

Abstract

Using Sorafenib as a starting point, a series of potent and selective inhibitors of CDK8 was developed. When cocrystallized with CDK8 and cyclin C, these compounds exhibit a Type-II (DMG-out) binding mode.
Keywords: Sorafenib, CDK8, inhibitor, DMG-out
In recent literature, CDK8 has been identified as playing a role in the control of transcription regulating cellular proliferation and differentiation. CDK8 is a cyclin-dependent kinase that forms part of the mediator complex,1 which itself regulates the transcriptional activity of RNA polymerase II. A number of studies have shown that CDK8 modulates the transcriptional output from distinct transcription factors involved in oncogenic control. These factors include the Wnt/β-catenin pathway, Notch, p53, and TGF-β.2,3
The means by which CDK8 activity accomplishes the regulation of these various pathways remains an active area of investigation. However, CDK8 has been found to be amplified and overexpressed in colon cancer,4 acting through the Wnt pathway and β-catenin, its key signaling molecule.5 Suppression of CDK8 expression in colon cancer cell lines expressing high levels of both CDK8 and β-catenin led to reduced proliferation of these cell lines.6 Furthermore, in a survey of colorectal cancer tissue samples, CDK8 was associated with β-catenin activation and with reduced rates of survival.7 Two series of chemical probes targeting CDK8 have recently been published. CCT2515458,9 was identified and optimized through its suppression of the Wnt pathway. It was shown to be a type I inhibitor of CDK8 and CDK19 with excellent selectivity over a panel of 291 kinases and exhibited activity in a Wnt pathway-dependent mouse tumor model.
We recently reported the independent development of azabenzothiophene-derived CDK8 inhibitors that are potent and selective and suppress phosphorylation of STAT1 at Ser727 similarly to CCT251545.10 Both series of compounds interact with the protein in the same fashion, principally through a hydrogen bond from Lys52 to a carbonyl on the ligand, a hydrogen bond accepted from the hinge residue Ala100 to a pyridine nitrogen and through a cation–pi interaction with Arg356.
As work on the azabenzothiophene inhibitors was underway, we targeted the development of a potent and selective series of type II kinase inhibitors. In many kinases, displacement of the Asp-Phe-Gly (DFG) sequence that precedes the activation loop defines type I and type II modes of inhibition, with type II generally showing a displaced DFG motif from a buried position in the protein to a DFG-out position, opening a “deep pocket” that can be occupied by inhibitors. In the case of CDK8, this trio of residues is Asp-Met-Gly (DMG). A DMG-out class of inhibitors was sought to explore this mode of inhibition. We began with Sorafenib (1),11 a known type II kinase inhibitor. Sorafenib did not, however, meet the selectivity requirements we had established for tool compounds to study the role of CDK8. We used a Sorafenib-bound CDK8 structure (PDB: 3RGF) to model design ideas that could lead to type II inhibitors.
Our initial efforts were aimed at altering the hinge binding element of Sorafenib, and the 2,4-diaminopyrimidine seen in 2 appeared most favorable, making additional backbone hydrogen bonds with the hinge residues Asp98 and Ala100 from its amino groups. We hypothesized that these additional contacts would improve CDK8 binding, as the pyrimidine ring nitrogen was predicted to nearly superimpose with the pyridine ring nitrogen in 1. We were encouraged to see that this modification improved the CDK8 IC50 by about 2-fold in a TR-FRET based binding assay (Table 1) and simultaneously improved the kinase selectivity. When compound 2 was screened against a panel of 220 kinases, only four of 220 kinases (1.8%) exhibited >50% inhibition at 0.1 μM, with one of them being CDK8, an improvement over Sorafenib, where 10 out 220 kinases (4.5%) exhibited >50% inhibition at 0.1 μM (Figure 1).
Table 1. Potency Comparison between Sorafenib (1) and 2.
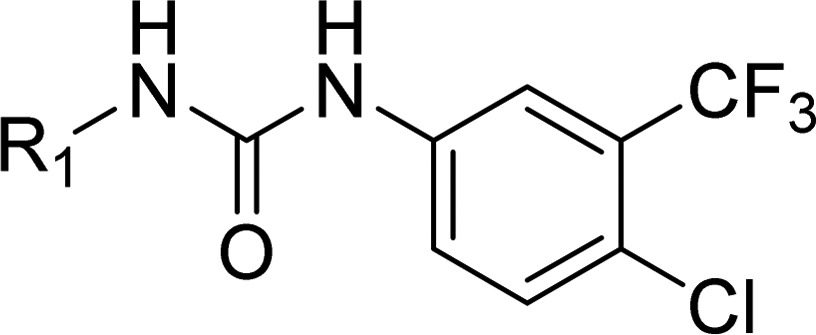
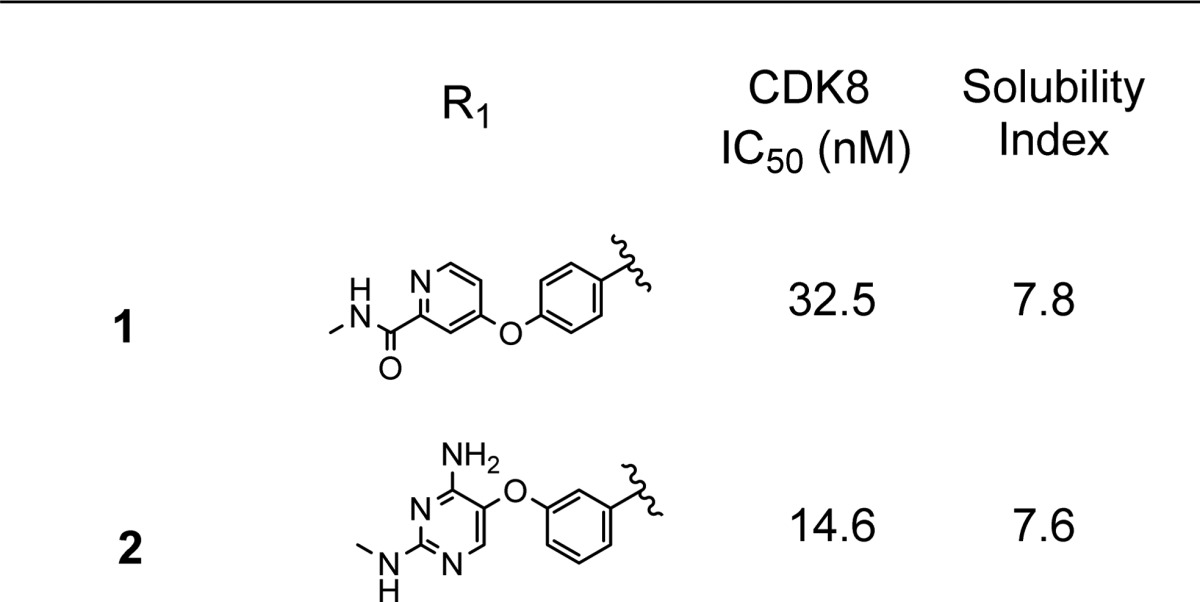
Figure 1.

