

Abstract
Activation of the mammalian target of rapamycin complex 1 (mTORC1) subunit Raptor induces cell growth and is a downstream target of Akt. Elevated levels of aldosterone activate Akt, and, in pulmonary arterial hypertension (PAH), correlate with pulmonary arteriole thickening, which suggests that mTORC1 regulation by aldosterone may mediate adverse pulmonary vascular remodeling. We hypothesized that aldosterone-Raptor signaling induces abnormal pulmonary artery smooth muscle cell (PASMC) survival patterns to promote PAH. Remodeled pulmonary arterioles from SU-5416/hypoxia-PAH rats and monocrotaline-PAH rats with hyperaldosteronism expressed increased levels of the Raptor target, p70S6K, which provided a basis for investigating aldosterone-Raptor signaling in human PASMCs. Aldosterone (10−9 to 10−7 M) increased Akt/mTOR/Raptor to activate p70S6K and increase proliferation, viability, and apoptosis resistance in PASMCs. In PASMCs transfected with Raptor–small interfering RNA or treated with spironolactone/eplerenone, aldosterone or pulmonary arterial plasma from patients with PAH failed to increase p70S6K activation or to induce cell survival in vitro. Optimal inhibition of pulmonary arteriole Raptor was achieved by treatment with Staramine-monomethoxy polyethylene glycol that was formulated with Raptor-small interfering RNA plus spironolactone in vivo, which decreased arteriole muscularization and pulmonary hypertension in 2 experimental animal models of PAH in vivo. Up-regulation of mTORC1 by aldosterone is a critical pathobiologic mechanism that controls PASMC survival to promote hypertrophic vascular remodeling and PAH.—Aghamohammadzadeh, R., Zhang, Y.-Y., Stephens, T. E., Arons, E., Zaman, P., Polach, K. J., Matar, M., Yung, L.-M., Yu, P. B., Bowman, F. P., Opotowsky, A. R., Waxman, A. B., Loscalzo, J., Leopold, J. A., Maron, B. A. Up-regulation of the mammalian target of rapamycin complex 1 subunit Raptor by aldosterone induces abnormal pulmonary artery smooth muscle cell survival patterns to promote pulmonary arterial hypertension.
Keywords: vascular remodeling, pulmonary vascular disease, kinase signaling
Pulmonary arterial hypertension (PAH) is characterized by concentric hypertrophy and intimal thickening of distal pulmonary arterioles that, in part, results from abnormal pulmonary artery smooth muscle cell (PASMC) survival and apoptosis resistance (1, 2). Inhibition of established proproliferative signaling pathways in PASMCs temporarily improves pulmonary hypertension in patients (3), but does not fully abrogate the angioproliferative pathophenotype of PAH or provide sustained benefits to cardiopulmonary hemodynamics (4). In contrast, converging lines of evidence indicate progressive vascular remodeling in PAH despite contemporary therapies (5), which suggests that unidentified mechanisms may exist that regulate PASMC growth to increase vascular resistance and promote PAH.
Mammalian target of rapamycin (mTOR) is a protein kinase that is composed of 2 functional multiprotein complexes, rapamycin-sensitive mTORC1, which promotes apoptosis-resistant cellular proliferation, and mTORC2, which is insensitive to acute rapamycin in PASMCs and controls cytoskeletal organization (6). In PASMCs that were harvested from patients with idiopathic PAH, mTOR levels were increased, and mTORC2 has been shown to mediate glycolysis-dependent cellular proliferation and survival (7). It is noteworthy that mTORC1-induced activation of kinases that target S6 ribosomal proteins, in particular p70S6K, is stimulated by a diverse range of intermediaries that are relevant to PAH, including mTORC2, hypoxia, and Akt (8, 9); however, the factors that regulate mTORC1 and the potential importance of mTORC1-dependent activation of PASMC proliferation to PAH pathobiology remain incompletely characterized.
Elevated circulating levels of the mineralocorticoid hormone, aldosterone, are observed in PAH, are clinically correlated with hemodynamic indices of pulmonary vascular remodeling, and contribute to pulmonary hypertension pathobiology in PAH in vivo (10–13). There is accumulating evidence to suggest that Akt-dependent mTOR activation by aldosterone contributes to the pathogenesis of human diseases that are characterized by cell proliferation, including adrenal adenoma (14) and tubulointerstitial nephritis (15). Although Akt activation by aldosterone is also reported in vascular cells and is linked to the development of systemic hypertension (16), the relevance of mTOR to aldosterone-induced pulmonary vascular remodeling is not known. Therefore, we hypothesized that aldosterone-induced mTORC1 up-regulation via Akt signal transduction is an unrecognized mechanism by which to account for abnormal PASMC survival and the hypertrophic vasculopathy of PAH.
MATERIALS AND METHODS
Cell culture and treatments
Human male PASMCs (male donors; Lonza, Basel, Switzerland) were grown to confluence by using SmGM-2 medium that was supplemented with 5% fetal bovine serum (FBS) and a Bulletkit (Lonza) containing the following growth supplements: human epidermal growth factor, insulin, human fibroblastic growth factor-B, and gentamicin. All cells were incubated at 37°C, 5.0% CO2, and passaged 2 times per wk by using 0.5% trypsin/EDTA; experiments were performed on cell passages 4–10. In selected experiments, culture medium was supplemented with 0.5–1.0% FBS for at least 12 h to assess the effect of serum starvation on mTOR signaling. Aldosterone (Steraloids, Newport, RI, USA) was dissolved in DMSO (10−6 M), which served as a vehicle control. Cells were treated with aldosterone (10−9–10−7 M) for 0, 15, 30, 60, or 120 min. To inhibit the mineralocorticoid receptor (MR), cells were coincubated with spironolactone (10 µM; Sigma-Aldrich, St. Louis, MO, USA) or pretreated for 15 min with eplerenone (10 µM; Sigma-Aldrich) unless specified otherwise.
Immunoblotting
Proteins were size-fractionated electrophoretically by using SDS-PAGE and were transferred to PVDF membranes according to methods reported previously (11). Membranes were incubated with anti-p70S6K and anti-P-p70S6K(Thr389) (Cell Signaling Technology, Danvers, MA, USA); anti-mTOR, anti-P-mTOR(Ser2448) (Cell Signaling Technology), anti-Raptor, and anti-P-Raptor (Ser792) (Abcam, Cambridge, MA, USA); anti-Akt and anti-P-Akt(Ser473) (Cell Signaling Technology); and anti-Rictor and anti-Rictor(Thr1105) (Cell Signaling Technology) antibodies overnight at 4°C and visualized by using an ECL detection system (GE Healthcare Life Sciences, Pittsburgh, PA, USA).
