical prerequisites to failure of β cells, including the activation of β cell-reactive T cells, the creation of an environment with pro-inflammatory cytokines and chemokines, and the failed control by immune regulatory function [2]. Growing evidence from animal models and patients with T1D points a central role of pro-inflammatory cytokines and cell-mediated immunity in the pathogenesis of β cell destruction. Extensive efforts have been made in clinical trials with targeted immunotherapy directed towards cytokines and adaptive lymphocytes, such as anti-IL-1 (canakinumab or anakinra), anti-TNF-α (etanercept), anti-CD3 (teplizumab or otelixizumab, to block T cell function), anti-CD20 (rituximab, for B cell depletion) and CTLA4-Ig (abatacept, to block T-cell activation) [3-9]. Although, some transient or short-term beneficial effects have been observed in preserving residual β cell function or reduction of insulin requirements, the overall effects have not been sustained. Recurrence of β cell loss was found in the majority of patients after treatment and pathogenic autoreactive lymphocytes have survived or recrudesced after immunomodulatory intervention. Thus, it is essential to revisit and elucidate the underlying pathogenic mechanisms and explore new strategies beyond the current concepts.
Both innate and adaptive immunity are involved in the development of T1D [4, 10]. The gut microbiota has emerged recently as an essential player in the development of autoimmune diseases including T1D [11, 12]. Altered gut microbiota have been shown to associate with different diseases in both rodent models and humans [13-16]. Innate immunity is evolutionarily conserved and serves as the first line of defense of the body in response to exogenous insults such as bacterial, viral and fungal infections [17]. Recent studies have demonstrated that gut microbiota and innate immunity act through highly conserved pattern-recognition receptors (PRR) to coordinate the innate inflammatory response to both endogenous and exogenous stimuli, and further shape adaptive immunity [13, 16, 18]. PRRs are a group of germline-encoded proteins expressed by cells of the innate immune system and some tissue cells, which can survey both the extracellular and intracellular surroundings for conserved microbial determinants that serve as indicators of infection. There are five major families of PRRs characterized so far: Toll-like receptors (TLRs), C-type lectin receptors (CLRs), nucleotide-binding domain, leucine-rich repeat-containing (or NOD-like) receptors (NLRs), RIG-I-like receptors (RLRs) and the AIM2-like receptors (ALRs) [19, 20]. By recognizing pathogen-associated molecular patterns (PAMPs), which are associated with microbial pathogens or cellular stress, as well as damage-associated molecular patterns (DAMPs), PRRs can induce either transcriptional or non-transcriptional innate immune responses to lead to the production of pro-inflammatory cytokines (IL-1β and TNF-α) and interferons or the induction of phagocytosis, autophagy and cell death, respectively [19]. The non-antigen (Ag)-specific innate immune response makes a crucial contribution to the activation of Ag-specific and more efficient adaptive immunity. However, altered innate immune responses and impaired B and T cell tolerance result in autoimmune diseases [21]. This review will focus on the association between innate immunity and adaptive immunity and discusses new findings regarding the effect of the innate immune response on β cell function and autoimmunity.
2. TLRs and T1D
The TLR family is one of most important and well-characterized PRRs, members of which selectively recognize a large number of PAMPs derived from microbes and activate innate immune responses. To date, there are 10 human TLRs and 13 mouse TLRs identified. Of these, TLR1, TLR2, TLR4, TLR5, TLR6 and TLR10 are located on the cell surface while TLR3, TLR7, TLR8 and TLR9 are located in intracellular endosomes [22]. TLRs are widely expressed in various immune cells [monocytes, macrophages, dendritic cells (DCs), and B cells] as well as non-immune cells (keratinocytes, epithelial cells or tissue cells) [22, 23]. Activation of most TLRs, after engaging with their ligands, transduces a signal through the MyD88-dependent pathway, except for TLR3 which is TRIF dependent, to activate the transcriptional factor NF-κB resulting in the production of pro-inflammatory cytokines [24, 25]. Data from T1D rodent models - non-obese diabetic (NOD) or Bio-Breeding (BB) rats and human studies have shown that TLR signaling plays a critical role in the development of T1D [12, 26-28].
2.1. TLR2 and T1D
TLR2 can recognize a variety of microorganism-derived components and certain endogenous ligands. Its main microbial ligands include the lipoproteins from bacteria and mycoplasma, the peptidoglycan moiety from bacteria, zymosan from some fungi and lipoteichoic acid from certain gram-positive microorganisms [29, 30]. TLR2 forms hydrophilic heterodimers with other structurally associated TLRs such as TLR1 and TLR6 to recognize distinct ligands [31]. Stimulation with TLR2 ligands, such as triacyl or diacyl lipoprotein can induce the production of various pro-inflammatory cytokines in macrophages or DCs [32].
Previous studies have shown that TLR2 signaling plays an important role in infectious and autoimmune diseases. Kim et al (2007) observed that the development of autoimmune diabetes (both spontaneous and streptozotocin-induced) was markedly inhibited in TLR2-deficient mice, suggesting that TLR2 plays an important role in the initiation of the disease [33]. TLR2 deficiency did not affect T cell functions. However, apoptotic β-cells primed diabetogenic T cells through a TLR2-dependent activation of antigen-presenting cells (APC) [33, 34]. These findings suggest that β-cell death and its sensing via TLR2 may be an initial event for the stimulation of APCs and the autoimmune cascade leading to the clinical onset of diabetes. Interestingly, a recent study by Burrows et al (2015) showed that the incidence of diabetes was not significantly different in TLR2-sufficient and deficient NOD mice when in germ-free (GF) conditions [35]. However, GF TLR2−/−NOD mice had a higher incidence of diabetes and more severe insulitis than specific pathogen-free (SPF) TLR2−/−NOD mice [35]. This suggests that commensal microbes regulate the pro-diabetic effect of TLR2.
However, other studies showed that ligation of TLR2 could prevent T1D [36, 37]. Zymosan, a fungal cell wall component, stimulated APCs from NOD mice by interacting with TLR2 and Dectin 1 (a CLR family member) to produce large amounts of IL-10, TGF-β1, IL-12 and TNF-α both in vitro and in vivo. The function and the expansion of CD4+CD25+ regulatory T cells (Tregs) were also promoted [38, 39]. NOD mice treated with either zymosan or β-cell-Ag-loaded zymosan-exposed DCs had less severe insulitis and delayed hyperglycemia [40]. Further study showed that β-cell-Ag, together with zymosan, gave superior protection from diabetes when administered to NOD mice at the pre-diabetic and early hyperglycemic stages than when zymosan was used alone [41]. This therapeutic strategy significantly promoted the expansion of Foxp3-positive Tregs and IL-10, IL-4 and IL-17-expressing CD4+ T cells, especially in the pancreatic lymph nodes (PLNs) [41]. Taken together, these data suggest that TLR2 may play both pathogenic and preventive roles in the development of T1D, depending on the stimuli that the innate immune system encounters. The successful intervention through the innate immune response by TLR2 and Dectin 1 signaling pathways could provide a promising therapeutic strategy to prevent or reverse T1D, but this has yet to be tested in humans.
2.2. TLR3 and TLR7 in T1D
TLR3 detects viral double-stranded (ds) RNA in the endolysosome [42]. The expression of TLR3 has been observed in a number of human tissues including placenta, pancreas, lung, liver, heart, lymph nodes and brain [43]. TLR3 is usually expressed intracellularly in various human and mouse immune cells including T cells, macrophages, DCs, natural killer cells and γδ T cells [44, 45]. TLR3 binds viral dsRNA and induces downstream innate immune responses through a MyD88-independent but TRIF-dependent pathway, unlike the other TLRs [46]. TLR3 is also a receptor for polyinosinic:polycytidylic acid [poly(I:C)], a synthetic analog of dsRNA. Previous studies showed that administration of poly (I:C) either prevents or accelerates diabetes development in NOD mice or BB rats depending on the doses or kinetics of poly (I:C) action [47-49]. However, administration of poly(I:C) can accelerate virus-induced diabetes in BBDR rats that are normally resistant to T1D development [50]. This suggests that dsRNA sensors, such as TLR3, as well as two RLRs, retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated gene-5 (MDA5), can promote diabetes in an IFN-dependent manner, as activation of all of these innate receptors induces strong type 1 interferon responses.
