

Abstract
Chronic exposures to arsenic had been associated with metabolism diseases. Peroxisome proliferator-activated receptor gamma (PPARγ) was found in the liver, regulated metabolism. Here, we found that the expression of PPARγ was decreased, the generation of reactive oxygen species (ROS) and autophagy were increased after treatment with As2O3 in offsprings’ livers. Taurine (Tau), a sulfur-containing β–amino acid could reverse As2O3-inhibited PPARγ. Tau also inhibit the generation of ROS and autophagy. We also found that As2O3 caused autophagic cell death and ROS accelerated in HepG2 cells. Before incubation with As2O3, the cells were pretreated with PPARγ activator Rosiglitazone (RGS), we found that autophagy and ROS was inhibited in HepG2 cells, suggesting that inhibition of PPARγ contributed to As2O3-induced autophagy and the generation of ROS. After pretreatment with Tau, the level of PPARγ was improved and the autophagy and ROS was inhibited in As2O3-treated cells, suggesting that Tau could protect hepatocytes against As2O3 through modulating PPARγ pathway.
Arsenic was a ubiquitous naturally occurring metalloid toxicant and carcinogen that found in groundwater, food, dust, and ambient air. It posed health risks to more than 2% of the world population. Chronic arsenic exposure increased the risk of a number of cancers and chronic noncancer diseases, including cardiovascular, pulmonary, and metabolic diseases1,2. Prenatal arsenic exposure had been associated with altered gene expression in human cord blood leukocytes and various target tissues in rodents3. Transplacental studies in mice showed that the offspring of dams who were given, 42.5 ppm and 85 ppm arsenic via drinking water from gestational day 8 to 18 (last two-thirds of pregnancy) had a dose-dependent increase in liver, lung, ovary, and adrenal tumors when they reached adulthood. Furthermore, mice that received arsenic exposure in utero and throughout their life course developed more frequent and aggressive tumors at much lower doses compared with mice that only received arsenic exposure during the gestational period4. Inorganic arsenicals were converted to monomethylarsenic acid (MMA) and dimethylarsenicacid (DMA) in the body, predominantly in the liver5. Therefore, exposure to arsenic might induce hepatotoxicity.
Autophagy occurred in all nucleated type cells and the process of autophagic flux was essential in animal, plant, and yeast cells6,7. It was a highly regulated lysosomal pathway involved in the recycling of cytosol and the removal of superfluous or damaged organelles. Autophagy was essential for the survival, differentiation, development and homeostasis of cells. Dysregulated autophagy had been suggested to play pathogenic roles in a variety of disease processes including cancer, neurodegeneration, diabetes, aging and heart disease8,9. In the process of autophagy, some autophagy-related (ATG) family members were transcriptionally induced and the conversion of LC3, a protein involved in autophagosome formation, from LC3-I to LC3-II can be facilitated10,11. LC3 was an autophagy biomarker that was lapidated during induction of autophagic flux and was required for autophagosome formation. Another autophagy marker was p62/SQSTM1. P62 played an important role in the degradation of polyubiquitinated substrates by autophagy, thus causing its own degradation12. Our previous studies had indicated that arsenic increased autophagosome formation and caused autophagic cell death in INS-1 cells13. Whether arsenic exerted hepatotoxicity and the precise molecular mechanisms of arsenic hepatotoxicity were not completely elucidated.
The peroxisome proliferator-activated receptors (PPARs) were a family of nuclear fatty acid receptors that regulated tissue specific cellular metabolism and differentiation14. In humans, there were three PPAR isoforms, α, β and γ, which respond to a discrete set of ligands15. Peroxisome proliferator-activated receptor-γ (PPARγ) was a ligand-activated transcription factor of the nuclear hormone receptor super family. PPARγ was highly expressed in adipocytes and the liver10,14,16. These nuclear receptors when activated directly bind to DNA and regulated gene expression through transcriptional activation (also called master regulators of transcriptional cascades). PPARγ was involved in a variety of physiological processes, including the regulation of the metabolism, inflammation, cellular growth and differentiation17,18. Previous study had found that the disruption of PPARγ might result the activation of autophagy19. In our study, we explored the relationship between PPARγ and the autophagy induced by arsenic in offsprings’ livers.
PPARγ was activated by the thiazolidinediones (TZDs), a group of drugs widely used in patients in the management of type 2 diabetes (T2D), as they regulate glucose metabolism, adiposeness, differentiation, and the expression of several genes including antioxidant defenses14,20. However, TZDs were once withdrawn from the market or had restricted prescription because they provoked adverse effects such as weight gain, edema, liver injury, cancer, and heart failure21. Taurine (Tau), a sulfur-containing β–amino acid, was a major free intracellular amino acid present in many tissues of human and animals22. It was mainly distributed in the brain, heart, kidney, and reproductive organs, with many physiological activities23. Biosynthesis and dietary intake were the only sources of Tau in our bodies. Tau was synthesized from methionine and cysteine mainly in the liver and the biosynthetic capacity of Tau was very low in human24,25. Its recognized metabolic function in liver was conjugation with bile acids, which was important for bile secretion and lipid digestion24,26. Recent studies reported that Tau supplementation could prevent diabetes mellitus, insulin resistance and its complications9,24. Tau was a key determinant of oxidative phosphorylation and it played an essential role in mitochondria. It was reported that mitochondrial ROS generation was enhanced in taurine-deficient heart27,28. Nrf2 played a critical role in defending against oxidative stress and inflammation29. When oxidative stress was stimulated, Nrf2 would bind to antioxidant response elements (ARE)30. Eventually, it led to the restoration of cellular redox homeostasis31. The thioredoxin (Trx) system also played the antioxidant role. The cysteine disulfide bridges in oxidized proteins were reduced by Trx system32. It was found that PPARγ could up-regulate Nrf233,34. Our previous study had found that Tau protected against As2O3-induced autophagy in pancreas of rat offsprings through Nrf2/Trx pathway35. Therefore, we investigated whether Tau executed the action of protecting hepatocytes against As2O3 through modulating PPARγ/Nrf2 pathway in this study.
Results
As2O3 changed offsprings’ livers morphology
Pathological analysis showed that the size of hepatocyte was decreased after treatment with As2O3 (Fig. 1). We found that hepacyte edema occurred in As2O3-treated offsprings’ livers. The size of As2O3-treatedhepatocyte was increased and the cellular edema was disappeared after pretreatment with Tau.