Immunohistochemistry in vitro
PASMCs were grown to confluence on glass chamber slides (4 chambers/slide) and fixed after treatment with vehicle control, aldosterone (10−7 M), or standard culture medium that contained PAH patient plasma for a total concentration of 10% for 1 h. Anti-Raptor and anti-P-Raptor(Ser792) immunohistochemistry (Abcam) was subsequently performed by using the 3,3′-diaminobenzidine substrate method (Vector Laboratories, Burlingame, CA, USA) as described previously (11).
Cell proliferation assay
Confluent PASMCs were exposed to culture medium that was supplemented with 1% or 5% FBS and were pulsed with bromodeoxyuridine (BrdU) for 12 h. Cell culture medium was then aspirated by negative pressure suction and replaced with medium containing 1% or 5% FBS and vehicle control or aldosterone (10−7 M) for 2 h. Cell proliferation was determined by using the BrdU incorporation assay according to manufacturer instructions (Roche, Basel, Switzerland), as published previously (17).
Cell viability assay
Human PASMCs were incubated by using standard culture medium in a 96-well plate at a concentration of ∼ 5000 cells/well for 24 h. After incubation, methyl thiazolyl tetrazolium (5 mg/ml; Sigma-Aldrich) was added to each well for 4 h, and cells were treated for 2 h with vehicle control or aldosterone (10−7 M) in the presence or absence of eplerenone (10 μM). Solubilized formazan products were quantified by spectrophotometry at 490 nm by using a 96-well absorbance plate reader (Bio-Rad, Hercules, CA, USA) (18).
Cell apoptosis assay
Human PASMCs were plated in a 96-well plate by using standard culture medium and treated with varying concentrations of H2O2 (0–1 mM) for 24 h to induce apoptosis (19). Culture medium was replaced with medium that contained vehicle control or aldosterone (10−7 M) for 2 h, and apoptosis was quantified by using an ELISA to measure cytoplasmic histone-associated DNA fragments (Roche), according to manufacturer instructions.
Immunofluorescence
Cells were grown to confluence on chamber slides and fixed with ice-cold acetone for 20 min. Cells were then blocked in 10% goat albumin (Life Technologies) in PBS for 1 h at room temperature. Cells were stained for p70S6K or P-p70S6K(Thr389) by using rabbit anti-rat p70S6K and rabbit anti-rat P-p70S6K antibodies (Thermo Fisher Scientific Life Sciences, Waltham, MA, USA). The secondary antibody was goat anti-rabbit (Abcam) that was conjugated with Alexa Fluor 488. Samples were mounted on glass slides with antifade mounting medium with DAPI (Vector Laboratories) and were imaged by using a Nikon TE300 fluorescence microscope.
Raptor small interfering RNA transfection in vitro and in vivo
Cells were transfected with Raptor small interfering RNA (siRNA; 40 nM) or siRNA scrambled (negative) control (si-Scc; Santa Cruz Biotechnology, Dallas, TX, USA) by using Lipofectamine 2000 (Thermo Fisher Scientific Life Sciences) for 5 h in OptiMEM I medium, which also served as vehicle control. Cells were then placed in full-growth medium for 24 h before treatment with aldosterone for 24 h. The Raptor siRNA (Santa Cruz Biotechnology) used for transfection consisted of a pool of the following sequences: Pool A: sense: 5′-CCAUGUAAUCAGAGCAUUAtt-3′; antisense: 5′-UAAUGCUCUGAUUACAUGGtt-3′; Pool B: sense: 5′- CAACCACAAUGAUGGUAUUtt-3′; antisense: 5′-AAUACCAUCAUUGUGGUUGtt-3′; and Pool C: sense: 5′-CCUAGAGGCUCAUUAUCUAtt-3′; antisense: 5′-UAGAUAAUGAGCCUCUAGGtt-3′. For experiments that analyzed the effect of Raptor inhibition in vivo, the Raptor siRNA (si-Raptor) sequence was sense: 5′-GAAUCAUGAGGUCGUAUAA-3′ (Dharmacon, Lafayette, CO, USA); si-Scc was from Dharmacon.
Generation of siRNA:Staramine-monomethoxy polyethylene glycol nanocomplexes
Methods for the preparation of siRNA:Staramine-monomethoxy polyethylene glycol (mPEG) nanocomplexes have been published previously (20, 21). In brief, Staramine synthesis and purification was verified by HPLC, mass spectrometry, and NMR. Chloroform solutions of 10:1 mixtures of Staramine and Star-mPEG11 were rotary evaporated to a film. The flask of liposome film was held under high vacuum for ≥ 12 h and a water injection was added to the film to achieve the goal Staramine-mPEG concentration. After sonication for 30 min, liposome solution was filtered, diluted with 5% dextrose, which generated an isotonic solution, and mixed with the desired amount of siRNA at 0.1 mg/ml. The formulation was concentrated further to 0.8 mg/ml by using Amicon ultra centrifugal filter units. Particle size and ζ potential of the complexes were measured by using the 90Plus/BI-MAS Particle sizer with BI-Zeta option (Brookhaven Instruments, Holtsville, NY, USA). The si-Raptor formulation had a mean particle size of 96.8 nm and a ζ potential of +30.61 mV, and the si-Scc formulation had a mean particle size of 96.8 nm and a ζ potential of 27.52 mV.
Animal models of PAH
Male Sprague-Dawley rats (age 12–14 wk; Charles River Laboratories, Wilmington, MA, USA) were handled in accordance with US National Institutes of Health guidelines, and all procedures were approved by the local committee at Brigham and Women’s Hospital, Harvard Medical School. Rats were fed standard chow unless otherwise indicated. A summary of each animal model with respect to time course, treatment protocol, and therapeutic intervention is provided in Fig. 1. For experiments in which spironolactone was added to the drinking water, drug dilution was calculated on the basis of publically available reports on the expected daily water consumption rate of male Sprague-Dawley rats. Water consumption was monitored during pilot studies, and it was confirmed that the experimental PAH models that were under investigation did not substantially influence the rate of water consumption in the rats. A similar approach was employed for experiments in which eplerenone was integrated into rat chow.
Figure 1.

