

Abstract

Malaria persists as one of the most devastating global infectious diseases. The pyrimidine biosynthetic enzyme dihydroorotate dehydrogenase (DHODH) has been identified as a new malaria drug target, and a triazolopyrimidine-based DHODH inhibitor 1 (DSM265) is in clinical development. We sought to identify compounds with higher potency against Plasmodium DHODH while showing greater selectivity toward animal DHODHs. Herein we describe a series of novel triazolopyrimidines wherein the p-SF5-aniline was replaced with substituted 1,2,3,4-tetrahydro-2-naphthyl or 2-indanyl amines. These compounds showed strong species selectivity, and several highly potent tetrahydro-2-naphthyl derivatives were identified. Compounds with halogen substitutions displayed sustained plasma levels after oral dosing in rodents leading to efficacy in the P. falciparum SCID mouse malaria model. These data suggest that tetrahydro-2-naphthyl derivatives have the potential to be efficacious for the treatment of malaria, but due to higher metabolic clearance than 1, they most likely would need to be part of a multidose regimen.
Introduction
Malaria is one of the most deadly infectious diseases in human history with 3.2 billion people in 97 countries at risk.1 An estimated 444,000 deaths from malaria were reported by the WHO in 2015 and ∼90% of these occurred in sub-Saharan Africa, mostly among children under the age of five. Human malaria, which is transmitted by the female Anopheles mosquito, can be caused by five species of Plasmodia; however, Plasmodium falciparum and Plasmodium vivax are the most signficant.2,3P. falciparum is dominant in Africa and accounts for most of the deaths, while P. vivax has a larger global distribution. To simplify treatment options it is desirable that new drugs be efficacious against all human infective species.4 Malaria is a treatable disease and malarial control programs depend on drug therapy for treatment and chemoprevention, and on insecticides (including insecticide impregnated bed nets) to prevent transmission.2 A large collection of drugs has been used for the treatment of malaria, but many of the most important compounds have been lost to drug resistance (e.g., chloroquine and pyrimethamine).4−6 Artemisinin combination therapies (ACT) replaced older treatments, becoming highly effective, crucial tools in global efforts that have led to the decline in malaria deaths over the past decade. However, resistance to the artemisinin components (associated with Kelch13 propeller protein mutations7−9) has been found in Southeast Asia putting at risk malaria treatment programs. To combat drug resistance a significant effort is underway to identify new compounds that can be used for the treatment of malaria, with several new entities currently in clinical development.4,5,10
The triazolopyrimidine DSM265 (1) (Figure 1) developed by our group is the first antimalarial agent that targets dihydroorotate dehydrogenase (DHODH) to reach clinical development, validating this target for the treatment of malaria.11,12 DHODH is a mitochondrial enzyme that is required for the fourth step of de novo pyrimidine biosynthesis, catalyzing the flavin-dependent oxidation of dihydroorotate to orotic acid with mitochondrially derived coenzyme Q (CoQ) serving as a second substrate.13 Pyrimidines are essential for both RNA and DNA biosynthesis, and because Plasmodia do not encode pyrimidine salvage enzymes, which are found in humans and other organisms, the de novo pyrimidine pathway and DHODH are essential to the parasite. We identified the triazolopyrimidine DHODH inhibitor series by a target-based high throughput screen, and the initial lead DSM1 (2) (Figure 1) was shown to selectively inhibit P. falciparum DHODH and to kill parasites in vitro, but it was ineffective in vivo due to poor metabolic properties.14−16 The series was subsequently optimized to improve its in vivo properties resulting in the identification of DSM74 (3), which while metabolically stable lacked potency.16 X-ray structures of 2 and 3 bound to PfDHODH were then used to guide the medicinal chemistry program in the search for more potent analogues, resulting in the identification of 1.11,12,17
Figure 1.

Structures of selected triazolopyrimidine PfDHODH inhibitors. Shown structures include, 2-(1,1-difluoroethyl)-5-methyl-N-[4-(pentafluoro-λ6-sulfanyl)phenyl]-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (1), (5-methyl[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)naphthalen-2-ylamine (2), and (5-methyl[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)(4-trifluoromethylphenyl)amine (3).
While 1 has progressed successfully to Phase II clinical trials,4 we sought to identify potential backup compounds if unforeseen issues arise during its clinical development. Importantly, 1 has potent activity on PfDHODH and P. falciparum parasites in vivo, and its pharmacokinetic properties support its use as a single dose treatment or once weekly prophylactic.12 However, while 1 has excellent selectivity against human DHODH and thus is not expected to show on target activity in humans, it does have some activity against rodent DHODHs that has complicated animal toxicity testing. We therefore sought to identify compounds that have broad selectivity against all mammalian DHODHs, while if possible identifying compounds with higher potency against PfDHODH that could potentially reduce the dose required for treatment. We describe herein a series of triazolopyrimidines where the SF5-aniline moiety of 1 has been replaced by either substituted 1,2,3,4-tetrahydro-2-naphthyl or 2-indanyl amines. Compounds from both series are inactive against a range of mammalian DHODHs, while several of the identified tetrahydronapthalenes are more potent than 1 against P. falciparum parasites in vitro and indeed are among the most potent compounds that have been identified in the series. However, these compounds are less metabolically stable than 1 in mouse liver microsomes, suggesting the likelihood of higher clearance in mice and explaining the requirement for higher doses to inhibit parasites in the in vivo mouse model of malaria.
Chemistry
The focus of the study was to examine the structure–activity relationships (SARs) of replacing the p-SF5-aniline of 1 with substituted 1,2,3,4-tetrahydro-2-naphthyl or 2-indanyl amines with the goal of eliminating activity on rodent DHODHs, while potentially improving potency against P. falciparum DHODH and thereby lowering the dose required for in vivo efficacy. The clinical candidate 1 is a potent inhibitor of PfDHODH with only minimal activity detected against human DHODH (selectivity window > 5000-fold; Table 1). However, 1 showed inhibitory activity against rodent and to a lesser extent dog DHODHs, although potency toward these enzymes remained considerably less than for PfDHODH.12 Because these species are required for toxicity testing in preclinical studies we sought to eliminate rodent and dog DHODH inhibitory activity for any potential candidate that might be developed as a backup to 1. We previously reported that substitution of the p-SF5-aniline with p-CF3 aniline in the context of either mono or difluoro groups at the meta position led to greater inhibition of the mammalian enzymes.18 X-ray structural data suggested that increasing hydrophobicity drove the interaction of these compounds with the mammalian enzymes. We hypothesized based on these structures that increasing the size of the aromatic amine might prevent binding to mammalian DHODHs, while increasing potency toward the parasite enzyme. We previously tested several tetrahydro-2-naphthyl and 2-indanyl amines as substitutes for the CF3-aniline of 3 but found these compounds lacked potency, and furthermore, the one analogue containing 1,2,3,4-tetrahydro-2-naphthyl also lacked metabolic stability in mouse microsomes.19 However, these amines were never tested within the context of haloalkyl groups (CF2CH3 or CF3) at the C2 position of the triazolopyrimidine ring, which was a key substitution required to boost the potency of the series.11 We therefore synthesized a series of tetrahydro-2-naphthyl or 2-indanyl analoues with halo alkyl groups at the C2 position. Substituents (halo, haloalkyl, Me, CF3, OMe, SO2Me, and SO2NMe2) were incorporated onto the tetrahydro-2-naphthyl or 2-indanyl aromatic rings to improve metabolic stability over the unsubstituted analogues.
Table 1. Structure–Activity Relationshipsa.

| IC50 (μM) |
||||||
|---|---|---|---|---|---|---|
| compd | R | Ar/R1 | PfDHODH | PvDHODH | hDHODH | EC50 (μM) Pf3D7 cellsd |
| 1 | CF2CH3 | 4-SF5–Ph | 0.0089 (0.0073–0.011) | 0.027 (0.023–0.032) | >100 | 0.0043–0.0078 |
| 2 | H | 2-naphthyl | 0.047 | 0.23 | >100 | 0.068 |
| 3 | H | 4-CF3–Ph | 0.28 | 2.6 | >100 | 0.34 |
| 5 | CF2CH3 | H | 0.021 (0.019–0.024) | 0.035 (0.030- 0.041) | ND | 0.0028 (0.0022–0.0037) |
| 6 | CF2CH3 | 6-F | 0.024 (0.021–0.027) | 0.056 (0.048–0.066) | >100 | 0.0075 (0.0063–0.0089) |
| 7 | CF2CH3 | 6-Cl (E-I) | 0.0037 (0.0021–0.0066) | 0.029 (0.019–0.042) | >100 | 0.0038 (0.0030–0.0049) |
| 8 | CF2CH3 | 6-Cl (E-II) | 1.34 (1.1–1.7) | ND | >100 | 0.42 (0.37–0.47) |
| 9 | CF2CH3 | 7-Cl (E-I, S) | 0.0063 (0.005–0.008) | 0.010 (0.0083- 0.013) | >100 | 0.0012 (0.0011–0.0013) |
| 10 | CF2CH3 | 7-Cl (E-II, R) | 0.79 (0.70–0.94) | ND | >100 | 2.8 (1.0–7.8) |
| 11 | CF2CH3 | 6-Br (E-I)b | 0.0046 (0.0038- 0.0056) | 0.024 (0.021–0.027) | >100 | 0.0026 (0.0016–0.0042) |
| 12 | CF2CH3 | 6-Br (E-II)c | 0.79 (0.59–1.1) | 2.1 (1.8–2.5) | >100 | 0.14 (0.090–0.21) |
| 13 | CF2CH3 | 7-Br (E-I) | 0.0046 (0.0036–0.006) | 0.012 (0.010–0.013) | >100 | 0.00039 (0.00034–0.00044) |
| 14 | CF2CH3 | 7-Br (E-II) | 0.64 (0.53–0.77) | ND | >100 | 1.4 (1.2–1.5) |
| 15 | CF2CH3 | 6-OMe | 0.14 (0.11–0.16) | 0.22 (0.20–0.25) | >100 | 0.017 (0.014–0.019) |
| 16 | CF2CH3 | 7-OMe (E-I) | 0.021 (0.018–0.024) | 0.13 (0.11–0.15) | >100 | 0.025 (0.023–0.026) |
| 17 | CF2CH3 | 7-OMe (E-II) | 1.0 (0.86–1.2) | ND | ND | 0.64 (0.56–0.75) |
| 18 | CF2CH3 | 6,7-di-F (E-I) | 2.3 (1.9–2.8) | ND | ND | >2.5 |
| 19 | CF2CH3 | 6,7-di-F (E-II) | 0.060 (0.049–0.072) | 0.071 (0.066–0.076) | ND | 0.022 (0.018–0.026) |
| 20 | CF2CH3 | 6-F,7- CF3 | 0.089 (0.078–0.10) | 0.39 (0.34–0.44) | >100 | 0.097 (0.080–0.12) |
| 21 | CF2CH3 | 6- CF3, 7-F | 0.087 (0.067–0.11) | 0.82 (0.69–0.96) | >100 | 3.3 (2.97–3.63) |
| 22 | CF3 | H | 0.17 (0.12–0.23) | ND | >100 | 0.021 (0.017–0.025) |
| 23 | CF3 | 6-F | 0.077 (0.066–0.089) | 0.28 (0.25–0.33) | ND | 0.066 (0.062–0.07) |
| 24 | CF3 | 7-Cl (E-I) | 0.015 (0.012–0.018) | ND | >50 | 0.033 (0.030–0.035) |
| 25 | CF3 | 7-Cl (E-II) | 1.1 (0.98–1.2) | ND | ND | 1.5 (1.2–1.7) |
| 26 | CF2CH3 | H | 0.048 (0.040–0.059) | ND | >100 | 0.036 (0.031–0.042) |
| 27 | CF2CH3 | 5-Br | 0.11 (0.083–0.14) | ND | ND | 0.59 (0.25–1.4) |
| 28 | CF2CH3 | 5-Cl | 0.085 (0.075–0.097) | ND | ND | 0.12 (0.0.045–0.20) |
| 29 | CF2CH3 | 5,6-di-Cl | 0.14 (0.11–0.17) | ND | ND | 0.58 (0.22–1.5) |
| 30 | CF2CH3 | 5-SO2Me | 4.0 (2.7–6.1) | ND | ND | 6.5 (4.9–8.1) |
| 31 | CF2CH3 | 5-SO2NMe2 | 8.0 (6.7–9.7) | ND | ND | ND |
| 32 | CF2CH3 | 4,7-di-Me | 0.014 (0.012- 0.017) | 0.024 (0.022- 0.027) | >100 | 0.00039 (0.00021–0.00075) |
| 33 | CF2CH3 | 4,7-di-F | 0.065 (0.057–0.075) | 0.15 (0.14–0.16) | >100 | 0.046 (0.002–0.0048) |
| 34 | CF2CH3 | 4-F | 0.052 (0.041–0.066) | 0.3 (0.26–0.34) | >100 | 0.064 (0.061–0.066) |
| 35 | CF2CH3 | 5,6-di-F | 0.052 (0.042–0.064) | 0.37 (0.28–0.50) | >100 | 0.040 (0.035–0.044) |
| 36 | CF2CH3 | 4-CF3 | 0.066 (0.054–0.080) | 0.28 (0.22–0.34) | >100 | 0.0043 (0.0029–0.0056) |
| 37 | CF3 | 5,6-di-F | 0.12 (0.11–0.14) | ND | ND | 0.15 (0.092–0.23) |
DHODH IC50 and P. falciparum EC50 values were determined from triplicate data points at each concentration in the dose titration. Values in parentheses represent the 95% confidence interval of the fit.
Enantiomer-I is the second eluted on the chiral column.
Enantiomer-II is the first eluted on the chiral column.