Figure 1. The morphology of As2O3-treated offsprings’ livers by hematein eosin (scale bar = 50 μM).

Arrows indicate hepatic cells.
The effects of As2O3 on offsprings’ livers weight and body weight
The offsprings’ liver weight gains were significantly increased after treatment with As2O3, but As2O3 did not affect the body weights (Table 1). After pretreatment with Tau, the liver weights were significantly decreased (Table 1). These changes might be related with the cellular edema in offsprings’ livers.
Table 1. Effects of As2O3 (2 mg/kg BW to 8 mg/kg BW) on liver and body weights at postnatal day 42 (PND 42) in offspring rats.
| Treatment | Body weight (g) | Liver weight (g) |
|---|---|---|
| 0 | 228.37 ± 14.32 | 6.83 ± 0.55 |
| 2 | 232.56 ± 17.08 | 7.23 ± 0.65* |
| 4 | 239.87 ± 16.62 | 7.95 ± 0.03* |
| 8 | 241.03 ± 17.12 | 8.27 ± 0.51* |
| 8+Tau | 232.13 ± 8.69 | 7.35 ± 0.07# |
Values are means ± SD (n = 7). *p < 0.05 vs control; #p < 0.05 vs 8 mg/kg BW.
As2O3 caused autophagosome accumulated in livers
To investigate whether autophagy was involved in As2O3-induced toxicity, we utilized transmission electron microscopy to observe the ultrastructure in As2O3-treatedlivers. Morphological hallmark of autophagy was the presence of autophagosomes (Fig. 2A). Quantification of the autophagosomes numbers per cell demonstrated that As2O3 increased autophagosomes number significantly and in a dose-dependent manner. The number of autophagosomes in As2O3-treated rats was obviously decreased by pretreatment with Tau (Fig. 2B).
Figure 2. Ultrastructural features of As2O3-treated offsprings’ livers by transmission electron microscope.

(A) The rats were treated with 2 mg/kg BW to 8 mg/kg BW As2O3. The rats of group 5 were pretreated with Tau before treatment with 8 mg/kg BW As2O3. Arrows indicate autophagosomes (scale bar = 20 μM). (B) Comparison of the numbers of autophagosomes per viable cell (n = 15). Bar represents mean ± SD. (**P < 0.01 vs. control; ##P < 0.01).
As2O3 activated autophagy in offsprings’ livers
In this study, we evaluated the expression of autophagy biomarkers, LC3-II and P62, by Western blot analysis (Fig. 3A,C). The level of LC3-II was increased dramatically and the level of P62 was decreased in As2O3-treated livers as shown in Western blot assay. After pretreatment with Tau, the expression of LC3-II was decreased and the expression of P62 was increased dramatically in As2O3-treated cells (Fig. 3B,D), this gave us a clue to investigate the correspondence of autophagy and cytotoxicity at different As2O3 concentration and whether Tau could protect against the autophagy induced by As2O3.
Figure 3. Expression of LC3-II and P62 in As2O3-treated offsprings’ livers.

(A) The rats were treated with 2 mg/kg BW to 8 mg/kg BW As2O3. The rats of group 5 were pretreated with Tau before treatment with 8 mg/kg BW As2O3. The protein fraction was analyzed by Western blot. β-actin was taken as internal control. (B) Densitometric analyses of LC3-II expressed in livers (**P < 0.01 vs. control; ##P < 0.01). (C) Expression of P62 in As2O3-treated offsprings’ livers. (D) Densitometric analyses of P62 expressed in livers (**P < 0.01 vs. control; ##P < 0.01).
As2O3 reduced the expression of PPARγ in offsprings’ livers
PPARγ was a metabolism related protein in livers. The expression of PPARγ was decreased in As2O3-treated offsprings’ livers (Fig. 4A). Tau could protect against the reduction of PPARγ induced by As2O3 in offsprings’ livers (Fig. 4B).
Figure 4. PPARγ protein levels in As2O3-treated livers.

(A) The rats were treated with 2 mg/kg BW to 8 mg/kg BW As2O3. The rats of group 5 were pretreated with Tau before treatment with 8 mg/kg BW As2O3. The protein fraction was analyzed by Western blot. β-actin was taken as internal control. (B) Densitometric analyses of PPARγ levels in offsprings’ livers (**P < 0.01 vs. control; ##P < 0.01).
As2O3 reduced the expression of PPARγ gene in offsprings’ livers
Because of the reduction of PPARγ protein, we used RT-PCR to measure the changes of PPARγ gene levels. The data showed that the expression of PPARγ was decreased significantly in a dose-dependent manner in As2O3-treated livers (Fig. 5A). After pretreatment with Tau, the expression of PPARγ was accelerated significantly in As2O3-treated offsprings’ livers (Fig. 5B).
Figure 5. PPARγ gene levels in offsprings’ livers treated with As2O3.
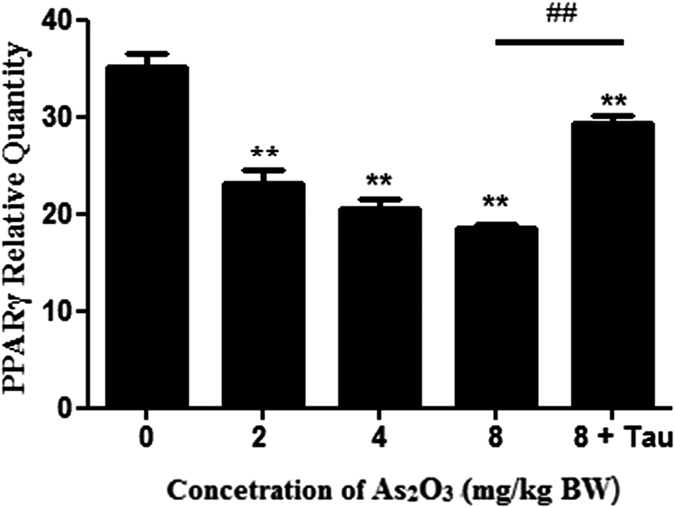
(A) The rats were treated with 2 mg/kg BW to 8 mg/kg BW As2O3. The rats of group 5 were pretreated with Tau before treatment with 8 mg/kg BW As2O3. The gene fraction was analyzed by RT-PCR. GADPH was taken as internal control. (B) Densitometric analyses of PPARγ levels in offsprings’ livers. Relative levels of PPARγ was expressed as a percentage of GADPH (**P < 0.01 vs. control; ##P < 0.01).