Experimental models of PAH and their respective treatment protocols. A, B) To determine the effect of MR inhibition on Raptor and Raptor-associated signaling intermediates in remodeled pulmonary arterioles, male Sprague-Dawley (SD) rats were administered 0.9% normal saline (NS) as vehicle control (V) or MCT (50 mg/kg) by intaperitoneal (IP) injection and treated with spironolactone (25 mg/kg/d) in the drinking water (A), or SU-5416 (20 mg/kg) by subcutaneous (SQ) injection followed by chronic hypoxia (10% fraction of inspired oxygen) for 21 d and treated with eplerenone (EPL; 0.6 mg/g chow) (B). C) In a disease prevention protocol, male SD rats were administered V or MCT (50 mg/kg) and initiated treatment with Staramine-mPEG (2 mg/kg) formulated with si-Scc, Staramine-mPEG formulated with si-Raptor, or si-Raptor plus spironolactone (SPIRO; 25 mg/kg/d) that continued until completion of the protocol. D) In a disease treatment protocol, male SD rats were administered V or SU-5416 (20 mg/kg) and exposed to hypoxia (10% fraction of inspired oxygen) for 21 d followed by exposure to normoxia for 21 d. In this model, si-Raptor or si-Raptor plus SPIRO (25 mg/kg/d) was initiated on day 21 and continued until the completion of the protocol. IV, intravenous; Un, untreated.
SU-5416/hypoxia PAH
We used an angioproliferative model of PAH to assess the contribution of aldosterone to pulmonary arterial expression of the Raptor target p70S6K in PAH in vivo. Rats (∼ 225 g) were administered a single s.c. injection of VEGF-2 inhibitor, SU-5416 (20 mg/kg; Sigma-Aldrich), and were exposed immediately to chronic hypoxia (10% fraction of inspired oxygen) (22, 23). Rats were randomly assigned to treatment with the selective MR antagonist eplerenone (0.6 mg/1 g standard chow; Test Diet, St. Louis, MO, USA) or standard chow as a control, as previously published by our laboratory (11, 23). Cardiac catheterization and tissue analyses were performed on all rats 21 d after the administration of SU-5416.
Monocrotaline PAH.
Two series of monocrotaline (MCT)-PAH experiments were used to address the effect of aldosterone-Raptor signaling on pulmonary arterial remodeling and pulmonary hypertension in vivo. In the first protocol, we assessed the contribution of aldosterone to pulmonary arterial p70S6K expression. To accomplish this, male rats were administered by i.p. injection 0.5 ml MCT (50 mg/ml; Sigma-Aldrich) or vehicle control (0.9% normal saline) and were immediately randomly assigned to treatment with vehicle or spironolactone (Henry Schein, Melville, NY, USA) in the drinking water (25 mg/kg/d) that was continued until tissue analysis was performed 23–25 d later, as published previously by our laboratory (11, 23).
In a second MCT-PAH experiment, we used a disease prevention protocol to determine if combination Raptor plus MR inhibition is superior to Raptor inhibition alone for the prevention of vascular remodeling and pulmonary hypertension in PAH in vivo. In this protocol, male rats that were administered 0.5 ml MCT (50 mg/ml; Sigma-Aldrich) by i.p. injection were immediately randomly assigned to the following treatment conditions: Staramine-mPEG–formulated si-Scc, Staramine-mPEG–formulated si-Raptor, or Staramine-mPEG–formulated si-Raptor plus spironolactone (25 mg/kg/d) (11). The Staramine-mPEG nanocomplexes were administered by tail-vein injection (2.0 mg/kg) on protocol days 1, 6, 10, and 16. Cardiac catheterization and tissue harvest experiments were performed 21–23 d after treatment with MCT.
SU-5416/hypoxia-normoxia
Owing to prior reports indicating that severe arteriole thickening is achieved in SU-5416/hypoxia-treated rats returned to normoxia (22), an additional animal model of PAH was selected to study the effects of MR and/or Raptor inhibition on pulmonary hypertension. Male Sprague-Dawley rats (∼ 225 g) were administered a single s.c. injection of SU-5416 (20 mg/kg; Sigma-Aldrich), exposed immediately to chronic hypoxia (10% fraction of inspired oxygen) for 21 d, and then returned to normoxia until completion of the protocol 21 d later (SU-5416/hypoxia-normoxia). In a disease treatment protocol, si-Raptor or si-Raptor plus spironolactone (25 mg/kg/d) was initiated at day 21 of the protocol, which is a time point after pulmonary vascular injury and pulmonary hypertension in this model (11).
Hemodynamic analyses
All experiments were performed with rats sedated by using ketamine (100 mg/kg) and xylazine (10 mg/kg) according to methods previously published by our laboratory (11, 23).
Right heart catheterization
The anterior triangle of the right neck was dissected to expose the right internal jugular vein. A 0.04 × 0.023 inch–sized polyvinylchloridine catheter with a J-end was flushed with 10% heparinized saline and connected to a Grass pressure transducer and Grass model 79 polygraph. A surgical knot that used a 4.0-proline suture was placed at the distal end of the jugular vein to achieve hemostasis before the insertion of the catheter. The catheter was advanced to the right atrium, and the systolic pressure was recorded. The catheter was then advanced further, and right ventricle (RV) systolic pressure was recorded, which was assumed to be equal to the pulmonary artery systolic pressure (PASP) in the setting of a normal pulmonic valve (11, 23). All right heart catheterizations were completed within 30 min of sedation induction.
Left heart catheterization and hemodynamics
Immediately after the completion of the right heart catheterization, dissection medial to the jugular vein was performed to expose the right carotid artery. Without disrupting the carotid sinus or vagus nerve, the distal and proximal aspects of the carotid artery were cross-clamped, and a 4.0-proline suture was positioned between the clamps to achieve hemostasis after the insertion of the catheter and subsequent release of the distal clamp. A 2 F high fidelity Millar catheter (Model SPR-869; Millar Instruments, Houston, TX, USA) was inserted into the carotid artery. The distal clamp was released and the suture was fastened sufficiently to prevent significant blood loss. The catheter was advanced, and central aortic blood pressure was recorded. The catheter was then advanced further across the aortic valve, and left ventricular (LV) end-diastolic pressure was recorded. Cardiac index (CI) was derived from pressure-volume loop analysis as described previously (11, 23). The vascular resistance index was calculated by using the standard equation: indexed pulmonary vascular resistance was calculated as [(mean pulmonary artery pressure − LV end-diastolic pressure)/CI)], and systemic vascular resistance index was calculated as [(mean arterial pressure − mean right atrial pressure)/CI] (11, 23).
Right ventricular weight
The superior vena cava, inferior vena cava, and ascending aorta were ligated and the heart was separated. The heart was removed from the rat, and the RV was dissected. All residual intracavitary blood volume was exsanguinated before weighing the RV and the LV plus septum. Data are expressed as the ratio of RV weight (g)/LV + septum weight (g) (11, 23).
Tissue histology
Lung tissue
A 1-cm incision was made in the rat RV free wall to allow for extravasation of retrograde perfusate and to avoid RV injury during the lung perfusion process. Lung vessels were perfused with saline via the trachea and were inflated with 10% phosphate-buffered formalin at a pressure of 20 cmH2O through the trachea as described previously (11, 23). After fixation with formalin for ≥ 24 h at room temperature, lung tissue was processed and embedded in paraffin by using a Hypercenter XP System and Embedding Center (Shandon, Pittsburgh, PA, USA) (11, 23). The paraffin-embedded lung tissue was cut into 5-µm sections. Hematoxylin and eosin staining was performed according to previously published methods (11, 23). The 3,3′-diaminobenzidine substrate method was used for P-p70S6K(Thr389), p70S6K, Rictor, Raptor, α-smooth muscle actin, and IgG (negative control) immunohistochemical staining.