The synthetic strategy to generate the appropriately substituted triazolopyrimidines has previously been described.11,15,16 For the studies described herein, 7-chloro-2-(1,1-difluoroethyl)-5-methyl[1,2,4]triazolo[1,5-a] pyrimidine (4a) and 7-chloro-2-(trifluoromethyl)-5-methyl[1,2,4]triazolo[1,5-a] pyrimidine (4b) were prepared using these methods (Scheme 1) and were reacted with the requisite amines to afford the final products containing either the 1,2,3,4-tetrahydro-2-naphthyl (5–25) or 2-indanyl (26–37) amines in place of the p-SF5-aniline of 1. Most amines utilized for the study were commercially available with the exception that amines for compounds 29–37 were synthesized as described in Scheme 2 and Supplementary Scheme S1. The 1,2,3,4-tetrahydro-2-naphthyls contained a chiral center, and for select compounds, individual enantiomers were purified using a chiral column as described in the Experimental Section.
Scheme 1. General Synthetic Method.

Scheme 2. Synthesis of 5-(Methylsulfonyl)-2,3-dihydro-1H-inden-2-amine·HCl (43).

Reagents and conditions: (i) Ac2O, NaOAc, AcOH, RT, 12 h; (ii) ClSO3H, CHCl3, RT, 1 h; (iii) (a) SnCl2·2H2O, AcOH, HCl, RT, 2 h; (b) NaOMe, MeI, MeOH; (iv) mCPBA, CHCl3; (v) 3 N HCl.
The amine precursor 5-(methylsulfonyl)-2,3-dihydro-1H-inden-2-amine·HCl 43 was prepared in five steps (Scheme 2). First, 2-aminoindane 38 was acetylated with acetic anhydride-sodium acetate to yield the acetamido derivative 39. Further, regioselective chlorosulfonylation of 39 led to the sulfonyl chloride intermediate 40. Reduction of the sulfonyl chloride to the thiol was achieved by tin chloride, which was converted to a methylthio derivative 41 using sodium methoxide and methyl iodide. Next, oxidation of methylthio intermediate 41 with mCPBA gave the acetyl protected methanesulfonyl derivative 42. Upon deprotection of this acetyl group using 3 N HCl, the required amine precursor 43 was obtained.
Results
Optimization of Inhibitor Potency and Selectivity
Compounds were first analyzed for potency against P. falciparum, P. vivax, and human DHODH, and for activity on P. falciparum parasites in vitro (Table 1). Compounds in the series showed IC50s against PfDHODH in the range of 0.005–8 μM and EC50s against P. falciparum 3D7 parasites in whole cell assays between 0.00039–6.5 μM (Table 1). Generally, the tetrahydro-2-naphthyl compounds (5–25) were more potent in both assays than the 2-indanyls (26–37). The most potent analogues based on the whole cell P. falciparum assay were 9 and 13, both of which contain a halogen (chloro or bromo, respectively) in the 7 position. Compound 13 is the most potent analogue identified in the triazolopyrimidine series that has been reported. Overall the trend was for 7-substituted tetrahydro-2-naphthyls to be more potent than those with substitutions at the 6 position. Compounds with Br and Cl substituents (7, 9, 11, 13) were more potent than those with fluoro 6, OMe (15, 16) or with disubstitutions (19, 20, 21), with the least active tetrahydro-2-naphthyl 21 containing a 6-CF3, 7-F. Potency of 21 in the whole cell parasite assay was worse than expected based on the inhibitory activity against PfDHODH, suggesting that the compound may be poorly transported into the parasite. Analogues with CF3 at C2 (22, 23, 24) on the triazolopyrimidine ring were less active than those with CF2CH3 (5, 6, 9) at this position. Within the indanyl series the most potent compounds were 32 and 36 containing 4,7-dimethyl or 4-CF3 substituents, respectively, while the least active compounds were 30 and 31 containing 5-SO2Me or 5-SO2NMe2 substituents, respectively.
For the tetrahydro-2-naphthyls where individual enantiomers were characterized, differences in potency between the active enantiomer (7, 9, 11, 13, 16, 19, 24) and the inactive enantiomer (8, 10, 12, 14, 17, 18, 25) ranged from 20- to 360-fold toward PfDHODH and from 30- to 2000-fold against P. falciparum 3D7 parasites. The small molecule X-ray structure of the active enantiomer 9 was solved demonstrating that it was in the S configuration (Figure S1 and Table S1). We did not isolate the individual enantiomers for compounds from the indane series (27, 28, 30, 31, 34, and 36), and it is likely that the purified active enantiomers would also show higher activity for these compounds as well. Thus, the reported potency data for the racemic mixture likely overestimates the true IC50 by ∼2-fold.
Overall we observed a good correlation between potency on PfDHODH and potency on 3D7 P. falciparum parasites (Figure S2). There was a tendency for the most potent compounds on the parasite to show lower potency against PfDHODH, but this difference is likely caused by the complication of stoichiometric binding for compounds with IC50s at or below the concentration of enzyme used in the assay (PfDHODH = 5 nM). In order to confirm that, for the most potent compounds, parasite killing is due to DHODH inhibition, we tested select compounds (9 and 13) for their ability to inhibit growth of a genetically engineered parasite strain that expresses yeast DHODH (D10 yDHODH20). This strain is resistant to DHODH inhibitors if their mechanism of action is on target. The EC50s (D10 yDHODH EC50 = 8.6 and 7.4 μM for 9 and 13, respectively) for both compounds were increased by >600-fold against this parasite strain relative to the wild-type 3D7 parasites, supporting DHODH inhibition as their mechanism of action (Table 1 and Figure S3). The addition of proguanil did not reverse the resistance of this strain, which is also consistent with DHODH as the mechanism of parasite killing. In contrast, it has been previously reported that proguanil reverses the atovaquone resistance phenotype of the D10 yDHODH parasite strain, providing a mechanism to distinguish between DHODH and bc1 targeted parasite growth inhibition.20 The weak but detectable activity observed on the D10 yDHODH parasites for both compounds suggests that at high concentrations there is a secondary target. For a couple of select compounds (9 and 35) whole cell activity was also tested on several additional cell lines including chloroquine and pyrimethamine resistant parasites (Table S2). Both compounds were equally active on all tested strains.
Species selectivity was first evaluated by testing against human and P. vivax DHODH (Table 1) and then for a selection of the more potent compounds, inhibitor activity was also measured for mouse, rat, and dog DHODH (Table 2). None of the compounds showed any inhibition of human DHODH up to the highest tested concentration (100 μM), which is similar to results for 1. However, unlike 1, none of the compounds showed any activity against either the rodent or dog DHODHs (Table 2), demonstrating that we had achieved our objective of eliminating mammalian DHODH activity. Measured IC50s for P. vivax DHODH ranged from 1.5–10-fold higher than for PfDHODH. The 7-position substituted tetrahydro-2-naphthyls, 9 and 13 showed minimal differences (1.5–3-fold) between the two enzymes suggesting that they would show good activity against both P. falciparum and P. vivax parasites, whereas the 6 position compounds (7 and 11) had 5–8-fold lower activity on P. vivax than P. falciparum DHODH.
Table 2. Activity of Select Triazolopyrimidines on Various Mammalian DHODHs.

| DHODH IC50 (μM) |
||||
|---|---|---|---|---|
| compd | human | rat | mouse | dog |
| 1a | ∼100 | 2.6 ± 0.39 | 2.3 ± 0.64 | 16 ± 6.5 |
| 35 | >100 | >100 | >100 | >100 |
| 11 | >100 | >100 | >100 | >100 |
| 13 | >100 | >100 | >100 | >100 |
| 7 | >100 | >100 | >100 | >100 |
| 9 | >100 | >100 | >100 | >100 |
Errors represent standard deviation for four independent replicates using different batches. Each replicate IC50 was determined from triplicate data points at each concentration in the dose titration.
X-ray Structure of 13 Bound to PfDHODH
In order to understand the structural basis for the superior potency of 13 we solved its X-ray structure bound to PfDHODH. The structure was solved to 2.32 Å resolution at an Rwork and Rfree of 0.18 and 0.21, respectively (Figure 2 and Table S3). Strong electron density was observed for the entirety of 13 (Figure S4A). Compound 13 was oriented in the pocket similarly to other triazolopyrimidines (e.g., 1–3(12,17,18)) with the triazolopyrimidine ring bound adjacent to the flavin cofactor in position to form H-bond interactions with Arg-265 and His-185 (Figures 2 and S4B). The tetrahydro-2-naphthyl moiety bound in a hydrophobic pocket forming edge-to-face stacking interactions with Phe-227 and Phe-188 and was in a very similar orientation to what we previously observed for the naphthyl moiety of 2 (Figure 2). Some modest conformational differences in ring geometry were observed reflecting the difference between the fully planar and aromatic naphthyl and the puckered configuration of the nonaromatic ring of the tetrahydro-2-naphthyl. A halogen bond was observed between the 7-bromo group on the tetrahydro-2-naphthyl of 13 and Cys-233 SH, which likely provides significant binding energy to the enzyme inhibitor interaction. This is a novel interaction that has not been previously observed.
Figure 2.
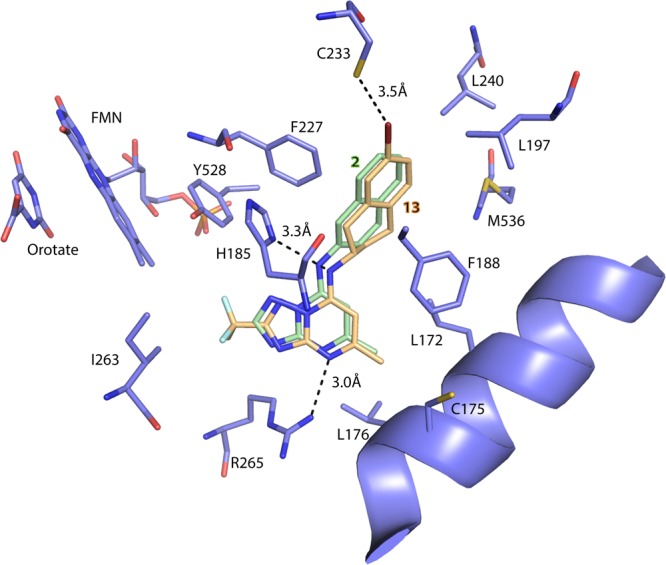
X-ray structure of PfDHODH bound to 13 (PfDHODH-13). Limited residues from the 4 Å shell around 13 are shown, and the structure has been aligned to the PfDHODH structure bound to 2 (PDB 3I65) to allow comparison of the binding modes. Only the inhibitor 2 from 3I65 is displayed. PfDHODH amino acid, FMN, and orotate carbons are shown in purple, the carbons of 13 are shown in tan, and the carbons of 2 are shown in green. Nitrogens are blue, oxygens are red, sulfur is light yellow, fluorines are light blue, and bromine is deep red. Protein residues are labeled with their amino acid number.
Physicochemical Properties and Plasma Protein Binding
Selected compounds were evaluated for their physicochemical properties to determine if they had good drug-like properties. Analysis included in silico calculations, chromatographic estimation of Log D (pH 7.4), aqueous solubility, and plasma protein binding (Tables 3 and 4). Tested compounds had physicochemical properties that are suggestive of good oral absorption (MW < 430, H bond donors ≤ 2, H bond acceptors ≤ 6, polar surface area < 70 Å2). Log D ranged from 3.1–4.1, and the more potent analogues tended to have higher Log D values. Aqueous solubility (pH 6.5) was poor (1.6–6.3 μg/mL) to moderate (12.5–25 μg/mL), and in general, compounds in this series had lower kinetic solubility than 1 (Table 3). More extensive solubility studies in simulated gastric and intestinal fluids was conducted on a selection of the most potent analogues from the 2-indanyl (35) and the tetrahydro-2-naphthyl (7, 9, 11, 13) series (Table 4). All five compounds had better solubility than 1 in 0.1 N HCl. Solubility in fasted-state simulated intestinal fluid was similar to or slightly improved compared to 1. The tetrahydro-2-naphthyl compounds substituted at the 6 position (11 and 7) showed considerably better solubility in fed-state simulated intestinal fluid than either 1 or the 7 position (9 and 13) substituted compounds.
Table 3. Physicochemical Properties, in Vitro Metabolism, and hERG Activity.
| compd | log DpH 7.4 | kinetic solubility pH 6.5 (μg/mL)a | PPBb (H/R/M, % bound) | blood to plasma ratio (Rat) | in vitro CLint (H/R/M, μL/min/mg protein)c | predicted in vivo CLint (H/R/M, mL/min/kg)d | hERG (μM) |
|---|---|---|---|---|---|---|---|
| 1e | 3.6 | 12.5–25 | 99.9/97.7/99.7 | 0.8 | 4.3/1.8/2.8 | 3.5/3.1/7.2 | 1.6, 6.2 |
| 2e | 3.2 | 12.5–25 | NDf | ND | 96/ND/229 | 79/ND/591 | ND |
| 3e | 2.5 | 6.3–12.5 | ND | ND | 7.5/ND/ND | 6.2/ND/ND | ND |
| 6 | 3.5 | 6.3–12.5 | ND | ND | 14/39/122 | 12/67/315 | 0.7 |
| 7 | 3.9 | 3.1–6.3 | 98.8/99.2/ND | 0.7 | <7/33/44 | <5.8/57/114 | 1.4 |
| 9 | 3.9 | 3.1–6.3 | 96.7/96.4/97.4 | 1.5 | <7/38/31 | <5.8/65/80 | 1.5 |
| 11 | 4.0 | 3.1–6.3 | 99.0/99.7/99.2 | 0.7 | <7/11/26 | <5.8/19/67 | 0.5 |
| 12 | 4.0 | 3.1–6.3 | ND | ND | <7/13/40 | <5.8/22/103 | ND |
| 13 | 4.0 | 1.6–3.1 | 96.3/97.2/ND | 1.5 | <7/39/32 | 5.8/67/83 | 1.1 |
| 16 | 3.3 | 12.5–25 | ND | ND | 18/19/25 | 15/33/65 | 1.8 |
| 19 | ND | ND | ND | ND | 11/24/59 | 9.1/41/152 | ND |
| 20 | 4.0 | 3.1–6.3 | ND | ND | <7/13/17 | <5.8/22/44 | ND |
| 21 | 4.1 | 1.6–3.1 | ND | ND | <7/<7/<7 | <5.8/<12/<18 | ND |
| 24 | 4.1 | 1.6–3.1 | ND | ND | 9/ND24 | 7.4/ND/62 | ND |
| 26 | 3.1 | 25–50 | ND | ND | 24/245/1004 | 20/422/2590 | ND |
| 28 | 3.5 | 6.3–12.5 | ND | ND | 10/54/43 | 8.2/93/111 | ND |
| 32 | 3.8 | 6.3–12.5 | ND | ND | 168/178/1000 | 138/306/2580 | ND |
| 33 | 3.2 | 6.3–12.5 | ND | ND | 33/1013/1500 | 27/1740/3870 | ND |
| 34 | 3.3 | 12.5–25 | ND | ND | 11/72/76 | 9.1/124/196 | ND |
| 35 | 3.4 | 6.3–12.5 | 93.4/94.2/95.3 | 1.2 | <7/<7/21 | <5.8/<12/54 | 1.7 |
| 36 | 3.7 | 6.3–12.5 | ND | ND | 30/61/119 | 25/105/307 | ND |
Kinetic solubility range following 30 min at room temperature.