Kinase selectivity for compound 2 and Sorafenib (1) measured at a concentration of 0.1 μM in a panel of 220 kinases. Each vertical line represents a single kinase and is colored by the percent inhibition seen. Selected potently inhibited kinases are highlighted.
Having improved our enzymatic potency and selectivity through this alteration, we focused on improving the physical properties of our molecules. We utilized a calculation of compounds’ solubility index12 (SI = cLogD7.4 + aromatic rings) to drive this process. Lower values of the solubility index predict improved solubility in aqueous buffer. The most straightforward way to reduce the solubility index is to decrease the number of aromatic rings, as it has been shown that disrupting molecular planarity is beneficial for aqueous solubility.13 We therefore sought to eliminate the central phenyl ring in our initial set of compounds. In order to find a suitable replacement, we utilized a pharmacophore similarity search program called FastROCS,14 which performs a rapid three-dimensional shape and pharmacophore match to a database of conformers generated for an internal library of 110,000 compounds. We used a docked pose of compound 2 as a query structure and examined the output for structures that matched the pharmacophore of 2 both at the point of hinge binding and the biaryl urea, which forces the DFG-out binding mode, but did not have an aromatic ring linking the two regions. Among those structures with the highest score, we focused on the pyrrolidine 3 (Figure 2). The structure overlapped very well with compound 2, notably in the aromatic urea region, which is essential for DFG-out binding, and had a saturated pyrrolidine linker in place of a phenyl ring. Assayed as a racemic mixture, 3 exhibited comparable potency to the input compound 2.
Figure 2.
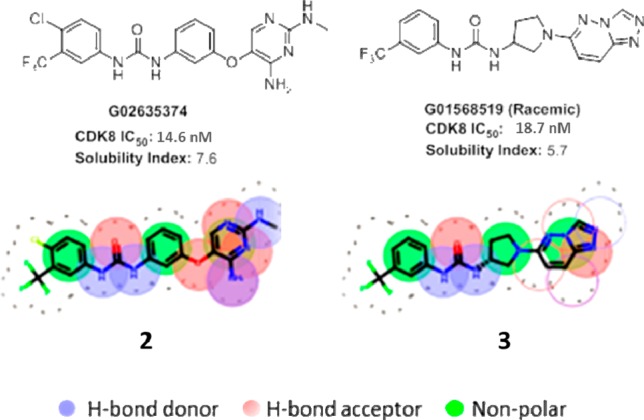
Two-dimensional comparison of shape and volume between compound 2 and the new pyrrolidine series (3).
We then synthesized one of the analogues in both enantiomeric series, showing that the S isomer was more potent (Table 2). While the solubility index improved, we did not see a significant improvement in measured solubility. However, we were pleased to see further improvement in potency (Table 2).
Table 2. Activity and Kinetic Solubility Data for 1-(1-([1,2,4]Triazolo[4,3-b]pyridazin-6-yl)pyrrolidin-3-yl)-3-ureas 4 and 5.