As2O3 reduced the expression of Nrf2 and Trx proteins in offsprings’ livers
The expression of Nrf2 and Trx proteins were decreased in As2O3-treated offsprings’ livers (Fig. 6A). Tau could increase the reduction of Nrf2 and Trx induced by As2O3 in offsprings’ livers (Fig. 6B,C).
Figure 6. Nrf2 and Trx protein levels in As2O3-treated livers.

(A) The rats were treated with 2 mg/kg BW to 8 mg/kg BW As2O3. The rats of group 5 were pretreated with Tau before treatment with 8 mg/kg BW As2O3. The protein fraction was analyzed by Western blot. β-actin was taken as internal control. (B) Densitometric analyses of Nrf2 levels in offsprings’ livers (**P < 0.01 vs. control; ##P < 0.01). (C) Densitometric analyses of Trx levels in offsprings’ livers (**P < 0.01 vs. control; ##P < 0.01).
As2O3 accelerated Malonic Dialdehyde (MDA) level in offsprings’ livers
As2O3 could induce autophagy through ROS generation in vitro13. To investigate the level of oxidative stress, we used MDA test kit to measure the level of MDA. We found that the level of MDA was increased significantly after treatment with As2O3. After pretreatment with Tau, the level of MDA was decreased in the livers of As2O3-treated rats (Fig. 7). It suggested that treatment with As2O3 could accelerate the generation of ROS, and pretreatment with Tau could withstand the oxidative stress induced by As2O3.
Figure 7. Effects of As2O3 on MDA level.
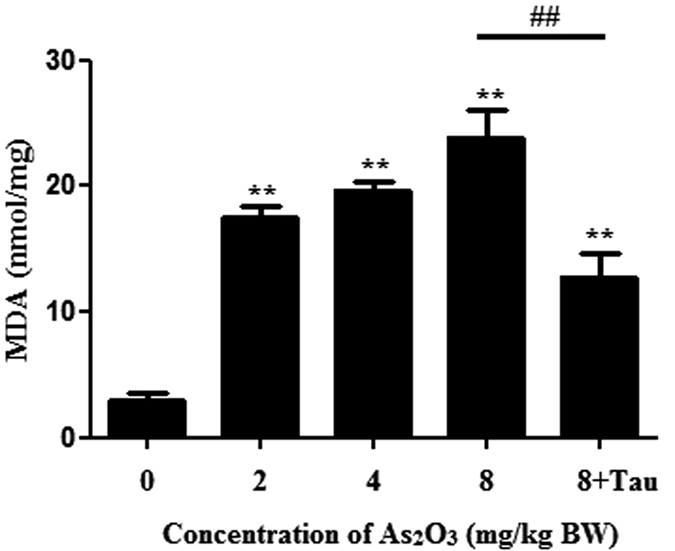
The rats were treated with 2 mg/kg BW to 8 mg/kg BW As2O3. The rats of group 5 were pretreated with Tau before treatment with 8 mg/kg BW As2O3. The level of MDA was measured by MDA test kit (#P < 0.05; **P < 0.01 vs. control).
As2O3 caused autophagic cell death in HepG2 cells
The treatment of HepG2 cells with 1 μM As2O3 for 24 h resulted in cell death in this study36. The cell viability of As2O3-treated HepG2 cells was increased by knockdown of Atg5 with Atg5 siRNA (Fig. 8). This suggested that autophagy was the major cause of the cell death induced by As2O3.
Figure 8. Effect of autophagy on cell viability of As2O3-treated cells.
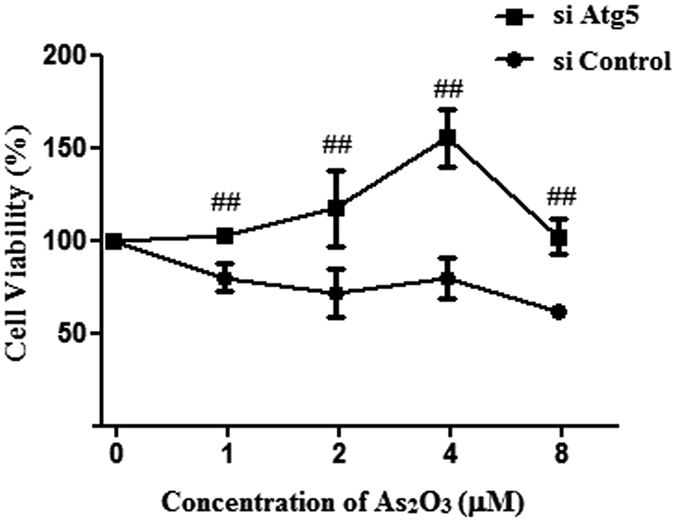
HepG2 cells were transfected with either 50 nM siRNA against human Atg5 (si Atg5) or scrambled control siRNA (si Control), and then treated with 1 μM–8 μM As2O3 for 24 h. The cell viability was assessed using the MTT assay. The trace represents the means ± SD. (##P < 0.01 vs. cells transfected with si Control and treated with the respective As2O3 concentration, n = 2).
The inhibition of PPARγ contributed to As2O3-induced cell death
The treatment of HepG2 cells with 1 μM As2O3 for 24 h resulted in cell death in this study. The cell viability of As2O3-treated HepG2 cells was elevated significantly after pretreatment with RGS, at a concentration of 100 μM (Fig. 9). It suggested that inhibition of PPARγ contributed to As2O3-induced cell death.
Figure 9. Effect of RGS on the cell viability of As2O3-treated HepG2 cells.