Although the precise role of viruses in the pathogenesis of T1D remains controversial, previous studies from both mouse models and human patients have revealed that viral infection might be associated with T1D development [50-55]. The induction of prion-inflammatory cytokines by viral infection could affect islet autoimmunity and β-cell decay. In fulminant human diabetes, which may be triggered by enteroviral infection, there is increased expression of TLR3 in mononuclear cells that infiltrate islets and strong up-regulation of RIG-1 and MDA5 in β cells [56]. It is conceivable that, in fulminant T1D, the innate immune responses induced by enterovirus and sensed by TLR3 initiate pro-inflammatory signals and up-regulate RIG-I and MDA5 in β cells. These innate immune responses enhance antigen-specific adaptive immune responses, not only fighting enteroviral infection, but also leading to aggressive β cell destruction [57, 58 ]. Enhanced expression of TLR3, RIG-I and MDA5 was also observed in human pancreatic islets in vitro when they were infected with Coxsackie B5 virus or challenged with IFN-α or IFN-γ with IL-1β [58, 59]. These data demonstrated that TLR3 could recognize both intracellular and extracellular dsRNAs and trigger the production of the pro-inflammatory cytokines, resulting in β-cell apoptosis. Thus, TLR3 could play a role in human T1D. To further examine this possibility, studies have shown an association of TLR3 and T1D in different populations from South Africa, UK, Finland and Brazil [43, 60-62]. Most of the data indicated a strong association of SNPs in the TLR3 pathway or polymorphisms of the TLR3 gene with T1D [43, 60-62]. However, recent studies in Polish and Norwegian patients with T1D and healthy controls did not find an association between TLR3 polymorphisms and the risk for autoimmune destruction of β cells [63, 64]. Furthermore, deficiency of TLR3 did not affect spontaneous T1D development in the NOD mouse [65]. However, TLR3 plays an important role in virus-induced T1D in NOD mice and C57BL/6 mice [66, 67]. Therefore, further studies and comprehensive approaches will be needed to help elucidate the precise role of TLR3 signaling in T1D.
TLR7 recognizes single strand RNA (ssRNA), and unlike TLR3, which is independent of MyD88, downstream signaling of TLR7 is dependent on MyD88 [23]. Activation of TLR7 by RNA virus or synthetic stimulator can result in lymphocyte activation and promote accelerated diabetes in NOD mice [68, 69]. In another mouse model, RIP-GP mice expressing lymphocytic choriomeningitis virus (LCMV) glycoprotein (GP) as a transgene under the control of the rat insulin promoter, immunization with LCMV-GP derived peptide promoted large numbers of autoreactive cytotoxic CD8+ T cells but did not induce autoimmune diabetes. However, subsequent treatment with a TLR7 agonist (R-848) elicited overt autoimmune disease, with enhanced serum IFN-α production and pancreatic β-cell MHC I expression [66]. To study the role of TLR7 in the pathogenesis of spontaneous T1D development, we have generated TLR7-deficient NOD mice. Our preliminary data has shown that TLR7 deficiency confers protection in T1D development [Peng et al, unpublished data]. These data suggest that TLR7 plays a critical role in the pathogenesis of T1D development and blockade of the TLR7 pathway may provide a potential therapeutic application.
2.3. TLR4 and T1D
TLR4 can specifically bind to lipopolysaccharide (LPS) of Gram-negative bacteria together with myeloid differentiation factor 2 (MD-2). Furthermore, TLR4 can be stimulated by several other PAMPs, including the fusion (F) protein from respiratory syncytial virus, the envelope protein from mouse mammary tumor virus and endogenous molecules (heat-shock proteins, hyaluronic acid and β-defensin 2) [19]. LPS is an important structural component of the outer membrane of Gram-negative bacteria and has been one of the most studied stimulatory components of bacteria. LPS binds to TLR4 to initiate the cascade of immune responses through both MyD88-dependent and TRIF-dependent pathways to induce the production of type 1 interferons and pro-inflammatory cytokines (such as IL-1β and TNF-α). TLR4 is widely expressed in various tissues and cells, especially in monocytes.
Previous studies have demonstrated increased TLR4 mRNA in monocytes from both diabetic NOD mice and patients with T1D compared to non-diabetic control subjects, indicating that the TLR4 signaling pathway is tightly associated with diabetes development [70, 71]. Garay-Malpartida et al (2011) found that human β cells express TLR4 mRNA and surface protein [72]. The increased TLR4 expression was accompanied by decreased insulin secretion in an LPS concentration-dependent manner, in both human and mouse β-cells [72]. Li et al (2012) demonstrated higher expression of high-mobility group box 1 (HMGB1), an inflammatory trigger in a number of autoimmune diseases, in the cytoplasm of islets in diabetic NOD mice compared with non-diabetic mice [73]. HMGB1 may signal through TLR4 and selectively damage β cells during T1D development. Administration of monoclonal antibody to TLR4/MD-2 (TLR4-Ab) reversed new-onset diabetes in NOD mice, mediated by induction of tolerogenic APCs, and promoted the expansion of Tregs in both the periphery and the pancreatic islets [74]. These results indicate that TLR4 plays an important role in the pathogenesis of T1D. Interestingly, TLR4−/−NOD mice develop accelerated diabetes compared to wild-type (WT) NOD mice [12, 35, 75]. Thus, TLR4 may also function as a tolerogenic signal in T1D development. This was confirmed by Burrows et al (2015) using GF TLR4−/− NOD mice that do not have accelerated diabetes [12, 35, 75]. This study suggests that the protective effect of TLR4 signaling is, again, modulated by commensal microbiota. Tian and colleagues reported over a decade ago that LPS can protect NOD mice from T1D development [76]. Thus, TLR4 is associated with protection and may have promise as a tolerogenic adjuvant, which could potentially be exploited in therapy to induce immunoregulation.
2.4. TLR5 and T1D
TLR5 mainly recognizes flagellin from flagellated bacteria and is highly expressed by DCs of the lamina propria in the small intestine as well as neutrophils, monocytes and epithelial cells. After ligation with flagellin, TLR5 in lamina propria DCs can induce B cells to differentiate into IgA-producing plasma cells and induce the differentiation of naive T cells into antigen-specific Th17 and Th1 cells [77]. TLR5 signals through MyD88 to induce inflammatory cytokine production, but additional recruitment of TRIF by TLR5 in intestinal epithelial cells can lead to the activation of NF-κB rather than IFN-regulatory factor-3 (IRF3) [78]. A previous study has shown that TLR5-deficient C57BL/6 mice have altered gut microbiota leading to the development of obesity and insulin resistance [79]. However, the role of TLR5 in T1D development is not clear. Weile et al (2011) observed significantly increased TLR5 mRNA and protein levels in isolated islets of Langerhans from male Lewis rats or male BALB/c mice upon stimulation with glucose [80]. The up-regulation of TLR5 was accompanied by the reduction of insulin secretion and increase of nitric oxide, pro-inflammatory cytokines and heat-shock protein [80]. We have generated TLR5-deficient NOD mice and our unpublished data suggest that TLR5 is likely to be dispensable for T1D development as TLR5−/−NOD mice developed a similar incidence of diabetes to WT NOD littermates (Hu, et al, manuscript in preparation). In line with our results, TLR5 gene expression was not significantly associated with T1D in the studies using The Australia Childhood Diabetes DNA Repository (http://www.cdr.net.au/projects/type1.html). Thus, at this time, these studies indicate that TLR5 does not appear to play a major role in the development of T1D.