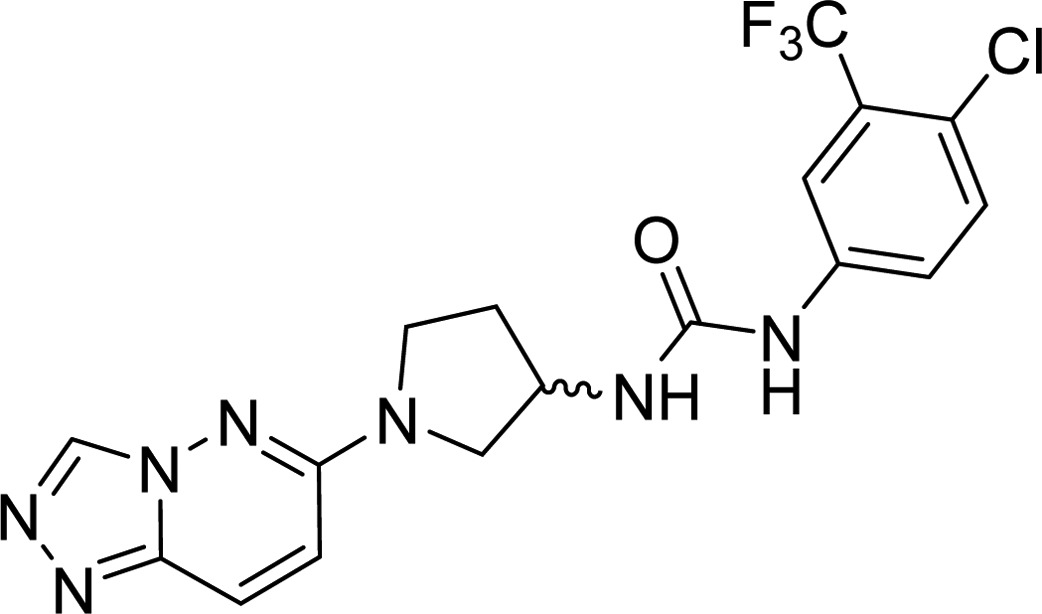
| stereochemistry | CDK8 IC50 (nM) | solubility index | kinetic solubility (μM) | |
|---|---|---|---|---|
| 4 | S | 4.2 | 5.7 | 1.0 |
| 5 | R | 166 | 5.7 | 1.0 |
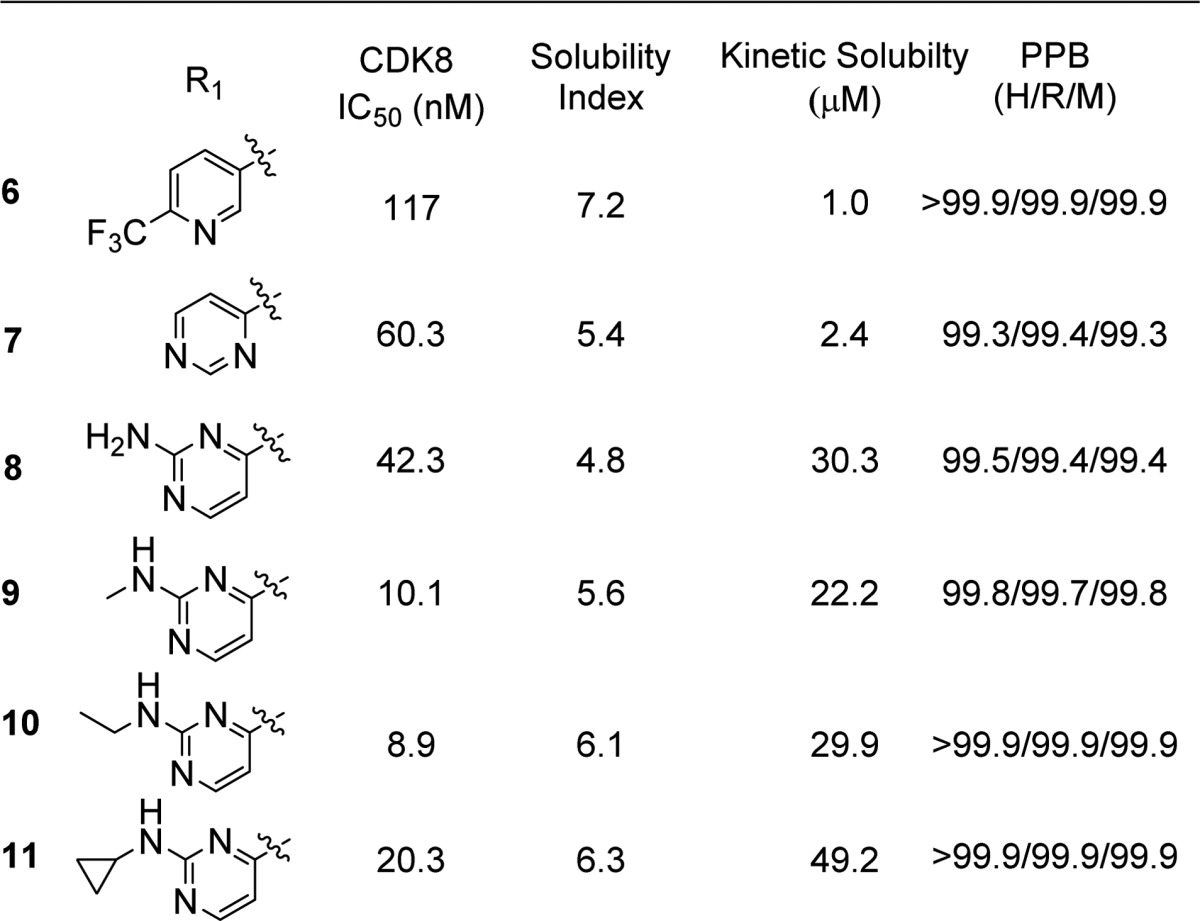
In order to address the solubility issue, we focused our attention on the 6-substituted triazolopyridazine, which was predicted to act as the hinge binder in our molecules. Since the docking protocol and the Sorafenib crystal structure we utilized predicted that this bicycle only makes one contact with the kinase hinge backbone, we attempted to achieve the same key contacts using a single functionalized ring, again improving the solubility index and hopefully the measured solubility. Compounds 6–11 were therefore prepared and evaluated for CDK8 inhibitory activity as well as kinetic solubility. Replacement of the triazolopyridazine with a 2-(trifluoromethyl)pyridine (6) or a pyrimidine (7) resulted in compounds with greatly reduced activity and did not significantly improve solubility. Addition of an amino group at the 2-position of the pyrimidine (8) restored some activity and improved solubility. Substitutions on the amino group further improved potency, with methyl and ethyl being the optimal groups discovered in an initial investigation (Table 3).
Table 3. Potency, Kinetic Solubility, and Plasma Protein Binding Data for (S)-1-(4-Chloro-3-(trifluoromethyl)phenyl)-3-(pyrrolidin-3-yl)ureas 6–11.