The number of muscularized arteries with a diameter of 20–50 µm that were located distal to terminal bronchioles were counted in 10 consecutive fields (×200) per section (11). The cross-sectional area of the lumen and blood vessel wall was assessed in α-actin–stained samples (n = 8–10 vessels/rat) by using ImageJ software (NIH, Bethesda, MD, USA), and changes in hypertrophic remodeling of arterioles was calculated by analyzing the difference between the area of the larger and small curvatures divided by the area of the lesser curvature × 100 (11). Immunohistochemistry was performed by using the 3,3′-diaminobenzidine substrate method (Vector Laboratories), and luminosity was calculated in ≥ 5 vessels/rat. For anti-Raptor and anti-α1-smooth muscle actin double immunohistochemistry experiments, 2 consecutive 5-μm lung sections were selected for analysis, and pulmonary arterioles (20–50 µm in diameter) that appeared in both sections were analyzed to determine colocalization of the target proteins.
RV
The LV, interventricular septum, and RV from control and SU-5416/hypoxia-normoxia-PAH rats were visualized under high magnification and separated carefully at the insertion point of the RV. The mid-to-superior aspects of the RV free wall were dissected and fixed with formalin for at least 24 h. The paraffin-embedded RV tissue was then cut longitudinally into 5-µm sections, and anti-Raptor immunohistochemistry was performed by using the 3,3′-diaminobenzidine substrate method.
Kidney tissue
After the completion of lung perfusion, dissection, and fixation, the whole left kidney was resected and formalin fixed. Hematoxylin and eosin and anti-Raptor immunohistochemistry was performed on renal arterioles that measured 20–80 μm in diameter (n = 5 vessels/rat).
TUNEL assay
Paraffin-embedded lung sections that measured 5 μm were prepared from control rats and MCT-PAH rats that were untreated or treated with si-Raptor or si-Raptor plus spironolactone. The sections were deparaffinized in xylenes and rehydrated in 100% and 95% ethanol. TUNEL assay was performed by using the TUNEL apoptosis detection kit (EMD Millipore, Billerica, MA, USA) according to manufacturer instructions, and images were acquired by using a Nikon TE300 fluorescence microscope. Image exposure and brightness were adjusted by confirming similar background luminosity and setting the total RGB channel to the base of the signal distribution, respectively, in all images (Adobe Photoshop CS3; Adobe Systems, San Jose, CA, USA) as reported previously (24). The effect of MCT, si-Raptor, and si-Raptor plus spironolactone on pulmonary arteriole apoptosis was determined by calculating the % TUNEL-positive nuclei for ∼ 20 vessels per treatment condition and controls (25).
Patient aldosterone level measurement by ELISA
The study was approved by the Partners Human Research Committee and complies with the Declaration of Helsinki. For all patients, the following hemodynamic criteria (4) were used to diagnose World Health Organization Group 1 pulmonary hypertension (PAH): mean pulmonary artery pressure ≥ 25 mmHg, pulmonary vascular resistance ≥ 3.0 Wood units, and pulmonary artery wedge pressure < 15 mmHg. The accuracy of the diagnosis was confirmed by 2 pulmonary vascular disease clinical experts (A.R.O. and A.B.W.). Plasma from patients was acquired from the pulmonary artery or pulmonary artery wedge position at the time of supine right heart catheterization, as published previously (10). Patients who were referred for right heart catheterization to assess unexplained dyspnea (n = 3) and who did not achieve these hemodynamic criteria and did not have a history of PAH that was confirmed through review of the medical record were considered controls. Whole blood samples were centrifuged immediately after acquisition at 1200 rpm for 10 min at 4°C, and the plasma was stored at −80°C (10). Aldosterone levels were analyzed by ELISA (detection limit 21 pg/ml; mean intra-assay variation, 10.7%; interassay variation, 15.4%; Cayman Chemical, Ann Arbor, MI, USA) according to manufacturer instructions.
Statistical analysis
Data are expressed as mean ± sem. Comparison between 2 groups was performed by the Student unpaired 2-tailed t test. One-way ANOVA was used to examine differences in the response to treatment between groups, with post hoc analysis performed by the Tukey method. P < 0.05 was considered significant.
RESULTS
Aldosterone increases activated p70S6K in 2 experimental PAH models in vivo
By using 2 experimental animal models of PAH, we first analyzed the relationship between aldosterone and pulmonary arteriole expression of p70S6K, which is the principal mTORC1 target kinase that mediates cell growth and proliferation. We measured by immunohistochemistry the levels of P-p70S6K(Thr389), which is a phosphorylated form of p70S6K that is associated with kinase activation in vivo (26), and total p70S6K in pulmonary arterioles (located distal to terminal bronchioles with diameters of 20–50 μm) that were harvested from male rats after treatment with SU-5416 (20 mg/kg) and exposure to hypoxia for 21 d (SU-5416/hypoxia). Compared with controls, SU-5416/hypoxia rats with confirmed PAH and increased aldosterone levels (11, 23) demonstrated increased p70S6K [104.0 ± 2.6 vs. 138.7 ± 3.3 arbitrary units (a.u.); P < 0.001; n = 4–6 rats/condition] and P-p70S6K(Thr389) (6.2 ± 1.4 vs. 26.4 ± 1.8 a.u.; P < 0.001; n = 4–6 rats/condition). Similar findings were observed in a second animal model of PAH: compared with controls, rats with MCT-PAH and confirmed hyperaldosteronism (11, 23) demonstrated a significant increase in pulmonary arteriole p70S6K (86 ± 14 vs. 144 ± 6.5 a.u.; P = 0.02; n = 3 rats/condition) and P-p70S6K(Thr389) (114 ± 9.4 vs. 188 ± 14.4 a.u.; P = 0.01; n = 3 rats/condition).
We next assessed the effect of pharmacological MR inhibition with spironolactone (25 mg/kg/d) or eplerenone (0.6 mg/g chow) treatment initiated at the time of MCT or SU-5416/hypoxia, respectively, on p70S6K expression. In remodeled pulmonary arterioles, eplerenone decreased levels of p70S6K and P-p70S6K(Thr389) in SU-5416/hypoxia-PAH rats by 47% (P < 0.01) and 37% (P < 0.01), respectively (Fig. 2A). Directionally similar findings were also observed for MCT-PAH rats that were treated with spironolactone in which pulmonary arteriole p70S6K and P-p70S6K were decreased by 16% (P = 0.15) and 48% (P < 0.02), respectively, compared with MCT-PAH rats (Fig. 2B).