2.5. TLR9 in T1D
TLR9 recognizes unmethylated CpG DNA that is found in bacteria and viruses and is also present in mammalian cells. TLR9 signaling is MyD88-dependent. TLR9 is mainly located in endoplasmic reticulum (ER) membrane, endosomes, lysosomes, and endolysosomes of different immune cells including macrophages, DCs, B cells and T cells, but is also highly expressed in plasmacytoid DCs (pDCs) [24, 81-85]. TLR9 is an essential PRR along with TLR7 for virus-induced type 1 IFN production by pDCs [86]. We have shown that TLR9−/−NOD mice were significantly protected from T1D development, which is mediated by the immune regulatory molecule CD73 (see below) [87]. Zhang et al (2010) found that TLR9−/−NOD mice expressed lower levels of IFN-α in PLNs and reduced frequencies of diabetogenic CD8+ T cells, both of which may contribute to diabetes protection seen in TLR9−/−NOD mice [88]. A recent human study by Kayserova et al (2014) analyzed the frequency of myeloid (mDCs) and pDCs in 97 T1D patients (69 new onset, 28 long-term diabetic patients), 67 first-degree relatives, and 64 controls in the Czech Republic [89]. The authors observed a lower number of mDCs and pDCs in patients with T1D and their first-degree relatives. Of all the tested TLR ligands, only stimulation with CpG 2216 induced IFN-α production and the highest level was found in the first-degree relatives of patients with T1D who were positive for islet autoantibodies [89]. However, it is likely that other mechanisms are also involved in diabetes protection in addition to reduction in IFN-α in TLR9−/− NOD mice. The role of IFN-α in T1D is complex; our previous studies showed that over-expression of IFN-α in islet β cells promoted diabetes development in one T1D model but it protected against diabetes development in another [90]. Our recent study revealed that CD73 was significantly up-regulated in immune cells from peripheral lymphoid tissues in TLR9−/−NOD mice [87]. The elevated CD73 expression appeared to be specific to TLR9 deficiency and MyD88-independent. Moreover, the elevation of CD73 expression was limited to the NOD background. Increased CD73 expression was also associated with lower levels of pro-inflammatory cytokines and more anti-inflammatory cytokine production in CD4+ T cells in TLR9−/−NOD mice. Elevation of CD73 expression was also associated with improved β cell function. These data indicate an important immune regulatory role of CD73 in regulation of diabetes development and may offer a new therapeutic strategy for specific intervention to prevent T1D using TLR9 antagonists.
Second to pDC, TLR9 is also highly expressed in B cells. Gianchecchi et al (2014) investigated the effect of the C1858T (Lyn) polymorphism in the protein tyrosine phosphatase non-receptor type 22 (PTPN22) on innate and adaptive immunity in patients with T1D and healthy control subjects [91]. The authors found that the presence of the Lyn variant was associated with a significant increase in the percentage of transitional B cells in C/T carriers of patients with T1D and control subjects compared to patients and control subjects carrying the C/C polymorphism [91]. Further stimulation with CpG resulted in a significant increase of IgM+ memory B cells in C/T carrier patients, suggesting that altered B cell homeostasis mediated by increased TLR9 signaling could contribute to the pathogenesis of T1D. Blockade of TLR9 signaling with the TLR9 antagonist, chloroquine, protected NOD mice from diabetes [87, 88]. Taken together, both mouse and human studies strongly support the pathogenic role of TLR9 in T1D and, therefore, TLR9 may serve as an important and effective target for immunotherapeutic intervention in human T1D. Non-specific agents are available that antagonize TLR9, but it is likely that more specific therapies would be required to target TLR9 for an immunoregulatory role in adjunct immunoregulatory therapy in T1D
2.6. MyD88, TRIF and T1D
MyD88 is the major adaptor protein of TLRs, except TLR3, and plays a central role in the innate and adaptive immune response. This protein functions as an essential signal transducer in the IL-1 and TLR signaling pathways [23]. These pathways regulate the activation of numerous pro-inflammatory genes. Previous studies have demonstrated that MyD88 plays a critical role in regulating the homeostasis of gut microbiota [12, 92]. Our previous study also showed that deficiency of MyD88 completely abrogated diabetes development in NOD mice that were kept in SPF conditions; however, the protection was abolished when the mice were housed in GF conditions [12]. Deficiency of MyD88 also resulted in a reduction of antimicrobial peptides in the mouse intestine, thus altering gut microbiota composition and further shaping adaptive immunity [93, 94]. TRIF is another key adaptor protein for TLR3 (complete) and TLR4 (partial)-mediated signaling pathways. TRIF associates with TRAF6 and TBK1 independently, and can activate two distinct transcription factors, NF-κB and IRF3, respectively, to induce the production of type 1 interferons [23, 95]. Furthermore, TRIF-dependent type I IFN signaling in T cells is essential to Th1 lineage differentiation and re-activation of memory T cells in response to bacterial infection, indicating the importance of TRIF as a mediator of the innate and adaptive immune interactions in generating protective memory immunity against pathogens [96, 97]. Although TRIF deficiency does not seem to affect diabetes development in NOD mice housed in SPF conditions [our unpublished data], yet, TRIF deficiency partially, but significantly, reversed diabetes protection in SPF MyD88-deficient NOD mice [35]. This suggests that TRIF signaling may provide a protective role in MyD88−/−NOD mice and the protection is diminished when TRIF is not present. It is clear that both MyD88 and TRIF, as the major downstream adaptor proteins to the TLRs, play a critical role in mediating diabetes development.
The roles of the remaining TLRs, including TLR1, TLR6 and TLR8, have not been well studied in T1D development. Both TLR1 and TLR6 can form heterodimers with TLR2 respectively to recognize different microbial ligands and initiate innate immune responses [29, 31]. A recent human study found that SNPs and haplotypes of TLR1 and TLR6 gene were closely associated with T1D in a Chinese Han population, indicating a potential role of TLR1 and TLR6 in the etiology of T1D [98]. TLR8 recognizes single-stranded RNA derived from virus, bacteria or self-antigens [19, 23]. A recent study from the Type I Diabetes Genetics Consortium assessed, in affected sib-pair families of two parents and two affected offspring, whether established T1D susceptibility SNPs and candidate SNPs in innate immune genes are associated with T1D. A significant association was observed between the Xp22 SNP (rs5979785) and T1D [99]. The Xp22 SNP is located 30 kb centromeric of the functional candidate TLR7 and TLR8 genes, suggesting that SNP rs5979785, or variants in linkage disequilibrium with it, could alter TLR7 and/orTLR8 gene expression and may be associated with risk of T1D [99]. More studies will be needed to address the precise role of these TLRs in the development of T1D.
3. NLRs and T1D
The NLR family of proteins is a group of intracellular PRRs of the innate immune system that play a vital role in the recognition of a wide spectrum of PAMPs and DAMPs. The NLR proteins are composed of a central nucleotide-binding domain termed NACHT domain (also referred to as NOD domain), N-terminal effector domains (CARDs, a pyrin domain, and baculovirus inhibitor of apoptosis protein repeat BIR domain) and C-terminal leucine-rich repeats (LRR) [18, 20, 29, 100-102]. In humans, the NLR family comprises 22 genes, whereas the mouse genome contains at least 34 NLR-encoding genes.
3.1. NOD2 and T1D
NOD1 and NOD2 belong to the CARD-containing subfamily and recognize the structures of bacterial peptidoglycans, g-D-glutamyl-mesodiaminopimelic acid and muramyl dipeptide, respectively [20, 101]. After sensing peptidoglycans, NOD1 and NOD2 can activate NF-κB pathways to induce transcriptional up-regulation of pro-inflammatory cytokines (IL-1β, IL-6, TNF and IL-18), chemokines (CCL5 and CXCL5) and also recruit neutrophils to the site of infection [103-105]. NOD1 and NOD2 have been implicated in the initiation of the adaptive immune response. The role of NOD1 in T1D development is currently unknown. We have generated NOD2-deficient NOD mice and observed spontaneous diabetes development. We found that NOD2-deficient NOD mice are protected from diabetes development and the protection is most likely mediated by altered gut microbiota [Li, et al, manuscript in preparation]. This suggests that NOD2 is involved in the pathogenesis of T1D. However, further studies are needed to elucidate the underlying mechanisms of NOD2 and gut microbiota in T1D development.
3.2. NALP3 and T1D
The NLRP family is another subfamily of NLRs containing pyrin domain and has 14 members identified so far. Previous studies have shown that some NLRP members participate in the induction of the inflammatory response mediated by IL-1 family, such as IL-1β, IL-18 and IL-33 [19, 23, 29]. NLRP3 may function as an activator of NF-κB signaling and is an essential component of the inflammasome, a protein complex including NLRP3, apoptosis-associated speck-like protein and caspase 1 [106, 107]. Activation of NLRP3 leads to oligomerization and recruitment of apoptosis-associated speck-like protein and caspase 1, which promotes autocleavage and activation of caspase 1 [108]. Active caspase 1 cleaves pro-IL-1β to active IL-1β, which, when secreted, can exert direct cytotoxic effects as well as recruit other inflammatory cells.