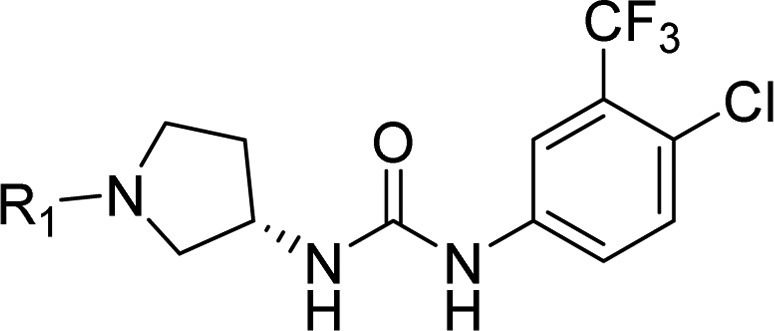
As a path forward, methylaminopyrimidine was selected due to the combination of improved solubility and potency while being less lipophilic than the ethylaminopyrimidine. We synthesized a series of analogues using this headgroup, aiming to further improve solubility and plasma protein binding by removing the highly lipophilic CF3 substituent (Table 4). While the solubility did improve through removal of this group it became apparent that having the trifluoromethyl group in the meta position of the phenyl urea was necessary for optimal potency. All the compounds that were made lacking this group were significantly weaker binders of CDK8, with one notable exception being compound 12. In addition to having acceptable potency and improved solubility, compound 12 exhibited exquisite selectivity, inhibiting only CDK8 > 30% at 1 μM, in a panel of 220 kinases (Figure 3).
Table 4. Potency, Kinetic Solubility, and Plasma Protein Binding Data for (S)-1-(1-(2-(Methylamino)pyrimidin-4-yl)pyrrolidin-3-yl)ureas 12–18.

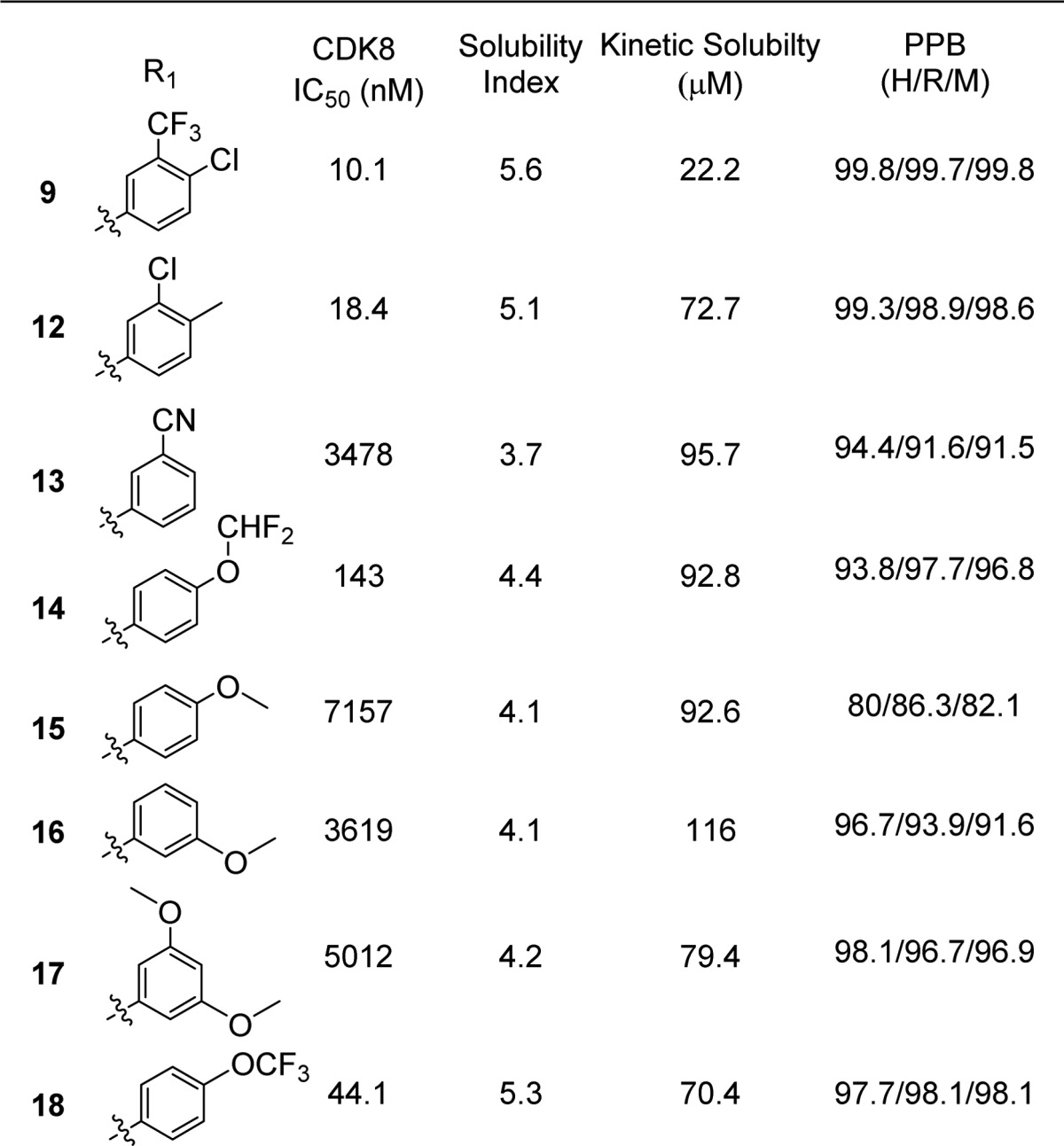
Figure 3.