Figure 2.

The effect of MR receptor antagonism on p70S6K expression in 2 models of experimental PAH. A) Male Sprague-Dawley (SD) rats were injected with SU-5416 and exposed to chronic hypoxia for 21 d. Immediately after exposure to hypoxia, rats were randomly assigned to receive standard chow or eplerenone (EPL; 0.6 gm/1 gm chow) until completion of the study (n = 4–6 rats/condition). B) In a second model, SD rats were randomly assigned to receive vehicle control (V) or spironolactone (SPIRO; 25 mg/kg/d) immediately after the i.p. administration of V or MCT (50 mg/kg) (n = 3 rats/condition). Immunohistochemistry was performed on distal pulmonary arterioles measuring 20–50 μm in diameter to assess pulmonary arteriole levels of p70S6K or P-p70S6K(Thr389), which is the activated form of p70S6K associated with increased ribosomal activity and cell growth. The effect of MR inhibition on expression levels of Rictor, which is the active subunit of mTORC2, was also assessed. IgG served as negative control. Data are presented as mean ± sem. All images were obtained at 400× magnification. Representative micrographs are shown.
On the basis of prior reports that demonstrated cross-talk between mTORC1 and mTORC2 in PAH (7), we next explored the effect of aldosterone inhibition on expression of Rictor, which is the functional subunit of mTORC2. Although Rictor expression was increased in MCT-PAH and SU-5416/hypoxia-PAH rats by 4.9-fold (P < 0.05) and 5.4-fold (P < 0.05), respectively, this effect was significantly attenuated by treatment with spironolactone or eplerenone (Fig. 2).
Aldosterone activates p70S6K via Akt/mTOR signaling in PASMCs in vitro
Our findings indicate an association between aldosterone, activated p70S6K, and hypertrophic remodeling of pulmonary arterioles in 2 animal models of PAH in vivo; therefore, we next explored PASMC involvement in aldosterone-p70S6K in vitro. Serum-replete PASMCs were treated with vehicle control or aldosterone (10−9–10−7 M) for 60 min, and levels of p70S6K and P-p70S6K(Thr389) were assessed by immunoblot. We first confirmed that vehicle control for 60 min did not affect the P-p70S6K-to-p70S6K ratio significantly compared with untreated cells (86 ± 11 vs. 91 ± 14 a.u.; P = 0.79; n = 3; Supplemental Fig. 1A). However, we observed that, compared with vehicle-treated cells, aldosterone (10−7 M) increased P-p70S6K(Thr389) (12 ± 0.9 vs. 37 ± 5.9 a.u.; P < 0.02; n = 3) without affecting total p70S6K (Fig. 3A).
Figure 3.

Acute aldosterone (ALDO) treatment up-regulates Akt/Raptor /p70S6K in PASMCs in vitro. A) Cultured human PASMCs were treated with aldosterone 10−9–10−7 M) for 1 h, and levels of P-p70S6K(Thr389), P-Akt(Ser473), P-mTOR(Ser2448,) P-Raptor(Ser792), and P-Rictor(Thr1105) were measured by immunoblot and compared with levels of the respective unmodified proteins (n = 3 for each blot). B) To support these findings, cultured PASMCs were treated with vehicle (V) control or aldosterone (10−7 M) for 1 h, and anti-Raptor and anti-P-Raptor(Ser792) immunohistochemistry was performed (n = 3). Data are presented as mean ± sem. Representative micrographs (×400 magnification) and blots are shown.
In systemic vascular cells, aldosterone is reported to stimulate Akt signaling, which, in turn, is an established upstream mediator of mTOR-induced p70S6K activation in tumor cells and in cell lines that are characterized by dysregulated growth (27, 28). To determine whether aldosterone affected p70S6K via a similar mechanism in PASMCs, we explored the effect of aldosterone on Akt and mTOR protein expression. Although no significant difference was observed between vehicle control– and aldosterone (10−7 M)-treated PASMCs for total levels of Akt or mTOR, we found that aldosterone increased levels of P-Akt(Ser473) and P-mTOR(Ser2448), which are the activated forms of these proteins (29), by 3.2-fold (P < 0.001; n = 3) and 1.3-fold (P < 0.05; n = 3), respectively (Fig. 3A).
We next explored whether up-regulation of Akt/mTOR in PASMCs was associated with activation of the functional mTORC1 subunit, Raptor, which induces p70S6K-mediated cellular ribosomal production (30). Consistent with findings for Akt and mTOR, we observed that aldosterone (10−7 M) treatment for 60 min increased P-Raptor(Ser792) expression by 3.0-fold (P < 0.01; n = 3) as assessed by immunoblot and by 2.4-fold (P < 0.02; n = 3) as assessed by immunohistochemistry compared with vehicle-treated cells, which was not contingent on changes to total levels of Raptor or activation of Rictor (Fig. 3). Furthermore, under serum-starved conditions, aldosterone (10−9–10−7 M) for 60 min induced a concentration-dependent increase in the ratio of P-Raptor(Ser792)-to-Raptor that was similar in magnitude to our findings in serum-replete PASMCs (Supplemental Fig. 1B). We also observed in serum-replete cells a time course (0, 15, 30, and 60 min) effect in which the ratios of P-Akt(Ser473)-to-/Akt, P-mTOR(Ser2448)-to-mTOR, P-Raptor(Ser792)-to-Raptor, and P-p70S6K(Thr389)-to-p70S6K were increased significantly by aldosterone (10−7 M) at 30 and 60 min (Supplemental Fig. 2A). Collectively, these data indicate that at concentrations akin to those observed in the pulmonary circulation of patients with PAH, aldosterone up-regulates the Akt/mTOR/p70S6K axis via nongenomic pathways in PASMCs in vitro, and that the effect of aldosterone on Raptor did not depend on culture medium nutrition status or concomitant activation of Rictor.
Raptor is required for aldosterone-induced up-regulation of p70S6K in PASMCs
We next tested the hypothesis that Raptor is critical for stimulation of p70S6K activation by aldosterone. Raptor expression in cultured PASMCs was inhibited by using si-Raptor (40 nM). Compared with vehicle-treated cells, si-Raptor–transfected cells demonstrated a significant decrease in Raptor protein levels (63 ± 2.3 vs. 36 ± 3.8 a.u.; P < 0.01; n = 3) that was not observed in cells treated with lipofectamine alone or si-Ssc (Fig. 4A). Molecular inhibition of Raptor was also associated with a significant decrease (34%) in the ratio of P-p70S6K(Thr389)-to-p70S6K (P < 0.05; n = 3) in aldosterone-treated cells relative to vehicle control (Fig. 4B).