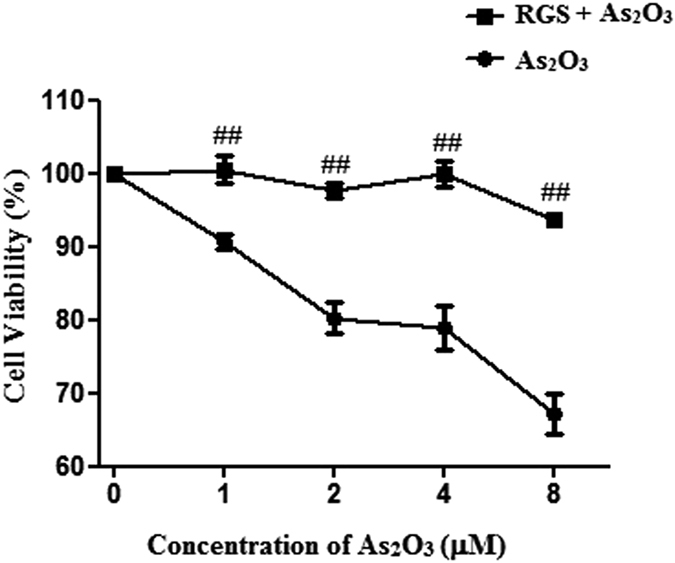
Before incubation with 1 μM As2O3 for 24 h, the cells were pretreated with 100 μM RGS for 6 h. The cell viability was assessed using the MTT assay. The trace represents the means ± SD. (##P < 0.01 vs. cells not treatment with RGS and treatment with the respective As2O3 concentration, n = 2).
Tau reduced cell death of As2O3-treated HepG2 cells
We found that the autophagy in livers was decreased significantly after pretreatment of Tau. To investigate the action of Tau in vitro, the As2O3-treated HepG2 cells were pretreated with Tau at a concentration of 20 μM. The cell viability was elevated significantly (Fig. 10). It suggested that Tau reduced the cell death in As2O3-treated HepG2 cells.
Figure 10. Effect of Tau on the cell viability of As2O3-treated HepG2 cells.
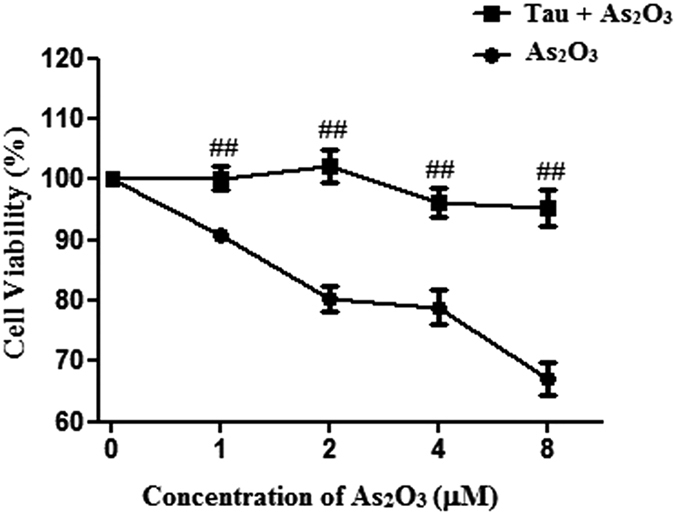
Before incubation with 1 μM As2O3 for 24 h, the cells were pretreated with 20 μM Tau for 6 h. The cell viability was assessed using the MTT assay. The trace represents the means ± SD. (##P < 0.01 vs. cells not treated with Tau and treated with the respective As2O3 concentration, n = 2).
RGS and Tau increased the expression of PPARγ in As2O3–treated HepG2 cells
The expression of PPARγ level was decreased significantly after treatment with 1 μM As2O3 for 24 h in this study (Fig. 11A). After pretreatment of RGS and Tau respectively, the level of PPARγ was increased significantly in As2O3-treated HepG2 cells (Fig. 11B).
Figure 11. PPARγ protein levels in As2O3-treated HepG2 cells.

(A) Before incubation with 1 μM As2O3 for 24 h, the cells were pretreated with 100 μM RGS and 20 μM Tau respectively for 6 h. The protein fraction was analyzed by Western blot. β-actin was taken as internal control. (B) Densitometric analyses of PPARγ levels in HepG2 cells (**P < 0.01 vs. control; ##P < 0.01).
The activation of PPARγ and Tau reduced the generation of ROS in As2O3–treated HepG2 cells
The generation of ROS was increased significantly after treatment with 1 μM As2O3 for 24 h in this study (Fig. 12A). After pretreatment with RGS, the generation of ROS was decreased significantly in As2O3-treated HepG2 cells (Fig. 12A). It suggested that the activation of PPARγ could reduce the generation of ROS induced by As2O3. After pretreatment with Tau, the generation of ROS was also decreased significantly in As2O3-treated HepG2 cells (Fig. 12B). It suggested that Tau had the same effect with RGS. The up-regulation of PPARγ could withstand the generation of ROS induced by As2O3.
Figure 12. The generation of ROS in As2O3-treated HepG2 cells.

(A) Before incubation with 1 μM As2O3 for 24 h, the cells were pretreated with 100 μM RGS and 20 μM Tau respectively for 6 h. Representative fluorescence microscopic images showing the fluorescence of DCF oxidized by intracellular ROS in HepG2 cells. After treatment, the cells were incubated with 5 μM DCFH-DA for 15 min in the dark and assessed by fluorescence microscope (scale bar = 20 μM). (B) The fluorescence intensity of the generation of ROS in As2O3-treated HepG2 cells (n = 6, **P < 0.01 vs. control; ##P < 0.01).
The activation of PPARγ and Tau accelerated the expression of Nrf2 and Trx proteins in As2O3-treated HepG2 cells
The expression of Nrf2 and Trx proteins were decreased significantly after treatment with 1 μM As2O3 for 24 h in this study (Fig. 13A). After pretreatment of RGS, the expression of Nrf2 and Trx were increased significantly in As2O3-treated HepG2 cells (Fig. 13B,C). It suggested that the activation of PPARγ could accelerate the expression of Nrf2 and Trx. After pretreatment with Tau respectively, the expression of Nrf2 and Trx were also increased significantly (Fig. 13B,C). It suggested that Tau had the effect of up-regulating PPARγ and Tau could withstand the reduction of Nrf2 and Trx induced by As2O3.
Figure 13. Nrf2 and Trx protein levels in As2O3-treated HepG2 cells.