In addition to the well-documented role of host response to PAMPs and DAMPs, NLRP3 has also been shown to play an important role in a number of autoimmune diseases [109, 110]. Increased NLRP3 expression was observed in the spinal cord during experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis [109]. Significantly delayed EAE progress and reduced severity of disease were found in NLRP3−/− mice, accompanied by a significant reduction of the immune cell infiltrate including macrophages, DCs, CD4+ and CD8+ T cells in the spinal cords of the NLRP3−/− mice [109]. It was shown that NLRP3 plays a pathogenic role in the development of EAE by mediating Th1 and Th17 responses. Although deficiency of caspase-1 or IL-1β did not protect NOD mice from T1D [111, 112], our recent study demonstrated that NLRP3 plays an important role in the immune-pathogenesis of T1D development in NOD mice [113]. NLRP3 deficiency not only altered T cell activation and Th1 differentiation, but also affected the migration of pathogenic T cells to the pancreatic islet. The expression of the chemokine receptors CCR5 and CXCR3 was significantly down-regulated on T cells of NLRP3−/−NOD mice. Furthermore, NLRP3 ablation reduced the expression of CCL5 and CXCL10 on pancreatic islet cells in an IRF-1-dependent manner [113]. Our data strongly support a role for NLRP3 in the pathogenesis of T1D. Interestingly, a human study to characterize the association of NLRP3 SNPs and T1D in a north-eastern Brazilian population found that NLRP3 rs10754558 SNP was associated specifically with T1D and NLRP3 rs358294199 SNP with celiac disease in Brazilians, indicating that variations in NLRP3 could be a predisposing genetic factor that contributes to the development of autoimmune diseases [114]. These data strongly suggest that NLRP3 plays a very important role in the pathogenesis of T1D and may serve as a target for immunological intervention, which should be further explored.
4. Summary and perspective
The incidence of type 1 diabetes worldwide is increasing and currently, exogenous insulin therapy is the only treatment for patients with T1D, apart from the relatively few who have had successful pancreas transplants. However, insulin is not a cure, and the majority of patients with type 1 diabetes currently have no alternatives to administration of insulin to maintain life, but a significant proportion will suffer from chronic macrovascular and microvascular complications. Developing therapy to maintain endogenous insulin production would be a major boon.
Great efforts have been made in the studies using murine models and human samples in the past three decades to understand the pathogenesis of T1D. The underlying mechanisms have been extensively investigated and our understanding of this disease has improved. Genetic factors predispose to the disease and environmental factors can modify the development of the disease. Innate immunity plays an important role in the pathogenesis of T1D by cross-talk with adaptive immunity. In this review, we have highlighted the recent progress in the role of innate immunity, especially PRRs, in the development of T1D (Figure 1). It is clear that TLR7, TLR9, MyD88 and NLPR3 play a disease predisposing role in the T1D, in animal models and this needs to be consolidated by further investigation in the human disease (Tables 1 and 2). Controversial results have been found in other PRRs, such as TLR2, TLR3, TLR4, TLR5 and others. These PRRs play a critical role in defending against infection by sensing specific ligands derived from exogenous microorganisms to induce innate immune responses and shape adaptive immunity. Furthermore, gut microbiota play a critical role in modifying diabetes incidence by interacting with the innate immune response, complicating the simplicity of the role of these PRRs in disease initiation and progress. Further studies are clearly required to fully elucidate the underlying mechanisms and to discover how manipulation of pathways fundamental to defense against infection may be used to reduce pathological immune responses.
Figure 1. The innate immune response affects adaptive immunity and β cells.
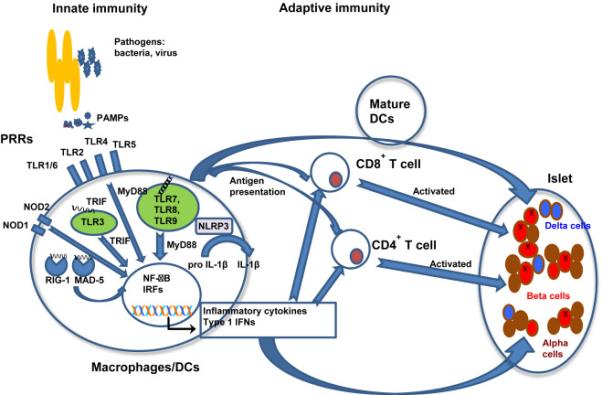
Immature macrophages or dendritic cells (DCs) express pattern-recognition receptors (PRRs) either on the outer membranes or in the internal endosomes. Recognition of pathogen-associated molecular patterns (PAMPs) associated with groups of pathogens such as bacteria, virus or fungi, or danger-associated molecular pattern molecules (DAMPs) by PRRs, results in the activation of downstream transcription factors NF-κB or IRFs, which induce the production of inflammatory cytokines or type 1 interferons (IFN). Activation of the NLRP3 inflammasome triggers innate immune responses and releases active IL-1β. These secreted cytokines and type 1 IFNs can cause islet beta cell stress and also facilitate the activation of naïve T cells that are primed with the islet beta cell autoantigens presented by macrophages or DCs. The primed T cells will differentiate into different types of effective cell subsets in draining lymph nodes and pancreatic islets. This event attracts more mature macrophages, DCs and effector T cells to migrate to the pancreatic islets to both directly damage the β cells and indirectly via the pro-inflammatory cytokines.
Table 1.
Summary of animal studies to elucidate the role of PRRs in the development of T1D
| PRR | Mouse Strain | Spontaneous Disease or Administered Treatment | Outcome | Mechanisms | References |
|---|---|---|---|---|---|
| TLR | |||||
| TLR2 | TLR2-/-B6 TLR2-/-NOD |
STZ-induction or spontaneous | Protection | Apoptotic β-cell injury could stimulate the priming of diabetogenic T cells through a TLR2-dependent activation of antigen-presenting cells | [33] |
| TLR2-/-NOD | Spontaneous | Higher incidence of diabetes and more severe insulitis in GF mice than SPF mice | Commensal microbes may regulate the pro-diabetic effect | [35] | |
| NOD | Treated prediabetic mice with agonist Pam3CSK | Protection | Increased number and function of CD4+CD25+ Tregs, also endowing dendritic cells with tolerogenic properties | [37] | |
| NOD | Zymosan and β-cell Ag at prediabetic or early hyperglycemic stages | Protection | Promoted Foxp3+ Tregs and IL-10, IL-4 and Il-17 expressing CD4+T cells | [41] | |
| TLR3 | NOD or BB rats | Administration of low-dose poly (I:C) | Protection | Possible recruitment of regulatory cells or induction of suppressor cell activity. | [47,49] |
| BBDR rats | Administration of high-dose poly (I:C) | Induction of diabetes | Induced the expansion of peripheral blood NK cells | [48] | |
| TLR3-/-NOD | Spontaneous | No effect | [65] | ||
| RIP-GP mice (mixed background of 129Sv × C57BL/6) | Immunized with gp33 and adoptively transferred with 107 splenocytes derived from LCMV-gp33/H-2Db-specific TCR-transgenic 318 mice. Then treated with 200 μg of poly(I:C). | Diabetes induction | MHC I up-regulated in beta cells. | [66] | |
| TLR3-/- NOD | CVB4 inoculation | Protection | Islets less infiltrated with T cells compared to WT | [67] | |
| TLR4 | NOD | TLR4/MD-2 antibody | Reverse new-onset diabetes | Induced APC tolerance and expansion of Tregs. | [72] |
| TLR4-/-NOD | Spontaneous | Acceleration | Reduced capacity of Tregs to inhibit T cell proliferation. Alteration of gut microbiota. | [12,35,75] | |
| NOD/scid | Co-transfer of LPS-activated B cells with diabetogenic splenocytes | Protection | Down-regulated Th1 autoimmunity and more secreted TGFp to inhibit APC activity | [76] | |
| TLR5 | TLR5-/-NOD | Spontaneous | No effect | Unpublished data | |
| TLR7 | NOD or NY8.3NOD | Administration of TLR7 agonist with or without anti-CD40 | Acceleration | Activated lymphocytes and promoted the production of pro-inflammatory cytokines | [68] |
| RIP-GP mice (mixed background of 129Sv × C57BL/6) | Immunized with gp33 and adoptively transferred with 107 splenocytes derived from LCMV-gp33/H-2Db-specific TCR-transgenic 318 mice, followed by administration of R-848. | Diabetic induction | Enhanced production of IFN-α and upregulation of pancreatic MHC II. | [66] | |
| TLR7-/-NOD | Spontaneous | Protection | Unpublished data | ||
| TLR9 | TLR9-/-NOD | Spontaneous | Protection | Upregulated CD73 in immune cells accompanying with lower levels of pro-inflammatory cytokines and higher levels of anti-inflammatory cytokines in CD4+ T cells. | [65,87] |
| TLR9-/-NOD | Spontaneous | Protection | Reduced levels of IFNα in PLNs and reduced frequencies of diabetogenic CD8+ T cells | [88] | |
| MyD88 | MyD88-/-NOD | Spontaneous | Complete protection in SPF | Altered gut microbiota | [12] |
| TRIF | TRIF-/-NOD | Spontaneous | No significant effect | Unpublished data | |
| TRIF-/-MyD88-/-NOD | Spontaneous | Reversed protection from diabetes in SPF conditions. | [35] | ||
| NLR | |||||
| NOD2 | NOD2-/-NOD | Spontaneous | Protection | Altered gut microbiota | Unpublished data |
| NLRP3 | NLRP3-/-NOD | Spontaneous | Protection | Altered T cell activation, Th1 differentiation and pathogenic T cell migration to the pancreatic islet. | [113] |
Table 2.
