

Abstract
The tubulin-microtubule system is a common target of many anticancer drugs. However, the use of chemotherapeutics frequently leads to the development of a clinically relevant phenomenon of multidrug resistance (MDR). One of the basic mechanisms involved in MDR involves elevated expression and/or activity of several ATP-binding cassette superfamily members (ABC transporters) which are normally responsible for the efflux of xenobiotics or secondary metabolites outside the cell. Here we present the synthesis and biological characteristics of ferrocenyl analogues of plinabulin, i.e. one of the so-called “spindle poisons”. We found that replacement of the phenyl group of plinabulin by the ferrocenyl moiety turns this compound into a potent inhibitor of ABCB1 and ABCG2, thus making it possible to overcome the multidrug resistance phenomenon. We also demonstrated that the alkyl group attached to the imidazole moiety of ferrocenyl analogues of plinabulin strongly affects their potency to inhibit tubulin polymerization.
Keywords: Ferrocene, cancer, ABCB1 inhibitor, ABCG2 inhibitor, multidrug resistance, bioorganometallic chemistry
Microtubules are cytoskeleton elements composed of α- and β-tubulin heterodimers which are involved in a number of cellular process, e.g. cell motility, intracellular transport, and mitosis. Compounds interacting with the dynamic tubulin-microtubule system are used as chemotherapeutic drugs against many types of neoplasms, including ovarian, breast, head and neck, and pancreatic cancers.1 Widely used are taxanes, e.g. paclitaxel2 and its semisynthetic analogue docetaxel,3 which can induce polymerization of tubulins and stabilize microtubules. Another example are the Vinca alkaloids vincristine and vinblastine, which promote depolymerization of the microtubule back to α- and β-tubulin.4 Some other naturally occurring inhibitors of tubulin polymerization, e.g. colchicine,5 are not used in therapy due to their high toxicity.
Because the use of taxanes or other anticancer drugs leads to many side effects,6 there is an ongoing search for natural or synthetic compounds which could preserve their anticancer activity while being less threatening to normal cells and tissues. One of the most promising potent anticancer drug-candidates is plinabulin,7 a synthetic analogue of naturally occurring 2,5-piperazinedione–phenylahistin (Figure 1).8,9 It binds to the colchicine binding site of tubulin and thus induces a collapse of the tumor vasculature, reducing the blood supply to the cancer cells and, consequently, leading to the death of tumor cells. These effects are accompanied by its direct cytotoxic action on cancer cells..10 Plinabulin is currently being studied in a phase II clinical trial on nonsmall cell lung cancer.11
Figure 1.
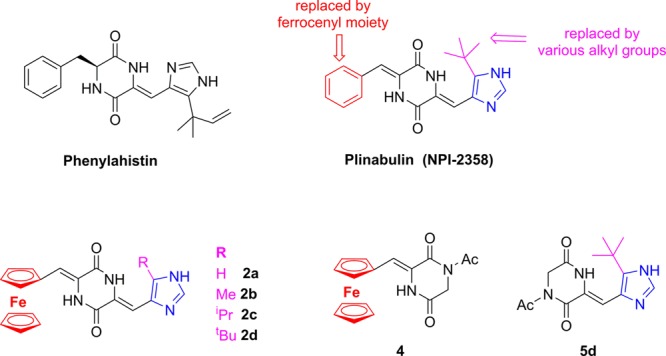
Structure of phenylahistin, plinabulin (top), and its ferrocenyl analogues (2a–d) and half-plinabulin analogues (4, 5d) synthesized herein.
One of the biggest problems in cancer chemotherapy is the development of resistance to anticancer drugs. During the first course of chemotherapy, drugs kill only drug-sensitive cancer cells, while some cancer cells survive and the tumor begins to grow again. Additionally, the mechanism by which these survivor cells have escaped the therapy is enhanced by negative selection, so all tumor cells become drug-resistant and the treatment fails. Although there are multiple mechanisms underlying this phenomenon, one of the most important of these seems to involve the transmembrane transport proteins of the ATP-binding cassette superfamily (ABC transporters).12 In normal cells, these proteins are responsible for the efflux of xenobiotics or secondary metabolites outside the cytoplasm. Their overexpression in cancer cells leads to resistance to many anticancer drugs, e.g. cisplatin, colchicine, vinblastine, paclitaxel, doxorubicin and other small molecules, such as protein kinase inhibitors.13 Thus, it is important to develop new compounds which will be able to not only kill cancer cells at low concentrations but also to overcome the multidrug-resistant phenotype (MDR) of cancer cells.