Protein binding by ultracentrifugation in human (H), rat (R), and mouse (M) plasma.
In vitro intrinsic clearance in human (H), rat (R), and mouse (M) liver microsomes.
Predicted in vivo intrinsic clearance obtained using physiological scaling factors.21
ND, not determined.
Table 4. Solubility in Physiologically Relevant Mediaa.
| solubility
(μg/mL) |
||||||
|---|---|---|---|---|---|---|
| media | 1b | 35 | 11 | 13 | 7 | 9 |
| 0.1 N HCl | 6.8 | 180 | 189 | 52 | 429 | 105 |
| FeSSIF pH 5.0 | 27.6c | 65c | 211c | 39 | 211 | 36 |
| FaSSIF pH 6.5 | 5.1 | 15 | 13 | 8.3 | 47 | 12 |
Solubility after 5–6 h at 37 °C. FaSSIF = fasted state simulated intestinal fluid. FeSSIF = fed state simulated intestinal fluid.
Data from ref (12).
pH 5.8.
Human, rat, and mouse plasma protein binding (PPB) was assessed for the more potent compounds that were progressed to in vivo pharmacokinetic testing (7, 9, 11, 13, 35). The tetrahydro-2-naphthyl compounds substituted at the 6 position (7 and 11) showed higher protein binding compared to the compounds substituted at the 7 position (9 and 13). Binding was similar across the species tested for each of the compounds. By comparison, 1 exhibited high protein binding in human and mouse plasma but lower binding in rat plasma. The indanyl analogue 35 had considerably lower protein binding compared to either 1 or the tetrahydro-2-naphthyl compounds.
In Vitro Metabolism
To obtain a preliminary indication of the likelihood that compounds would show good in vivo pharmacokinetic properties, selected compounds were analyzed for metabolic stability in vitro using human, rat, and mouse liver microsomes. We previously found that the in vitro microsomal stability of compounds from the triazolopyrimidine series provided a good rank order estimation of which compounds would have the highest in vivo exposure.11,15,16,19 We report in Table 3 the in vitro intrinsic clearance (CLint, μL/min/mg microsomal protein) and the predicted in vivo intrinsic clearance (mL/min/kg) obtained using physiologically based scaling factors as previously described.21 Given the early stage of optimization for the series, protein binding was not determined for all compounds, and therefore, no attempt was made to predict the in vivo blood clearance (which requires corrections for both plasma and microsomal binding). In vitro CLint values of <20 μL/min/mg protein were taken as being suggestive of good metabolic stability, assuming that clearance only occurs via hepatic metabolism. Overall, the substituted tetrahydro-2-naphthyls showed good metabolic stability in human liver microsomes, but they were less stable in mouse microsomes (Table 3). Chloro-, bromo-, and CF3-substituted compounds (7, 9, 11, 12, 13, 20, 21) were marginally more metabolically stable than the fluoro- (6, 19) or OMe-substituted (16) compounds.
With the exception of 35, the indanyl analogues were generally metabolically unstable showing high intrinsic clearance in mouse liver microsomes and moderate to high values in human and rat microsomes. The least stable compounds were the unsubstituted 2-indanyl 26 and the 4,7 dimethyl substituted derivative 32. Replacement of both methyl groups with fluorine 33 did not improve stability, although perhaps surprisingly the single 4-F derivative 34 was significantly more stable. Substantial improvement in metabolic stability was obtained by moving the fluorine to the 5 and 6 positions of the ring 35, leading to low intrinsic clearance in human and rat microsomes and moderate clearance in mouse microsomes. These data suggested that 35 would show good plasma exposure in both mice and rats.
hERG Channel Activity and CYP Inhibition
In order to evaluate any potential cardiac risks, select compounds were tested for inhibition of the human ether-a-go-go-related gene (hERG) K+ channel in a standard patch clamp assay. hERG channel inhibition has been associated with QT prolongation and arrhythmias, and patch clamp assays have become a routine method to provide an initial analysis that a compound may potentially be associated with QT syndromes in humans.22 Compounds in both the tetrahydro-2-naphthyl and 2-indanyl series inhibited the hERG channel with IC50s in the range of 0.5–1.8 μM (Table 3).
In order to test for the potential for drug–drug interactions, select compounds were tested for their ability to inhibit cytochrome P450 isoforms in human liver microsomes (Table 5). All tested compounds showed moderate inhibition of isoforms 2C9 and 2D6. The most significant inhibition was observed for 9 and 13 as both inhibited these two isoforms in the 2–4 μM range.
Table 5. Inhibition of Cytochrome P450 Enzymes in Human Liver Microsomes.
| IC50 (μM) |
|||||
|---|---|---|---|---|---|
| CYP | 35 | 11 | 13 | 7 | 9 |
| 1A2 | >20 | >20 | >20 | >20 | >20 |
| 2B6 | >20 | 17.7 | >20 | 18.4 | >20 |
| 2C8 | ND | 18.7 | 16.9 | >20 | >20 |
| 2C9 | 18.5 | 8.7 | 3.1 | 10.2 | 2.1 |
| 2C19 | >20 | >20 | >20 | >20 | >20 |
| 2D6 | 5.9 | >20 | 3.9 | 20 | 2.7 |
| 3A4 | >20 | >20 | >20 | >20 | >20 |
In Vivo Pharmacokinetic Studies in Rats and Mice
Five of the most potent compounds were selected for assessment of their in vivo pharmacokinetic properties based on their good in vitro metabolic stability. Mice were dosed orally while rats were dosed orally, and intravenously to allow determination of the clearance, volume of distribution, and oral bioavailability. Compounds included one from the indanyl series (35) and four from the tetrahydro-2-naphthyl series (7, 9, 11, 13).
All five compounds showed good exposure over 24 h in both mice and rats after oral administration (Tables 6 and 7; Figures 3 and 4). Compounds were well tolerated at the administered doses, and no adverse reactions were observed. In mice, Cmax and AUC24h values were highest for 35 and lower for the tetrahydro-2-naphthyl compounds. Compared to 1 and taking into account the differences in dose, total Cmax and AUC24h values were similar or lower for the tetrahydro-2-naphthyl compounds, whereas unbound Cmax and AUC24h were similar (for 7) or somewhat higher (3–4-fold for 9, 11, and 13). The indanyl (35) had the highest plasma exposure in mice in terms of both total and unbound Cmax and AUC24h and significantly higher exposure compared to 1. Similar to 1, the terminal elimination half-life (t1/2) in mice was ∼2 h for all five compounds (Table 6 and Figure 3).
Table 6. Mouse Plasma Pharmacokinetic Parameters after a Single Oral Dosea.
| parameter | 1b | 35 | 11 | 13 | 7 | 9 |
|---|---|---|---|---|---|---|
| dose (mg/kg) | 10 | 20 | 20 | 20 | 20 | 20 |
| mouse PPB (% bound) | 99.7 | 95.3 | 99.2 | 97.2c | 99.2c | 97.4 |
| Tmax (h) | 1.5 | 1 | 1 | 1 | 1 | 1 |
| apparent T1/2 (h) | 2.5 | 2.3 | 2.1 | 2.1 | 1.9 | 1.5 |
| Cmax (μM) | 3.1 (0.01) | 29 (1.4) | 8.6 (0.07) | 4.5 (0.13)c | 7.0 (0.06)c | 5.0 (0.13) |
| AUC0-inf (μM·h) | 22.6 (0.07) | 169 (7.9) | 51 (0.41) | 16.7 (0.47)c | 19.7 (0.16)c | 22.7 (0.59) |
Values in parentheses have been corrected for plasma protein binding and represent unbound values.
Average from two studies as previously reported.12
Protein binding in mouse plasma is an approximation based on rat and human plasma protein binding data. Calculated unbound parameters (in italics) are therefore approximations only.
Table 7. Rat Plasma Pharmacokinetic Parameters after Intravenous (IV) or Oral (PO) Administrationa.
| intravenous administration | ||||||
|---|---|---|---|---|---|---|
| parameter | 1b | 35 | 11 | 13 | 7 | 9 |
| dose (mg/kg) | 2.6 | 1.8 | 2.0 | 1.1 | 1.9 | 1.6 |
| rat PPB (% bound) | 97.7 | 94.2 | 99.7 | 97.2 | 99.2 | 96.4 |
| blood/plasma | 0.8 | 1.2 | 0.7 | 1.5 | 0.7 | 1.5 |
| apparent T1/2 (h) | 12.6 | 11.2 | 6.2 | 6.1 | 7.1 | 5.9 |
| AUC0-inf (μM·h) | 17.4 (0.40) | 27.1 (1.57) | 30 (0.09) | 2.0 (0.05) | 24 (0.19) | 3.5 (0.12) |
| CLp (mL/min/kg) | 6.0 (263) | 3.0 (52) | 2.8 (933) | 22.4 (800) | 3.5 (438) | 21.2 (589) |
| Vss (L/kg) | 5.9 (257) | 2.7 (47) | 1.1 (367) | 8.7 (311) | 1.5 (188) | 6.7 (186) |
| oral administration | ||||||
|---|---|---|---|---|---|---|
| parameter | 1b | 35 | 11 | 13 | 7 | 9 |
| dose (mg/kg) | 18.8 | 21.3 | 24.4 | 18.3 | 18.2 | 17.5 |
| Tmax (h) | 7.0 | 4.0 | 2.5 | 3.3 | 4.0 | 3.3 |
| Cmax (μM) | 3.9 (0.09) | 12.5 (0.72) | 37 (0.11) | 2.7 (0.07) | 23.8 (0.19) | 3.3 (0.12) |
| AUC0-inf (μM·h) | 122 (2.80) | 202 (11.7) | 318 (0.95) | 28.1 (0.79) | 227 (1.82) | 24.6 (0.89) |
| oral BA (%) | 57 | 63 | 86 | 86 | 100 | 67 |
Values represent the average n = 2 rats per dose group. Values in parentheses have been corrected for plasma protein binding and represent unbound values.
Data from ref (12).
Figure 3.
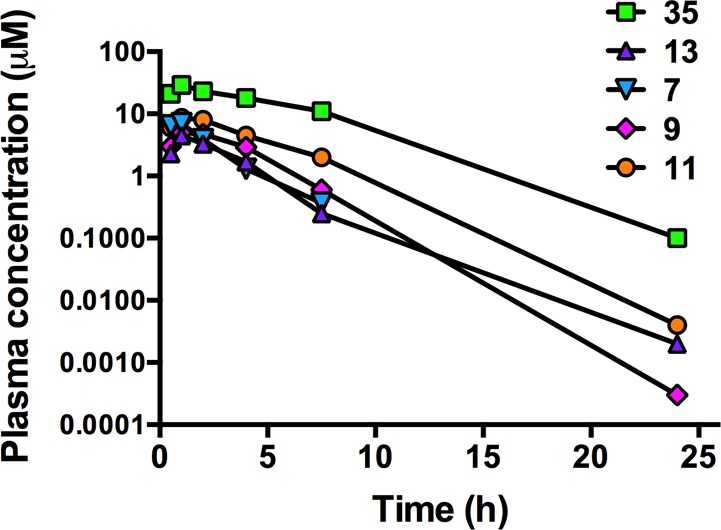
Plasma concentration versus time profiles following a single oral dose of 20 mg/kg to male Swiss outbred mice. Data represent the mean concentrations for two mice at each time point.
Figure 4.

Plasma concentration versus time profiles following a single intravenous (IV) or oral (PO) dose to male Sprague–Dawley rats. Dose levels are shown in Table 7. Each data point represents the mean of data from two rats, and error bars represent the range.
In rats, the oral bioavailability was high for all compounds tested, ranging from 63–100% compared to 57% for 1 (Table 7 and Figure 4). The plasma clearance and volume of distribution were significantly lower for the tetrahydro-2-naphthyl compounds substituted on the 6 position (7 and 11) than for those substituted on the 7 position (9 and 13); however, when corrected for differences in plasma protein binding, values across the four tetrahydro-naphthyl compounds were comparable (within 2-fold). The 6 position compounds also had lower blood to plasma partitioning consistent with their higher plasma protein binding compared to the 7 position analogues. Unbound clearance values for the tetrahydro-2-naphthyl compounds were somewhat lower for the two chloro-substituted compounds (438 and 589 mL/min/kg for 7 and 9, respectively) compared to the bromo compounds (933 and 800 mL/min/kg for 11 and 13, respectively), and all were higher than for 1 (263 mL/min/kg). As a result, unbound AUC values for the tetrahydro-2-naphthyls were approximately 2–4-fold lower than 1 after oral dosing at approximately 20 mg/kg. The t1/2 for the tetrahydro-2-naphthyl compounds ranged from 6 to 7 h in comparison to 13 h for 1. The indanyl compound (35) had low unbound clearance and volume of distribution, and overall unbound oral exposure was superior to 1 and to the tetrahydro-2-naphthyls (Table 7).