Summary of human studies to elucidate the role of PRRs in the development of T1D
| PRR | Experimental subjects | Main findings | References |
|---|---|---|---|
| TLR | |||
| TLR2 | PBMC from T1D patients and healthy controls in USA | Increased TLR2 expression in monocytes | [70] |
| TLR3 | Pancreata from patients with fulminant diabetes in Japan | Confirmation of TLR3 expression in islet-infiltrating mononuclear cells | [56] |
| Pancreata from human donors from Sweden or Finland infected with Coxsackie B5 virus or challenged with IFNα or IFN-γ with IL-1β | Enhanced expression of TLR3 | [58,59] | |
| T1D patients and control subjects were recruited from South Africa, UK, Finland or Brazil respectively for the polymorphism screening | Significant association of SNP in theTLR3 pathways or polymorphism of the TLR3 gene with T1D. | [43,60-62] | |
| T1D patients and control objects were recruited from Poland or Norway respectively to test for polymorphism screening | No significant association of TLR3 polymorphism with T1D | [63,64] | |
| TLR4 | PBMC from T1D patients and healthy control subjects in the USA | Increased TLR4 expression in monocytes | [70] |
| Isolated human islets obtained from non-diabetic brain-dead donors | TLR4 mRNA and surface expression were restricted to β-cells | [72] | |
| TLR7 and TLR8 | DNA samples from the sib-pair families of two parents and two affected offspring from Asia Pacific, North America and Europe. | A significant association was observed between the Xp22 SNP (rs5979785) and T1D where the Xp22 SNP is located 30 kb centromeric of the functional candidate TLR8 and TLR7 genes | [99] |
| TLR9 | PBMC from T1D patients, their first-degree relatives and healthy controls in Czech Republic | Stimulation with CpG 2216 induced IFN-α production that was highest in relatives of T1D patients, with the exception of autoantibody-negative relatives bearing the protective haplotypes. | [89] |
| NLR | |||
| NLRP3 | Genomic DNAs extracted from PBMCs from 196 Brazilian children and adolescents with T1D, 59 with CD, and 165 with AD. | NLRP3 rs10754558 SNP was significantly associated with T1D | [114] |
The main immunotherapeutic strategies to intervene in T1D have attempted to either induce antigen-specific tolerance with known antigens or target specific inflammatory cytokines or immune cell subsets based on successful preclinical data in murine models. Although some clinical trials have shown promising results, such as short-term prolonged β cell survival and reduced insulin intake, most of these trials have so far failed to deliver long-term benefits [53, 115, 116]. The fact that T1D is a complex disease requires new strategies for immunotherapy and combination therapy may be a promising approach to advance our fight with T1D. Including selective targeting of innate immune responses may provide additional adjuvants or alternative components for consideration in combination therapy.
Highlights.
Toll-like receptors (TLRs) and nucleotide-binding domain, leucine-rich repeat-containing (or NOD-like) receptors (NLRs) are the most studied receptors of innate immunity.
TLRs and NLRs play an important role in the immunopathogenesis of type 1 diabetes (T1D) in both animal models and human T1D.
Selective targeting of innate immune responses could provide novel therapeutic strategies to prevent and/or treat the disease.
Acknowledgements
This work was supported by grants MRC G0901155, MR/K021141/1 DUK, 08/3719, 11/4319 EFSD/NN 2014 to FSW and NIH grants DK088181, DK092882 and the Yale Diabetes Research Center (P30-DK-45735) to LW and JDRF grant (1-INO-2015-136) to LW.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature. 2010;464:1293–300. doi: 10.1038/nature08933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wallberg M, Cooke A. Immune mechanisms in type 1 diabetes. Trends Immunol. 2013;34:583–91. doi: 10.1016/j.it.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 3.Aronson R, Gottlieb PA, Christiansen JS, Donner TW, Bosi E, Bode BW, et al. Low-dose otelixizumab anti-CD3 monoclonal antibody DEFEND-1 study: results of the randomized phase III study in recent-onset human type 1 diabetes. Diabetes Care. 2014;37:2746–54. doi: 10.2337/dc13-0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lehuen A, Diana J, Zaccone P, Cooke A. Immune cell crosstalk in type 1 diabetes. Nat Rev Immunol. 2010;10:501–13. doi: 10.1038/nri2787. [DOI] [PubMed] [Google Scholar]
- 5.Moran A, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, et al. Interleukin-1 antagonism in type 1 diabetes of recent onset: two multicentre, randomised, double-blind, placebo-controlled trials. Lancet. 2013;381:1905–15. doi: 10.1016/S0140-6736(13)60023-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Orban T, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, et al. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial. Lancet. 2011;378:412–9. doi: 10.1016/S0140-6736(11)60886-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, Becker DJ, Gitelman SE, Goland R, et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med. 2009;361:2143–52. doi: 10.1056/NEJMoa0904452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sherry N, Hagopian W, Ludvigsson J, Jain SM, Wahlen J, Ferry RJ, Jr., et al. Teplizumab for treatment of type 1 diabetes (Protege study): 1-year results from a randomised, placebo-controlled trial. Lancet. 2011;378:487–97. doi: 10.1016/S0140-6736(11)60931-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vudattu NK, Herold KC. Treatment of new onset type 1 diabetes with teplizumab: successes and pitfalls in development. Expert Opin Biol Ther. 2014;14:377–85. doi: 10.1517/14712598.2014.881797. [DOI] [PubMed] [Google Scholar]
- 10.Diana J, Gahzarian L, Simoni Y, Lehuen A. Innate immunity in type 1 diabetes. Discov Med. 2011;11:513–20. [PubMed] [Google Scholar]
- 11.Tlaskalova-Hogenova H, Stepankova R, Kozakova H, Hudcovic T, Vannucci L, Tuckova L, et al. The role of gut microbiota (commensal bacteria) and the mucosal barrier in the pathogenesis of inflammatory and autoimmune diseases and cancer: contribution of germ-free and gnotobiotic animal models of human diseases. Cell Mol Immunol. 2011;8:110–20. doi: 10.1038/cmi.2010.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature. 2008;455:1109–13. doi: 10.1038/nature07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chu H, Mazmanian SK. Innate immune recognition of the microbiota promotes host-microbial symbiosis. Nat Immunol. 2013;14:668–75. doi: 10.1038/ni.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kamada N, Seo SU, Chen GY, Nunez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol. 2013;13:321–35. doi: 10.1038/nri3430. [DOI] [PubMed] [Google Scholar]
- 15.Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9:313–23. doi: 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu HJ, Wu E. The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes. 2012;3:4–14. doi: 10.4161/gmic.19320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Medzhitov R, Janeway C., Jr. Innate immunity. N Engl J Med. 2000;343:338–44. doi: 10.1056/NEJM200008033430506. [DOI] [PubMed] [Google Scholar]
- 18.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–26. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 19.Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol. 2015;33:257–90. doi: 10.1146/annurev-immunol-032414-112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawai T, Akira S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol. 2009;21:317–37. doi: 10.1093/intimm/dxp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuchroo VK, Ohashi PS, Sartor RB, Vinuesa CG. Dysregulation of immune homeostasis in autoimmune diseases. Nat Med. 2012;18:42–7. doi: 10.1038/nm.2621. [DOI] [PubMed] [Google Scholar]