Tremendous progress in the bioorganometallic chemistry of anticancer, antimalarial and antibacterial compounds has been observed in recent years. The presence of an metal atom which can exist in various oxidation states allows to synthesize a large number of new types of molecules with properties that are different from those of typical organic compounds.14 Depending on the metal–ligand bond nature, it is possible to develop bioactive organometallic compounds which contain covalent metal–carbon bonds and/or labile ligands.15−20 Pioneering studies in this field were carried out by Jaouen and co-workers17 for ferrocene, which is one of the metallocenes. Ferrocene alone exhibits only slight cytotoxic activity, whereas conjugated with biologically active compounds it provides them with added value, e.g. which leads to some promising anticancer agents.17,21 One of the possible explanations for its activity is based on reversible cellular oxidation of the ferrocenyl moiety to a cytotoxic ferrocenium ion accompanied by the formation of reactive oxygen species (ROS), e.g. hydroxyl radicals. ROS are capable of damaging DNA and other important biological molecules, thus leading to cellular injury and death.22−24 It has been showed that organometallic compounds are, in some cases, able to overcome drug resistance (this was observed, for example, in cancer25 and parasite [Plasmodium] cells26). Vock et al. showed that the ruthenium half-sandwich complexes of a known MDR modulator are able to inhibit the activity of the ABCB1 protein more efficiently than the purely organic form of the MDR modulator.27
Herein we report the synthesis of ferrocenyl analogues of plinabulin and the systematic study of their cytotoxic activity against a panel of human cancer lines, including their ABC-transporter overexpressing multidrug resistance variants. We also studied the ability of novel compounds to interfere with tubulin polymerization and stability as well as their influence on the cell cycle. Finally, we demonstrated here that some of these compounds are potent inhibitors of MDR proteins.
To test the influence of plinabulin phenyl group replacement by the ferrocenyl moiety on biological activities toward cancer cells, we synthesized ferrocene-plinabulin analogues with various alkyl groups attached to imidazole moieties 2a–d. We also synthesized a ferrocene-containing conjugate which is structurally similar to plinabulin but is devoid of imidazole moiety 4, as well as an imidazole conjugate without ferrocenyl moiety 5d (Figure 1). Synthesis of the desired compounds was performed by modified standard procedure (Scheme 1). First, glycine anhydride was refluxed with an excess of acetic anhydride to give 1,4-diacetylpiperazine-2,5-dione 3. Due to the instability of the crude product (catalyzed by traces of acetic acid), it was purified by silica gel chromatography. In the next step, a condensation reaction of 3 with ferrocenecarboaldehyde in a mixture of tert-butanol and DMF, using potassium tert-butanolate as a base, gave the desired 4 as dark red crystals in 79% yield. Unfortunately, further condensation with commercially available 1H-imidazole-4-carboaldehyde under various reaction conditions failed, and only the products of decomposition of the starting 4 or a trace amount of product 2a were obtained (e.g., DMF as a solvent, cesium carbonate as a base, room temperature or 50 °C, reaction time 2–24h–only a trace of 4 were detected (by TLC); when the condensation reaction was performed at 90 °C, instead of RT, for 2.5 h, the desired product was obtained in 3% yield). We assumed that either no reaction occurred or the reaction yield was low due to the presence of an acidic amide proton in 4. Thus, we decided to reacetylate the nitrogen atom of the amide. Similarly to the synthesis of 3, we refluxed 4 with an excess of acetic anhydride but only unreacted 4 was recovered. The deprotonation reaction of 4 with potassium bis(trimethylsilyl)amide followed by reaction with acetyl chloride also resulted in recovery of an unreacted substrate. Finally, we tried to reacetylate 4 in reaction with an excess of acetic acid and DIC as the coupling agent in the presence of DMAP. Depending on the reaction time, we were able to isolate either a desired reacetylated 6 in 40% yield (the reaction stopped after 1 h) or its rearrangement product (the reaction stopped after 24 h).28 Unfortunately, compound 6 was not stable enough to perform the next condensation reaction. Therefore, we decided to reverse the order of the condensation steps. First, in the reaction of 3 with 5-alkyl-1H-imidazole-4-carboaldehydes in DMF in the presenceof cesium carbonate at 35 °C for 4 h, 5a–d were prepared in moderate yield (38–41%). The next condensation step of 5a–d with ferrocenecarboaldehyde in the presence of cesium carbonate carried out at 90 °C for 2 h gave the required ferrocenyl analogues of plinabulin 2a–d in a moderate to good yield (40–61%; and 15.6–25% overall yield after two condensation steps). The structures of the synthesized compounds were confirmed by 1D and 2D NMR spectra, mass spectrometry and by elemental analysis (See Supporting Information for more detailed information).