In Vivo Efficacy in the SCID Mouse Model of P. falciparum
Based on the observation of good oral exposure after dosing in mice, we tested three compounds in the SCID mouse P. falciparum efficacy model, which has become the standard model for evaluating the efficacy of new antimalarial compounds.23 The tetrahydro-2-naphthyl compounds substituted at the 7 position (9 and 13) were more potent against P. falciparum in the in vitro parasite assay than those substituted on the 6 position (7 and 11) and showed fairly equivalent exposure in mice after oral dosing. We therefore decided to test both a 6- and 7-substituted tetrahydro-2-naphthyl (7 and 9) in the P. falciparum SCID mouse model. Because 35 showed significantly higher plasma exposure in mice than the remaining compounds, we also selected this compound for an efficacy study despite the 10-fold lower potency in the P. falciparum parasite assay.
All three compounds were dosed orally once a day for 4 days and tested for efficacy and blood concentrations at two doses (10 and 30 mg/kg). Each was well tolerated, and there were no adverse reactions. The effective dose that led to 90% parasite clearance (ED90) was calculated 24 h after the last dose (Table 8 and Figure 5A). Pharmacokinetic sampling was performed on day 1 to allow the total blood AUC at the ED90 to be assessed (Tables 8 and S4; Figure 5B). Because of the complicated nature of blood in the SCID mouse model (i.e., mice contain a mixture of human and mouse plasma and erythrocytes and a hematocrit of 70–80%), it was not possible to accurately convert blood concentrations to plasma concentrations or to correct concentrations for differences in plasma protein binding.
Table 8. SCID Mouse Efficacy Data Based on Once Daily (QD) Dosing for 4 Daysa.
| compd | ED90 (mg/kg/day) | AUCED90 (μM·h/day) |
|---|---|---|
| 9b | ∼19 | 10.3 |
| 35b | ∼35 | ∼137 |
| 7b | >30 | >23 |
| 1 QDc | 8.1 | 45.7 |
| 1 BIDd | 3.0 | 30.8 |
| chloroquined | 4.3 | 3.1 |
ED90 was calculated from parasitemia levels measured 24 h after the last dose. AUC data are based on Day 1 pharmacokinetic data.
Based on only two dose levels administered.
1 QD data were taken from ref (11) and converted to different units for consistency.
1 BID and chloroquine data were taken from ref (12) and converted to different units for consistency.
Figure 5.

SCID mouse in vivo efficacy data. Mice were infected with P. falciparum Pf3D70087/N9 parasites by intravenous injection on day zero, and dosing was once daily for 4 days starting on day 3 postinfection. (A) Blood parasitemia levels were monitored daily by flow cytometry. (B) Pharmacokinetic analysis was performed after the first dose. Pharmacokinetic parameters are reported in Table S4.
All three compounds showed efficacy in this model; however, 9 significantly outperformed the other two compounds based on ED90 (Tables 8 and S4; Figure 5). The ED90 was higher for all three compounds than for 1 ranging from ∼19 mg/kg for 9 to >30 mg/kg for 7 and 35. The pharmacokinetic data indicated that, as expected, 35 exhibited higher total blood concentrations than the other two compounds. Both 7 and 9 were approximately dose linear for both Cmax and AUC, whereas 35 showed significant nonlinearity and evidence of a second peak at 8 h at the lower dose (Table S4). Despite requiring a higher dose, 9 showed higher apparent efficacy compared to 1 based on the total blood AUC at the ED90, which was 5-fold lower than that required for 1 in the once daily dosing regimen (Table 8). However, when accounting for differences in the plasma unbound fractions for 1 and 9 (approximately 0.002 for 1 and 0.03 for 9, Table 3), 1 likely has lower unbound concentrations at the ED90 and hence higher intrinsic efficacy. In contrast, 35 was significantly less effective than 1 suggesting that the lower intrinsic potency of 35 could not be overcome by higher exposure levels. The pharmacokinetic/pharmacodynamic relationship for 7 could not be evaluated in detail as the ED90 was not reached in this study.
Discussion
Malaria is one of the most serious global infectious diseases, and while a number of effective drugs have been used to treat malaria, drug resistance has led to the loss of most agents that have been in widespread use.2 A robust drug pipeline is therefore key to malaria control and elimination efforts.4 DHODH has emerged as a key new target in efforts to identify antimalarial drugs that work on clinical isolates resistant to standard therapies, and as noted, the triazolopyrimidine 1 is currently in clinical development.12 In order to identify potential backup compounds to 1 in case issues arise in its clinical development, we sought to identify additional compounds within the triazolopyrimidine series with the potential to advance into preclinical development. One identified potential liability of 1 has been the finding that, while it does not inhibit human DHODH, it does show moderate inhibition of the mouse and rat enzymes, which complicates toxicity testing in animals. We therefore sought to identify new derivatives that showed broad selectivity against all of the key mammalian enzymes. Herein we describe a series of tetrahydro-2-naphthyl and 2-indanyl triazolopyrimidine analogues, which display a range of potencies against Plasmodium DHODH but importantly lack activity against human DHODH or any of the key mammalian enzymes from species that are important for toxicological profiling (mouse, rat, and dog). Pharmacologic profiling and in vivo efficacy assays suggest that several of the identified analogues have potential, with 9 showing the most promising profile.
In evaluating the tetrahydro-2-naphthyl and 2-indanyl triazolopyrimidine analogues we found that the tetrahydro-2-naphthyls (e.g., 7, 9, 11, 13) had significantly greater potency on PfDHODH and Plasmodium parasites in whole cell assays than the 2-indanyl analogues (e.g., 35). The most potent of the tetrahydro-2-naphthyl derivatives also showed good activity (within 1.5-fold) against P. vivax DHODH suggesting that they would be useful for the treatment of both P. falciparum and P. vivax. In contrast, the best of the 2-indanyl analogues (35) was 7-fold less active on P. vivax DHODH suggesting it might not have good activity against the P. vivax parasite.
The X-ray structure of the 7-bromo analogue 13 showed that, in addition to the previously observed H-bonds and stacking interactions observed for other analogues in the series,11,12,17,18 a good halogen bond was formed between the bromo group of 13 and Cys-233 that is likely to provide considerable binding energy to the interaction. Indeed halogen bonding to protein residues that include Lewis bases (e.g., sulfur) are commonly observed in protein structures deposited in the PDB.24 The overall binding mode of 13 is very similar to that of 2, which was expected based on their close structural similarity. Like the naphthyl of 2, the tetrahydro-2-naphthyl of 13 better fills the available binding pocket than the SF5-aniline of 1, which supports our initial hypothesis that compounds with improved potency could be identified by substitution of the SF5-aniline with bulkier aromatic groups.
Comparison of the tetrahydro-2-naphthyl compounds containing 7 position (9 and 13) versus 6 position (7 and 11) substitutions identified a number of interesting differences. Analogues substituted in the 7 position were more potent than 6 position analogues, and indeed 13 with subnanomolar activity against the parasite in whole cell assays is the most potent analogue that has been identified in the series. However, these assays did not take into account potential differences in protein binding. The 6 position analogues exhibited consistently higher plasma protein binding than the 7 position compounds and a similar trend is also likely for binding to albumax present in the in vitro test medium. Assessment of the pharmacologic properties showed that while the 7 position tetrahydro-2-naphthyls had greater apparent antimalarial potency, analogues substituted at the 6 position had somewhat improved physicochemical properties. The 6 position analogues 7 and 11 were more soluble (in 0.1 N HCl and FeSSIF media) than either of the 7 position analogues (9 and 13). While the total plasma exposure (AUC and Cmax) in rats was higher for the 6 position analogues, correction of the data for protein binding indicated that the unbound exposure and unbound clearance were similar (within 2-fold) for the 6 and 7 position analogues with the chloro-substituted compounds (7 and 9) showing about 2-fold lower unbound clearance than the bromo-substituted compounds (11 and 13). So while total exposure appears greater for the 6 position analogues, unbound concentrations are comparable across the tetrahydro-naphthyl compounds tested. Protein binding data in mouse plasma was only available for 9, 11, and 35, and binding for each of these compounds was similar in each of the three species tested. Mouse plasma protein binding was therefore estimated for 7 and 13 using the average of the human and rat values, suggesting that unbound concentrations for each of the compounds are likely to be within a similar range (Table 6). The best of the 2-indanyl analogues (35) had considerably higher unbound plasma exposure in rats compared to the tetrahydro-2-naphthyls and to 1 and had similarly low unbound clearance.
The in vitro data generated in liver microsomes provided a mechanism to rank compounds based on their relative metabolic stability. The compounds selected for in vivo testing were expected to have good exposure, and this was seen following dosing to both rats and mice indicating that, qualitatively, the in vitro data were informative. Converting the in vitro intrinsic clearance determined in rat microsomes to a plasma clearance using physiologically based scaling factors and taking into account binding to rat plasma and microsomal proteins (as per ref (21)) and blood to plasma partitioning ratio led to an under prediction of the actual in vivo clearance by a factor of 3- to 16-fold for the five compounds tested. These trends are similar to what had been observed previously for 1(16) and suggest that in vivo clearance mechanisms for the series also involve additional non-CYP pathways not fully represented by the microsomal test system. In the case of 1, hepatocyte assays also led to an under prediction of in vivo clearance highlighting the difficulties in obtaining accurate clearance predictions based on in vitro data alone. Further studies with the current compounds using hepatocytes may have provided additional insight into their clearance mechanisms.
In vivo efficacy was assessed in the P. falciparum SCID mouse model. Of the three tested compounds, 9 (7 position) had the most potent in vivo activity. The total blood AUC required to reach ED90 was lower than for 1; however, it took a higher dose (∼19 mg/kg versus 8.1 mg/kg for QD dosing) to achieve this activity, possibly due to a higher unbound clearance of 9 compared to 1 as seen in rats. The binding trends in human and mouse plasma would suggest that unbound concentrations of 9 in the SCID mouse are likely to be considerably higher compared to those for 1 at the same total blood concentration. Therefore, the true in vivo potency of 1 based on unbound concentrations is likely to be greater than that for 9. In spite of the high blood exposure, the 2-indanyl (35) lacked sufficient intrinsic potency to provide good activity in the in vivo model. In order to improve patient compliance, single dose treatments are being sought by the international organizations that are promoting new drug discovery for malaria.4 While all three tested compounds (7, 9, and 35) showed in vivo antimalarial activity, it is unlikely that any of these compounds have the necessary properties to support a single dose treatment regimen at a practical dose level in humans.
Preliminary safety analysis included characterizing hERG channel activity to assess potential cardiac effects and CYP inhibition studies to determine the potential for drug–drug interactions. All five of the profiled compounds showed some CYP inhibition, with CYP isoform 2D6 being inhibited to the greatest extent. CYP2D6 inhibition was modest with the most potent inhibition observed in the 3–6 μM range, but does suggest some potential for drug–drug interactions with compounds that are substrates for this enzyme. Tested compounds in these series also showed some hERG inhibition (0.5–1.5 μM range). This inhibitory activity is similar to what was observed for 1, where subsequent studies using the rabbit wedge model and dog cardiovascular studies showed no evidence for QT prolongation, arrhythmias, or cardiac effects.12 Additionally, for 1 the unbound plasma concentration at likely therapeutic doses is 70–320-fold lower than the IC50 for hERG channel inhibition, suggesting it would be very unlikely that 1 would inhibit the channel in vivo. Insufficient efficacy data is available for compounds 7 and 35 to determine the safety margin relative to unbound concentrations at an efficacious dose; however, for 9 the Cmax at the ED90 in the SCID study (unbound concentration of approximately 0.05 μM) is ∼30-fold lower than the IC50 on the hERG channel, again suggesting a low likelihood the channel would be inhibited in vivo. However, given the hERG channel activity, careful evaluation of cardiac safety would be an important component of preclinical development if any of these compounds were to be advanced.
Several potential avenues remain feasible for additional optimization of the triazolopyrimidine series. First, only the chloro-substituted tetrahydro-2-naphthyls were tested in the SCID efficacy model, leaving open the possibility that one of the bromo analogues might have shown better in vivo efficacy. Additional optimization of the series might focus on reducing lipophilicity as this could lead to reduced intrinsic clearance and better in vivo exposure and also could lead to reduced hERG activity. Reduced lipophilicity would also likely result in improved aqueous solubility thereby simplifying formulation approaches for oral administration. Decreasing lipophilicity will likely require the SF5-anline of 1 to be replaced with more hydrophilic aromatic amines, provided potency can be maintained. Within the tetrahydro-2-naphthyls, it might yet be possible to identify more metabolically stable analogues by identifying the specific sites of metabolism and attempting to block them.
Conclusion
We have described the identification of several tetrahydro-2-naphthyl and 2-indanyl triazolopyrimidine analogues that are potent inhibitors of Plasmodium DHODH and that have improved species selectivity over the mammalian enzymes in comparison to the clinical candidate 1. The compounds have potent activity against P. falciparum in parasite assays and showed antimalarial activity in the P. falciparum SCID mouse model of disease. These compounds showed good plasma exposure after oral dosing in mice and rats and were well tolerated in the in vivo studies that were performed. Several of the identified compounds, most notably 9, have the potential to be further developed for treatment of malaria. However, the findings that 9 is less intrinsically potent than 1in vivo (based on expected unbound concentrations) and that the unbound clearance in rat is 2-fold higher than for 1 suggest that this compound is unlikely to meet the strict criteria required for a single dose cure at a reasonable dose.
Experimental Section
DHODH Purification
Plasmodium and mammalian DHODHs were expressed in E. coli as N-terminal truncations (lacking the mitochondrial transmembrane domains) fused to His6-purification tags as previously described.11,17,18,25 Protein (PfDHODHΔ384–413) for crystallization studies was further truncated to remove a proteolytically sensitive loop (amino acids 384–421) that prevents crystallization as previously described.17,26 Proteins were expressed in E. coli BL21 phage-resistant cells (Novagen), and they were purified by Ni2+-agarose column chromatography (GE Healthcare Life Sciences, HisTrap HP) and gel-filtration column chromatography (GE Healthcare Life Sciences, HiLoad 16/600 Superdex 200 pg) as previously described.11,17,18 Protein was concentrated to 10–30 mg/mL and stored at −80 °C.