Figure 4.

Raptor is required for up-regulation of p70S6K by aldosterone. A) Raptor expression was inhibited in cultured PASMCs using si-Raptor (40 nM; n = 3). B) PASMCs transfected with si-Raptor and stimulated with aldosterone (10−7 M) for 1 h demonstrated a decrease in the ratio of P-p70S6K(Thr389)-to-p70S6K compared with aldosterone (ALDO)-stimulated cells treated with vehicle control (V; n = 3) or transfected with si-Scc. Un, untreated; V, lipofectamine transfection reagent. Data are presented as mean ± sem. Representative immunoblots are shown.
MR blockade inhibits Akt/Raptor/p70S6K up-regulation by aldosterone in PASMCs
To determine whether MR stimulation is required to mediate the acute effects of aldosterone on mTORC1 signaling, we treated PASMCs with vehicle control or aldosterone (10−7 M) for 60 min in the presence or absence of spironolactone (10 μM), and we assessed changes in the expression levels of Akt/Raptor/p70S6K by immunoblot. We observed no significant effect from spironolactone on the ratio of activated-to-total levels of Akt, Raptor, or p70S6K in the basal state; however, compared with aldosterone alone, coincubation of aldosterone-treated cells with spironolactone decreased expression levels of P-Akt(Ser473)/Akt (173.5 ± 13.5 vs. 136.6 ± 5.7% control; P < 0.01; n = 3) and P-Raptor(Thr389)/Raptor (28 ± 3 vs. 21 ± 3 a.u.; P = 0.02; n = 3), which corresponded to a significant decrease in P-p70S6K(Thr389)/p70S6K (190 ± 10 vs. 120 ± 10 a.u.; P < 0.001; n = 3; Fig. 5). Similar observations were made for PASMCs that were coincubated with the selective MR antagonist eplerenone (10 μM) and aldosterone (10−7 M) for 30 and 60 min in which P-Akt(Ser473)/Akt and P-Raptor(Thr389)/Raptor were decreased significantly compared with treatment for 30 min with aldosterone alone (Supplemental Fig. 2B).
Figure 5.

Pharmacologic MR antagonism inhibits aldosterone (ALDO)-induced up-regulation of Akt/Raptor/p70S6K in PASMCs in vitro. Human PASMCs were treated with vehicle control or aldosterone (10−7 M) for 1 h in the presence or absence of spironolactone (SP; 10 μM), and expression levels of P-Akt(Ser473) (A), P-Raptor(Ser792) (B), and P-p70S6K(Thr389) (C) were measured by immunoblot and compared with levels of the respective unmodified proteins (n = 3 for each blot). V, vehicle. Data are presented as mean ± sem. Representative blots are shown.
Acute aldosterone treatment promotes apoptosis resistance and cell growth in PASMCs in vitro
We next investigated the functional significance of Akt/Raptor/p70S6K axis up-regulation by aldosterone on PASMC survival and growth patterns. The effect on apoptosis by treatment with aldosterone (10−7 M) for 2 h was assessed by quantifying cytoplasmic histone-associated DNA fragments in cells that were exposed to vehicle control or H2O2 (250, 500, and 1000 μM) for 24 h to promote cell death (19). Compared with vehicle-treated cells, a dose-dependent increase in apoptosis was observed for H2O2-treated cells (1.05 ± 0.4 vs. 12.3 ± 5.4 vs. 17.2 ± 3.9 vs. 27.7 ± 4.3 enrichment units/106 cells; P < 0.05 by ANOVA; n = 3–6). However, compared with vehicle control, aldosterone treatment decreased apoptosis in cells that were stimulated with H2O2 at 500 μM (−5.3 ± 3.3 enrichment units/106 cells; P = 0.08; n = 6) and at 1000 μM (−16.5 ± 2.8 enrichment units/106 cells; P < 0.001; n = 3; Fig. 6A).
Figure 6.

Aldosterone (ALDO) induces PASMC growth, survival, and apoptosis resistance. A) Human PASMCs were grown in standard culture medium and exposed to H2O2 (0, 250, 500, and 1000 μM) for 24 h to induce apoptosis. Cells were then treated with vehicle control (V) or ALDO (10−7 M) for 2 h, and apoptosis was assessed by quantifying cytoplasmic histone-associated DNA fragments (n = 3). B) Cells were treated with V or ALDO (10−7 M) in the presence or absence of the selective MR antagonist eplerenone (EPL; 10 μM) for 1 h, and cell viability was assessed by quantifying solubilized formazan products measured by spectrophotometric analysis at 490 nm (n = 3). C) Cells were grown to confluence and then standard culture medium was replaced with serum-replete (5% FBS) or serum-starved (1% FBS) medium that contained V or ALDO (10−7 M). After incubation of cells for 2 h, cell proliferation was determined by measuring BrdU incorporation (n = 3). Data are presented as mean ± sem.
Next, PASMCs were treated with vehicle control or aldosterone (10−7 M) for 2 h in the presence or absence of eplerenone (10 μM), and we measured the conversion of tetrazolium salt to formazan by colometric analysis to quantitate cell viability. Compared with vehicle control, aldosterone significantly increased viability (100.5 ± 2.9 vs. 123.7 ± 5.6% control; P = 0.01; n = 3), which was fully inhibited by cotreatment with eplerenone (123.7 ± 5.6 vs. 101.8 ± 1.3% control; P = 0.01; n = 3; Fig. 6B). We used the BrdU incorporation assay to measure DNA synthesis as a method by which to assess the effect of aldosterone on PASMC proliferation. In serum-replete PASMCs (5% FBS), aldosterone significantly increased BrdU incorporation compared with vehicle control (0.91 ± 0.03 vs. 0.72 ± 0.06 a.u.; P < 0.05; n = 3–5) (Fig. 6C). To confirm our earlier findings that suggested that serum starvation does not affect aldosterone-induced activation of the Akt/Raptor/p70S6K axis differentially compared with serum-replete conditions, we also assessed BrdU incorporation in serum-starved PASMCs (1% FBS). We observed no significant difference in BrdU incorporation in aldosterone-treated PASMCs relative to vehicle-treated cells by serum-starved or serum-replete status (Fig. 6C).
