

Abstract
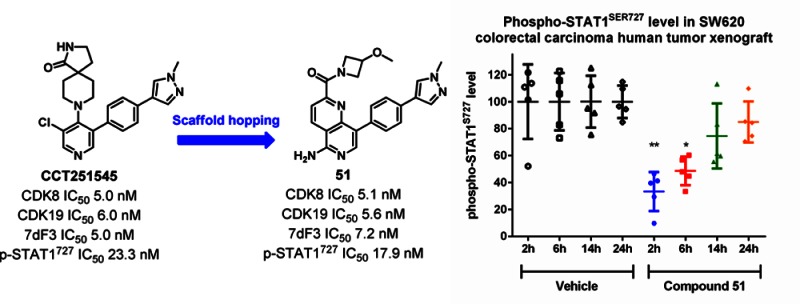
We demonstrate a designed scaffold-hop approach to the discovery of 2,8-disubstituted-1,6-naphthyridine- and 4,6-disubstituted-isoquinoline-based dual CDK8/19 ligands. Optimized compounds in both series exhibited rapid aldehyde oxidase-mediated metabolism, which could be abrogated by introduction of an amino substituent at C5 of the 1,6-naphthyridine scaffold or at C1 of the isoquinoline scaffold. Compounds 51 and 59 were progressed to in vivo pharmacokinetic studies, and 51 also demonstrated sustained inhibition of STAT1SER727 phosphorylation, a biomarker of CDK8 inhibition, in an SW620 colorectal carcinoma human tumor xenograft model following oral dosing.
Keywords: CDK8, CDK19, mediator complex, kinase inhibitor, aldehyde oxidase
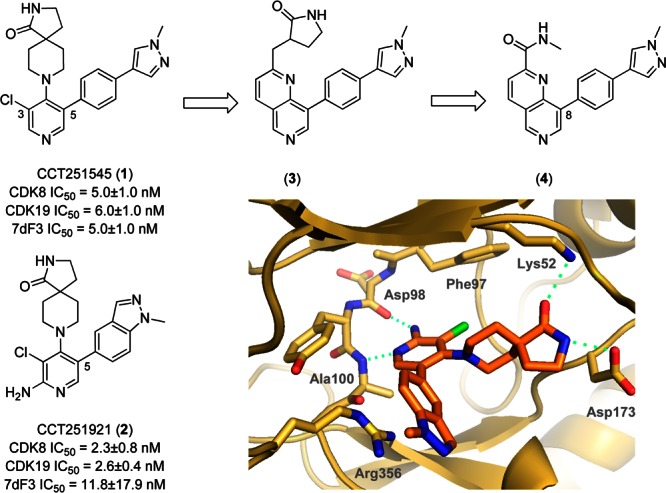
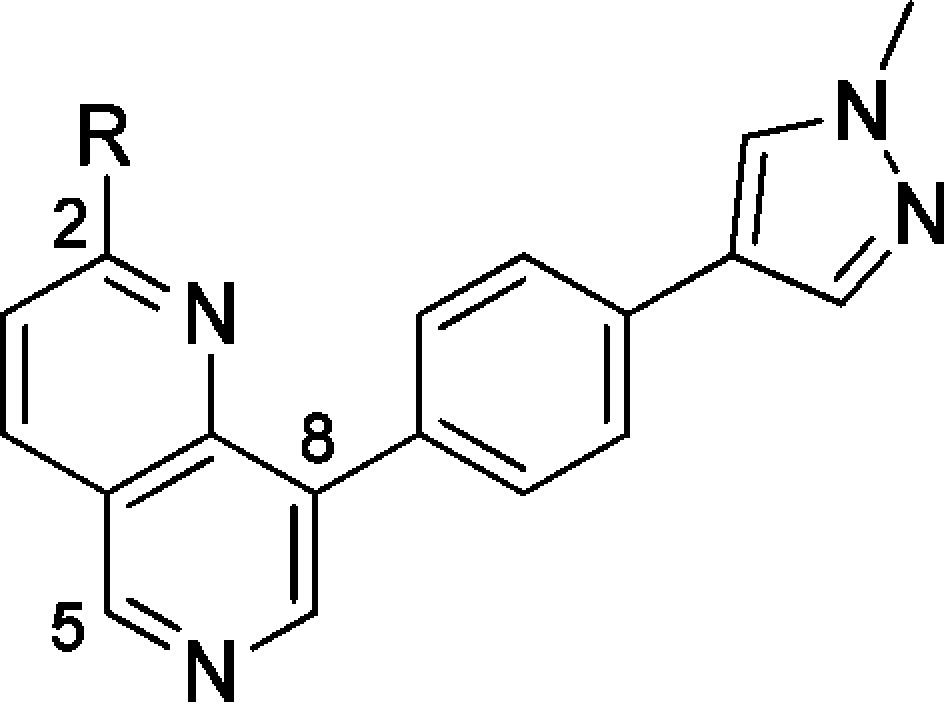
Mediator complex-associated kinases CDK8 and CDK19 are involved in the regulation of multiple transcription pathways; notably, CDK8 has been implicated as an oncogene in colorectal and gastric cancers through activation of WNT signaling.1−4 There has been increasing interest in small molecule modulators of CDK8 including Sorafenib,5 Senexin A,6 Cortistatin A,7 and a recently reported series of 6-aza-benzothiophenes.8 In addition, we have reported the discovery and optimization of a 3,4,5-trisubstituted pyridine series to give CCT251545 (1) and CCT251921 (2), potent, selective, and orally bioavailable dual CDK8/19 ligands (Figure 1).9−11 In the course of this work, we were keen to expand the range of CDK8/19 inhibitors and sought a structurally differentiated backup series. Our efforts focused upon replacement of the pyridine core, cognizant of our previously reported observation that both in vitro CDK8/19 affinity and potent cell-based activity of this series benefited from a twisted conformation about the planes of the piperidine and pyridine rings; such a twisted conformation is also observed in CDK8-ligand cocrystal structures enabling the lactam amide to bridge Lys52 and Asp173 in CDK8 (Figure 1).10,11 We hypothesized that construction of a 6-membered ring across the 3- and 4-positions of the pyridine, while breaking the piperidine ring, would provide a scaffold hop with sufficient flexibility to maintain key lactam amide interactions with CDK8 (Figure 1). This rigidification–derigidification tactic generated target 3 that we simplified by retrosynthetic analysis and considerations