(A) Before incubation with 1 μM As2O3 for 24 h, the cells were pretreated with 100 μM RGS and 20 μM Tau respectively for 6 h. The protein fraction was analyzed by Western blot. β-actin was taken as internal control. (B) Densitometric analyses of Nrf2 protein levels in HepG2 cells (**P < 0.01 vs. control; ##P < 0.01). (C) Densitometric analyses of Trx protein levels in HepG2 cells (**P < 0.01 vs. control; ##P < 0.01)”.
RGS and Tau reduced autophagy in As2O3-treated HepG2 cells
In the study, the level of LC3-II was increased significantly and the expression of P62 was decreased in As2O3-treated HepG2 cells (Fig. 14A,C). After pretreatment with RGS, we found that autophagy was inhibited in As2O3-treated cells (Fig. 14C,D), suggesting that inhibition of PPARγ contributed to As2O3-induced autophagy. The level of autophagy was decreased after pretreatment with Tau in As2O3-treated cells (Fig. 14C,D), suggesting that Tau could protect hepatocytes against As2O3 through modulating PPARγ–autophagy pathway.
Figure 14. Expression of LC3-II and P62 in HepG2 cells with As2O3.

(A) Before incubation with 1 μM As2O3 for 24 h, the cells were pretreated with 100 μM RGS and 20 μM Tau respectively for 6 h. The protein fraction was analyzed by Western blot. β-actin was taken as internal control. (B) Densitometry analyses of LC3-II expressed in As2O3-treated HepG2 cells (**P < 0.01 vs. control; ##P < 0.01). (C) The expression of P62 in HepG2 cells with As2O3. Before incubation with 1 μM As2O3 for 24 h, the cells were pretreated with 100 μM RGS and 20 μM Tau respectively for 6 h. The P62 protein fraction was analyzed by Western blot. β-actin was taken as internal control. (D) Densitometry analyses of P62 expressed in As2O3-treated HepG2 cells (*P < 0.05, **P < 0.01 vs. control; ##P < 0.01).
Discussion
Drinking water contaminated with inorganic arsenic was a major threat to human health with more than 100 million people worldwide exposed to levels that exceed the World Health Organization’s (WHO) recommended limit of 10 μg As/L37. Arsenic affected a multitude of biological systems, however, the mechanism by which arsenic elicits its toxic effects remains largely unknown. Numerous studies had been conducted to elucidate the molecular events associated with arsenic exposure, and resulting data suggested multiple mechanisms. Previous studies showed that mice might be less susceptible than human to arsenic toxicity, partly due to a faster metabolism and clearance of arsenic38. Therefore, it was necessary to use higher exposure concentration of arsenic than the environmentally relevant concentrations in mouse experiment39. The As2O3 concentration used in this study ranged from 2 mg/kg BW to 8 mg/kg BW.
Autophagy was a gatekeeping mechanism for stabilizing cell homeostasis. Studies had shown that autophagy played important roles in physiology and pathophysiology40. Our previous study found that arsenic accelerated autophagosome formation and caused autophagic cell death in INS-1 cells13. Autophagy had recently been shown to be involved in metabolism, triggering great interests in elucidating the underlying mechanism and testing the feasibility of targeting autophagy for the prevention and treatment of obesity and related metabolic disorders41. In addition to LC3-II, P62 was another autophagy specific substrate. P62 was degraded in autolysosomes42. In this study, Western blot analysis revealed that As2O3 accelerated the expression of LC3-II in offsprings’ livers and the level of P62 was decreased significantly in HepG2 cells after treatment with As2O3. It was a reliable indicator for the activation of autophagy in As2O3-treated offsprings’ livers. As2O3-induced autophagy was prevented by Tau, suggesting that As2O3-induced injury of livers might have the connection with autophagy and Tau might prevent livers from As2O3-induced autophagy.
PPARγ, which belonged to a family of nuclear hormone receptors that regulated the function and expression of complex gene networks, especially involved in cell proliferation and differentiation, glucose metabolism and homeostasis, insulin sensitivity and lipid metabolism43. It was reported that after treatment with RSG observably attenuated GCI-induced elevation of the LC3-II and Beclin-1 hippocampus of brain, these observations suggested that RSG might exert its inhibitory effect by inactivating neuronal autophagy through decreasing Beclin-1 and LC3-II, and thus achieve a cerebral protective effect44. Prenatal arsenic exposure had been associated with altered gene expression in human cord blood leukocytes and various target tissues in rodents3. We found that As2O3 reduced the expression of PPARγ protein and inhibited the expression of PPARγ gene in offsprings’ livers. It suggested that the reduction of PPARγ expression was involved in As2O3-induced autophagy in offsprings’ livers. After pretreatment with RGS, the expression of PPARγ was increased dramatically and the level of autophagy was decreased in HepG2 cells. It suggested that the inhibition of PPARγ contributed to As2O3-induced autophagy.
Arsenic was able to induce the generation of ROS12. In our previous studies, we found that arsenic could induce autophagic cell death through ROS pathway. Nrf2 was the key factor of the oxidative stress reaction. The Trx system was composed of NADPH, TrxR and Trx. It was also a crucial line of defense against ROS through its activity of disulfide reductase45. In this study, we found that after treatment with As2O3, the expression of Nrf2 and Trx were decreased obviously. The level of MDA was increased significantly in As2O3-treated livers. We used the activator of PPARγ to investigate whether the up-regulation of PPARγ could accelerate the expression of Nrf2 and Trx. We found that after pretreatment with RGS, the levels of Nrf2 and Trx were both increased obviously. It suggested that the inhibitor of PPARγ contributed to ROS-dependent autophagic injury in As2O3-treated offsprings’ livers.