DHODH Kinetic Analysis
Steady-state kinetic analysis was performed using a dye-based spectrophotometric method in assay buffer (100 mM HEPES, pH 8.0, 150 mM NaCl, 10% glycerol, 0.05% Triton X-100) as previously described.11 All buffers were degassed prior to use. Enzyme and substrate concentrations were DHODH (E = 5 nM), substrates (0.2 mM l-dihydroorotate, 0.02 mM CoQd, and 0.12 mM 2,5-dichloroindophenol (DCIP)). Inhibitor stock solutions (100 mM) were made in DMSO and protected from light in dark amber vials. Serial dilutions were than performed to generate a 3-fold dilution series of 100× stocks also in DMSO that were used in the final assay (final inhibitor concentration range was 0.01–100 μM). Data were collected in triplicate for each concentration in the titration. Data were fitted to the log[I] vs response (three parameters) equation to determine the concentration that gave 50% enzyme inhibition (IC50) except for compounds where the IC50 > 10 μM, which were instead fitted to the standard IC50 equation (Y = 1/(1 + X/IC50)) in Graphpad Prism. Error represents the 95% confidence interval of the fit.
P. falciparum Growth Assays
P. falciparum parasites were grown in RPMI-1640 containing 0.5% Albumax-II as previously described.11 Inhibitor stock solutions were prepared as described above for DHODH kinetic analysis except that a 500× dilution series prepared in DMSO was used to generate a 10× dilution series in RPMI so that final DMSO concentration in the media was 0.2%. Growth assays for P. falciparum 3D7 cells and drug-resistant parasites were performed using the SYBR-green 72 h growth assay.18 Data were collected in triplicate to quadruplicate for each concentration in the titration. Data were fitted to the log[I] vs response variable slope (four parameter) equation in graph pad prism to determine the effective concentration (EC50) that led to 50% reduction in parasitemia. Error represents the 95% confidence interval of the fit.
In Vitro Metabolism and Physicochemical Assessment
In Vitro Metabolism
Selected compounds were incubated at a concentration of 1 μM with mouse, rat, or pooled human liver microsomes (BD Gentest, Woburn, MA or XenoTech LLC, Lenexa, Kansas City) at a microsomal protein concentration of 0.4 mg/mL.11 Substrate depletion was assessed by LC–MS. Measured in vitro intrinsic clearance values (μL/min/mg microsomal protein) were converted to predicted in vivo intrinsic clearances (mL/min/kg) using published physiologically-based scaling factors.21
Solubility
Aqueous solubility of compounds was assessed by nephelometry as described previously.27 Compound stock solutions were prepared in DMSO, which was then spiked into pH 6.5 phosphate buffer giving a final DMSO concentration of 1%. Solubility measurements in physiologically relevant media were conducted for selected compounds. Media included 0.1 N HCl to represent a simulated gastric condition, fasted (FaSSIF, pH 6.5) and fed (FeSSIF, pH 5.0 or 5.8) state simulated intestinal fluids prepared as described previously.28 Solid material was incubated in media for a period of 5–6 h at 37 °C with periodic mixing and sample supernatant concentrations determined by HPLC with UV detection following two separate centrifugations to remove undissolved compound.
Plasma Protein Binding and Partitioning
Select compounds were analyzed for plasma protein binding (PPB) in human, rat, or mouse plasma using an ultracentrifugation method at 37 °C as previously described11 followed by LC–MS analysis as described below. Protein binding was calculated by comparing the unbound concentration in the ultracentrifuged samples to the total plasma concentration in control samples incubated at 37 °C for the same time period. For compounds progressing to rat pharmacokinetic studies, blood to plasma partitioning ratios in rat blood were also assessed as described previously.16
Pharmacokinetic Analysis in Mice and Rats
Pharmacokinetic studies for select compounds were performed in mice and rats in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes, and the study protocols were approved by the Monash Institute of Pharmaceutical Sciences Animal Ethics Committee.
Oral doses were administered as a suspension in a vehicle containing 0.5% w/v carboxymethylcellulose, 0.5% v/v benzyl alcohol (as a preservative), and 0.4% v/v Tween 80, and intravenous doses were administered in cosolvent vehicle containing 40% propylene glycol, 10% ethanol, 0.4% Tween 80 in water. IV doses to rats were administered as a 10 min constant rate infusion of 1 mL into an indwelling jugular vein cannula, and oral doses were administered by gavage as 10 mL/kg (both rats and mice). Blood was collected from rats via a cannula inserted into the carotid artery on the day prior to dosing, and from mice via either submandibular bleed or terminal cardiac puncture (maximum of two samples per mouse) into tubes containing heparin as an anticoagulant, and plasma was separated by centrifugation. Compound concentrations were quantitated by LC–MS using a Waters Xevo TQ mass spectrometer coupled to a Waters Acquity UPLC. Chromatography was conducted using a Supelco Ascentis Express RP Amide column (50 × 2.1 mm, 2.7 μm) with a mobile phase consisting of 0.05% formic acid in water and 0.05% formic acid in methanol mixed via a linear gradient with a cycle time of 4 min. The flow rate was 0.4 mL/min, and the injection volume was 3 μL. Detection was via positive electrospray ionization mass spectrometry with multiple-reaction monitoring using a cone voltage of 30–40 V and collision energy of 25–35 V. Calibration curves were prepared using blank rat or mouse plasma, and validation studies confirmed the accuracy, precision, and limit of quantitation to be within acceptable ranges.
Cyp Inhibition Assays
Inhibition of cytochrome P450 (CYP) enzymes was assessed in human liver microsomes using a substrate specific approach where the formation of metabolites specific to a particular CYP isoform was monitored. Microsomes were suspended in phosphate buffer and incubated at 37 °C in the presence of probe substrates,29 and reactions were initiated by the addition of an NADPH-regenerating system. Reactions were quenched at appropriate times using acetonitrile. Samples were centrifuged and concentrations of the metabolites assessed by LC–MS. The IC50 value for each compound or control inhibitor was the concentration at which there was a 50% reduction in the amount of metabolite formed relative to the maximum metabolite formation in the absence of inhibitor.
hERG Analysis
Select compounds were tested for activity against the human ether-a-go-go related gene (hERG) K+ channel in a IonWorks patch clamp electrophysiology assay under contract to Essen BioScience (Welwyn Garden City Hertfordshire, UK).
SCID Mouse Efficacy Studies
NOD-scid IL-2Rγnull (NSG) mice (Jackson Laboratory, USA) (23–36 g) were engrafted with human erythrocytes and then infected with P. falciparum Pf3D70087/N9 parasites (20 × 106) by intravenous injection as described.23 Compounds (7, 9, and 35) were administered by oral gavage once daily (QD) for four consecutive days starting on day 3 postinfection in vehicle (0.5% w/v sodium carboxymethylcellulose, 0.5% v/v benzyl alcohol, 0.4% v/v Tween 80 in water). Parasitemia was monitored by flow cytometry. To verify the actual doses administered, formulation concentrations were measured. Compound levels in the formulations or in the blood of infected mice were measured by LC–MS/MS as described above. Human biological samples were sourced ethically, and their research use was in accordance with the terms of the informed consents.
Small Molecule X-ray Structure Determination and Refinement of 9
Compound 9 was crystallized from ethanol. A colorless prism, measuring 0.20 × 0.13 × 0.10 mm3 was mounted on a loop with oil. Data was collected at −173 °C on a Bruker APEX II single crystal X-ray diffractometer, Mo-radiation. Crystal-to-detector distance was 40 mm, and exposure time was 15 s per frame for all sets. The scan width was 0.5°. Data collection was 99.9% complete to 25° in θ. A total of 113483 reflections were collected covering the indices, h = −11 to 11, k = −38 to 38, l = −18 to 18. Of that, 16915 reflections were symmetry independent and the Rint = 0.0703 indicated that the data was of average quality (0.07). Indexing and unit cell refinement indicated a primitive monoclinic lattice. The space group was found to be P21 (No. 4). The data were integrated and scaled using SAINT aand SADABS within the APEX2 software package by Bruker.30 Solution by direct methods (SHELXS, SIR97)31,32 produced a complete heavy atom phasing model consistent with the proposed structure. The structure was completed by difference Fourier synthesis with SHELXL97.33,34 Scattering factors are from Waasmair and Kirfel.35 Hydrogen atoms were placed in geometrically idealized positions and constrained to ride on their parent atoms with C···H distances in the range 0.95–1.00 Å. Isotropic thermal parameters Ueq were fixed such that they were 1.2Ueq of their parent atom Ueq for CH’s and 1.5Ueq of their parent atom Ueq in the case of methyl groups. All non-hydrogen atoms were refined anisotropically by full-matrix least-squares.
PfDHODH Crystallization, Data Collection, Structure Determination and Refinement for the PfDHODH-13 Bound Structure
Preliminary crystallization conditions were found using the random crystallization screen Cryos suite (Nextal). Then the condition was refined by variation of pH, precipitant, and protein concentrations to find optimal conditions. Crystallization was setup with hanging drop vapor diffusion at 20 °C. Crystals of PfDHODHΔ384–413-13 complex grew at condition with 0.16 M ammonium sulfate, 0.1 M sodium acetate, pH 4.2, 9.5% PEG4000 (w/v), 24% glycerol (v/v), and 10 mM DTT. The crystallization drop was mixed with an equal volume of reservoir solution and PfDHODHΔ384–413 (27 mg/mL) pre-equilibrated with 1 mM 13 (0.1 M stock solution in DMSO) and 2 mM dihydroorotate (DHO, 0.1 M stock solution in DMSO). All commercially available reagents were obtained from Sigma. Crystals typically grew in 1 week.
Diffraction data were collected at 100 K on beamline 19ID at Advanced Photon Source (APS) using an ADSC Q315 detector. The crystal of PfDHODHΔ384–413-13 diffracted to 2.32 Å and has a space group of P64 with the cell dimension of a = b = 86.1, c = 139.1 (Table S3). The structure contains only one molecule of PfDHODH in the asymmetric unit. Diffraction data were integrated, and intensities were scaled with HKL2000 package.36
Crystallographic phases for PfDHODH inhibitors were solved by molecular replacement with Phaser37 using the previously reported structure of PfDHODHΔ384–413 bound to DSM1 (PDB ID 3I65(17)) as a search model (ligands were removed from the search model). Structures were rebuilt with COOT38 and refined in PHENIX39 to Rwork and Rfree of 0.18 and 0.21, respectively (Table S3). Electron density for residues 159–160, 344–354, and 567–569 were missing and thus not built into the final structure. The final structure contained 2972 atoms and 73 water molecules. Atomic representations were created using PyMOL Molecular Graphics System (Version 1.7, Schrödinger).
Chemistry General Methods
All reagents and starting materials were obtained from commercial suppliers and used without further purification unless otherwise stated. Reaction progress was monitored by thin layer chromatography (TLC) on preloaded silica gel 60 F254 plates. Visualization was achieved with UV light and iodine vapor. Flash chromatography was carried out using prepacked Teledyne Isco Redisep Rf silica-gel columns as the stationary phase and analytical grade solvents as the eluent unless otherwise stated. Yields were of purified product and were not optimized. 1H NMR spectra were recorded on Bruker Avance 300 and 500 MHz spectrometer at ambient temperature. Chemical shifts are reported in parts per million (δ) and coupling constants in Hz. 1H NMR spectra were referenced to the residual solvent peaks as internal standards (7.26 ppm for CDCl3, 2.50 ppm for DMSO-d6, and 3.34 ppm for MeOD). Spin multiplicities are described as s (singlet), brs (broad singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublets), dt (doublet of triplets), and m (multiplet). Total ion current traces were obtained for electrospray positive and negative ionization (ES+/ES−) on a Bruker Esquire Liquid Chromatograph-Ion trap mass spectrometer. Analytical chromatographic conditions used for the LC–MS analysis are as follows: Column, Zorbax Extend C18 from Agilent technologies, 2.1 × 100 mm. The stationary phase particle size is 3.5 μM. Solvents were A, aqueous solvent = water + 5% acetonitrile + 1% acetic acid; B, organic solvent = acetonitrile + 1% acetic acid; Methods, 14 min run time (0–10 min 20–100% B, flow rate 0.275 mL/min; 10–12 min 100% B, flow rate 0.350 mL/min; 12–12.50 min 100–20% B, flow rate 0.350 mL/min; 12.50–14.0 min 20% B, flow rate 0.350 mL/min). The following additional parameters were used: injection volume (10 μL), column temperature (30 °C), UV wavelength range (254–330 nm). Analytical HPLC analyses were performed on a SupelcoSIL LC18 column (5 μm, 4.6 mm × 25 cm) with a linear elution gradient ranging from 0 to 100% ACN over 27 min, using a SupelcoSIL LC18 column (5 μm, 4.6 mm × 25 cm) at a flow rate of 1 mL/min. A purity of >95% has been established for all reported compounds (Table 1). The two enantiomers were separated for selected compounds on a semipreparative chiral HPLC on Chiralcel OD-H (250 × 20) mm column eluting with 0.1% diethylamine in hexane (A)/ethanol (B) in 36 min run time (0–10 min, 20% B; 10–20 min, 20–35% B; 20–30 min, 35% B; 30.01–36 min 20% B; flow rate 5 mL/min).
Experimental Data
2-(1,1-Difluoroethyl)-5-methyl-N-(1,2,3,4-tetrahydronaphthalen-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (5)
White solid (89% yield). 1H NMR (400 MHz, DMSO-d6): δ 8.24 (d, J = 7.1 Hz, 1H), 7.13- 7.09 (m, 4H), 6.67 (s, 1H), 4.16–4.01 (br, 1H), 3.10–3.04 (m, 2H), 3.02–2.85 (m, 2H), 2.45 (s, 3H), 2.15–2.06 (m, 4H), 2.04–1.91 (m, 1H). ESIMS m/z: 344 (MH)+.
2-(1,1-Difluoroethyl)-N-(6-fluoro-1,2,3,4-tetrahydronaphthalen-2-yl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (6)
White solid (75% yield). 1H NMR (500 MHz, DMSO-d6): δ 8.29 (d, J = 8.8 Hz, 1H), 7.28–7.07 (m, 1H), 7.06–6.88 (m, 2H), 6.69 (s, 1H), 4.17–3.94 (m, 1H), 3.14–2.92 (m, 4H), 2.47 (s, 3H), 2.12 (t, J = 18.8, 3H), 2.03–1.90 (m, 2H). ESIMS m/z: 362.1(MH)+.