of synthetic tractability to a 2,8-disubstitued-1,6-naphthyridine scaffold exemplified by 4. We were concerned that the aromatic 1,6-naphthyridine scaffold precludes the twisted conformation observed in ligand-bound structures of the 3,4,5-trisubstituted pyridine series; furthermore, preparation of amides such as 4, while synthetically feasible, may not adequately mimic the precedented interaction of spiroamides 1 and 2 with Lys52. With these caveats in mind, we set out to explore the 1,6-naphthyridine scaffold by incorporating substituents at the C-8 position that had proved optimal at C-5 in the 3,4,5-trisubstituted pyridine series.11 Pleasingly, the hypothesized combination of a methylpyrazolylphenyl substituent at C-8 and methyl amide at C-2 of the 1,6-naphthyridine scaffold (compound 4, Table 1) led to potent CDK8 affinity that translated to inhibition of WNT signaling in our previously described luciferase reporter assay in HEK293 7dF3 cells.11 Similarly, compound 5 and isomer 6 bearing a methylindazole at the C-8 position were also potent; however, compounds 4–6 were all metabolically unstable (Table 1). Gratifyingly, introduction of an N-methyl or NH sultam (7 and 8, Table 1) improved metabolic stability while maintaining potency.
Figure 1.
3,4,5-Trisubstituted pyridine series and scaffold-hop to the 1,6-naphthyridine series.
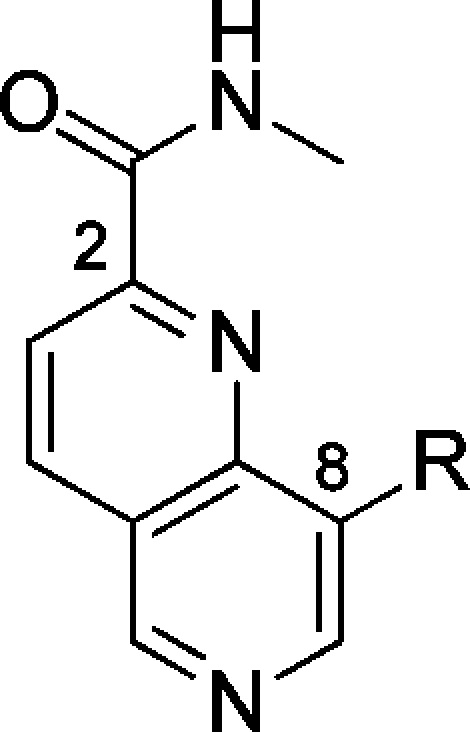
Table 1. Introduction of Optimal Substituents at C-8 of the 1,6-Naphthyridine Scaffold12.

M, mouse; R, rat; H, human.
To compare the binding mode of the 1,6-naphthyridine series with the 3,4,5-trisubstituted pyridine series, we solved the crystal structure of 7 in complex with the kinase domain of CDK8 and cyclin C (Pdb code: 5I5Z). Consistent with our hypothesis, 7 binds in the ATP binding site with the naphthyridine N-6 nitrogen interacting with the backbone NH of the hinge residue Ala100 (Figure 2A). Notably, the C2-amide carbonyl interacts with Lys52, and the C-8 phenyl substituent forms a pi–cation interaction with Arg356 consistent with our previous observations in the 3,4,5-trisubsitiuted pyridine series.10,11 Overlay of 7 with 1 (CCT251545) confirmed our design strategy; most notably, both the methylamide of 7 and spirolactam of 1 interact with Lys52. Additionally, C-3 and C-4 of the naphthyridine ring are 3.7 and 3.9 Å from the closest gatekeeper heavy atom indicating that the hydrophobic interactions of the C3–Cl in 1 are maintained in 7 (Figure 2B).
Figure 2.