Scheme 1. Synthesis of the Ferrocenyl Analogues of Plinabulin.

The cytotoxic properties of the synthesized compounds were determined in MCF7 (human mammary adenocarcinoma), HepG2 (human hepatocellular cancer) and SW620 (human colon adenocarcinoma) cell lines (Table 1). We also employed a panel of drug-resistant cell lines obtained by a gradual selection of SW620 sensitive cells with common chemotherapeutics. Depending on the selecting agent, they were designed as SW620C – selected with cisplatin, SW620D – doxorubicin, SW620E – etoposide, SW620M – methotrexate and SW620V – vincristine. We previously characterized these cells with respect to multidrug-resistant protein phenotype (at mRNA, protein and activity level, submitted) and found that ABCG2 is the transporter overexpressed in the SW620C line, ABCC1 is the main MDR protein of SW620M, while SW620D, -E, and -V cells highly overexpress ABCB1. We found that plinabulin is highly toxic (at a nanomolar range) to the SW620 and SW620M lines, while its toxicity is rather moderate (IC50 values at tens of micromolar levels in SW602C, SW620E and MCF7 cells) or even negligible in other cell lines. This was somehow unexpected, as NPI-2358 was shown to be highly active against several cell lines of different background, including also multidrug-resistant cells.29
Table 1. Cytotoxic Properties of Plinabulin, Its Ferrocenyl Analogues 2a–2d, and Half-Analogues 4 and 5a Expressed as IC50a.
| Cell line | NPI-2358 | 2a | 2b | 2c | 2d | 4 | 5d |
|---|---|---|---|---|---|---|---|
| SW620 | 0.034 (0.021–0.056) | >100 | 31.43 (21.59–45.76) | 9.65 (7.52–12.38) | 16.65 (11.42–24.26) | >100 | >100 |
| SW620C | 34.11 (12.96–89.77) | >100 | 67.74 (47.79–96.02) | 31.42 (25.24–39.10) | 19.83 (16.11–24.40) | >100 | >100 |
| SW620D | >100 | >100 | 72.04 (51.25–101.30) | 24.23 (19.14–30.68) | 21.77 (16.38–28.94) | >100 | >100 |
| SW620E | 52.45 (23.90–115.10) | >100 | 64.05 (42.79–95.88) | 24.57 (20.34–29.69) | 18.64 (11.67–29.78) | >100 | >100 |
| SW620M | 0.132 (0.024–0.744) | >100 | 43.53 (32.52–58.27) | 23.54 (13.04–42.48) | 32.69 (20.48–52.17) | >100 | >100 |
| SW620V | >100 | >100 | >100 | 27.95 (9.99–78.24) | 13.68 (5.26–35.60) | >100 | >100 |
| MCF7 | 23.79 (7.33–77.29) | >100 | >100 | 43.50 (17.83–106.10) | 42.61 (10.41–174.40) | >100 | >100 |
| HepG2 | >100 | >100 | >100 | 92.95 (46.33–186.50) | >100 | >100 | >100 |
Mean values and 95%-confidence intervals (parentheses) are given in μM. Data of at least 3 independent experiments.
The ferrocenyl analogues of plinabulin 2c and 2d – both with a bulky alkyl group at the imidazole moiety–were found to be equally effective against all cell lines that were tested, although their toxicity (IC50 in the ten micromolar range) differed from line to line, being the highest for SW620 and the lowest for HepG2 cells. What is interesting to note is that none of the MDR lines was less sensitive to any of these compounds. Compound 2b was moderately toxic to almost all the cell lines except for SW620V, MCF7 and HepG2, while compound 2a and both half analogues were nontoxic in the range studied. It seemed feasible that both the ferrocene and imidazole moieties (the latter was substituted by a bulky alkyl group) are required for 2,5-piperazinedione to be biologically active, thus we discontinued further analyses of 4 and 5d at this point.