Pulmonary arterial plasma aldosterone levels in PAH are sufficient to stimulate Raptor activation in PASMCs
To investigate the potential translational relevance of our findings to patients, we analyzed the effect of pulmonary arterial plasma that was harvested from PAH patients or controls (Table 1) on Raptor phosphorylation in PASMCs. Compared with controls (n = 3), pulmonary arterial plasma aldosterone levels were increased significantly in patients with PAH (n = 4; 1176 ± 114 vs. 2609 ± 351 ng/dL; P = 0.02), as reported previously by our laboratory (Fig. 7A) (10). We performed anti-Raptor immunohistochemistry in cultured PASMCs that were treated for 60 min with standard culture medium (CM) that was supplemented with plasma from patients with PAH to a final concentration of 10% (PAH-CM). We observed that the P-Raptor(Ser792)-to-Raptor ratio was increased significantly in PASMCs treated with PAH-CM compared with untreated cells, which was inhibited fully by pretreatment of PAH-CM for 4 h with spironolactone (10 μM; 50 ± 2 vs. 70 ± 1 vs. 50 ± 1 a.u.; P < 0.001; n = 3; Fig. 7B). These findings were supported by immunofluorescence staining that demonstrated a similar effect of spironolactone on P-p70S6K(Thr389) relative to p70S6K in PAH-CM–treated cells (Fig. 7C).
Table 1.
Clinical and hemodynamic characteristics of patients
| Characteristic | Controls (n = 3) | PAH (n = 4) | P |
|---|---|---|---|
| Male (n) | 1 | 0 | |
| Age (yr) | 64 ± 6.1 | 61 ± 5.6 | 0.65 |
| Comorbidities | |||
| Essential hypertension | 1 | 3 | |
| Dyslipidemia | 2 | 2 | |
| Systemic sclerosis | 0 | 1 | |
| Obstructive sleep apnea | 0 | 1 | |
| Fibromyalgia | 0 | 1 | |
| Depression | 0 | 1 | |
| Cervical spondylosis | 0 | 1 | |
| Unexplained dyspnea | 1 | 0 | |
| Bronchopulmonary dysplasia | 1 | 0 | |
| Tetralogy of Fallot | 1 | 0 | |
| Hemodynamics | |||
| RA pressure (mmHg) | 8.6 ± 3.1 | 9.0 ± 1.9 | 0.92 |
| PASP (mmHg) | 35 ± 6.0 | 82 ± 2.8 | <0.01 |
| PADP (mmHg) | 14 ± 3.0 | 34 ± 1.9 | <0.01 |
| mPAP (mmHg) | 23 ± 1.9 | 52 ± 1.6 | <0.0001 |
| PAWP (mmHg) | 11 ± 3.0 | 11 ± 1.2 | 0.95 |
| Cardiac output (L/min) | 6.1 ± 0.5 | 5.6 ± 0.6 | 0.58 |
| CI (L/min/m2) | 3.4 ± 0.3 | 2.7 ± 0.3 | 0.16 |
| PVR (Wood units) | 1.9 ± 0.1 | 8.7 ± 1.1 | <0.01 |
Data are presented as mean ± sem. mPAP, mean pulmonary artery pressure; PADP, pulmonary artery diastolic pressure; PAWP, pulmonary artery wedge pressure; RA, right atrial.
Figure 7.

Pulmonary arterial plasma aldosterone (ALDO) levels from patients with PAH ex vivo are sufficient to stimulate activation of Raptor and p70S6K in PASMCs in vitro. A) Plasma was acquired from the pulmonary artery or wedge position in patients with PAH (n = 4) or controls (n = 3) referred for right heart catheterization, and aldosterone levels were measured by ELISA. B, C) Anti-Raptor and anti-P-Raptor(Ser792) immunohistochemistry (B) and anti-P-p70S6K(Thr389) (green), anti-p70S6K (green), and DAPI (blue) immunofluorescence staining (C) was performed on untreated normal human PASMCs or PASMCs treated with CM that was supplemented with pulmonary artery plasma (10% final concentration) from patients with PAH in the presence or absence of spironolactone (SP; 10 μM; n = 3–4). Data are presented as mean ± sem. Representative micrographs and immunofluorescence images are shown.
MR antagonism inhibits Raptor to improve pulmonary arteriole remodeling and PAH in vivo
To support our observations in PASMCs in vitro, we assessed the effect of aldosterone and/or Raptor inhibition on the prevention of vascular remodeling and pulmonary hypertension in PAH in vivo. First, we demonstrated that Raptor expression was inhibited in PASMCs by using an siRNA (40 nM) oligonucleotide sequence that also served as si-Raptor for formulation with Staramine-PEG for use in vivo (Fig. 8A). We then performed double immunohistochemistry to demonstrate the colocalization of Raptor and α1-smooth muscle actin in hypertrophic pulmonary arterioles (Supplemental Fig. 3A). We observed that, compared with controls, anti-Raptor immunohistochemistry demonstrated that male rats treated with MCT expressed increased Raptor in muscularized distal pulmonary arterioles (56.2 ± 7.4 vs. 105.6 ± 4.2 a.u.; P < 0.001; n = 4–5 rats/condition; Fig. 8B, C), which was associated with significantly increased PASP, indexed pulmonary vascular resistance (PVRi), and RV weight/LV plus septum weight (Fulton index) that was observed without significant differences between conditions for body weight or mean arterial pressure (Table 2).
Figure 8.

Combination therapy with si-Raptor plus MR antagonism prevents PAH in vivo. A) Raptor expression was inhibited in cultured PASMCs by using a si-Raptor (40 nM; n = 3) oligonucleotide sequence that also served as the si-Raptor for formulation with Staramine-PEG. B) In a prevention protocol, male Sprague-Dawley rats were administrated MCT (50 mg/kg) and treated with Staramine-mPEG formulated with si-Scc, Staramine-mPEG formulated with si-Raptor, or si-Raptor plus spironolactone (SP; 25 mg/kg/d). Staramine (2 mg/kg) was administered by i.v. tail vein injection in 4 doses at protocol days 1, 6, 10, and 16. At the completion of the protocol 21–23 d after MCT administration, paraffin-embedded lung sections were harvested from rats and hematoxylin and eosin (H/E) staining was performed. B, C) Anti-Raptor immunohistochemistry was performed to quantify the effect of treatment on Raptor expression levels in distal pulmonary arterioles measuring 20–50 μm in diameter (n = 4–5 rats/condition). To determine if differences in pulmonary arteriole Raptor levels influenced vascular remodeling patterns by treatment condition, anti-α1 smooth muscle actin (SMA) immunohistochemistry was performed. B, D, E) The number of muscularized pulmonary arterioles (red arrows) per high-powered field (h.p.f.) (B, D) and hypertrophic remodeling severity, expressed as the % vessel wall/luminal area (B, E), was calculated (n = 4–5 rats/condition). F) The percent of TUNEL-positive nuclei in remodeled pulmonary arterioles was calculated in each treatment condition (n ≅ 20 vessels/condition). DIC, differential interference contrast; Un, untreated; V, vehicle. Data are presented as mean ± sem. Representative blots and micrographs are shown.
Table 2.