- 22.Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. 2014;5:461. doi: 10.3389/fimmu.2014.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 24.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–84. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 25.O'Neill LA, Golenbock D, Bowie AG. The history of Toll-like receptors - redefining innate immunity. Nat Rev Immunol. 2013;13:453–60. doi: 10.1038/nri3446. [DOI] [PubMed] [Google Scholar]
- 26.Zipris D. Innate immunity and its role in type 1 diabetes. Curr Opin Endocrinol Diabetes Obes. 2008;15:326–31. doi: 10.1097/MED.0b013e3283073a46. [DOI] [PubMed] [Google Scholar]
- 27.Roesch LF, Lorca GL, Casella G, Giongo A, Naranjo A, Pionzio AM, et al. Culture-independent identification of gut bacteria correlated with the onset of diabetes in a rat model. ISME J. 2009;3:536–48. doi: 10.1038/ismej.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gulden E, Wen L. Toll-Like Receptor Activation in Immunity vs. Tolerance in Autoimmune Diabetes. Front Immunol. 2014;5:119. doi: 10.3389/fimmu.2014.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–20. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 30.Liu Y, Yin H, Zhao M, Lu Q. TLR2 and TLR4 in autoimmune diseases: a comprehensive review. Clin Rev Allergy Immunol. 2014;47:136–47. doi: 10.1007/s12016-013-8402-y. [DOI] [PubMed] [Google Scholar]
- 31.Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik SG, et al. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell. 2007;130:1071–82. doi: 10.1016/j.cell.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 32.Oliveira-Nascimento L, Massari P, Wetzler LM. The Role of TLR2 in Infection and Immunity. Front Immunol. 2012;3:79. doi: 10.3389/fimmu.2012.00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim HS, Han MS, Chung KW, Kim S, Kim E, Kim MJ, et al. Toll-like receptor 2 senses beta-cell death and contributes to the initiation of autoimmune diabetes. Immunity. 2007;27:321–33. doi: 10.1016/j.immuni.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 34.Lee MS, Kim DH, Lee JC, Kim S, Kim HS. Role of TLR2 in the pathogenesis of autoimmune diabetes and its therapeutic implication. Diabetes Metab Res Rev. 2011;27:797–801. doi: 10.1002/dmrr.1231. [DOI] [PubMed] [Google Scholar]
- 35.Burrows MP, Volchkov P, Kobayashi KS, Chervonsky AV. Microbiota regulates type 1 diabetes through Toll-like receptors. Proc Natl Acad Sci U S A. 2015;112:9973–7. doi: 10.1073/pnas.1508740112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Filippi CM, Ehrhardt K, Estes EA, Larsson P, Oldham JE, von Herrath MG. TLR2 signaling improves immunoregulation to prevent type 1 diabetes. Eur J Immunol. 2011;41:1399–409. doi: 10.1002/eji.200939841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim DH, Lee JC, Kim S, Oh SH, Lee MK, Kim KW, et al. Inhibition of autoimmune diabetes by TLR2 tolerance. J Immunol. 2011;187:5211–20. doi: 10.4049/jimmunol.1001388. [DOI] [PubMed] [Google Scholar]
- 38.Liu H, Komai-Koma M, Xu D, Liew FY. Toll-like receptor 2 signaling modulates the functions of CD4+ CD25+ regulatory T cells. Proc Natl Acad Sci U S A. 2006;103:7048–53. doi: 10.1073/pnas.0601554103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sutmuller RP, den Brok MH, Kramer M, Bennink EJ, Toonen LW, Kullberg BJ, et al. Toll-like receptor 2 controls expansion and function of regulatory T cells. J Clin Invest. 2006;116:485–94. doi: 10.1172/JCI25439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karumuthil-Melethil S, Perez N, Li R, Vasu C. Induction of innate immune response through TLR2 and dectin 1 prevents type 1 diabetes. J Immunol. 2008;181:8323–34. doi: 10.4049/jimmunol.181.12.8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karumuthil-Melethil S, Sofi MH, Gudi R, Johnson BM, Perez N, Vasu C. TLR2- and Dectin 1-associated innate immune response modulates T-cell response to pancreatic beta-cell antigen and prevents type 1 diabetes. Diabetes. 2015;64:1341–57. doi: 10.2337/db14-1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–8. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 43.Assmann TS, Brondani Lde A, Boucas AP, Canani LH, Crispim D. Toll-like receptor 3 (TLR3) and the development of type 1 diabetes mellitus. Arch Endocrinol Metab. 2015;59:4–12. doi: 10.1590/2359-3997000000003. [DOI] [PubMed] [Google Scholar]
- 44.Dar AA, Patil RS, Chiplunkar SV. Insights into the Relationship between Toll Like Receptors and Gamma Delta T Cell Responses. Front Immunol. 2014;5:366. doi: 10.3389/fimmu.2014.00366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wesch D, Beetz S, Oberg HH, Marget M, Krengel K, Kabelitz D. Direct costimulatory effect of TLR3 ligand poly(I:C) on human gamma delta T lymphocytes. J Immunol. 2006;176:1348–54. doi: 10.4049/jimmunol.176.3.1348. [DOI] [PubMed] [Google Scholar]
- 46.Jiang Z, Mak TW, Sen G, Li X. Toll-like receptor 3-mediated activation of NF-kappaB and IRF3 diverges at Toll-IL-1 receptor domain-containing adapter inducing IFN-beta. Proc Natl Acad Sci U S A. 2004;101:3533–8. doi: 10.1073/pnas.0308496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Serreze DV, Hamaguchi K, Leiter EH. Immunostimulation circumvents diabetes in NOD/Lt mice. J Autoimmun. 1989;2:759–76. doi: 10.1016/0896-8411(89)90003-6. [DOI] [PubMed] [Google Scholar]
- 48.Sobel DO, Newsome J, Ewel CH, Bellanti JA, Abbassi V, Creswell K, et al. Poly I:C induces development of diabetes mellitus in BB rat. Diabetes. 1992;41:515–20. doi: 10.2337/diab.41.4.515. [DOI] [PubMed] [Google Scholar]
- 49.Sobel DO, Goyal D, Ahvazi B, Yoon JW, Chung YH, Bagg A, et al. Low dose poly I:C prevents diabetes in the diabetes prone BB rat. J Autoimmun. 1998;11:343–52. doi: 10.1006/jaut.1998.0203. [DOI] [PubMed] [Google Scholar]
- 50.Blankenhorn EP, Cort L, Greiner DL, Guberski DL, Mordes JP. Virus-induced autoimmune diabetes in the LEW.1WR1 rat requires Iddm14 and a genetic locus proximal to the major histocompatibility complex. Diabetes. 2009;58:2930–8. doi: 10.2337/db09-0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wen L, Peng J, Li Z, Wong FS. The effect of innate immunity on autoimmune diabetes and the expression of Toll-like receptors on pancreatic islets. J Immunol. 2004;172:3173–80. doi: 10.4049/jimmunol.172.5.3173. [DOI] [PubMed] [Google Scholar]
- 52.Hober D, Sauter P. Pathogenesis of type 1 diabetes mellitus: interplay between enterovirus and host. Nat Rev Endocrinol. 2010;6:279–89. doi: 10.1038/nrendo.2010.27. [DOI] [PubMed] [Google Scholar]
- 53.Coppieters KT, Boettler T, von Herrath M. Virus infections in type 1 diabetes. Cold Spring Harb Perspect Med. 2012;2:a007682. doi: 10.1101/cshperspect.a007682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Laitinen OH, Honkanen H, Pakkanen O, Oikarinen S, Hankaniemi MM, Huhtala H, et al. Coxsackievirus B1 is associated with induction of beta-cell autoimmunity that portends type 1 diabetes. Diabetes. 2014;63:446–55. doi: 10.2337/db13-0619. [DOI] [PubMed] [Google Scholar]
- 55.Oikarinen S, Tauriainen S, Hober D, Lucas B, Vazeou A, Sioofy-Khojine A, et al. Virus antibody survey in different European populations indicates risk association between coxsackievirus B1 and type 1 diabetes. Diabetes. 2014;63:655–62. doi: 10.2337/db13-0620. [DOI] [PubMed] [Google Scholar]