To confirm the cytotoxicity results and to have some insight into the mode of action of the synthesized compounds, we performed a cell cycle analysis in cells exposed to NPI-2358 and its organometallic analogues 2a–2d (Table S1). We observed a high pre-G0/G1 (apoptotic) peak and significantly elevated G2/M peak values for plinabulin, thus indicating that cells exposed to these compounds are unable to proceed with division (due to their inability to form a mitotic spindle with impaired microtubules) and die. Exposure to compounds 2c and 2d resulted in a similar pattern of DNA distribution in the analyzed cells. Compounds 2a and 2b exhibited a slight, albeit significant, increase of the G2/M peak value, although it was unaccompanied by the presence of the apoptotic peak.
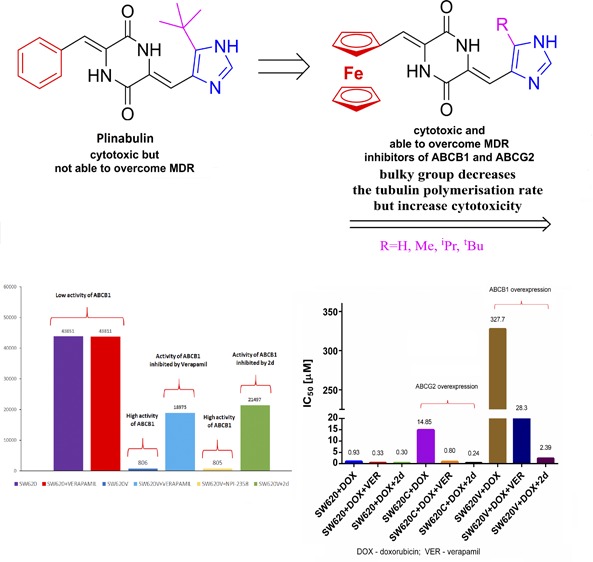
To check whether chemical modifications alter the tubulin-binding properties of ferrocenyl plinabulin analogues, we performed a tubulin polymerization inhibition study (Figure 2). In the range studied (between 1 and 100 μM), both compounds with the bulky group at the imidazole moiety (2c and 2d) tended to decrease the tubulin polymerization rate, while compounds 2a and 2b were rather moderate modulators of this process. The overall potency of plinabulin ferrocenyl analogues was at least an order of magnitude lower than that of vincristine and NPI-2358 itself. When analyzing the toxicity profile of 2d (Table 1), we found that it was significantly active in ABCB1-overexpressing SW620D, -E and -V lines, especially when taking into account the moderate or even lack of effect of plinabulin on these lines. Thus, we decided to check the potential of 2d to modulate MDR protein activity in real-time transport experiments. We compared the results obtained in SW620 cell line panel with commercially available hABCB1- and hABCG2-transfected MDCKII cells. The results are summarized in Table 2. Compound 2d clearly exhibited the properties of a low-specificity inhibitor of MDR proteins. It inhibited ABCB1 transport activity (measured by calcein accumulation assay30) to the same extent as verapamil, which is an established inhibitor of this protein.31 Its activity against ABCG2 was also notable, as accumulation of pheophorbide A increased in 2d-treated SW620C or MDCKII-BCRP cells to the same level as in the presence of Ko143 - a specific inhibitor of ABCG2.32 No effect on the ABCC1 transport properties was observed as the BCECF efflux was undisturbed by the presence of 2d. It is worth mentioning that NPI-2358 showed none of these modulatory activities (except for some effect in MDCKII-MDR1 cells), which supports previous reports.29
Figure 2.
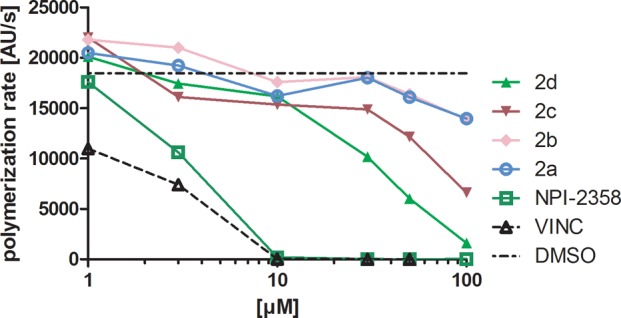
Influence of NPI-2358 and its ferrocenyl analogues 2a–2d on tubulin polymerization rate. Data of a representative experiment. VINC – vincristine used as a positive control. DMSO is shown for reference (its concentration was constant in all samples).