2-(1,1-Difluoroethyl)-N-(6-chloro-1,2,3,4-tetrahydronaphthalen-2-yl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (7) (Enantiomer-I)
White solid. 1H NMR (400 MHz, MeOD): δ 7.17–7.12 (m, 3H), 6.55 (s, 1H), 4.15–4.13 (m, 1H), 3.26–3.20 (m, 1H), 3.05–2.94 (m, 3H), 2.55 (s, 3H), 2. Twenty-nine −2.25 (m, 1H), 2.11 (t, J = 18.72 Hz, 3H), 2.01–1.97 (m, 1H). ESIMS m/z: 378 (MH)+.
2-(1,1-Difluoroethyl)-N-(6-chloro-1,2,3,4-tetrahydronaphthalen-2-yl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (8) (Enantiomer-II)
White solid. 1H NMR (400 MHz, MeOD): δ 7.17–7.12 (m, 3H), 6.55 (s, 1H), 4.18–4.12 (m, 1H), 3.26–3.21 (m, 1H), 3.05–2.95 (m, 3H), 2.55 (s, 3H), 2.29–2.25 (m, 1H), 2.11 (t, J = 18.72 Hz, 3H), 2.01–1.95 (m, 1H). ESIMS m/z: 378 (MH)+.
(S)-N-(7-Chloro-1,2,3,4-tetrahydronaphthalen-2-yl)-2-(1,1-difluoroethyl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (9) (Enantiomer-I)
White solid. 1H NMR (400 MHz, MeOD): δ 7.16–7.13 (m, 3H), 6.55 (s, 1H), 4.15–4.12 (m, 1H), 3.26–3.21 (m, 1H), 3.01–2.97 (m, 3H), 2.55 (s, 3H), 2.29–2.25 (m, 1H), 2.11 (t, J = 18.75 Hz, 3H), 2.01–1.94 (m, 1H). ESIMS m/z: 378(MH)+.
(R)-N-(7-Chloro-1,2,3,4-tetrahydronaphthalen-2-yl)-2-(1,1-difluoroethyl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (10) (Enantiomer-II)
White solid. 1H NMR (400 MHz, MeOD): δ 7.16–7.13 (m, 3H), 6.55 (s, 1H), 4.16–4.12 (m, 1H), 3.26–3.21 (m, 1H), 3.03–2.97 (m, 3H), 2.55 (s, 3H), 2.29–2.25 (m, 1H), 2.11 (t, J = 18.72 Hz, 3H), 2.01–1.95 (m, 1H). ESIMS m/z: 378(MH)+.
2-(1,1-Difluoroethyl)-N-(6-bromo-1,2,3,4-tetrahydronaphthalen-2-yl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (11) (Enantiomer-I Is the Second Eluted on the Chiral Column)
White solid. 1H NMR (400 MHz, CDCl3): δ 7.32–7.30 (m, 2H), 6.99 (d, J = 8.04 Hz,1H), 6.17 (d, J = 8.32 Hz, 1H), 6.14 (s, 1H), 4.02–3.99 (m, 1H), 3.29–3.23 (m, 1H), 3.02–2.99 (m, 2H), 2.91–2.84 (m, 1H), 2.60 (s, 3H), 2.31–2.28 (m, 1H), 2.15 (t, J = 18.76 Hz, 3H), 1.99–1.94 (m, 1H). ESIMS m/z: 424 (M + 2)
2-(1,1-Difluoroethyl)-N-(6-bromo-1,2,3,4-tetrahydronaphthalen-2-yl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (12) (Enantiomer-II Is the First Eluted on the Chiral Column)
White solid. 1H NMR (400 MHz, CDCl3): δ 7.32–7.29 (m, 2H), 6.99 (d, J = 8.04 Hz, 1H), 6.18 (d, J = 7.56 Hz, 1H), 6.15 (s, 1H), 4.01 (brs, 1H), 3.29–3.24 (m, 1H), 3.02–2.99 (m, 2H), 2.91–2.85 (m, 1H), 2.60 (s, 3H), 2.31–2.29 (m, 1H), 2.15 (t, J = 18.76 Hz, 3H), 1.99–1.95 (m, 1H). ESIMS m/z: 424 (M + 2).
2-(1,1-Difluoroethyl)-N-(7-bromo-1,2,3,4-tetrahydronaphthalen-2-yl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (13) (Enantiomer-I)
White solid. 1H NMR (400 MHz, DMSO-d6): δ 8.28 (d, J = 8.76 Hz, 1H), 7.34–7.30 (m, 2H), 7.11 (d, J = 8.00 Hz, 1H), 6.67 (s, 1H), 4.07 (brs, 1H), 3.09–3.05 (m, 2H), 2.93–2.89 (m, 2H), 2.45 (s, 3H), 2.29–2.25 (m, 1H), 2.11 (t, J = 19.12 Hz, 3H), 1.98–1.94 (m, 1H). ESIMS m/z: 422 (MH)+.
2-(1,1-Difluoroethyl)-N-(7-bromo-1,2,3,4-tetrahydronaphthalen-2-yl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (14) (Enantiomer-II)
White solid. 1H NMR (400 MHz, DMSO-d6): δ 8.28 (d, J = 8.00 Hz, 1H), 7.34–7.30 (m, 2H), 7.11 (d, J = 8.16 Hz, 1H), 6.68 (s, 1H), 4.07 (brs, 1H) 3.09–3.05 (m, 2H), 2.90–2.88 (m, 2H), 2.45 (s, 3H), 2.29–2.25 (m, 1H), 2.11 (t, J = 19.02 Hz, 3H), 1.98–1.94 (m, 1H). ESIMS m/z: 424 (M + 2).
2-(1,1-Difluoroethyl)-N-(6-methoxy-1,2,3,4-tetrahydronaphthalen-2-yl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (15)
White solid (85% yield). 1H NMR (300 MHz, CDCl3) δ 7.06 (d, J = 8.3 Hz, 1H), 6.86–6.63 (m, 2H), 6.31–6.07 (m, 2H), 4.15–3.92 (m, 1H), 3.82 (s, 3H), 3.27 (dd, J = 15.7, 4.2 Hz, 1H), 3.13–2.79 (m, 3H), 2.61 (s, 3H), 2.44–2.24 (m, 1H), 2.16 (t, J = 18.8 Hz, 3H), 2.07–1.88 (m, 1H). ESIMS m/z: 374.4 (MH)+.
2-(1,1-Difluoroethyl)-N-(7-methoxy-1,2,3,4-tetrahydronaphthal-en-2-yl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (16) (Enantiomer-I)
1H NMR (400 MHz, MeOD): δ 7.05 (d, J = 8.4, 1H), 6.75–6.55 (m, 2H), 6.55 (s, 1H), 4.15–4.12 (m, 1H), 3.76 (s, 3H), 3.25–3.19 (m, 1H), 3.01–2.94 (m, 3H), 2.56 (s, 3H), 2.28–2.25 (m, 1H), 2.11 (t, J = 18.75 Hz, 3H), 2.16–1.94 (m, 1H). ESI-MS m/z: 374.2 (MH)+.
2-(1,1-Difluoroethyl)-N-(7-methoxy-1,2,3,4-tetrahydronaphthal-en-2-yl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (17) (Enantiomer-II)
White solid. 11H NMR (400 MHz, MeOD): δ 7.05 (d, J = 8.4, 1H), 6.75–6.69 (m, 2H), 6.55 (s, 1H), 4.14–4.12 (m, 1H), 3.76 (s, 3H), 3.25–3.19 (m, 1H), 3.01–2.94 (m, 3H), 2.56 (s, 3H), 2.28–2.24 (m, 1H), 2.14 (t, J = 18.68 Hz, 3H), 2.03–1.96 (m, 1H). ESI-MS m/z: 374.2(MH)+.
2-(1,1-Difluoroethyl)-N-(6,7-difluoro-1,2,3,4-tetrahydronaphthalen-2-yl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (18) (Enantiomer-I)
White solid. 1H NMR (500 MHz, CDCl3): δ 7.05–6.92 (m, 2H), 6.43–6.25 (m, 2H), 4.13–3.97 (m, 1H), 3.36–3.16 (m, 1H), 3.12–2.85 (m, 3H), 2.64 (s, 3H), 2.43–2.28 (m, 1H), 2.16 (t, J = 18.92 Hz, 3H), 2.08–1.91 (m, 1H). ESIMS m/z: 380.2 (MH)+.
2-(1,1-Difluoroethyl)-N-(6,7-difluoro-1,2,3,4-tetrahydronaphthalen-2-yl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (19) (Enantiomer-II)
White solid. 1H NMR (300 MHz, CDCl3): δ 7.08–6.86 (m, 2H), 6.26–6.13 (s, 2H), 4.13–3.93 (m, 1H), 3.33–3.20 (m, 1H), 3.11–2.83 (m, 3H), 2.63 (s, 3H), 2.38–2.25 (m, 1H), 2.08 (t, J = 18.8 Hz, 3H), 2.06–1.89 (m, 1H). ESIMS m/z: 380.2 (MH)+.
2-(1,1-Difluoroethyl)-N-(6-fluoro-7-(trifluoromethyl)-1,2,3,4-tetrahydronaphthalen-2-yl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (20)
White solid (80% yield). 1H NMR (500 MHz, CDCl3): δ 7.39 (d, J = 7.0 Hz, 1H), 7.04 (d, J = 10.8 Hz, 1H), 6.36–6.06 (m, 2H), 4.11–4.01 (m, 1H), 3.35 (dd, J = 16.2, 4.7 Hz, 1H), 3.18–2.92 (m, 3H), 2.63 (s, 3H), 2.43–2.31 (m, 1H), 2.17 (t, J = 18.8 Hz, 3H), 2.06–1.95 (m, 1H). ESIMS m/z: 430.2 (MH)+.
2-(1,1-Difluoroethyl)-N-(7-fluoro-6-(trifluoromethyl)-1,2,3,4-tetrahydro-naphthalen-2-yl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (21)
White solid (83% yield). 1H NMR (300 MHz, CDCl3): δ 7.42 (d, J = 7.0 Hz, 1H), 6.99 (d, J = 10.6 Hz, 1H), 6.26–6.10 (m, 2H), 4.10–3.98 (m, 1H), 3.37 (dd, J = 16.9, 4.9 Hz, 1H), 3.15–2.89 (m, 3H), 2.64 (s, 3H), 2.47–2.28 (m, 1H), 2.16 (t, J = 18.8 Hz, 3H), 2.05–1.95 (m, 1H). ESIMS m/z: 452.3 (M+Na)+.
5-Methyl-N-(1,2,3,4-tetrahydronaphthalen-2-yl)-2-(trifluoromethyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (22)
White solid (90% yield). 1H NMR (300 MHz, CDCl3): δ 7.25–7.09 (m, 4H), 6.23 (s, 1H), 6.17 (d, J = 8.0 Hz, 1H), 4.15–3.98 (m, 1H), 3.35 (dd, J = 16.4, 5.0 Hz, 1H), 3.15–2.84 (m, 3H), 2.64 (s, 3H), 2.41–2.26 (m, 1H), 2.12–1.95 (m, 1H); ESIMS m/z: 348.3 (MH)+.
N-(6-Fluoro-1,2,3,4-tetrahydronaphthalen-2-yl)-5-methyl-2-(trifluoromethyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (23)
White solid (80% yield). 1H NMR (500 MHz, CDCl3): δ 7.17–7.05 (m, 1H), 7.0–6.82 (m, 2H), 6.43–6.12 (m, 2H), 4.12–3.99 (m, 1H), 3.37–3.26 (m, 1H), 3.10–2.89 (m, 3H), 2.64 (s, 3H), 2.40–2.29 (m, 1H), 2.09–1.98 (m, 1H). ESIMS m/z: 366.3 (MH)+.
N-(7-Chloro-1,2,3,4-tetrahydronaphthalen-2-yl)-5-methyl-2-(trifluoromethyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (24) (Enantiomer-I)
1H NMR (400 MHz, MeOD): δ 7.17–7.10 (m, 3H), 6.62 (s, 1H), 4.19–4.09 (m, 1H), 3.27–3.19 (m, 1H), 3.05–2.90 (m, 3H), 2.56 (s, 3H), 2.30–2.20 (m, 1H), 2.05–1.94 (m, 1H). ESIMS m/z: 382.2 (MH)+.
N-(7-Chloro-1,2,3,4-tetrahydronaphthalen-2-yl)-5-methyl-2-(trifluoromethyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (25) (Enantiomer-II)
1H NMR (400 MHz, MeOD): δ 7.17–7.10 (m, 3H), 6.62 (s, 1H), 4.18–4.10 (m, 1H), 3.27–3.19 (m, 1H), 3.05–2.90 (m, 3H), 2.56 (s, 3H), 2.30–2.20 (m, 1H), 2.05–1.97 (m, 1H). ESIMS m/z: 382.0 (MH)+.
2-(1,1-Difluoroethyl)-N-(2,3-dihydro-1H-inden-2-yl)-5-methyl[1,2,4]triazolo[1,5-a]pyrim-idin-7-amine (26)
Pale yellow solid (85% yield). 1H NMR (400 MHz, CDCl3): δ 7.30–7.24 (m, 4H), 6.3 (d, J = 7.3 Hz, 1H), 6.18 (s, 1H), 4.57–4.49 (m, 1H), 3.51 (dd, J = 7.1 Hz, J = 16.2 Hz, 2H), 3.13–3.08 (m, 2H), 2.62 (s, 3H), 2.13 (t, J = 18.7 Hz, 3H). ESIMS m/z: 330 (MH)+.