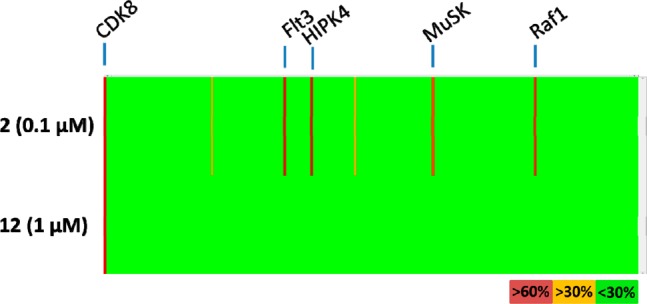
Kinase selectivity for compound 2, measured at a concentration of 0.1 μM in a panel of 220 kinases, and compound 12, measured at a concentration of 1 μM in a panel of 220 kinases. Each vertical line represents a single kinase and is colored by the percent inhibition seen. Selected potently inhibited kinases are highlighted.
Having come to the conclusion that, while being detrimental to the solubility and increasing plasma protein binding, the m-CF3 was essential for potency, we sought other ways to improve the properties of our molecules while retaining this functional group. Addition of a solubilizing group15 in the para position in our series of molecules proved to be very beneficial for solubility and plasma protein binding while retaining good potency (Table 5).
Table 5. Potency, Kinetic Solubility, and Plasma Protein Binding Data for (S)-1-(1-(2-(Methylamino)pyrimidin-4-yl)pyrrolidin-3-yl)-3-(3-(trifluoromethyl)phenyl)ureas 19–24.
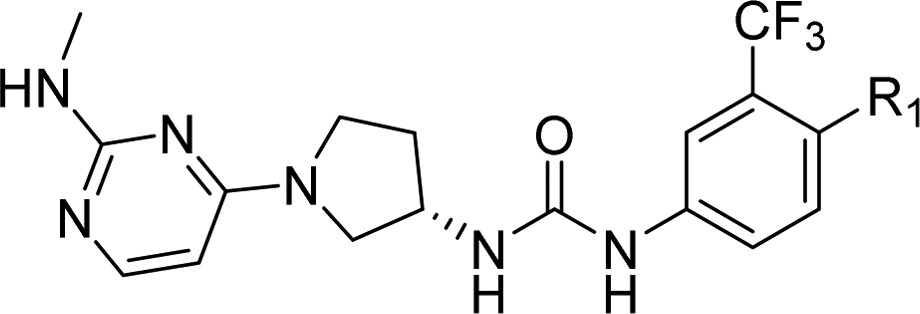
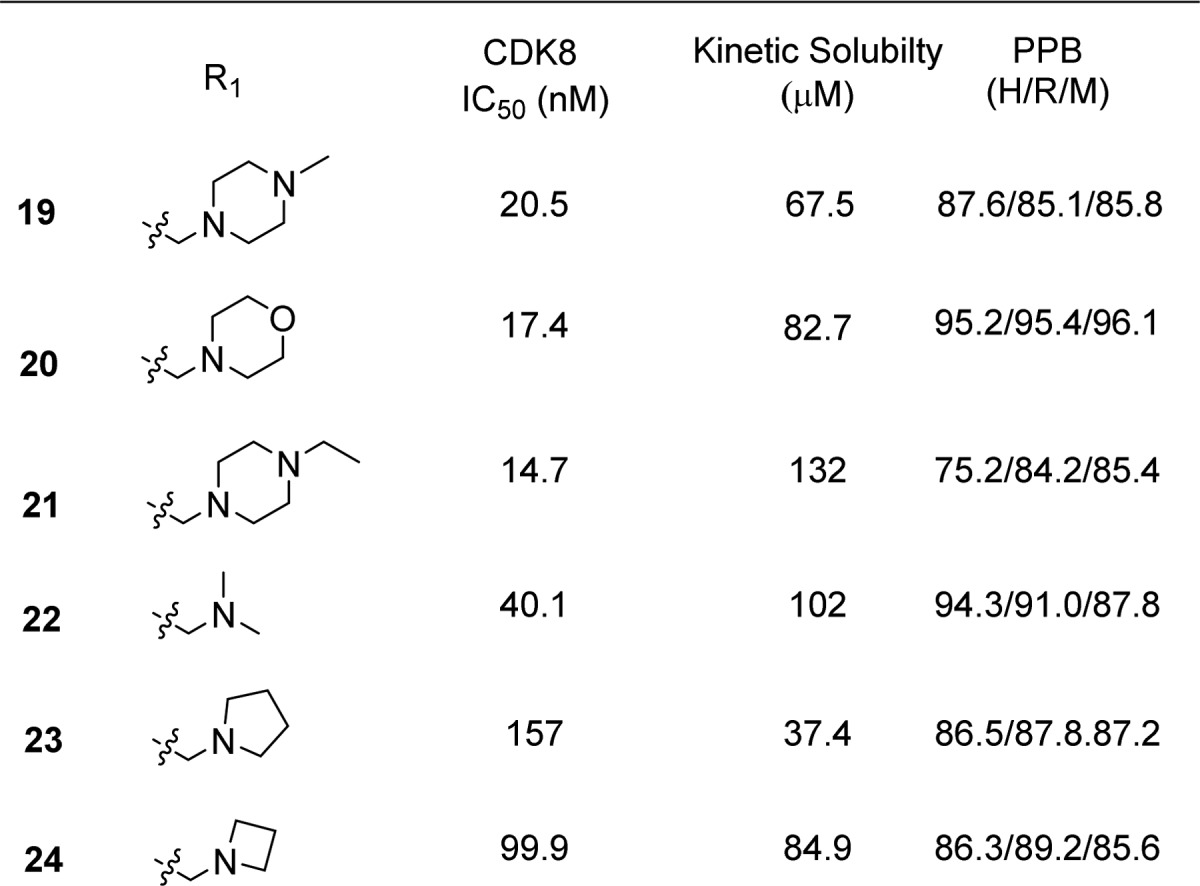
Compound 20 was tested for kinase selectivity and showed excellent selectivity with activity >30% against only one off-target kinase (Flt3) in a panel of 220 kinases (Figure 4).