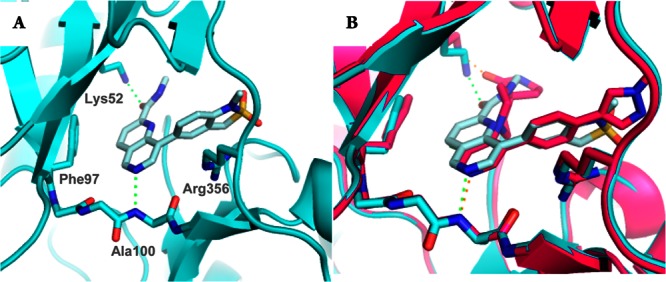
(A) X-ray crystal structure of 7 in CDK8/Cyclin C. (B) Overlay of 7 (cyan) with CCT251545 (pink).
We next explored optimization of the N-methylamide at C-2. The primary amide 9 (Table 2) proved equally potent and stable as methylamide 4 (Table 1). Compared to methylamide 4, a drop in cell-based potency was seen with the carboxylic acid 10 (Table 2), while introduction of a nitrile led to a drop in potency in both the biochemical- and cell-based assays (Table 2).
Table 2. Amide Variation at C-2 of the 1,5-Naphthyridine Series12.

Azetidine amide 12 proved potent but metabolically unstable, and introduction of methoxy or geminal difluoro substitution at C-3 of the azetidine (13 and 14) did not significantly reduce in vitro clearance. Consistent with azetidine amide 12, pyrrolidine amide 15 was potent but exhibited poor metabolic stability. The more sterically demanding 6-membered morpholino and piperazine amides 16 and 17 were less potent, and no improvement in in vitro clearance was observed. Reverse amide 18 and lactone 19 maintained biochemical potency relative to 4; however, both compounds proved 10-fold less active in the cell-based assay. Imidazolidinone 20, bearing a better hydrogen bond acceptor than lactone 19, demonstrated improved potency compared with 18 and 19 but suffered from suboptimal metabolic stability. Removal of the carbonyl group (21 and 22) led to a > 250-fold drop in CDK8 affinity consistent with the loss of a beneficial interaction with Lys52.
We next turned our attention to the introduction of heterocycles as amide isosteres at C-2 of the 1,6-naphthyridine scaffold. Pyrazoles 23 and 24 gave a significant drop in potency, whereas isoxazole 25 and N-methylimidazole 26 restored CDK8 affinity and cell-based inhibition of WNT signaling. We hypothesize that N-methylimidazole at C-2 may optimally bridge Lys52 and Asp173; however, 26 proved to be metabolicallly unstable and was not pursued further.
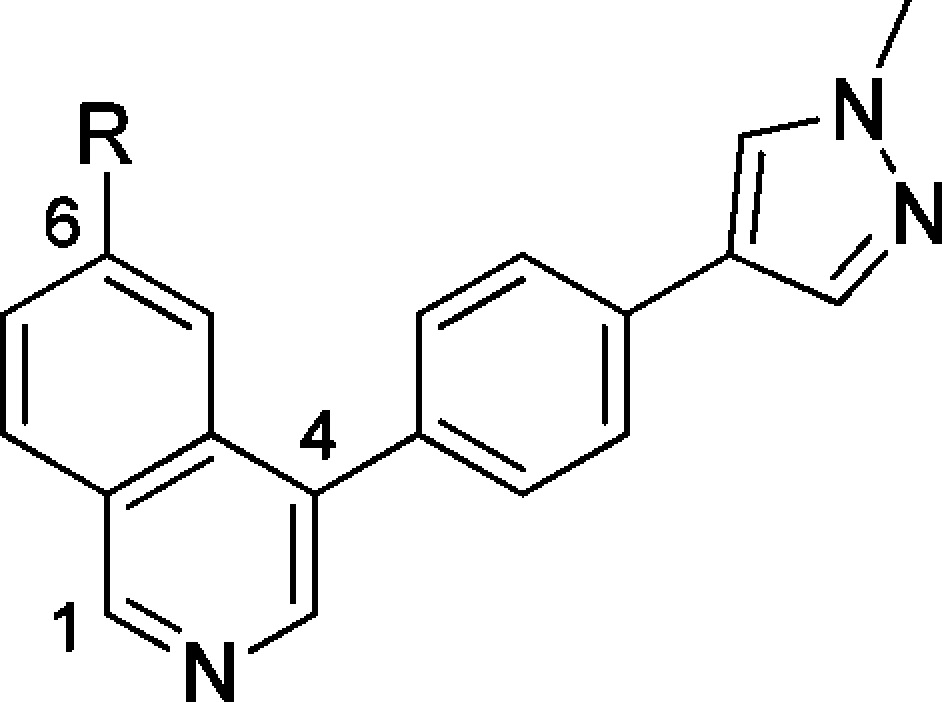
The X-ray crystal structure of 7 in CDK8 indicates that the N-1 nitrogen of the 1,6-napthyridine scaffold does not interact directly with the protein, and we therefore investigated the corresponding isoquinoline analogues. Compound 27 (Table 3), the matched pair of 4 (Table 1), confirmed that isoquinolines may have potential. Similarly, the secondary and tertiary amides (28–30, Table 3), reverse amide 31, oxazole 32, and N-methylimidazole 33 all demonstrated potent affinity for CDK8 but also proved metabolically unstable. Unsurprisingly, we observed similar SAR trends between the 1,6-naphthyridine and isoquinoline series. Additional functionality was tested at the C-6 position; the cyclic carbamate 34 had potent CDK8 affinity and cell-based activity but suffered from a suboptimal Clint profile, whereas sulphonamides 35 and 36 proved less active in the cell-based assay (7- and 12-fold drop off, respectively) without improvement in metabolic stability.