N-(5-Bromo-2,3-dihydro-1H-inden-2-yl)-2-(1,1-difluoroethyl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (27)
White foam (57% yield). 1H NMR (400 MHz, DMSO-d6): δ 8.55–8.49 (br, 1H), 7.46–7.44 (br, 1H), 7.37–7.35 (m, 1H), 7.22–7.20 (m, 1H), 6.63 (s, 1H), 4.68–4.58 (br, 1H), 3.32–3.28 (br, 1H), 3.21–3.12 (m, 3H), 2.47 (s, 3H), 2.09 (t, J = 19.2 Hz, 3H). ESIMS m/z: 408 (MH)+.
N-(5-Chloro-2,3-dihydro-1H-inden-2-yl)-2-(1,1-difluoroethyl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (28)
White solid (82% yield). 1H NMR (400 MHz, DMSO-d6): δ 8.57–8.48 (m, 1 H), 7.34–7.29 (m, 1 H), 7.29–7.19 (m, 2 H), 6.63 (s, 1 H), 4.72–4.57 (m, 1 H), 3.41–3.35 (m, 2 H), 3.23–3.07 (m, 2 H), 2.09 (t, J = 19.1 Hz, 3 H). ESIMS m/z: 364 (MH)+.
N-(5,6-Dichloro-2,3-dihydro-1H-inden-2-yl)-2-(1,1-difluoroethyl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (29)
White solid (41% yield). 1H NMR (400 MHz, CDCl3): δ 7.37 (s, 2H), 6.28 (d, J = 7.6 Hz, 1H), 6.16 (s, 1H), 4.60–4.52 (m, 1H), 3.51–3.45 (m, 2H), 3.07 (dd, J = 4.5 Hz, J = 16.4 Hz, 2H), 2.62 (s, 3H), 2.13 (t, J = 18.7 Hz, 3H). ESIMS m/z: 398 (MH)+.
2-(1,1-Difluoroethyl)-5-methyl-N-(5-(methylsulfonyl)-2,3-dihydro-1H-inden-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (30)
White solid (58% yield). 1H NMR (400 MHz, DMSO-d6): δ 8.57 (d, J = 7.6 Hz, 1 H), 7.82–7.78 (m, 1 H), 7.78–7.69 (m, 1 H), 7.57–7.47 (m, 1 H), 6.65 (s, 1 H), 4.79–4.63 (m, 1 H), 3.52–3.38 (m, 2 H), 3.30–3.18 (m, 2H), 3.19 (s, 3 H), 2.48 (s, 3 H), 2.09 (t, J = 19.2 Hz, 3 H); ESIMS m/z: 408 (MH)+.
2-(2-(1,1-Difluoroethyl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-ylamino)-N,N-dimethyl-2,3-dihydro-1H-indene-5-sulfonamide (31)
White solid (62% yield). 1H NMR (400 MHz, DMSO-d6): δ 8.59 (d, J = 7.8 Hz, 1 H), 7.61 (br s, 1 H), 7.60–7.54 (m, 1 H), 7.53–7.48 (m, 1 H), 6.65 (s, 1 H), 4.76–4.64 (m, 1 H), 3.52–3.39 (m, 2 H), 3.30–3.19 (m, 2 H), 2.61 (s, 6 H), 2.48 (s, 3 H), 2.09 (t, J = 19.2 Hz, 3 H). ESIMS m/z: 437 (MH)+.
N-(4,7-Dimethyl-2,3-dihydro-1H-inden-2-yl)-2-(1,1-difluoroethyl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (32)
White solid (74% yield). 1H NMR (400 MHz, CDCl3): δ 6.99 (s, 2H), 6.31 (d, J = 7.6 Hz, 1H), 6.19 (s, 1H), 4.58–4.50 (m, 1H), 3.45 (dd, J = 7.3 Hz, J = 16.2 Hz, 2H), 3.05–2.99 (m, 2H), 2.63 (s, 3H), 2.26 (s, 6H), 2.14 (t, J = 18.7 Hz, 3H). ESIMS m/z: 358 (MH)+.
N-(4,7-Difluoro-2,3-dihydro-1H-inden-2-yl)-2-(1,1-difluoroethyl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (33)
White solid (65% yield). 1H NMR (300 MHz, CDCl3): δ 6.94 (t, J = 5.8 Hz, 2H), 6.42 (brs, 1H), 6.21 (s, 1H), 4.73–4.53 (m, 1H), 3.70–3.44 (m, 2H), 3.35–3.08 (m, 2H), 2.67 (s, 3H), 2.14 (t, J = 18.8 Hz, 3H). ESIMS m/z: 366.3 (MH)+.
2-(1,1-Difluoroethyl)-N-(4-fluoro-2,3-dihydro-1H-inden-2-yl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (34)
White solid (58% yield). 1H NMR (300 MHz, CDCl3): δ 7.24 (dd, J = 8.1, 5.2 Hz, 1H), 7.04–6.88 (m, 2H), 6.33 (d, J = 7.3 Hz, 1H), 6.19 (s, 1H), 4.68–4.48 (m, 1H), 3.50 (dt, J = 15.4, 7.7 Hz, 2H), 3.20–3.00 (m, 2H), 2.63 (s, 3H), 2.15 (t, J = 18.8 Hz, 3H). ESIMS m/z: 348.5 (MH)+.
N-(5,6-Difluoro-2,3-dihydro-1H-inden-2-yl)-2-(1,1-difluoroethyl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (35)
White solid (75% yield). 1H NMR (500 MHz, CDCl3): δ 7.10 (t, J = 8.6 Hz, 2H), 6.31 (d, J = 7.5 Hz, 1H), 6.18 (s, 1H), 4.71–4.48 (m, 1H), 3.50 (dd, J = 16.2, 7.0 Hz, 2H), 3.17–2.97 (m, 2H), 2.64 (s, 3H), 2.16 (t, J = 18.8 Hz, 3H). ESIMS m/z: 366.3 (MH)+.
2-(1,1-Difluoroethyl)-5-methyl-N-(4-(trifluoromethyl)-2,3-dihydro-1H-inden-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (36)
White solid (70% yield). 1H NMR (500 MHz, CDCl3): δ 7.56 (d, J = 7.8 Hz, 1H), 7.50 (d, J = 7.5 Hz, 1H), 7.40 (t, J = 7.7 Hz, 1H), 6.32 (d, J = 7.4 Hz, 1H), 6.22 (s, 1H), 4.71–4.57 (m, 1H), 3.71 (dd, J = 17.2, 7.2 Hz, 1H), 3.60 (dd, J = 16.5, 7.3 Hz, 1H), 3.30 (dd, J = 17.1, 4.1 Hz, 1H), 3.19 (dd, J = 16.5, 4.6 Hz, 1H), 2.66 (s, 3H), 2.16 (t, J = 18.8 Hz, 3H). ESIMS m/z: 398.3 (MH)+.
N-(5,6-Difluoro-2,3-dihydro-1H-inden-2-yl)-5-methyl-2-(trifluoromethyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (37)
1H NMR (400 MHz, MeOD): δ 7.17 (t, J = 9.04 Hz, 2H), 6.62 (s, 1H), 4.78–4.62 (m, 1H), 3.45 (dd, J = 16.01, 7.52 Hz, 2H), 3.15 (dd, J = 15.85, 6.4 Hz, 2H), 2.59 (s, 3H). ESIMS m/z: 370.0 (MH)+.
N-(2,3-Dihydro-1H-inden-2-yl)acetamide (39)
2,3-Dihydro-1H-inden-2-amine hydrochloride (38) (50 g, 295 mmol), 45 mL of acetic anhydride, and 25 g of sodium acetate were suspended in 100 mL of acetic acid and stirred overnight at RT. The mixture was poured into 3 vols of ice–water and neutralized with ammonium hydroxide. The mixture was extracted with chloroform, and the combined organic layers were washed with water, diluted HCl, water, dried, and evaporated. The residue was recrystallized from CHCl3–Et2O to yield N-(2,3-dihydro-1H-inden-2-yl)acetamide (39) (43 g, 66% yield).
2-Acetamido-2,3-dihydro-1H-indene-5-sulfonyl Chloride (40)
To a stirred, cooled solution of N-(2,3-dihydro-1H-inden-2-yl)acetamide (39) (24.4 g, 139 mmol) in 400 mL of chloroform was added dropwise 55 mL of chlorosulfonic acid below 0 °C. The solution was stirred while cooling and then allowed to reach RT slowly by stirring overnight. The excess of chlorosulfonic acid decomposed by adding the mixture dropwise to stirred iced water. The layers were separated, and the aqueous layer was extracted several times with chloroform. The chloroform layers were washed, dried, and evaporated. The concentrate was heated and diluted with additional chloroform but was still containing some undissolved gum. It was filtered, washed with Et2O, and dried to yield 2-acetamido-2,3-dihydro-1H-indene-5-sulfonyl chloride (40) as crystals (25.6 g, 67% yield).
N-(5-(Methylthio)-2,3-dihydro-1H-inden-2-yl)acetamide (41)
A solution of 2-acetamido-2,3-dihydro-1H-indene-5-sulfonyl chloride (40) (13.7 g, 50 mmol) in 250 mL of acetic acid was stirred and warmed to 75 °C whereupon a solution of 50 g of tin(II) chloride dihydrate in 45 mL of conc. HCl was added in one portion. The solution was stirred 90–120 min at r.t. The solution was poured into several vols of water containing some conc. HCl and a yellowish-white solid precipitated. The mixture was filtered (became rapidly gummy when dried on a clay plate), washed with cold water, and dissolved in 250 mL of MeOH. To the solution, under nitrogen, was added 3 g of sodium methoxide in 4 mL (>0.05 mol) of methyl iodide. The mixture was stirred 2 h at RT and left standing overnight. The mixture was concentrated and then partitioned between water and chloroform. After drying and evaporation, 8.6 g of oil were obtained, which was purified by chromatography on neutral alumina eluting with EtOAc. N-(5-(Methylthio)-2,3-dihydro-1H-inden-2-yl)acetamide (41) was obtained as a white solid after washing with Et2O and recrystallization from chloroform-Et2O (3.7 g, 33% yield).
N-(5-(Methylsulfonyl)-2,3-dihydro-1H-inden-2-yl)acetamide (42)
A solution of N-(5-(methylthio)-2,3-dihydro-1H-inden-2-yl)acetamide (41) (2.2 g, 9.9 mmol) and m-chloroperbenzoic acid (5 g) in 900 mL of chloroform was stirred at RT for 2 h. The solution was washed with aq. 5% Na2CO3 and water, dried, and evaporated. The residue was triturated with Et2O, filtered, and dried to yield the N-(5-(methylsulfonyl)-2,3-dihydro-1H-inden-2-yl)acetamide (42) as a white solid (1.87g, 75% yield).
5-(Methylsulfonyl)-2,3-dihydro-1H-inden-2-amine hydrochloride (43)
A mixture of 1.9 g (7.5 mmol) of N-(5-(methylsulfonyl)-2,3-dihydro-1H-inden-2-yl)acetamide (42) and 20 mL of 3 N HCl was stirred under reflux for 2.5 h. The solution was cooled, and the residue was azeotroped with EtOH. Recrystallization from MeOH-EtOH yielded 5-(methylsulfonyl)-2,3-dihydro-1H-inden-2-amine hydrochloride (43) as crystals (0.87g, 55% yield).
1H NMR (500 MHz, DMSO-d6) δ 8.17 (br. s., 2H), 7.84 (s, 1H), 7.79–7.71 (m, 1H), 7.54 (d, J = 8.0 Hz, 1H), 4.06 (ddd, J = 2.5, 5.0, 7.6 Hz, 1H), 3.40–3.34 (m, 2H), 3.17 (s, 3H), 3.10–2.98 (m, 2H). ESIMS m/z: 212.1 (MH)+.
Acknowledgments
The authors would like to thank Michael Palmer for critical reading of the manuscript. This work was supported by funds from the United States National Institutes of Health grants R01AI103947 (to M.A.P. and P.K.R.) and U01AI075594 (to I.B., M.A.P., P.K.R., and S.A.C.) and from Medicines for Malaria Venture (MMV). M.A.P. acknowledges the support of the Welch Foundation (I-1257). M.A.P. holds the Beatrice and Miguel Elias Distinguished Chair in Biomedical Science and the Carolyn R. Bacon Professorship in Medical Science and Education.
Glossary
Abbreviations Used
- DHODH
dihydroorotate dehydrogenase
- PfDHODH
P. falciparum DHODH
- PvDHODH
P. vivax DHODH
- hDHODH
human DHODH
- DSM
code name assigned to compounds from our team and stands for Dallas, Seattle, and Melbourne, reflecting the location of the three project groups on the team
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.6b00275.
Accession Codes
Coordinates for the 13-PfDHODH X-ray structure have been submitted to the pdb (5FI8) and coordinates for the nine-small-molecule structure have been submitted to CCDC (1454120).
Author Contributions
S.K., J.M.C., M.M., L.D.L.H., K.M., and K.R.R. synthesized compounds described in the study; S.K. and W.K. performed small molecule X-ray crystallography; X.D., M.A.P., and D.T. performed, analyzed, or supervised protein X-ray crystallography, and D.M. contributed molecular modeling insight; K.W., G.C., J.M., E.R., and S.A.C. performed, analyzed, or supervised the ADME and pharmacokinetic studies; F.E.M. and M.A.P. performed, analyzed, or supervised the enzymology studies; P.K.R., D.W., and J.N.B. supervised chemistry studies; M.B.J., S.F.B., and I.A.B. performed, analyzed, or supervised in vivo efficacy studies; S.K., S.A.C., P.K.R., and M.A.P. wrote the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- WHO. World Malaria Report. http://wwwwhoint/malaria/media/world-malaria-report-2015/en/, 2015. (access date: Feb. 1, 2016).