It was reported that Tau was a non-essential free amino acid, which was one of the chemical components abundantly present in Lyciumbarbarum and crosses the blood–retinal barrier. A dietary source of Tau was essential for those animals (e.g. cat and humans), which cannot synthesize sufficient Tau and where greater consumption of Tau was required, such as in diabetes. Furthermore, several studies had reported that Tau potentiates the effect of insulin and possibly affected the insulin receptor. In addition, one study had indicated that high concentrations (20 mM) of Tau are capable of enhancing the phosphatidylinositide 3 (PI3)-kinase/Akt signaling pathway responsible for insulin-mediated stimulation of glucose transporter activity and glucose uptake9,16,26. Tau recognized metabolic function in liver was conjugation with bile acids, which was important for bile secretion and lipid digestion46. Tau supplementation might prevent alterations or restore endocrine pancreatic mass in malnutrition, obesity, T1D and T2D9. In this study, Tau prevented the stimulation of autophagy and the inhibition of PPARγ in As2O3-treated offsprings’ livers. Tau also reduced the generation of ROS by accelerating the expression of Nrf2 and Trx. The expression of PPARγ was improved dramatically and the level of autophagy was decreased after pretreatment of Tau in As2O3-treated HepG2 cells. After pretreatment with Tau, the generation of ROS was decreased obviously. It suggested that Tau could accelerate the expression of PPARγ and withstand the hepatotoxicity induced by As2O3.
Conclusion
In this study, we found that arsenic induced autophagy in offsprings’ livers through ROS pathway. The reduction of PPARγ level played a crucial role in this process. Tau could reverse arsenic-inhibited PPARγ. Tau could inhibit the generation of ROS and autophagy in arsenic-treated offsprings’ livers. We found that As2O3 caused autophagic cell death through ROS pathway in HepG2 cells. The inhibition of PPARγ contributed to As2O3-induced autophagy and generation of ROS. Tau could protect hepatocytes against As2O3 hepatotoxicity through modulating PPARγ–ROS-autophagy pathway.
Methods
Ethics statement
The Animal Ethics Committee of the Institute of Zoology, Dalian Medical University, approved this study. The institute issued an ID [SCKK (Liao) 2002–002] to this animal study and the ethical committee guided the animal use and conduct. All the experimental methods were carried out in accordance with the approved guidelines.
Experimental groups
To investigate the effects of As2O3 on offspring, groups of five healthy adult 90 Wistar rats (230 g–250 g) were purchased from Model Animal Research Center of Dalian Medical University (China). Experimental manipulation were described in our previous study. Briefly, the pregnant rats were randomly divided into five groups, each of 10 animals:
Group 1: Control group. Rats were treated with distilled water. The offsprings were treated as their mothers.
Group 2: Rats were treated with 2 mg/kg BW As2O3. The offsprings were treated as their mothers.
Group 3: Dams were treated with 4 mg/kg BW As2O3. The offsprings were treated as their mothers.
Group 4: Dams were treated with 8 mg/kg BW As2O3. The offsprings were treated as their mothers.
Group 5: Dams were treated with 8 mg/kg BW As2O3 and 150 mg/kg Tau. The offsprings were treated as their mothers.
The rats were given by gavage once a day from GD 6 until PND 42.
Cell culture and treatment
Human heptoma cell line HepG2 was purchased from the American Type Culture Collection. HepG2 cells were cultured in MEM/EBSS (Hyclone) medium supplemented with 10% fetal bovine serum (Biological Industries) and antibiotics (100 U/ml penicillin and 100 μg/ml streptomycin, Sigma) under a humidified atmosphere with 5% CO2 at 37 °C. As2O3 was purchased from Sigma Aldrich (CAS#: 1327-53-3), and 3.96 mg of As2O3 was dissolved in 1 ml phosphate buffer saline to prepare a stock solution of 20 μM. The cells were pretreated with 20 μM Tau and 100 μM RGS for 6 h.
Pathological analysis
Parts of liver were taken and fixed in 10% formalin solution. After 24 h–28 h, the livers were dehydrated in a grade alcohol series and embedded in paraffin wax. Sections of 4 mm–5 mm thickness were stained with hematoxylin-eosin and taken photos by microscope for pathological analysis.
Transmission electron microscopy
Parts of liver were fixed with 2% glutaraldehyde for 2 h, and then post fixed in 1% osmium tetroxide for 1 h. Dehydration was done in increasing concentration of ethanol followed by propylene oxide. While incubated in 70% ethanol, the pellet was stained en bloc with 1% uranyl acetate. Finally the pellet was embedded in Epon resin. Ultrathin sections were post stained with uranyl acetate and Reynold’s lead citrate routinely. Electron micrographs were taken with JEM 1400 transmission electron microscope at 80 kV.
Western blot
At the end of the designated treatments, the tissues were washed twice with ice-cold PBS and completely lysed in lysis buffer of a protein extraction kit (Keygen Biotech). The tissues lysate was centrifuged at 14000 rpm for 15 min at 4 °C, and the supernatant containing the total protein was collected. The concentration of total protein was quantified using BCA method. SDS-polyacrylamide gel electrophoresis was performed, and the proteins were then transferred to a nitrocellulose membrane. After blocking with 10% non-fat milk, the blots were incubated with primary antibodies against LC3-II (Sigma), PPARγ (Proteintech), Nrf2 (Proteintech), Trx (Abcam) or internal control β-actin (Santa). Blots were then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (Sigma) followed by detection with a SuperSignal West Pico Kit (Thermo Scientific) according to the manufacturer’s instructions. The expected protein bands were detected using Bio-Rad ChemiDocTMMP imaging system. Relative abundance was measured with Gel-Pro Analyzer 4.0 software. The results were representative of three independent experiments.
RT-PCR
Total RNA was isolated using RNAiso Plus (TaKaRa). RT-PCR was performed using PrimeScript™ RT reagent Kit (TaKaRa) and Trans Start Top Green Qpcr Super Mix (TRANSGEN BIOTECH). Relative expression of target gene was calculated with ΔΔCT method.
MTT assay
The cytotoxicity of As2O3 was detected by MTT assay. HepG2 cells (1 × 105/ml) were seeded in 96-well plates and treated with 1 μM-8 μM As2O3 for 24 h. After treatment, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was added, and the cells were incubated for 4 h at 37 °C. The supernatant was discarded, and 1% DMSO was added. The plate was gently agitated until the blue formazan crystals were fully dissolved. The absorbance at 570 nm was read using a Bio-Rad Microplate Reader, and the cell viability (%) was calculated using the following equation: (A570 of treated sample/A570 of control) × 100.
RNA interference
HepG2 cells were transfected with either 50 nM siRNA against Atg5 or scrambled control siRNA (Gene Pharma) using transfection reagent Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions.