Table 3. Isoquinoline Analogues12.

At this stage, compounds 7, 8, and 17, which exhibited the best in vitro mouse microsomal stability, were progressed to in vivo PK studies in Balb/C mice (Table S1). Unfortunately, all compounds revealed a clearance that exceeded liver blood flow. Although compound 8 is an efflux substrate (Table S1), this finding could not explain the observed high in vivo clearance across all three compounds.
Cognizant of the propensity for bicyclic heterocyclic aromatic systems to be aldehyde oxidase (AO) substrates,13 we examined metabolic turnover for exemplar 1,6-naphthyridines and isoquinolines in an AO assay. Disappointingly, we observed rapid turnover for all compounds examined (Table S2). Met-ID studies of 8 in human hepatocytes, followed by isolation and NMR analysis, suggested oxidation to give the C5-OH compound 37 as the main metabolite, which we subsequently confirmed by independent synthesis (Figure S1). Unfortunately, 37 proved inactive versus CDK8 indicating that a C5-hydroxyl group is not tolerated.
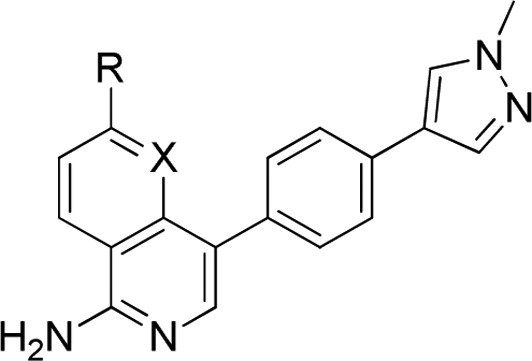
We, therefore, turned our attention to the design of compounds that have the potential to block the AO metabolism and maintain affinity for CDK8 and CDK19. First, we checked if pendant polar groups on the terminal pyrazole would reduce the rate of AO-mediated metabolism. In this context, several compounds in the 1,6-naphthyridine and isoquinoline series were prepared (Table S3); however, in all cases, pendant polar functionality failed to reduce AO metabolism. On the basis of these results, we next attempted the introduction of alternative substituents at C5 of the napthyridine scaffold, which had the potential to block AO metabolism while also being compatible with the known binding mode to the hinge region of CDK8 (Figure 2). Introduction of a methoxy, methyl, or difluoromethyl group into the optimized exemplars of the 1,6-naphthyridine and 2-isoquinoline series led to a significant drop in CDK8 affinity (Table S4). However, introduction of an amino group maintained potency consistent with our previously exemplified progression from a 3,4,5-trisubstituted pyridine series to a 3,4,5-trisubstituted 2-aminopyridine series;11 gratifyingly, this tactic successfully blocked AO-mediated metabolism for compounds 48 and 49 (Table S4). Compounds 48 and 49 (Table 4), direct amino analogues of 7 (Table 1) and 17 (Table 2), demonstrated acceptable in vitro metabolic stability, and in vivo PK properties were determined in Balb/C mice. In the case of 49, lower in vivo clearance compared with 17 was observed (Table 4); however, low oral bioavailability consistent with low passive permeability was also noted (Table 5).
Table 4. Pharmacokinetics of Compounds 7, 17, 48, and 49(12).
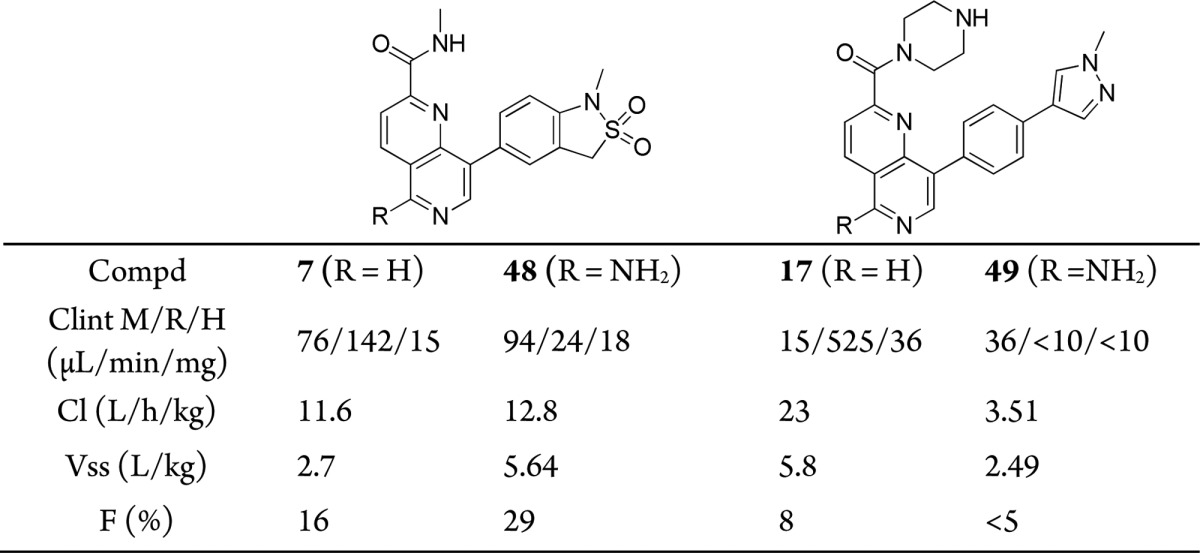
Table 5. Amino Derivatives12.