- White N. J.; Pukrittayakamee S.; Hien T. T.; Faiz M. A.; Mokuolu O. A.; Dondorp A. M. Malaria. Lancet 2014, 383, 723–735. 10.1016/S0140-6736(13)60024-0. [DOI] [PubMed] [Google Scholar]
- Miller L. H.; Ackerman H. C.; Su X. Z.; Wellems T. E. Malaria biology and disease pathogenesis: insights for new treatments. Nat. Med. 2013, 19, 156–167. 10.1038/nm.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells T. N.; Hooft van Huijsduijnen R.; Van Voorhis W. C. Malaria medicines: a glass half full?. Nat. Rev. Drug Discovery 2015, 14, 424–442. 10.1038/nrd4573. [DOI] [PubMed] [Google Scholar]
- Burrows J. N.; Hooft van Huijsduijnen R.; Mohrle J. J.; Oeuvray C.; Wells T. N. Designing the next generation of medicines for malaria control and eradication. Malar. J. 2013, 12, 187. 10.1186/1475-2875-12-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrows J. N.; Chibale K.; Wells T. N. The state of the art in anti-malarial drug discovery and development. Curr. Top. Med. Chem. 2011, 11, 1226–1254. 10.2174/156802611795429194. [DOI] [PubMed] [Google Scholar]
- Straimer J.; Gnadig N. F.; Witkowski B.; Amaratunga C.; Duru V.; Ramadani A. P.; Dacheux M.; Khim N.; Zhang L.; Lam S.; Gregory P. D.; Urnov F. D.; Mercereau-Puijalon O.; Benoit-Vical F.; Fairhurst R. M.; Menard D.; Fidock D. A. Drug resistance. K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science 2015, 347, 428–431. 10.1126/science.1260867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok S.; Ashley E. A.; Ferreira P. E.; Zhu L.; Lin Z.; Yeo T.; Chotivanich K.; Imwong M.; Pukrittayakamee S.; Dhorda M.; Nguon C.; Lim P.; Amaratunga C.; Suon S.; Hien T. T.; Htut Y.; Faiz M. A.; Onyamboko M. A.; Mayxay M.; Newton P. N.; Tripura R.; Woodrow C. J.; Miotto O.; Kwiatkowski D. P.; Nosten F.; Day N. P.; Preiser P. R.; White N. J.; Dondorp A. M.; Fairhurst R. M.; Bozdech Z. Drug resistance. Population transcriptomics of human malaria parasites reveals the mechanism of artemisinin resistance. Science 2015, 347, 431–435. 10.1126/science.1260403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miotto O.; Amato R.; Ashley E. A.; MacInnis B.; Almagro-Garcia J.; Amaratunga C.; Lim P.; Mead D.; Oyola S. O.; Dhorda M.; Imwong M.; Woodrow C.; Manske M.; Stalker J.; Drury E.; Campino S.; Amenga-Etego L.; Thanh T. N.; Tran H. T.; Ringwald P.; Bethell D.; Nosten F.; Phyo A. P.; Pukrittayakamee S.; Chotivanich K.; Chuor C. M.; Nguon C.; Suon S.; Sreng S.; Newton P. N.; Mayxay M.; Khanthavong M.; Hongvanthong B.; Htut Y.; Han K. T.; Kyaw M. P.; Faiz M. A.; Fanello C. I.; Onyamboko M.; Mokuolu O. A.; Jacob C. G.; Takala-Harrison S.; Plowe C. V.; Day N. P.; Dondorp A. M.; Spencer C. C.; McVean G.; Fairhurst R. M.; White N. J.; Kwiatkowski D. P. Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nat. Genet. 2015, 47, 226–234. 10.1038/ng.3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrows J. N.; Burlot E.; Campo B.; Cherbuin S.; Jeanneret S.; Leroy D.; Spangenberg T.; Waterson D.; Wells T. N.; Willis P. Antimalarial drug discovery - the path towards eradication. Parasitology 2014, 141, 128–139. 10.1017/S0031182013000826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coteron J. M.; Marco M.; Esquivias J.; Deng X.; White K. L.; White J.; Koltun M.; El Mazouni F.; Kokkonda S.; Katneni K.; Bhamidipati R.; Shackleford D. M.; Angulo-Barturen I.; Ferrer S. B.; Jimenez-Diaz M. B.; Gamo F. J.; Goldsmith E. J.; Charman W. N.; Bathurst I.; Floyd D.; Matthews D.; Burrows J. N.; Rathod P. K.; Charman S. A.; Phillips M. A. Structure-guided lead optimization of triazolopyrimidine-ring substituents identifies potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitors with clinical candidate potential. J. Med. Chem. 2011, 54, 5540–5561. 10.1021/jm200592f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips M. A.; Lotharius J.; Marsh K.; White J.; Dayan A.; White K. L.; Njoroge J. W.; El Mazouni F.; Lao Y.; Kokkonda S.; Tomchick D. R.; Deng X.; Laird T.; Bhatia S. N.; March S.; Ng C. L.; Fidock D. A.; Wittlin S.; Lafuente-Monasterio M.; Benito F. J.; Alonso L. M.; Martinez M. S.; Jimenez-Diaz M. B.; Bazaga S. F.; Angulo-Barturen I.; Haselden J. N.; Louttit J.; Cui Y.; Sridhar A.; Zeeman A. M.; Kocken C.; Sauerwein R.; Dechering K.; Avery V. M.; Duffy S.; Delves M.; Sinden R.; Ruecker A.; Wickham K. S.; Rochford R.; Gahagen J.; Iyer L.; Riccio E.; Mirsalis J.; Bathhurst I.; Rueckle T.; Ding X.; Campo B.; Leroy D.; Rogers M. J.; Rathod P. K.; Burrows J. N.; Charman S. A. A long-duration dihydroorotate dehydrogenase inhibitor (DSM265) for prevention and treatment of malaria. Sci. Transl. Med. 2015, 7, 296ra111. 10.1126/scitranslmed.aaa6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips M. A.; Rathod P. K. Plasmodium dihydroorotate dehydrogenase: a promising target for novel anti-malarial chemotherapy. Infect. Disord.: Drug Targets 2010, 10, 226–39. 10.2174/187152610791163336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips M. A.; Rathod P. K.; Baldwin J.; Gujjar R.. Dihydroorotate dehydrogenase inhibitors with selective anti-malarial activity. WO Patent 2007149211 A1, 2007.
- Phillips M. A.; Gujjar R.; Malmquist N. A.; White J.; El Mazouni F.; Baldwin J.; Rathod P. K. Triazolopyrimidine-based dihydroorotate dehydrogenase inhibitors with potent and selective activity against the malaria parasite, Plasmodium falciparum. J. Med. Chem. 2008, 51, 3649–3653. 10.1021/jm8001026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gujjar R.; Marwaha A.; El Mazouni F.; White J.; White K. L.; Creason S.; Shackleford D. M.; Baldwin J.; Charman W. N.; Buckner F. S.; Charman S.; Rathod P. K.; Phillips M. A. Identification of a metabolically stable triazolopyrimidine-based dihydroorotate dehydrogenase inhibitor with antimalarial activity in mice. J. Med. Chem. 2009, 52, 1864–1872. 10.1021/jm801343r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X.; Gujjar R.; El Mazouni F.; Kaminsky W.; Malmquist N. A.; Goldsmith E. J.; Rathod P. K.; Phillips M. A. Structural plasticity of malaria dihydroorotate dehydrogenase allows selective binding of diverse chemical scaffolds. J. Biol. Chem. 2009, 284, 26999–27009. 10.1074/jbc.M109.028589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X.; Kokkonda S.; El Mazouni F.; White J.; Burrows J. N.; Kaminsky W.; Charman S. A.; Matthews D.; Rathod P. K.; Phillips M. A. Fluorine modulates species selectivity in the triazolopyrimidine class of Plasmodium falciparum dihydroorotate dehydrogenase inhibitors. J. Med. Chem. 2014, 57, 5381–5394. 10.1021/jm500481t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gujjar R.; El Mazouni F.; White K. L.; White J.; Creason S.; Shackleford D. M.; Deng X.; Charman W. N.; Bathurst I.; Burrows J.; Floyd D. M.; Matthews D.; Buckner F. S.; Charman S. A.; Phillips M. A.; Rathod P. K. Lead-optimization of aryl and aralkyl amine based triazolopyrimidine inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase with anti-malarial activity in mice. J. Med. Chem. 2011, 54, 3935–3949. 10.1021/jm200265b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesan S. M.; Morrisey J. M.; Ke H.; Painter H. J.; Laroiya K.; Phillips M. A.; Rathod P. K.; Mather M. W.; Vaidya A. B. Yeast dihydroorotate dehydrogenase as a new selectable marker for Plasmodium falciparum transfection. Mol. Biochem. Parasitol. 2011, 177, 29–34. 10.1016/j.molbiopara.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring B. J.; Chien J. Y.; Adkison K. K.; Jones H. M.; Rowland M.; Jones R. D.; Yates J. W.; Ku M. S.; Gibson C. R.; He H.; Vuppugalla R.; Marathe P.; Fischer V.; Dutta S.; Sinha V. K.; Bjornsson T.; Lave T.; Poulin P. PhRMA CPCDC initiative on predictive models of human pharmacokinetics, part 3: Comparative assessement of prediction methods of human clearance. J. Pharm. Sci. 2011, 100, 4090–4110. 10.1002/jps.22552. [DOI] [PubMed] [Google Scholar]
- Kannankeril P.; Roden D. M.; Darbar D. Drug-induced long QT syndrome. Pharmacol. Rev. 2010, 62, 760–781. 10.1124/pr.110.003723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Diaz M. B.; Mulet T.; Viera S.; Gomez V.; Garuti H.; Ibanez J.; Alvarez-Doval A.; Shultz L. D.; Martinez A.; Gargallo-Viola D.; Angulo-Barturen I. Improved murine model of malaria using Plasmodium falciparum competent strains and non-myelodepleted NOD-scid IL2Rgammanull mice engrafted with human erythrocytes. Antimicrob. Agents Chemother. 2009, 53, 4533–4536. 10.1128/AAC.00519-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirimulla S.; Bailey J. B.; Vegesna R.; Narayan M. Halogen interactions in protein-ligand complexes: implications of halogen bonding for rational drug design. J. Chem. Inf. Model. 2013, 53, 2781–2791. 10.1021/ci400257k. [DOI] [PubMed] [Google Scholar]
- Baldwin J.; Farajallah A. M.; Malmquist N. A.; Rathod P. K.; Phillips M. A. Malarial dihydroorotate dehydrogenase. Substrate and inhibitor specificity. J. Biol. Chem. 2002, 277, 41827–41834. 10.1074/jbc.M206854200. [DOI] [PubMed] [Google Scholar]
- Booker M. L.; Bastos C. M.; Kramer M. L.; Barker R. H. jr; Skerlj R.; Bir Sdhu A.; Deng X.; Celatka C.; Cortese J. F.; Guerrero Bravo J. E.; Krespo Llado K. N.; Serrano A. E.; Angulo-Barturen I.; Belén Jiménez-Díaz M.; Viera S.; Garuti H.; Wittlin S.; Papastogiannidis P.; Lin J.; Janse C. J.; Khan S. M.; Duraisingh M.; Coleman B.; Goldsmith E. J.; Phillips M. A.; Munoz B.; Wirth D. F.; Klinger J. D.; Wiegand R.; Sybertz E. Novel inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase with anti-malarial activity in the mouse model. J. Biol. Chem. 2010, 285, 33054–33064. 10.1074/jbc.M110.162081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan C. D.; Lloyd R. S. A high-throughput screening method for the determination of aqueous drug solubility using laser nephelometry in microtiter plates. Anal. Chem. 2000, 72, 1781–1787. 10.1021/ac9912247. [DOI] [PubMed] [Google Scholar]
- Jantratid E.; Janssen N.; Reppas C.; Dressman J. B. Dissolution media simulating conditions in the proximal human gastrointestinal tract: an update. Pharm. Res. 2008, 25, 1663–1676. 10.1007/s11095-008-9569-4. [DOI] [PubMed] [Google Scholar]
- Nilsen A.; LaCrue A. N.; White K. L.; Forquer I. P.; Cross R. M.; Marfurt J.; Mather M. W.; Delves M. J.; Shackleford D. M.; Saenz F. E.; Morrisey J. M.; Steuten J.; Mutka T.; Li Y.; Wirjanata G.; Ryan E.; Duffy S.; Kelly J. X.; Sebayang B. F.; Zeeman A. M.; Noviyanti R.; Sinden R. E.; Kocken C. H.; Price R. N.; Avery V. M.; Angulo-Barturen I.; Jimenez-Diaz M. B.; Ferrer S.; Herreros E.; Sanz L. M.; Gamo F. J.; Bathurst I.; Burrows J. N.; Siegl P.; Guy R. K.; Winter R. W.; Vaidya A. B.; Charman S. A.; Kyle D. E.; Manetsch R.; Riscoe M. K. Quinolone-3-diarylethers: a new class of antimalarial drug. Sci. Transl. Med. 2013, 5, 177ra37. 10.1126/scitranslmed.3005029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruker. APEX2 (version 2.1–4), SAINT (version 7.34A), SADABS (version 2007/4); BrukerAXS Inc: Madison, WI. 2007. [Google Scholar]
- Altomare A.; Burla C.; Camalli M.; Cascarano L.; Giacovazzo C.; Guagliardi A.; Moliterni A.; Polidori G.; Spagna R. SIR97: a new tool for crystal structure determination and refinement. J. Appl. Crystallogr. 1999, 32, 115–119. 10.1107/S0021889898007717. [DOI] [Google Scholar]
- Altomare A.; Cascarano G.; Giacovazzo C.; Guagliardi A. Completion and refinement of crystal structures with SIR 92. J. Appl. Crystallogr. 1993, 26, 343–350. 10.1107/S0021889892010331. [DOI] [Google Scholar]
- Mackay S.; Edwards C.; Henderson A.; Gilmore C.; Stewart N.; Shankland K.; Donald A.. MaXus, a computer program for the solution and refinement of crystal structures from diffraction data; University of Glasgow: Scotland, 1997. [Google Scholar]
- Sheldrick G.SHELXL97, Program for the Refinement of Crystal Structures; University of Gottingen: Germany, 1997. [Google Scholar]
- Waasmaier D.; Kirfel A. New analytical scattering-factor functions for free atoms and ions. Acta Crystallogr., Sect. A: Found. Crystallogr. 1995, 51, 416–431. 10.1107/S0108767394013292. [DOI] [Google Scholar]
- Otwinowski Z.; Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- McCoy A. J. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2007, 63, 32–41. 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.