Figure 4.
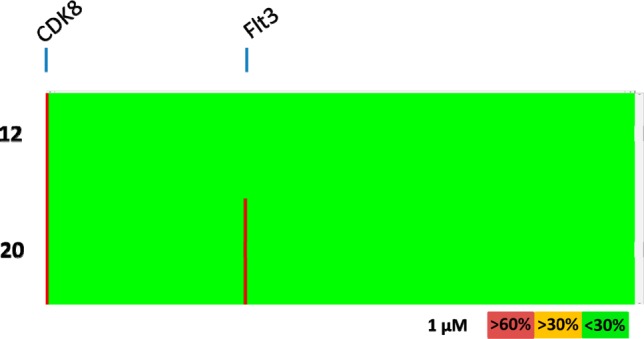
Kinase selectivity for compounds 12 and 20 measured at a concentration of 1 μM in a panel of 220 kinases. Each vertical line represents a single kinase and is colored by the percent inhibition seen. Selected potently inhibited kinases are highlighted.
In order to understand the binding interactions we had optimized more completely, we obtained a cocrystal structure of compound 20 in complex with CDK8:Cyclin C at 2.4 Å resolution (Figure 5 and Supporting Information). The compound binds to the kinase active site with the DMG motif in the “out” conformation, with the urea through morpholine moieties nominally filling the space vacated by the DMG loop. Two direct and two water-mediated hydrogen bonds are formed between inhibitor and the kinase. The methylpyrimidine group forms a single H-bond with the backbone nitrogen of Ala100 (2.9 Å) on the hinge region. The carbonyl oxygen of the central urea functionality forms the second direct H-bond to the backbone nitrogen of Asp173 (2.9 Å) of the DMG motif. Both of the urea nitrogens of 20 form hydrogen bonds with a solvent molecule, but neither nitrogen directly interacts with Glu66, unlike what is observed in the Sorafenib10 structure. This solvent molecule (water 1 in Figure 5) in turn indirectly links to the protein through H-bonds to two other waters. This bridging of the inhibitor across two water layers to the protein suggests that the urea group may not be necessary for potency, opening room for further improvement in physical properties by replacing the urea with a carbonyl containing isostere.
Figure 5.
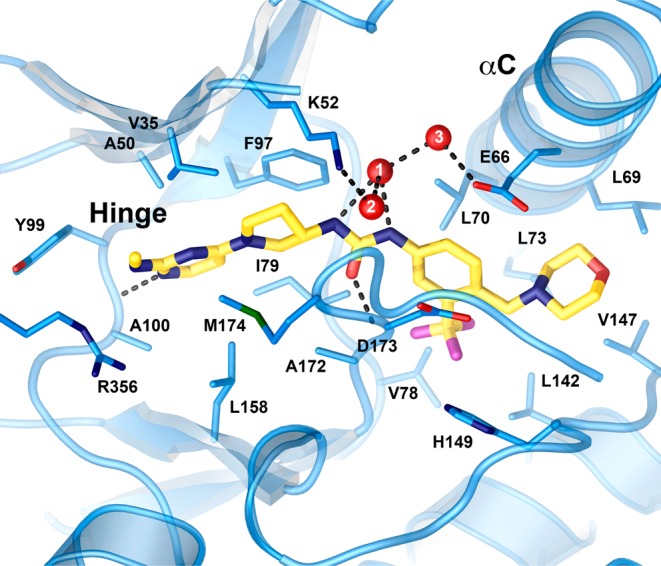
Crystal structure of compound 20 (gold carbons) at the CDK8 active site. The DMG loop (center, D173 and M174 shown) is in the “out” conformation. A network of three solvent molecules (red, numbered spheres link the center of the compound to the protein). Water 2 makes an additional H-bond to the backbone carbonyl of D173 that was omitted for clarity. The hinge and C-helix (αC) are labeled.
Having generated highly potent and selective binders of CDK8 we evaluated their antiproliferative effects in the colorectal carcinoma HCT-116 cell line, which had previously been shown to exhibit sharply reduced proliferation response to CDK8 protein depletion using targeted shRNA.1 Cells were treated with compounds for 72 h in media containing fetal bovine serum (FBS) at a concentration of either 0.625% or 10%. The lower concentration of FBS was used to mitigate the effects of the low free fraction of some of our compounds. Throughout the course of the experiment, the cells were in growth phase, and yet we did not observe significant growth inhibition (Table 6). Flavopiridol, a potent inhibitor of multiple CDKs16 with known antiproliferative properties17 was used as a positive control. With the Km for ATP measured at approximately 300 μM using an ADP generation activity assay (data not shown), the lack of antiproliferative activity could not be explained by competition from endogenous ATP. In addition, our compounds have moderate to good permeability so poor cell penetration likely did not account for lack of activity. Concerned that the compounds examined were acting weakly on CDK8 in the cellular context, we examined phosphorylation of S727 of STAT1, a known CDK8 substrate, under conditions of IFNγ stimulation using an in-cell western assay.18 The results were similar to those obtained in the proliferation assay. These potent inhibitors of CDK8/cyclin C in biochemical assays showed only weak if any inhibition of STAT1 phosphorylation in cells, while a control type I inhibitor from a previous publication inhibited with nanomolar potency.10 Our results corroborate the findings of Dale et al. that type II inhibitors of CDK8 exhibit weak activity in cellular assays (reporter, CETSA, and STAT1 phosphorylation) and support their assertion that the inactive form of CDK8 that is targeted by type II inhibitors is poorly accessible in cells.8
Table 6. Antiproliferative Activity on HCT-116 Cells and STAT1 S727 Phosphorylation IC50 of Compounds 9, 12, and 19–21.