Table 2. Direct Effect of 2d on Multidrug Resistance Protein Transport Activitya.
| Calcein accumulation assay (ABCB1 activity) [AU/min] | |||||
|---|---|---|---|---|---|
| SW620 | SW620 + 10 μM VER | SW620 V (ABCB1-overexpressing) | SW620 V + 10 μM VER | SW620 V + 10 μM NPI-2358 | SW620 V + 10 μM 2d |
| 43,851 ± 7,175 | 43,811 ± 7,499 | 806 ± 484 | 18,975 ± 2,013 | 805 ± 467 | 21,497 ± 2,160 |
| MDCKII | MDCKII + 10 μM VER | MDCKII-MDR1 | MDCKII-MDR1 + 10 μM VER | MDCKII-MDR1 + 10 μM NPI-2358 | MDCKII-MDR1 + 10 μM 2d |
| 62,902 ± 1,959 | 62,837 ± 1,973 | 2,207 ± 198 | 32,691 ± 634 | 11,498 ± 351 | 24,328 ± 1,776 |
| PheA accumulation assay (ABCG2 activity) [AU/min] | |||||
|---|---|---|---|---|---|
| SW620 | SW620 + 30 μM KO143 | SW620C (ABCG2-overexpressing) | SW620C + 30 μM KO143 | SW620C + 10 μM NPI-2358 | SW620C + 10 μM 2d |
| 5,645 ± 856 | 6,167 ± 939 | 2,063 ± 273 | 7,586 ± 1,151 | 1,611 ± 161 | 6,080 ± 2,504 |
| ΜDCKII | MDCKII + 30 μM KO143 | MDCKII-BCRP | MDCKII-BCRP + 30 μM KO143 | MDCKII-BCRP + 10 μM NPI-2358 | MDCKII-BCRP + 10 μM 2d |
| 10,647 ± 326 | 11,675 ± 429 | 6,942 ± 273 | 12,682 ± 450 | 7,845 ± 257 | 12,216 ± 373 |
| BCECF extrusion assay (ABCC1 activity) [AU × min] | |||||
|---|---|---|---|---|---|
| SW620 | SW620 + 100 μM PRB | SW620 M (ABCC1-overexpressing) | SW620M + 100 μM PRB | SW620M + 10 μM NPI-2358 | SW620M + 10 μM 2d |
| 19,557 ± 98 | 19,517 ± 98 | 17,113 ± 121 | 19,187 ± 214 | 17,353 ± 123 | 17,210 ± 114 |
Mean values and standard deviations are presented. Averaged data of 3 independent experiments. Effects of NPI-2358 and 2d on the SW620 and MDCKII cell lines do not differ from the control; thus, they are not shown here for the sake of clarity.
We were also interested whether the inhibitory effect of 2d could also be observed in a toxicity assay. Therefore, we exposed MDR cells to increasing concentrations of doxorubicin or mitoxantrone at 100 nM of 2d (Table S2). Again, we used 10 μM verapamil and 20 μM KO143 as MDR protein-specific controls. Modulators exerted no effect on control cell lines while 2d consistently sensitized all MDR cell lines tested to doxorubicin (completely restoring the sensitivity level of parental cells in case of SW620C and MDCKII-BCRP). Effect of 2d on mitoxantrone resistance could be observed for both SW620C and MDCKII-BCRP (although here to a lesser extent) further supporting the hypothesis of the ability of this compound to modulate ABCG2 activity.
We found that a simple replacement of the phenyl group of plinabulin by a ferrocenyl moiety leads to new compounds of unexpected biological properties. Our results clearly showed that both the ferrocenyl and imidazole moieties are required to maintain the activity of the synthesized compounds. The bulkiness of the imidazole-bound alkyl moiety strongly influences the cytotoxic profile of these compounds, as the isopropyl and tert-butyl derivatives showed the highest cytotoxic/antiproliferative activities. The effect of the imidazole-bound alkyl type is important also in the aspect of tubulin polymerization inhibition. Additionally, replacement of the phenyl group of plinabulin by the ferrocenyl moiety not only significantly enhances its toxicity against multidrug-resistant cell lines but also, quite unexpectedly, turns the ferrocenyl analogues of plinabulin into potent inhibitors of ABCB1 and ABCG2. Co-administration of vincristine and the ferrocenyl analogue of plinabulin should exert a synergic effect not only in terms of microtubule-related cytotoxicity but also by overcoming the multidrug-resistance barrier formed by ABCB1 or ABCG2 proteins, thus opening up novel therapeutic possibilities. We are currently investigating such a hypothesis in in vitro systems.