MDA assay
Oxidative stress was determined by the measurement of MDA47. Parts of livers were homogenized in phosphate buffer. After centrifugation, the supernatants were collected for MDA analysis. The level of MDA was quantified using MDA test kit (Keygen Biotech). The MDA concentration was calculated from the absorption at 532 nm.
Measurement of intracellular ROS
Intracellular ROS generation was detected by fluorescence microscopy, using DCFH-DA. Before incubation with 1 μM As2O3 for 24 h and were pretreated with RGS and Tau for 6 h. The cells were incubated with DCFH-DA (Dichlorodihydrofluorescein Diacetate, Sigma) at a final concentration of 5 μM for 15 min in the dark, and then viewed under the fluorescence microscope (Olympus BX63). Inside cells, DCFH-DA was oxidized to fluorescent compound DCF by ROS. The intensity of DCF fluorescence was quantified by Image-Pro Plus 7.0 software.
Statistical analysis
The data were expressed as the means ± standard deviation (SD) from at least three independent experiments performed in triplicate and analyzed using the SPSS 13.0 statistical software. The comparisons between groups were analyzed using one-way ANOVA and Student–Newman–Keuls (SNK) test, and P < 0.05 was considered statistically significant.
Additional Information
How to cite this article: Bai, J. et al. Taurine protects against As2O3-induced autophagy in livers of rat offsprings through PPARγ pathway. Sci. Rep. 6, 27733; doi: 10.1038/srep27733 (2016).
Acknowledgments
This work was supported by the National Natural Science Foundation of China (NSFC, 30972562), the Natural Science Foundation of Liaoning Province (2014023050), Dalian Municipal Science and Technology Plan Project (2014E12SF070).
Footnotes
Author Contributions J.B., X.Y. and X.S. designed the studies. J.B., H.G., G.Y. and L.S. participated in the animal experiments and biochemical analysis. J.B., L.J., Q.Z., T.Q., N.G., L.Y. and W.W. participated in the cell experiments and biochemical analysis. J.B. wrote the manuscript. X.F. and X.S. revised the manuscript. All authors reviewed the manuscript. X.S. is the guarantor of the article.
References
- Garciafigueroa D. Y., Klei L. R., Ambrosio F. & Barchowsky A. Arsenic-stimulated lipolysis and adipose remodeling is mediated by G-protein-coupled receptors. Toxicol. Sci. 134, 335–344 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble M. O. et al. Arsenic exposure, diabetes prevalence, and diabetes control in the Strong Heart Study. Am. J. Epidemiol. 176, 865–874 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas D. et al. Prenatal Arsenic Exposure and the Epigenome: Identifying Sites of 5-methylcytosine Alterations that Predict Functional Changes in Gene Expression in Newborn Cord Blood and Subsequent Birth Outcomes. Toxicol. Sci. 143, 97–106 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kile M. L. et al. Prenatal arsenic exposure and DNA methylation in maternal and umbilical cord blood leukocytes. Environ. Health Perspect. 120, 1061–1066 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G. in arsenic lung carcinogenicity. Oncoimmunology . 4, 995566 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolt A. M., Byrd R. M. & Klimecki W. T. Autophagy is the predominant process induced by arsenite in human lymphoblastoid cell lines. Toxicol. Appl. Pharmacol. 244, 366–373 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y., Xie Y., Chai L., Wang S. & Cheng M. Autophagy modification augmented the treatment effects initiated by arsenic trioxide in NB4 cells. Med. Oncol. 28, 231–236 (2011). [DOI] [PubMed] [Google Scholar]
- Fang L. et al. Autophagy attenuates diabetic glomerular damage through protection of hyperglycemia-induced podocyte injury. PloS One 8, 60546 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K. S. et al. Taurine ameliorates hyperglycemia and dyslipidemia by reducing insulin resistance and leptin level in Otsuka Long-Evans Tokushima fatty (OLETF) rats with long-term diabetes. Exp. Mol. Med. 44, 665–673 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerito O. et al. WIN induces apoptotic cell death in human colon cancer cells through a block of autophagic flux dependent on PPARgamma down-regulation. Apoptosis 19, 1029–1042 (2014). [DOI] [PubMed] [Google Scholar]
- Qi Y. et al. Autophagy inhibition by sustained overproduction of IL6 contributes to arsenic carcinogenesis. Cancer Res. 74, 3740–3752 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau A. et al. Arsenic inhibits autophagic flux, activating the Nrf2-Keap1 pathway in a p62-dependent manner. Mol. Cell. Biol. 33, 2436–2446 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X. X. et al. Sodium arsenite induces ROS-dependent autophagic cell death in pancreatic beta-cells. Food Chem. Toxicol. 70, 144–150 (2014). [DOI] [PubMed] [Google Scholar]
- Strand D. W. et al. PPARgamma isoforms differentially regulate metabolic networks to mediate mouse prostatic epithelial differentiation. Cell Death Dis . 3, 361 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodway H. A., Hunt A. N., Kohler J. A., Postle A. D. & Lillycrop K. A. Lysophosphatidic acid attenuates the cytotoxic effects and degree of peroxisome proliferator-activated receptor gamma activation induced by 15-deoxyDelta12,14-prostaglandin J2 in neuroblastoma cells. Biochem. J. 382, 83–91 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy F. P., Delany A. C., Kenny L. C. & Walsh S. K. PPAR-gamma – a possible drug target for complicated pregnancies. Br. J. Pharmacol. 168, 1074–1085 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerquetti L. et al. Rosiglitazone induces autophagy in H295R and cell cycle deregulation in SW13 adrenocortical cancer cells. Exp. Cell Res. 317, 1397–1410 (2011). [DOI] [PubMed] [Google Scholar]