| compd | HCT116 EC50 10% FBS (μM) | HCT116 EC50 0.625% FBS (μM) | MDCK perm AB × 10–6 cm/sec | MDCK perm BA × 10–6 cm/sec | STAT1 S727 phosphorylation IC50 (μM) | CDK8 IC50 (nM) |
|---|---|---|---|---|---|---|
| 9 | 4.4 | 2.9 | 3.9 | 4.0 | >10 | 10.1 |
| 12 | >10 | 4.9 | 3.9 | 7.0 | 18.4 | |
| 19 | 8.2 | >10 | 4.1 | 4.5 | 4.2 | 20.5 |
| 20 | >10 | 7.7 | 12.4 | 12.2 | >10 | 17.4 |
| 21 | 4.8 | 4.3 | 0.3 | 4.2 | 4.0 | 14.7 |
| Koehler et al.10 | 6.7 | 5.4 | 18.2 | 17.2 | 0.0023 | 1.5 |
| flavopiridol | 0.034 | 0.059 |
To investigate whether the observed weak antiproliferative activity was due to CDK8 engagement, we used a set of HCT-116 cell lines in which CDK8 alone or in combination with CDK19 was targeted for genomic deletion using CRISPR guide RNAs. These cell lines were originally designed and demonstrated to validate STAT1 S727 as a substrate of CDK8.10 We incubated wild type HCT-116 cells along with the CDK8 and the CDK8/19 double deletion clones derived from them with varying concentrations of 21. As can be seen in Figure 6, each of these cell lines showed very similar growth inhibition curves, indicating that growth inhibition is likely not a function of the inhibition of CDK8, but rather an off-target effect.
Figure 6.
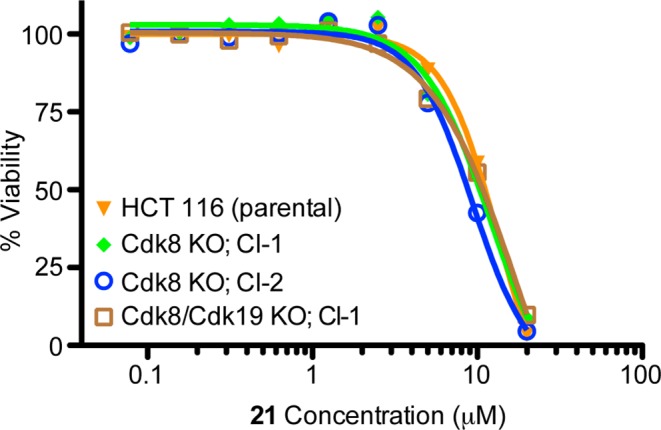
Growth inhibition of the wild type HTC-116, CDK8 deletion (clone 1 and 2), and CDK8/19 double deletion cells by varying concentrations of 21.
Structure-guided modification of the well-known kinase inhibitor Sorafenib led to a series of highly potent and selective type II inhibitors of CDK8. Despite their lack of cellular activity, we are confident that they are useful tool compounds to investigate the biology of the CDK8 kinase domain. Replacement of the phenyl ring linker by a saturated five-membered ring improved potency, selectivity, and solubility. Addition of a solubilizing group at the para position of the phenyl urea greatly improved solubility and PPB while retaining potency and selectivity. Similarly to what has been recently reported,8 our series of type II inhibitors did not significantly suppress phosphorylation of STAT1 at Ser727, while our series of type I inhibitors showed strong activity.10
Acknowledgments
We thank members of the DMPK and purification groups within Genentech Small Molecule Drug Discovery for purification and analytical support. The crystallographic experiments were performed on the X06SA beamline at the Swiss Light Source, Paul Scherrer Institut, Villigen, Switzerland.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00044. Coordinates and structure factors for the crystallographic experiment are deposited at the Protein Data Bank with accession code: 5HVY.