Glossary
Abbreviations
- BCECF
2′,7′-bis(2-carboxyethyl)-5-(and-6)-carboxyfluorescein
- Ko143
(3S,6S,12aS)-1,2,3,4,6,7,12,12a-octahydro-9-methoxy-6-(2-methylpropyl)-1,4-dioxopyrazino[1′,2′:1,6]pyrido[3,4-b]indole-3-propanoic acid 1,1-dimethylethyl ester
- MDR
multidrug resistance
- NPI-2358
plinabulin
- PheA
pheophorbide A
- PRB
probenecid
- ROS
reactive oxygen species
- VER
verapamil
- VINC
vincristine
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00046.
Experimental procedure for synthesis, cell culture, cytotoxicity testing, cell cycle analysis, flow cytometry assays of MDR protein activity, interference with tubulin polymerization, copies of 1H and 13C NMR spectra, and Tables S1 and S2, showing the DNA distribution and the resistance reversal potency (PDF)
Author Contributions
Anna Wieczorek was responsible for the synthesis of the investigated compounds and tubulin polymerization assay and participated in cytotoxicity, cell cycle, and transport assays. Andrzej Błauż was responsible for the supervision of cytotoxicity, cell cycle, and transport assays; he performed the resistance reversal assay. Contributions of both authors were equal and indispensable for this work; Błażej Rychlik was responsible for assay choice and design, analysis of the biological data, and overall supervision of the biological portion of this project; Janusz Zakrzewski and Damian Plażuk designed the project. All authors wrote their respective parts of the manuscript, which was finally compiled by Anna Wieczorek, Błażej Rychlik, and Damian Plażuk. All authors have given approval to the final version of the manuscript.
This work was supported by the National Science Centre Poland (contract No. UMO-2011/01/B/ST5/03933). Analysis of the multidrug-resistance protein activity of synthesized compounds was financed by the Polish POIG grant 01.01.02-10–005/08 TESTOPLEK, supported by the EU through the European Regional Development Fund.
The authors declare no competing financial interest.
Supplementary Material
References
- Seligmann J.; Twelves C. Tubulin: An example of targeted chemotherapy. Future Med. Chem. 2013, 5, 339. 10.4155/fmc.12.217. [DOI] [PubMed] [Google Scholar]
- Schiff P. B.; Fant J.; Horwitz S. B. Promotion of microtubule assembly in vitro by taxol. Nature 1979, 277, 665. 10.1038/277665a0. [DOI] [PubMed] [Google Scholar]
- Gueritte-Voegelein F.; Guenard D.; Lavelle F.; Le Goff M.-T.; Mangatal L.; Potier P. J. Med. Chem. 1991, 34, 992. 10.1021/jm00107a017. [DOI] [PubMed] [Google Scholar]
- Passarella D.; Giardini A.; Peretto B.; Fontana G.; Sacchetti A.; Silvani A.; Ronchi C.; Cappelletti G.; Cartelli D.; Borlak J.; Danieli B. Inhibitors of tubulin polymerization: Synthesis and biological evaluation of hybrids of vindoline, anhydrovinblastine and vinorelbine with thiocolchicine, podophyllotoxin and baccatin III. Bioorg. Med. Chem. 2008, 16, 6269. 10.1016/j.bmc.2008.04.025. [DOI] [PubMed] [Google Scholar]
- Jordan M. A. Mechanism of action of antitumor drugs that interact with microtubules and tubulin. Curr. Med. Chem. Anti-cancer agents 2002, 2, 1. [DOI] [PubMed] [Google Scholar]
- Ojima I. Guided Molecular Missiles for Tumor-Targeting Chemotherapy—Case Studies Using the Second-Generation Taxoids as Warheads. Acc. Chem. Res. 2008, 41, 108. 10.1021/ar700093f. [DOI] [PubMed] [Google Scholar]