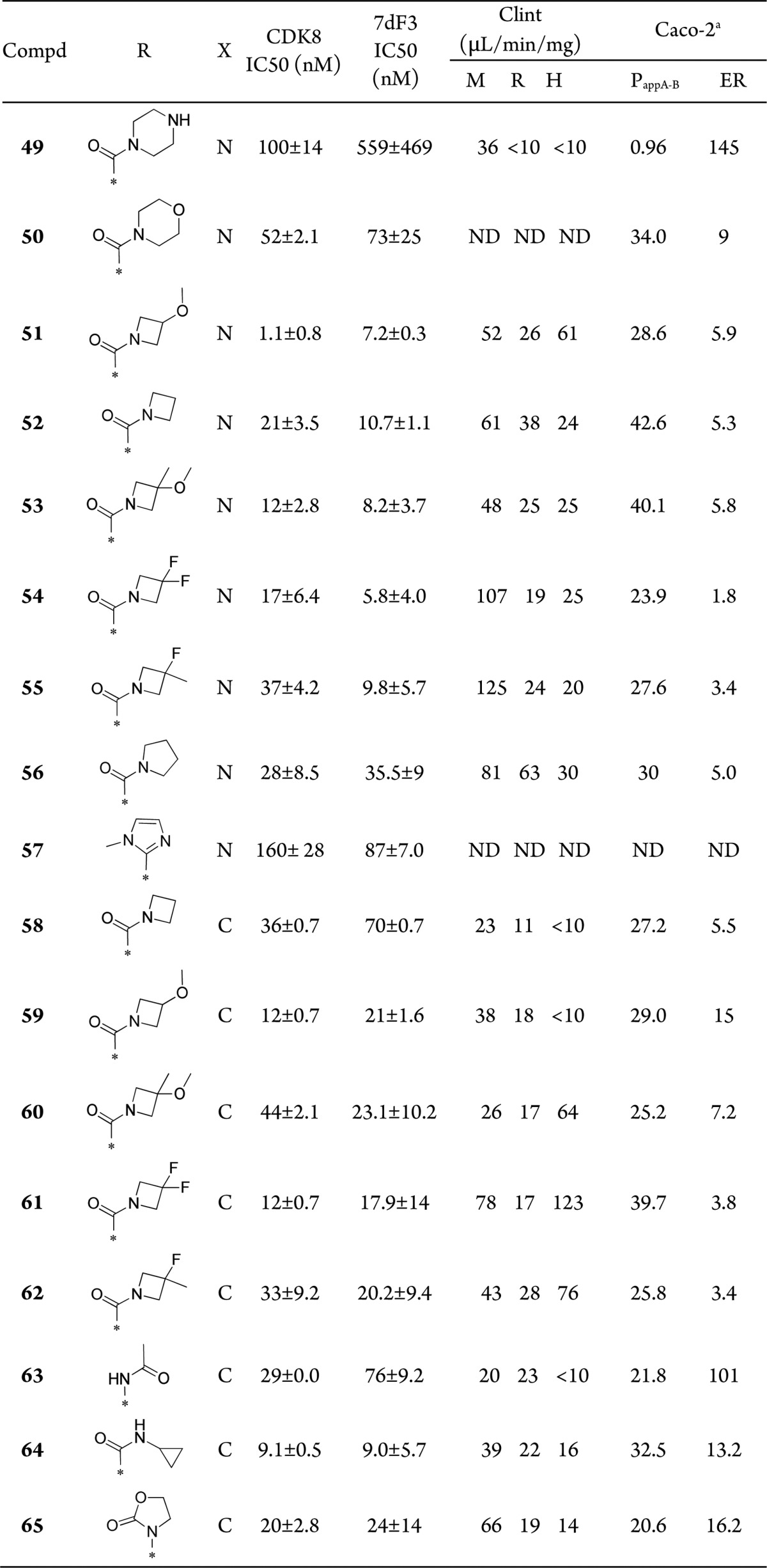
indicates PappA-B: × 10–6 cm/s.
On the basis of in vitro AO metabolism data and in vivo PK of 49, we sought additional C-5 amino derivatives. Pleasingly, replacement of piperazine by morpholine 50 or methoxyazetidine 51 to reduce the HBD count increased permeability and reduced efflux ratio (Table 5). We attempted to further reduce Caco-2 efflux liability by removing or masking HBAs (52 and 53); however, no improvement was observed. Installation of 3,3-difluoro or 3-fluoro-3-methylazetidine, 54 and 55, respectively, led to an acceptable efflux ratio; however, further profiling revealed mouse plasma instability, possibly due to amidase-mediated amide hydrolysis. Pyrrolidine amide 56 was prepared as an alternative to the unstable fluoroazetidines; disappointingly, no improvement in efflux ratio was observed, and affinity for CDK8 was eroded. We also prepared 57, the C5-amino analogue of 26 (Table 2), which resulted in a surprising and unexplained loss of CDK8 affinity (225-fold).