Synthetic schemes describing the preparation of 1–32 are provided along with kinase selectivity and crystallographic data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Conaway R. C.; Sato S.; Tomomori-Sato C.; Yao T.; Conaway J. W. The mammalian Mediator complex and its role in transcriptional regulation. Trends Biochem. Sci. 2005, 30, 250–255. 10.1016/j.tibs.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Nemet J.; Jelicic B.; Rubelj I.; Sopta M. The two faces of CDK8, a positive/negative regulator of transcription. Biochimie 2014, 97, 22–27. 10.1016/j.biochi.2013.10.004. [DOI] [PubMed] [Google Scholar]
- Li N.; Fassl A.; Chick J.; Inuzuka H.; Li X.; Mansour M. R.; Liu L.; Wang H.; King B.; Shaik S.; et al. Nat. Cell Biol. 2014, 16, 1080–1091. 10.1038/ncb3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestein R.; Bass A. J.; Kim S. Y.; Dunn I. F.; Silver S. J.; Guney I.; Freed E.; Ligon A. H.; Vena N.; Ogino S.; et al. CDK8 is a colorectal cancer oncogene that regulates β-catenin activity. Nature 2008, 455, 547–551. 10.1038/nature07179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo J.-O.; Song I. H.; Lim S.-C. Role of CDK8 and β-catenin in colorectal adenocarcinoma. Oncol. Rep. 2010, 24, 285–291. 10.3892/or_00000858. [DOI] [PubMed] [Google Scholar]
- Adler A. S.; McCleland M. L.; Truong T.; Lau S.; Modrusan Z.; Soukup T. M.; Roose-Girma M.; Blackwood E. M.; Firestein R. Cancer Res. 2012, 72, 2129–2139. 10.1158/0008-5472.CAN-11-3886. [DOI] [PubMed] [Google Scholar]
- Firestein R.; Shima K.; Nosho K.; Irahara N.; Baba Y.; Bojarski E.; Giovannucci E. L.; Hahn W. C.; Fuchs C. S.; Ogino S. CDK8 expression in 470 colorectal cancers in relation to β-catenin activation, other molecular alterations and patient survival. Int. J. Cancer 2010, 126, 2863–2873. 10.1002/ijc.24908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale T.; Clarke P. A.; Esdar C.; Waalboer D.; Adeniji-Popoola O.; Ortiz-Ruiz M.-J.; Mallinger A.; Samant R. S.; Czodrowski P.; Musil D.; Schwarz D.; Schenider R.; Stubbs M.; Ewan K.; Fraser E.; TePoele R.; Court W.; Box G.; Valenti M.; de Haven Brandon A.; Gowan S.; Rohdich F.; Raynaud F.; Schneider R.; Poeschke O.; Blaukat A.; Workman P.; Schiemann K.; Wienke D.; Blagg J. A selective chemical probe for exploring the role of CDK8 and CDK19 in human disease. Nat. Chem. Biol. 2015, 11, 973–980. 10.1038/nchembio.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallinger A.; Schiemann K.; Rink C.; Stieber F.; Calderini M.; Crumpler S.; Stubbs M.; Adeniji-Popoola O.; Poeschke O.; Busch M.; Czodrowski P.; Musil D.; Schwarz D.; Ortiz-Ruiz M.-J.; Schneider R.; Thai C.; Valenti M.; de Haven Brandon A.; Burke R.; Workman P.; Dale T.; Wienke D.; Clarke P. A.; Esdar C.; Raynaud F. I.; Eccles S. A.; Rohdich F.; Blagg J. Discovery of Potent, Selective and Orally Bioavailable Small-Molecule Modulators of the Mediator Complex-Associated Kinases, CDK8 and CDK19. J. Med. Chem. 2016, 59, 1078–1101. 10.1021/acs.jmedchem.5b01685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler M. F. T.; Bergeron P.; Blackwood E.; Bowman K.; Clark K. C.; Firestein R.; Kiefer J. R.; Maslos K.; McCleland M. L.; Orren L.; Salphati L.; Schmidt S.; Schneider E. V.; Wu J.; Beresini M. H. Development of a Potent, Specific CDK8 Kinase Inhibitor Which Phenocopies CDK8/19 Knockout Cells. ACS Med. Chem. Lett. 2016, 7, 223. 10.1021/acsmedchemlett.5b00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider E. V.; Bottcher J.; Blaesse M.; Neumann L.; Huber R.; Maskos K. Structure-kinetic relationship study of CDK8/CycC specific compounds. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 8081–8086. 10.1073/pnas.1305378110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill A. P.; Young R. J. Getting physical in drug discovery: a contemporary perspective on solubility and hydrophobicity. Drug Discovery Today 2010, 15, 648–655. 10.1016/j.drudis.2010.05.016. [DOI] [PubMed] [Google Scholar]
- Ishikawa M.; Hashimoto Y. Improvement in aqueous solubility in small molecule drug discovery programs by disruption of molecular planarity and symmetry. J. Med. Chem. 2011, 54, 1539–1554. 10.1021/jm101356p. [DOI] [PubMed] [Google Scholar]
- Feng J. A.; Aliagas I.; Bergeron P.; Blaney J. M.; Bradley E. K.; Koehler M. F. T.; Lee M.-L.; Ortwine D. F.; Tsui V.; Wu J.; Gobbi A. An integrated suite of modeling tool that empower scientists in structure- and property-based drug design. J. Comput.-Aided Mol. Des. 2015, 29, 511–523. 10.1007/s10822-015-9845-4. [DOI] [PubMed] [Google Scholar]
- Chao Q.; Sprankle K. G.; Grotzfeld R. M.; Lai A. G.; Carter T. A.; Velasco A. M.; Gunawardane R. N.; Cramer M. D.; Gardner M. F.; James J.; Zarrinkar P. P.; Patel H. K.; Bhagwat S. S. Identification of N-(5-tert-Butyl-isoxazol-3-yl)-N0-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo-[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea Dihydrochloride (AC220), a uniquely potent, selective and efficacious FMS-like tyrosine kinase-3 (FLT3) inhibitor. J. Med. Chem. 2009, 52, 7808–7816. 10.1021/jm9007533. [DOI] [PubMed] [Google Scholar]
- Karaman M. W.; Herrgard S.; Treiber D. K.; Gallant P.; Atteridge C. E.; Campbell B. T.; Chan K. W.; Ciceri P.; Davis M. I.; Edeen P. T.; et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- Jung C.; Motwani M.; Kortmansky J.; Sirotnak F. M.; She Y.; Gonen M.; Haimovitz-Friedman A.; Schwartz G. K. The cyclin-dependent kinase inhibitor flavopiridol potentiates γ-irradiation-induced apoptosis in colon and gastric cancer cells. Clin. Cancer Res. 2003, 9, 6052–6061. [PubMed] [Google Scholar]
- Bancerek J.; Poss Z. C.; Steinparzer I.; Sedlyarov V.; Pfaffenwimmer T.; Mikulic I.; Dölken L.; Strobl B.; Müller M.; Taatjes D. J.; et al. CDK8 kinase phosphorylates transcription factor STAT1 to selectively regulate the interferon response. Immunity 2013, 38, 250–262. 10.1016/j.immuni.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.