- Mita M. M.; Spear M. A.; Yee L. K.; Mita A. C.; Heath E. I.; Papadopoulos K. P.; Federico K. C.; Reich S. D.; Romero O.; Malburg L.; Pilat M.; Lloyd G. K.; Neuteboom S. T. C.; Cropp G.; Ashton E.; LoRusso P. M. Phase 1 first-in-human trial of the vascular disrupting agent plinabulin (NPI-2358) in patients with solid tumors or lymphomas. Clin. Cancer Res. 2010, 16, 5892. 10.1158/1078-0432.CCR-10-1096. [DOI] [PubMed] [Google Scholar]
- Kanoh K.; Kohno S.; Katada J.; Takahashi J.; Uno I. (−)-phenylahistin arrests cells in mitosis by inhibiting tubulin polymerization. J. Antibiot. 1999, 52, 134. 10.7164/antibiotics.52.134. [DOI] [PubMed] [Google Scholar]
- Kanoh K.; Kohno S.; Asari T.; Harada T.; Katada J.; Muramatsu M.; Kawashima H.; Sekiya H.; Uno I. -)-phenylahistin: A new mammalian cell cycle inhibitor produced by Aspergillus ustus. Bioorg. Med. Chem. Lett. 1997, 7, 2847. 10.1016/S0960-894X(97)10104-4. [DOI] [Google Scholar]
- Singh A. V.; Bandi M.; Raje N.; Richardson P.; Palladino M. A.; Chauhan D.; Anderson K. C. A novel vascular disrupting agent plinabulin triggers JNK-mediated apoptosis and inhibits angiogenesis in multiple myeloma cells. Blood 2011, 117, 5692. 10.1182/blood-2010-12-323857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer A. M. S.; Glaser K. B.; Cuevas C.; Jacobs R. S.; Kem W.; Little R. D.; McIntosh J. M.; Newman D. J.; Potts B. C.; Shuster D. E. The odyssey of marine pharmaceuticals: a current pipeline perspective. Trends Pharmacol. Sci. 2010, 31, 255. 10.1016/j.tips.2010.02.005. [DOI] [PubMed] [Google Scholar]
- Kunjachan S.; Rychlik B.; Storm G.; Kiessling F.; Lammers T. Multidrug resistance: Physiological principles and nanomedical solutions. Adv. Drug Delivery Rev. 2013, 65, 1852. 10.1016/j.addr.2013.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persidis A. Cancer multidrug resistance. Nat. Biotechnol. 1999, 17, 94. 10.1038/5289. [DOI] [PubMed] [Google Scholar]
- Hartinger C. G.; Dyson P. J. Bioorganometallic chemistry - From teaching paradigms to medicinal applications. Chem. Soc. Rev. 2009, 38, 391. 10.1039/B707077M. [DOI] [PubMed] [Google Scholar]
- Babak M. V.; Plażuk D.; Meier S. M.; Arabshahi H. J.; Reynisson J.; Rychlik B.; Błauż A.; Szulc K.; Hanif M.; Strobl S.; Roller A.; Keppler B. K.; Hartinger C. G. Half-Sandwich Ruthenium(II) Biotin Conjugates as Biological Vectors to Cancer Cells. Chem. - Eur. J. 2015, 21, 5110. 10.1002/chem.201403974. [DOI] [PubMed] [Google Scholar]
- Hartinger C. G.; Metzler-Nolte N.; Dyson P. J. Challenges and Opportunities in the Development of Organometallic Anticancer Drugs. Organometallics 2012, 31, 5677. 10.1021/om300373t. [DOI] [Google Scholar]
- Top S.; Tang J.; Vessieres A.; Carrez D.; Provot C.; Jaouen G. Ferrocenyl hydroxytamoxifen: a prototype for a new range of oestradiol receptor site-directed cytotoxics. Chem. Commun. 1996, 955. 10.1039/cc9960000955. [DOI] [Google Scholar]
- Ornelas C. Application of ferrocene and its derivatives in cancer research. New J. Chem. 2011, 35, 1973. 10.1039/c1nj20172g. [DOI] [Google Scholar]
- Liu Z.; Habtemariam A.; Pizarro A. M.; Fletcher S. A.; Kisova A.; Vrana O.; Salassa L.; Bruijnincx P. C. A.; Clarkson G. J.; Brabec V.; Sadler P. J. Organometallic half-sandwich iridium anticancer complexes. J. Med. Chem. 2011, 54, 3011. 10.1021/jm2000932. [DOI] [PubMed] [Google Scholar]