- Mahmood D. F. et al. Peroxisome proliferator-activated receptor gamma induces apoptosis and inhibits autophagy of human monocyte-derived macrophages via induction of cathepsin L: potential role in atherosclerosis. J. Biol. Chem. 286, 28858–28866 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M. et al. Disruption of PPARgamma signaling results in mouse prostatic intraepithelial neoplasia involving active autophagy. Cell Death Diffe . 17, 469–481 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corona J. C., de Souza S. C. & Duchen M. R. PPARgamma activation rescues mitochondrial function from inhibition of complex I and loss of PINK1. Exp. Neurol. 253, 16–27 (2014). [DOI] [PubMed] [Google Scholar]
- Liu C. et al. Identification of a novel selective agonist of PPARgamma with no promotion of adipogenesis and less inhibition of osteoblastogenesis. Sci. Rep . 5, 9530 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang N. et al. An integrated metabonomic and proteomic study on Kidney-Yin Deficiency Syndrome patients with diabetes mellitus in China. Acta Pharmacol. Sin. 36, 689–698 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacchiotti A. et al. Taurine rescues cisplatin-induced muscle atrophy in vitro: a morphological study. Oxid. Med. Cell Longev. 2014, 840951 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T., Schaffer S. W. & Azuma J. The potential usefulness of taurine on diabetes mellitus and its complications. Amino Acids. 42, 1529–1539 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirdah M. M. Protective and therapeutic effectiveness of taurine in diabetes mellitus: a rationale for antioxidant supplementation. Diabetes Metab. Syndr . 9, 55–64 (2015). [DOI] [PubMed] [Google Scholar]
- Song M. K., Salam N. K., Roufogalis B. D. & Huang T. H. Lycium barbarum (Goji Berry) extracts and its taurine component inhibit PPAR-gamma-dependent gene transcription in human retinal pigment epithelial cells: Possible implications for diabetic retinopathy treatment. Biochem. Pharmacol. 82, 1209–1218 (2011). [DOI] [PubMed] [Google Scholar]
- Jong C. J., Ito T. & Schaffer S. W. The ubiquitin-proteasome system and autophagy are defective in the taurine-deficient heart. Amino Acids. 47, 2609–2622 (2015). [DOI] [PubMed] [Google Scholar]
- Jong C. J., Azuma J. & Schaffer S. Role of mitochondrial permeability transition in taurine deficiency-induced apoptosis. Exp. Clin. Cardiol. 16, 125–128 (2011). [PMC free article] [PubMed] [Google Scholar]
- Saw C. L. et al. Nrf2 null enhances UVB-induced skin inflammation and extracellular matrix damages. Cell Biosci . 4, 39 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Li W. G. & Kong A. N. T. Anti-oxidative stress regulator NF-E2-related factor 2 mediates the adaptive induction of antioxidant and detoxifying enzymes by lipid peroxidation metabolite 4-hydroxynonenal. Cell Biosci . 2, 40–46 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kensler T. W. & Wakabayashi N. Nrf2: friend or foe for chemoprevention? Carcinogenesis 31, 90–99 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Piaz F., Braca A., Belisario M. A. & De Tommasi N. Thioredoxin system modulation by plant and fungal secondary metabolites. Curr. Med. Chem. 17, 479–494 (2010). [DOI] [PubMed] [Google Scholar]
- Luo Z., Aslam S., Welch W. J. & Wilcox C. S. Activation of nuclear factor erythroid 2-related factor 2 coordinates dimethylarginine dimethylaminohydrolase/PPAR-gamma/endothelial nitric oxide synthase pathways that enhance nitric oxide generation in human glomerular endothelial cells. Hypertension 65, 896–902 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X. R., Gonzales N. & Aronowski J. Pleiotropic role of PPARgamma in intracerebral hemorrhage: an intricate system involving Nrf2, RXR, and NF-kappaB. CNS Neurosci Ther. 21, 357–366 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J. et al. Taurine protects against As2O3-induced autophagy in pancreas of rat offsprings through Nrf2/Trx pathway. Biochimie 123, 1–6 (2016). [DOI] [PubMed] [Google Scholar]
- Jiang L. et al. As2O3 induces apoptosis in human hepatocellular carcinoma HepG2 cells through a ROS-mediated mitochondrial pathway and activation of caspases. Int. J. Clin. Exp. Med. 8, 2190–2196 (2015). [PMC free article] [PubMed] [Google Scholar]
- Martin E. et al. Metabolomic Characteristics of Arsenic-Associated Diabetes in a Prospective Cohort in Chihuahua, Mexico. Toxicol. Sci. 144, 338–346 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazumder D. N. G. Effect of chronic intake of arsenic-contaminated water on liver. Toxicol. Appl. Pharm . 206, 169–175 (2005). [DOI] [PubMed] [Google Scholar]
- Liu S. et al. Arsenic induces diabetic effects through beta-cell dysfunction and increased gluconeogenesis in mice. Sci. Rep . 4, 6894 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y. P., Liang Z. Q., Gao B., Jia Y. L. & Qin Z. H. Dynamic effects of autophagy on arsenic trioxide-induced death of human leukemia cell line HL60 cells. Acta. Pharmacol. Sin. 29, 123–134 (2008). [DOI] [PubMed] [Google Scholar]
- Zhang C. et al. Autophagy is involved in adipogenic differentiation by repressesing proteasome-dependent PPARgamma2 degradation. Am. J. Physiol. Endocrinol. Metab. 305, 530–539 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. N. et al. Citreoviridin induces ROS-dependent autophagic cell death in human liver HepG2 cells. Toxicon 95, 30–37 (2015). [DOI] [PubMed] [Google Scholar]
- Chen L. et al. Rosiglitazone ameliorates senescence-like phenotypes in a cellular photoaging model. J. Dermatol. Sci. 77, 173–181 (2015). [DOI] [PubMed] [Google Scholar]
- Shao Z. Q. & Liu Z. J. Neuroinflammation and neuronal autophagic death were suppressed via Rosiglitazone treatment: New evidence on neuroprotection in a rat model of global cerebral ischemia. J. Neurol. Sci 349, 65–71 (2015). [DOI] [PubMed] [Google Scholar]
- Lu J. & Holmgren A. The thioredoxin antioxidant system. Free Radical. Bio. Med . 66, 75–87 (2014). [DOI] [PubMed] [Google Scholar]
- Takatani T. et al. Taurine prevents the ischemia-induced apoptosis in cultured neonatal rat cardiomyocytes through Akt/caspase-9 pathway. Biochem. Biophys. Res. Commun. 316, 484–489 (2004). [DOI] [PubMed] [Google Scholar]
- Geng Z. W., X. Y. T. & Jia H. J. Reactive oxygen species (ROS) mediates non-freezing cold injury of rat sciatic nerve. Int. J. Clin. Exp. Med. 8, 15700–15707 (2015). [PMC free article] [PubMed] [Google Scholar]