- 56.Aida K, Nishida Y, Tanaka S, Maruyama T, Shimada A, Awata T, et al. RIG-I- and MDA5-initiated innate immunity linked with adaptive immunity accelerates beta-cell death in fulminant type 1 diabetes. Diabetes. 2011;60:884–9. doi: 10.2337/db10-0795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Le Bon A, Durand V, Kamphuis E, Thompson C, Bulfone-Paus S, Rossmann C, et al. Direct stimulation of T cells by type I IFN enhances the CD8+ T cell response during cross-priming. J Immunol. 2006;176:4682–9. doi: 10.4049/jimmunol.176.8.4682. [DOI] [PubMed] [Google Scholar]
- 58.Hultcrantz M, Huhn MH, Wolf M, Olsson A, Jacobson S, Williams BR, et al. Interferons induce an antiviral state in human pancreatic islet cells. Virology. 2007;367:92–101. doi: 10.1016/j.virol.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 59.Ylipaasto P, Kutlu B, Rasilainen S, Rasschaert J, Salmela K, Teerijoki H, et al. Global profiling of coxsackievirus- and cytokine-induced gene expression in human pancreatic islets. Diabetologia. 2005;48:1510–22. doi: 10.1007/s00125-005-1839-7. [DOI] [PubMed] [Google Scholar]
- 60.Pirie FJ, Pegoraro R, Motala AA, Rauff S, Rom L, Govender T, et al. Toll-like receptor 3 gene polymorphisms in South African Blacks with type 1 diabetes. Tissue Antigens. 2005;66:125–30. doi: 10.1111/j.1399-0039.2005.00454.x. [DOI] [PubMed] [Google Scholar]
- 61.Eleftherohorinou H, Wright V, Hoggart C, Hartikainen AL, Jarvelin MR, Balding D, et al. Pathway analysis of GWAS provides new insights into genetic susceptibility to 3 inflammatory diseases. PLoS One. 2009;4:e8068. doi: 10.1371/journal.pone.0008068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Assmann TS, Brondani Lde A, Bauer AC, Canani LH, Crispim D. Polymorphisms in the TLR3 gene are associated with risk for type 1 diabetes mellitus. Eur J Endocrinol. 2014;170:519–27. doi: 10.1530/EJE-13-0963. [DOI] [PubMed] [Google Scholar]
- 63.Fichna M, Zurawek M, Fichna P, Januszkiewicz-Lewandowska D, Ruchala M, Nowak J. Polymorphisms of the Toll-Like Receptor-3 Gene in Autoimmune Adrenal Failure and Type 1 Diabetes in Polish Patients. Arch Immunol Ther Exp (Warsz) 2015 doi: 10.1007/s00005-015-0360-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Witso E, Cinek O, Tapia G, Brorsson CA, Stene LC, Gjessing HK, et al. Genetic Determinants of Enterovirus Infections: Polymorphisms in Type 1 Diabetes and Innate Immune Genes in the MIDIA Study. Viral Immunol. 2015;28:556–63. doi: 10.1089/vim.2015.0067. [DOI] [PubMed] [Google Scholar]
- 65.Wong FS, Hu C, Zhang L, Du W, Alexopoulou L, Flavell RA, et al. The role of Toll-like receptors 3 and 9 in the development of autoimmune diabetes in NOD mice. Ann N Y Acad Sci. 2008;1150:146–8. doi: 10.1196/annals.1447.039. [DOI] [PubMed] [Google Scholar]
- 66.Lang KS, Recher M, Junt T, Navarini AA, Harris NL, Freigang S, et al. Toll-like receptor engagement converts T-cell autoreactivity into overt autoimmune disease. Nat Med. 2005;11:138–45. doi: 10.1038/nm1176. [DOI] [PubMed] [Google Scholar]
- 67.McCall KD, Thuma JR, Courreges MC, Benencia F, James CB, Malgor R, et al. Toll-like receptor 3 is critical for coxsackievirus B4-induced type 1 diabetes in female NOD mice. Endocrinology. 2015;156:453–61. doi: 10.1210/en.2013-2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee AS, Ghoreishi M, Cheng WK, Chang TY, Zhang YQ, Dutz JP. Toll-like receptor 7 stimulation promotes autoimmune diabetes in the NOD mouse. Diabetologia. 2011;54:1407–16. doi: 10.1007/s00125-011-2083-y. [DOI] [PubMed] [Google Scholar]
- 69.Pane JA, Webster NL, Coulson BS. Rotavirus activates lymphocytes from non-obese diabetic mice by triggering toll-like receptor 7 signaling and interferon production in plasmacytoid dendritic cells. PLoS Pathog. 2014;10:e1003998. doi: 10.1371/journal.ppat.1003998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Devaraj S, Dasu MR, Rockwood J, Winter W, Griffen SC, Jialal I. Increased toll-like receptor (TLR) 2 and TLR4 expression in monocytes from patients with type 1 diabetes: further evidence of a proinflammatory state. J Clin Endocrinol Metab. 2008;93:578–83. doi: 10.1210/jc.2007-2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mohammad MK, Morran M, Slotterbeck B, Leaman DW, Sun Y, Grafenstein H, et al. Dysregulated Toll-like receptor expression and signaling in bone marrow-derived macrophages at the onset of diabetes in the non-obese diabetic mouse. Int Immunol. 2006;18:1101–13. doi: 10.1093/intimm/dxl045. [DOI] [PubMed] [Google Scholar]
- 72.Garay-Malpartida HM, Mourao RF, Mantovani M, Santos IA, Sogayar MC, Goldberg AC. Toll-like receptor 4 (TLR4) expression in human and murine pancreatic beta-cells affects cell viability and insulin homeostasis. BMC Immunol. 2011;12:18. doi: 10.1186/1471-2172-12-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li M, Song L, Gao X, Chang W, Qin X. Toll-like receptor 4 on islet beta cells senses expression changes in high-mobility group box 1 and contributes to the initiation of type 1 diabetes. Exp Mol Med. 2012;44:260–7. doi: 10.3858/emm.2012.44.4.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bednar KJ, Tsukamoto H, Kachapati K, Ohta S, Wu Y, Katz JD, et al. Reversal of New-Onset Type 1 Diabetes With an Agonistic TLR4/MD-2 Monoclonal Antibody. Diabetes. 2015;64:3614–26. doi: 10.2337/db14-1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gulden E, Ihira M, Ohashi A, Reinbeck AL, Freudenberg MA, Kolb H, et al. Toll-like receptor 4 deficiency accelerates the development of insulin-deficient diabetes in non-obese diabetic mice. PLoS One. 2013;8:e75385. doi: 10.1371/journal.pone.0075385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tian J, Zekzer D, Hanssen L, Lu Y, Olcott A, Kaufman DL. Lipopolysaccharide-activated B cells down-regulate Th1 immunity and prevent autoimmune diabetes in nonobese diabetic mice. J Immunol. 2001;167:1081–9. doi: 10.4049/jimmunol.167.2.1081. [DOI] [PubMed] [Google Scholar]
- 77.Uematsu S, Fujimoto K, Jang MH, Yang BG, Jung YJ, Nishiyama M, et al. Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing Toll-like receptor 5. Nat Immunol. 2008;9:769–76. doi: 10.1038/ni.1622. [DOI] [PubMed] [Google Scholar]
- 78.Choi YJ, Im E, Chung HK, Pothoulakis C, Rhee SH. TRIF mediates Toll-like receptor 5-induced signaling in intestinal epithelial cells. J Biol Chem. 2010;285:37570–8. doi: 10.1074/jbc.M110.158394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, et al. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010;328:228–31. doi: 10.1126/science.1179721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Weile C, Josefsen K, Buschard K. Glucose activation of islets of Langerhans up-regulates Toll-like receptor 5: possible mechanism of protection. Clin Exp Immunol. 2011;166:251–7. doi: 10.1111/j.1365-2249.2011.04457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Applequist SE, Wallin RP, Ljunggren HG. Variable expression of Toll-like receptor in murine innate and adaptive immune cell lines. Int Immunol. 2002;14:1065–74. doi: 10.1093/intimm/dxf069. [DOI] [PubMed] [Google Scholar]