- Gasser G.; Ott I.; Metzler-Nolte N. Organometallic anticancer compounds. J. Med. Chem. 2011, 54, 3. 10.1021/jm100020w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plażuk D.; Vessieres A.; Hillard E. A.; Buriez O.; Labbe E.; Pigeon P.; Plamont M. A.; Amatore C.; Zakrzewski J.; Jaouen G. A [3]ferrocenophane polyphenol showing a remarkable antiproliferative activity on breast and prostate cancer cell lines. J. Med. Chem. 2009, 52, 4964. 10.1021/jm900297x. [DOI] [PubMed] [Google Scholar]
- Hillard E.; Vessières A.; Le Bideau F.; Plażuk D.; Spera D.; Huché M.; Jaouen G. A series of unconjugated ferrocenyl phenols: Prospects as anticancer agents. ChemMedChem 2006, 1, 551. 10.1002/cmdc.200500035. [DOI] [PubMed] [Google Scholar]
- Braga S. S.; Silva A. M. S. A new age for iron: Antitumoral ferrocenes. Organometallics 2013, 32, 5626. 10.1021/om400446y. [DOI] [Google Scholar]
- Vessières A. Metal carbonyl tracers and the ferrocifen family: Two facets of bioorganometallic chemistry. J. Organomet. Chem. 2013, 734, 3. 10.1016/j.jorganchem.2012.12.020. [DOI] [Google Scholar]
- Kater B.; Hunold A.; Schmalz H.-G.; Kater L.; Bonitzki B.; Jesse P.; Prokop A. Iron containing anti-tumoral agents: unexpected apoptosis-inducing activity of a ferrocene amino acid derivative. J. Cancer Res. Clin. Oncol. 2011, 137, 639. 10.1007/s00432-010-0924-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubar F.; Khalife J.; Brocard J.; Dive D.; Biot C. Ferroquine, an Ingenious Antimalarial Drug – Thoughts on the Mechanism of Action. Molecules 2008, 13, 2900. 10.3390/molecules13112900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vock C. A.; Ang W. H.; Scolaro C.; Phillips A. D.; Lagopoulos L.; Juillerat-Jeanneret L.; Sava G.; Scopelliti R.; Dyson P. J. Development of ruthenium antitumor drugs that overcome multidrug resistance mechanisms. J. Med. Chem. 2007, 50, 2166. 10.1021/jm070039f. [DOI] [PubMed] [Google Scholar]
- Wieczorek A.; Plażuk D.; Zakrzewski J.; Makal A.; Woźniak K. Synthesis and unusual ring transformation of 1-acyl-3-(ferrocenylmethylidene)-piperazine-2,5-diones. J. Organomet. Chem. 2013, 745–746, 373. 10.1016/j.jorganchem.2013.08.019. [DOI] [Google Scholar]
- Nicholson B.; Lloyd G. K.; Miller B. R.; Palladino M. A.; Kiso Y.; Hayashi Y.; Neuteboom S. T. C. NPI-2358 is a tubulin-depolymerizing agent: In-vitro evidence for activity as a tumor vascular-disrupting agent. Anti-Cancer Drugs 2006, 17, 25. 10.1097/01.cad.0000182745.01612.8a. [DOI] [PubMed] [Google Scholar]
- Holló Z.; Homolya L.; Davis C. W.; Sarkadi B. Calcein accumulation as a fluorometric functional assay of the multidrug transporter. Biochim. Biophys. Acta, Biomembr. 1994, 1191, 384. 10.1016/0005-2736(94)90190-2. [DOI] [PubMed] [Google Scholar]
- Cornwell M. M.; Pastan I.; Gottesman M. M. Certain calcium channel blockers bind specifically to multidrug-resistant human KB carcinoma membrane vesicles and inhibit drug binding to P-glycoprotein. J. Biol. Chem. 1987, 262, 2166. [PubMed] [Google Scholar]
- Allen J. D.; van Loevezijn A.; Lakhai J. M.; van der Valk M.; van Tellingen O.; Reid G.; Schellens J. H.; Koomen G. J.; Schinkel A. H. Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol. Cancer. Ther 2002, 1, 417. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.