To further explore the potential for blocking AO-metabolism, we prepared 1-aminoisoquinolines including selected matched pairs of the corresponding 1,6-naphthyridine series (Table 5). Removal of one HBA from the 1,6-naphthyridine to the isoquinoline scaffold did not result in a lower efflux ratio, presumably due to the buried location of the N-1 nitrogen atom, which renders its removal insignificant in terms of overall polarity and solvation potential (compounds 58–60). Consistent with the naphthyridine series, the 3,3-difluoro or 3-fluoro-3-methylazetidine amides, while providing lower efflux ratios, exhibited plasma instability (compounds 61 and 62). Additional exemplar amino-isoquinoline derivatives demonstrated improved metabolic stability versus their corresponding isoquinoline matched pairs; however, efflux ratios were suboptimal (63–65).
Selected C5-aminonaphthyridine and C1-amino-isoquinoline derivatives were tested in the AO-substrate assay and also in the chemical Litmus Test recently reported as a practical and convenient surrogate assay for AO-mediated metabolism.14 Consistent with expectation, all amino derivatives tested were stable in both the AO-substrate assay and Litmus Test, while all unsubstituted derivatives were unstable in both assays (Table S5). Thus, the Litmus Test proved a useful and accurate predictor for AO-mediated metabolism in this chemical series.
As a result of their favorable in vitro profiles, we selected 51 and 59 for further characterization and in vivo evaluation. Both compounds bind to CDK8 and CDK19 in the presence of cyclin C with similar affinity (5 nM and 6 nM, respectively, for 51; 33 nM and 19 nM, respectively, for 59; Table 6). We have previously reported that modulation of phospho-STAT1SER727 is a useful cell-based and in vivo biomarker for CDK8 inhibition.10 Both 51 and 59 demonstrated potent inhibition of phospho-STAT1SER727 in SW620 human colorectal carcinoma cells harboring an activating APC-mutation, indicating good translation to cell-based activity; both compounds were tested against a panel of >250 kinases and inhibited only a single kinase by more than 50% at 1 μM (FLT4: 54% and 62% inhibition, respectively) (Table S6). On the basis of these encouraging findings, naphthyridine 51 and isoquinoline 59 were studied in in vivo mouse and rat PK experiments. Both compounds demonstrated medium in vivo clearance and Vd but low bioavailability in mice, likely due to their affinity for efflux transporters. Indeed, we observed 20–40% of the parent compound excreted unchanged in feces after iv dosing indicative of hepatic uptake and biliary excretion.11
Table 6. Profile of 51 and 59(12).
| 51 | 59 | |
|---|---|---|
| CDK8 IC50 ± SD (nM) | 5.1 ± 0.3 | 32.7 ± 2.1 |
| CDK8 residence time (min) ± SD | 15 ± 2 | 23 ± 3 |
| CDK19 IC50 ± SD (nM) | 5.6 ± 0.6 | 18.8 ± 1.4 |
| CDK19 residence time (min) ± SD | 31 ± 3 | 42 ± 5 |
| pSTAT1SER727 IC50 ± SD (nM) | 17.9 ± 10.6 | 114.0 ± 20.6 |
| 7dF3 IC50 ± SD (nM) | 7.2 ± 3.1 | 21.0 ± 1.6 |
| Clint (μL/min/mg) M/R/H | 52/26/61 | 38/18/<10 |
| mousePKa | ||
| Cl (L/h/kg) | 1.94 | 1.57 |
| Vss (L/kg) | 1.87 | 1.62 |
| F (%) | 18 | 15 |
| % in feces (iv dosing) | 31 | 18 |
| ratPKa | ||
| Cl (L/h/kg) | 1.29 | |
| Vss (L/kg) | 2.13 | |
| F (%) | 57 | |
| % in feces (iv dosing) | 36 |
Dose 0.2 mg/kg (iv), 0.5 mg/kg (po).
Compound 51 was progressed to a PK/PD study in a SW620 colorectal carcinoma human tumor xenograft model. After a single 5 mg/kg po dose, we observed time-dependent inhibition of phospho-STAT1SER727 (Figure 3) consistent with plasma exposure (Figure S2) suggesting that a b.i.d. schedule would be required at this dose to maintain maximal target engagement.
Figure 3.
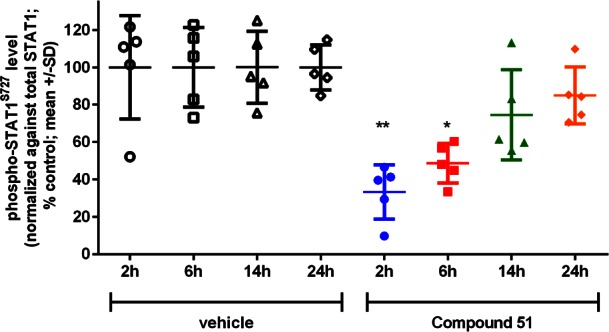
Phospho-STAT1SER727 level in SW620 colorectal carcinoma human tumor xenograft lysates following oral dosing with 51. Statistical analysis: nonparametric one-way ANOVA Kruskal–Wallis test (p = 0.0009) and Dunn’s post-test with 95% confidence interval against vehicle.