- 82.Wu O, Chen GP, Chen H, Li XP, Xu JH, Zhao SS, et al. The expressions of Toll-like receptor 9 and T-bet in circulating B and T cells in newly diagnosed, untreated systemic lupus erythematosus and correlations with disease activity and laboratory data in a Chinese population. Immunobiology. 2009;214:392–402. doi: 10.1016/j.imbio.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 83.Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity. 2010;32:305–15. doi: 10.1016/j.immuni.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 84.Hammond T, Lee S, Watson MW, Flexman JP, Cheng W, Fernandez S, et al. Toll-like receptor (TLR) expression on CD4+ and CD8+ T-cells in patients chronically infected with hepatitis C virus. Cell Immunol. 2010;264:150–5. doi: 10.1016/j.cellimm.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 85.Agudo J, Ruzo A, Tung N, Salmon H, Leboeuf M, Hashimoto D, et al. The miR-126-VEGFR2 axis controls the innate response to pathogen-associated nucleic acids. Nat Immunol. 2014;15:54–62. doi: 10.1038/ni.2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005;23:19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 87.Tai N, Wong FS, Wen L. TLR9 deficiency promotes CD73 expression in T cells and diabetes protection in nonobese diabetic mice. J Immunol. 2013;191:2926–37. doi: 10.4049/jimmunol.1300547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang Y, Lee AS, Shameli A, Geng X, Finegood D, Santamaria P, et al. TLR9 blockade inhibits activation of diabetogenic CD8+ T cells and delays autoimmune diabetes. J Immunol. 2010;184:5645–53. doi: 10.4049/jimmunol.0901814. [DOI] [PubMed] [Google Scholar]
- 89.Kayserova J, Vcelakova J, Stechova K, Dudkova E, Hromadkova H, Sumnik Z, et al. Decreased dendritic cell numbers but increased TLR9-mediated interferon-alpha production in first degree relatives of type 1 diabetes patients. Clin Immunol. 2014;153:49–55. doi: 10.1016/j.clim.2014.03.018. [DOI] [PubMed] [Google Scholar]
- 90.Wong FS, Wen L. IFN-alpha can both protect against and promote the development of type 1 diabetes. Ann N Y Acad Sci. 2008;1150:187–9. doi: 10.1196/annals.1447.031. [DOI] [PubMed] [Google Scholar]
- 91.Gianchecchi E, Crino A, Giorda E, Luciano R, Perri V, Russo AL, et al. Altered B cell homeostasis and toll-like receptor 9-driven response in type 1 diabetes carriers of the C1858T PTPN22 allelic variant: implications in the disease pathogenesis. PLoS One. 2014;9:e110755. doi: 10.1371/journal.pone.0110755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336:1268–73. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Brandl K, Plitas G, Schnabl B, DeMatteo RP, Pamer EG. MyD88-mediated signals induce the bactericidal lectin RegIII gamma and protect mice against intestinal Listeria monocytogenes infection. J Exp Med. 2007;204:1891–900. doi: 10.1084/jem.20070563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, Hooper LV. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc Natl Acad Sci U S A. 2008;105:20858–63. doi: 10.1073/pnas.0808723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Perry AK, Chen G, Zheng D, Tang H, Cheng G. The host type I interferon response to viral and bacterial infections. Cell Res. 2005;15:407–22. doi: 10.1038/sj.cr.7290309. [DOI] [PubMed] [Google Scholar]
- 96.Longhi MP, Trumpfheller C, Idoyaga J, Caskey M, Matos I, Kluger C, et al. Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J Exp Med. 2009;206:1589–602. doi: 10.1084/jem.20090247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sporri R, Reis e Sousa C. Inflammatory mediators are insufficient for full dendritic cell activation and promote expansion of CD4+ T cell populations lacking helper function. Nat Immunol. 2005;6:163–70. doi: 10.1038/ni1162. [DOI] [PubMed] [Google Scholar]
- 98.Sun C, Zhi D, Shen S, Luo F, Sanjeevi CB. SNPs in the exons of Toll-like receptors are associated with susceptibility to type 1 diabetes in Chinese population. Hum Immunol. 2014;75:1084–8. doi: 10.1016/j.humimm.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 99.Cooper JD, Walker NM, Smyth DJ, Downes K, Healy BC, Todd JA, et al. Follow-up of 1715 SNPs from the Wellcome Trust Case Control Consortium genome-wide association study in type I diabetes families. Genes Immun. 2009;10(Suppl 1):S85–94. doi: 10.1038/gene.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Claes AK, Zhou JY, Philpott DJ. NOD-Like Receptors: Guardians of Intestinal Mucosal Barriers. Physiology (Bethesda) 2015;30:241–50. doi: 10.1152/physiol.00025.2014. [DOI] [PubMed] [Google Scholar]
- 101.Geddes K, Magalhaes JG, Girardin SE. Unleashing the therapeutic potential of NOD-like receptors. Nat Rev Drug Discov. 2009;8:465–79. doi: 10.1038/nrd2783. [DOI] [PubMed] [Google Scholar]
- 102.Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol. 2012:4. doi: 10.1101/cshperspect.a006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Berrington WR, Iyer R, Wells RD, Smith KD, Skerrett SJ, Hawn TR. NOD1 and NOD2 regulation of pulmonary innate immunity to Legionella pneumophila. Eur J Immunol. 2010;40:3519–27. doi: 10.1002/eji.201040518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hasegawa M, Yamazaki T, Kamada N, Tawaratsumida K, Kim YG, Nunez G, et al. Nucleotide-binding oligomerization domain 1 mediates recognition of Clostridium difficile and induces neutrophil recruitment and protection against the pathogen. J Immunol. 2011;186:4872–80. doi: 10.4049/jimmunol.1003761. [DOI] [PubMed] [Google Scholar]
- 105.Werts C, le Bourhis L, Liu J, Magalhaes JG, Carneiro LA, Fritz JH, et al. Nod1 and Nod2 induce CCL5/RANTES through the NF-kappaB pathway. Eur J Immunol. 2007;37:2499–508. doi: 10.1002/eji.200737069. [DOI] [PubMed] [Google Scholar]
- 106.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13:397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.McCoy AJ, Koizumi Y, Higa N, Suzuki T. Differential regulation of caspase-1 activation via NLRP3/NLRC4 inflammasomes mediated by aerolysin and type III secretion system during Aeromonas veronii infection. J Immunol. 2010;185:7077–84. doi: 10.4049/jimmunol.1002165. [DOI] [PubMed] [Google Scholar]
- 108.Schroder K, Zhou R, Tschopp J. The NLRP3 inflammasome: a sensor for metabolic danger? Science. 2010;327:296–300. doi: 10.1126/science.1184003. [DOI] [PubMed] [Google Scholar]
- 109.Gris D, Ye Z, Iocca HA, Wen H, Craven RR, Gris P, et al. NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J Immunol. 2010;185:974–81. doi: 10.4049/jimmunol.0904145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kolly L, Karababa M, Joosten LA, Narayan S, Salvi R, Petrilli V, et al. Inflammatory role of ASC in antigen-induced arthritis is independent of caspase-1, NALP-3, and IPAF. J Immunol. 2009;183:4003–12. doi: 10.4049/jimmunol.0802173. [DOI] [PubMed] [Google Scholar]
- 111.Schott WH, Haskell BD, Tse HM, Milton MJ, Piganelli JD, Choisy-Rossi CM, et al. Caspase-1 is not required for type 1 diabetes in the NOD mouse. Diabetes. 2004;53:99–104. doi: 10.2337/diabetes.53.1.99. [DOI] [PubMed] [Google Scholar]
- 112.Wen L, Green EA, Stratmann T, Panosa A, Gomis R, Eynon EE, et al. In vivo diabetogenic action of CD4+ T lymphocytes requires Fas expression and is independent of IL-1 and IL-18. Eur J Immunol. 2011;41:1344–51. doi: 10.1002/eji.201041216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hu C, Ding H, Li Y, Pearson JA, Zhang X, Flavell RA, et al. NLRP3 deficiency protects from type 1 diabetes through the regulation of chemotaxis into the pancreatic islets. Proc Natl Acad Sci U S A. 2015;112:11318–23. doi: 10.1073/pnas.1513509112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pontillo A, Brandao L, Guimaraes R, Segat L, Araujo J, Crovella S. Two SNPs in NLRP3 gene are involved in the predisposition to type-1 diabetes and celiac disease in a pediatric population from northeast Brazil. Autoimmunity. 2010;43:583–9. doi: 10.3109/08916930903540432. [DOI] [PubMed] [Google Scholar]
- 115.Brooks-Worrell B, Palmer JP. Prevention versus intervention of type 1 diabetes. Clin Immunol. 2013;149:332–8. doi: 10.1016/j.clim.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 116.Herold KC, Vignali DA, Cooke A, Bluestone JA. Type 1 diabetes: translating mechanistic observations into effective clinical outcomes. Nat Rev Immunol. 2013;13:243–56. doi: 10.1038/nri3422. [DOI] [PMC free article] [PubMed] [Google Scholar]