In conclusion, starting from the 3,4,5-trisubstituted pyridine CCT251545, we designed a scaffold-hop that led to the discovery of 2,8-disubstituted-1,6-naphthyridine- and 4,6-disubstituted-isoquinolines-based dual CDK8/19 ligands.
Protein–ligand crystal structure determination in CDK8 confirmed a Type I binding mode and key interactions with Ala100 (hinge), Phe97 (gatekeeper), Lys52 (catalytic residue), as well as Arg356 from the CDK8 C-terminus. Both the naphthyridine and isoquinoline scaffolds exhibited site-specific AO-mediated metabolism, which could be blocked by the introduction of amino groups at C5 and C1, respectively. Notably, susceptibility to AO metabolism in both chemical series was entirely consistent with the chemical Litmus Test.14 Minimisation of efflux led to the synthesis of 51 and 59, both of which demonstrated acceptable oral pharmacokinetics; 51 also demonstrated inhibition of phospho-STAT1SER727, a biomarker of CDK8 inhibition, on oral dosing in an SW620 human colorectal carcinoma xenograft model. Compound 51 is an advanced chemical tool with which to further explore the efficacy, safety, and tolerability of dual CDK8/19 ligands.
Acknowledgments
We thank Dr. Amin Mirza, Meirion Richards, and Dr. Maggie Liu for their assistance with NMR, mass spectrometry, and HPLC; Ugo Zanelli and Dr. Katrin Georgi for the AO metabolism assay; Gary Nugent and Dr. Christian Herhaus for chemoinformatics support; and Kate Trotman for administrative support. We thank the team of Proteros Biostructures GmbH, Bunsenstrasse 7a, D-82152 Martinsried, Germany for the Reporter Displacement Assay and in particular Dr. Elisabeth V. Schneider and Dr. Alfred Lammens for the X-ray crystal structures with CDK8/CyclinC.
Glossary
Abbreviations
- AO
aldehyde oxidase
- CDK
cyclin-dependent kinase
- Cl
clearance
- Clint
intrinsic clearance
- ER
efflux ratio
- F
bioavailability
- HBA
hydrogen bond acceptor
- HBD
hydrogen bond donor
- Vd
volume of distribution
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00022.
In vivo mouse PK of 7, 8, and 17; AO data for 7, 8, 13, 19, 25–27, 29, 30, and 32; spectra of metabolite 37; AO data for 38–44, compounds with C-5 variation; AO data for 7, 13, 26, 28, 29, 31, 34, 48, 51, 59, and 63–65; kinase profile of 51 and 59; plasma concentration; preparation and spectra of 7, 8, 17, 37, 45, 48, 49, 51, and 59; crystallographic data analysis for 7 (PDF)
Author Contributions
The manuscript was written with contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by Cancer Research UK (Grant No. C309/A11566). We acknowledge NHS funding to the NIHR Biomedical Research Centre at The Institute of Cancer Research and The Royal Marsden. P.W. is a Cancer Research UK Life Fellow.
The authors declare the following competing financial interest(s): A.M., C.R., J.S., M.A.H., M.S., R.B., P.W., P.A.C., F.I.R., S.A.E., and J.B. are current or former employees of The Institute of Cancer Research, which has a commercial interest in the development of WNT pathway inhibitors. K.S., P.C., O.P., M.B., R.S., D.S., D.M., K.U., D.W., C.E., and F.R. are current employees of Merck KGaA.
Supplementary Material
References
- Morris E. J.; Ji J. Y.; Yang F.; Di Stefano L.; Herr A.; Moon N. S.; Kwon E. J.; Haigis K. M.; Naar A. M.; Dyson N. J. E2F1 represses beta-catenin transcription and is antagonized by both pRB and CDK8. Nature 2008, 455, 552–556. 10.1038/nature07310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestein R.; Bass A. J.; Kim S. Y.; Dunn I. F.; Silver S. J.; Guney I.; Freed E.; Ligon A. H.; Vena N.; Ogino S.; Chheda M. G.; Tamayo P.; Finn S.; Shrestha Y.; Boehm J. S.; Jain S.; Bojarski E.; Mermel C.; Barretina J.; Chan J. A.; Baselga J.; Tabernero J.; Root D. E.; Fuchs C. S.; Loda M.; Shivdasani R. A.; Meyerson M.; Hahn W. C. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature 2008, 455, 547–551. 10.1038/nature07179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestein R.; Shima K.; Nosho K.; Irahara N.; Baba Y.; Bojarski E.; Giovannucci E. L.; Hahn W. C.; Fuchs C. S.; Ogino S. CDK8 expression in 470 colorectal cancers in relation to beta-catenin activation, other molecular alterations and patient survival. Int. J. Cancer 2010, 126, 2863–2873. 10.1002/ijc.24908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S. C.; Kim M. Y.; Han S. I. Roles of cyclin-dependent kinase 8 and b-catenin in the oncogenesis and progression of gastric adenocarcinoma. Int. J. Oncol. 2011, 38, 1375–1383. 10.3892/ijo.2011.948. [DOI] [PubMed] [Google Scholar]
- Schneider E. V.; Böttcher J.; Blaesse M.; Neumann L.; Huber R.; Maskos K. The Structure of CDK8/CycC Implicates Specificity in the CDK/Cyclin Family and Reveals Interaction with a Deep Pocket Binder. J. Mol. Biol. 2011, 412, 251–266. 10.1016/j.jmb.2011.07.020. [DOI] [PubMed] [Google Scholar]
- Porter D. C.; Farmaki E.; Altilia S.; Schools G. P.; West D. K.; Chen M.; Chang B. D.; Puzyrev A. T.; Lim C.; Rokow-Kittell R.; et al. Cyclin-Dependent Kinase 8 Mediates Chemotherapy Induced Tumor-Promoting Paracrine Activities. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 13799–13804. 10.1073/pnas.1206906109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelish H. E.; Liau B. B.; Nitulescu I. I.; Tangpeerachaikul A.; Poss Z. C.; Da Silva D. H.; Caruso B. T.; Arefolov A.; Fadeyi O.; Christie A. L.; Du K.; Banka D.; Schneider E. V.; Jestel A.; Zou G.; Si C.; Ebmeier C. C.; Bronson R. T.; Krivtsov A. V.; Myers A. G.; Kohl N. E.; Kung A. L.; Armstrong S. A.; Lemieux M. E.; Taatjes D. J.; Shair M. D. Mediator kinase inhibition further activates superenhancer-associated genes in AML. Nature 2015, 526, 273–276. 10.1038/nature14904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler M. F. T.; Bergeron P.; Blackwood E. M.; Bowman K.; Clark K. R.; Firestein R.; Kiefer J. R.; Maskos K.; McCleland M. L.; Orren L.; Salphati L.; Schmidt S.; Schneider E. V.; Wu J.; Beresini M. H. Development of a Potent, Specific CDK8 Kinase Inhibitor Which Phenocopies CDK8/19 Knockout Cells. ACS Med. Chem. Lett. 2016, 7, 223. 10.1021/acsmedchemlett.5b00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallinger A.; Crumpler S.; Pichowicz M.; Waalboer D.; Stubbs M.; Adeniji-Popoola O.; Wood B.; Smith E.; Thai C.; Henley A. T.; Georgi K.; Court W.; Hobbs S.; Box G.; Ortiz-Ruiz M. J.; Valenti M.; De Haven Brandon A.; TePoele R.; Leuthner B.; Workman P.; Aherne W.; Poeschke O.; Dale T.; Wienke D.; Esdar C.; Rohdich F.; Raynaud F.; Clarke P. A.; Eccles S. A.; Stieber F.; Schiemann K.; Blagg J. Discovery of Potent, Orally Bioavailable, Small-Molecule Inhibitors of WNT Signaling from a Cell-Based Pathway Screen. J. Med. Chem. 2015, 58, 1717–1735. 10.1021/jm501436m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale T.; Clarke P. A.; Esdar C.; Waalboer D.; Adeniji-Popoola O.; Ortiz-Ruiz M.; Mallinger A.; Samant R. S.; Czodrowski P.; Musil D.; Schwarz D.; Schneider K.; Stubbs M.; Ewan K.; Fraser E.; TePoele R.; Court W.; Box G.; Valenti M.; de Haven Brandon A.; Gowan S.; Rohdich F.; Raynaud F.; Schneider R.; Poeschke O.; Blaukat A.; Workman P.; Schiemann K.; Eccles S. A.; Wienke D.; Blagg J. Identification of a potent and selective chemical probe for exploring the role of Mediator complex-associated protein kinases CDK8 and CDK19 in human disease. Nat. Chem. Biol. 2015, 11, 973–980. 10.1038/nchembio.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallinger A.; Schiemann K.; Rink C.; Stieber F.; Calderini M.; Crumpler S.; Stubbs M.; Adeniji-Popoola O.; Poeschke O.; Busch M.; Czodrowski P.; Musil D.; Schwarz D.; Ortiz-Ruiz M.-J.; Schneider R.; Thai C.; Valenti M.; De Haven Brandon A.; Burke R.; Workman P.; Dale T.; Wienke D.; Clarke P. A.; Esdar C.; Raynaud F. I.; Eccles S. A.; Rohdich F.; Blagg J. J. Med. Chem. 2016, 59, 1078–1101. 10.1021/acs.jmedchem.5b01685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protocols for the 7dF3 cell-based assay, CDK8 binding affinity, microsomal stability (Clint), Caco2, mouse and rat PK can be found in ref (11). IC50 data are reported as averages of at least two determinations with standard deviations.
- Pryde D. C.; Dalvie D.; Hu Q.; Jones P.; Obach R. S.; Tran T. D. Aldehyde oxidase: an enzyme of emerging importance in drug discovery. J. Med. Chem. 2010, 53, 8441–60. 10.1021/jm100888d. [DOI] [PubMed] [Google Scholar]
- O’Hara F.; Burns A. C.; Collins M. R.; Dalvie D.; Ornelas M. A.; Vaz A. D. N.; Fujiwara Y.; Baran P. S. A Simple Litmus Test for Aldehyde Oxidase Metabolism of Heteroarenes. J. Med. Chem. 2014, 57, 1616–1620. 10.1021/jm4017976. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.