The treatment effects of aldosterone-Raptor inhibition on pulmonary hypertension in MCT-PAH in vivo
| Variable | Control | MCT |
P | |||
|---|---|---|---|---|---|---|
| Untreated | si-Scc | si-Raptor | si-Raptor + Sp | |||
| Weight (g) | 251 (8.2) | 295 (34.4)* | 307 (8.1)* | 292 (8.5)* | 291 (8.5)* | < 0.0001 |
| HR (bpm) | 276 (8.6) | 271 (8.7) | 260 (7.6) | 288 (5.0) | 279 (7.8) | 0.10 |
| RAP (mmHg) | 4.1 (0.8) | 6.5 (1.3) | 7.5 (0.9)* | 6.2 (0.2) | 4.9 (0.4) | < 0.05 |
| PASP (mmHg) | 26 (1.9) | 77 (5.2)* | 47 (2.0)*,** | 46 (2.5)*,** | 38 (1.7)*,**,† | < 0.0001 |
| MAP (mmHg) | 79 (4.5) | 68 (3.6) | 72 (6.5) | 78 (6.9) | 86 (5.4) | 0.23 |
| LVEDP (mmHg) | 8.5 (0.6) | 10.4 (0.5) | 7.3 (1.6) | 10.0 (0.9) | 9.0 (1.3) | 0.36 |
| CI (L/min/g) | 218 (10.2) | 147 (5.3)* | 115 (16.6)* | 213 (15) | 210 (7.2) | < 0.0001 |
| PVRi (mmHg⋅min⋅g/ml) | 32 (3.4) | 70 (13.1)* | 82 (6)*,** | 26 (7.6)** | 24 (3.9)** | < 0.0001 |
| RV/LV + S | 0.24 (0.02) | 0.38 (0.09)* | 0.33 (0.07)* | 0.25 (0.04)** | 0.24 (0.05)** | < 0.0001 |
In a disease prevention experimental design, male Sprague-Dawley rats were injected with vehicle control (n = 4 rats/condition) or MCT (50 mg/kg) and treated with Staramine-mPEG (2 mg/kg i.v.; 4 total doses/rat) formulated with si-Scc, si-Raptor, or si-Raptor plus spironolactone (SP; 25 mg/kg/d; n = 6–7 rats/condition), and at days 21–23 hemodynamics were assessed by cardiac catheterization. HR, heart rate; LVEDP, LV end-diastolic pressure; LV + S, left ventricle + septum; MAP, mean arterial pressure; RAP, right atrial pressure. P values indicate results from 1-way ANOVA. Post hoc analyses using the Tukey method indicated the following: *P < 0.05 vs. control; **P < 0.05 vs. MCT; †P < 0.05 vs. MCT + si-Raptor. Data are presented as mean (sem). RV/LV + S ratio of weight is given in grams.
Compared with rats that were treated with MCT alone, no significant differences were observed in MCT rats treated with Staramine-mPEG formulated with si-Scc (2 mg/kg, 4 doses) on the levels of pulmonary vascular Raptor expression or the severity of pulmonary vascular remodeling, although the hemodynamic profile of rats in this condition was consistent with end-stage pulmonary hypertension as indicated by decreased PASP in the setting of increased PVRi and low CI (Table 2). In contrast, treatment of MCT rats with Staramine-mPEG formulated with si-Raptor (2 mg/kg, 4 doses) decreased pulmonary arteriole Raptor expression and hypertrophic remodeling (Fig. 8C–E) and increased apoptosis in pulmonary arterioles (Fig. 8F) compared with rats that were treated with MCT alone. Differences in vascular remodeling patterns in si-Raptor–treated rats corresponded to a significant improvement in PASP (77 ± 5.2 vs. 46 ± 2.5 mmHg; n = 5–6 rats/condition, P < 0.05) and RV/LV + septum weight (0.38 ± 0.09 vs. 0.25 ± 0.04, N = 5-6 rats/condition; P < 0.05), as well as a trend toward decreased PVRi (70 ± 13.1 vs. 26 ± 7.6 vs. mmHg⋅min⋅g/ml; n = 5–6 rats/condition; P = 0.08) and increased CI (147 ± 5.3 vs. 213 ± 15 L/min/g; n = 5–6 rats/condition; P = 0.06).
Effect of combination MR antagonism plus si-Raptor on PAH
We observed incomplete inhibition of pulmonary arterial Raptor in si-Raptor –treated male MCT rats. Owing to our observation indicating that aldosterone is a potent stimulator of Raptor-p70S6K signaling in PASMCs in vitro, we explored whether the addition of spironolactone (25 mg/kg/d) to si-Raptor –treated MCT-PAH rats affected pulmonary arterial Raptor expression, vascular remodeling, and hemodynamics in PAH (Fig. 8B–F). Compared with MCT-PAH + si-Raptor, a further decrement in pulmonary arteriole Raptor expression was observed in the MCT-PAH + si-Raptor + spironolactone condition (72.9 ± 5.4 vs. 58.4 ± 7.9 a.u.; n = 4–5 rats/condition; P < 0.05). Although no significant difference was observed between groups for the density of muscularized pulmonary arterioles, the combination si-Raptor + spironolactone decreased hypertrophic remodeling of vessels (670 ± 51.2 vs. 290 ± 25% vessel wall/luminal area; n = 4–5 rats/condition; P < 0.001; Fig. 8D), which corresponded to a further decrease in PASP (Table 2).
Next, to study the effect of aldosterone-Raptor inhibition on reversing pulmonary hypertension in an angioproliferative animal model of PAH, SU-5416/hypoxia-normoxia rats were treated with si-Raptor in the presence or absence of spironolactone (n = 3–6 rats/condition) that was initiated at the completion of the hypoxia phase of the protocol 21 d after the injection of SU-5416, which is a time point associated with histologic and hemodynamic evidence of PAH (11). Compared with controls, the number of remodeled distal pulmonary arterioles measuring 20–50 µm in diameter was increased in SU-5416/hypoxia-normoxia rats (1.4 ± 0.5 vs. 5.0 ± 0.3 arterioles/high-power field; n = 3 rats/condition;, P < 0.0001). Treatment with si-Raptor and si-Raptor plus spironolactone decreased the burden of remodeled arterioles in SU-5416/hypoxia-normoxia rats (3.0 ± 0.1 vs. 2.0 ± 0.4 vs. 5.0 ± 0.3 arterioles/high-power field; n = 3 rats/condition; P < 0.02), which corresponded to improvements in PASP, mean PAP, PVRi, and CI (Table 3). We explored the possibility that changes in RV Raptor expression could account for these findings, but no significant difference in levels was observed between control and SU-5416/hypoxia-normoxia rats.
Table 3.
The effect of aldosterone-Raptor inhibition on reversing pulmonary hypertension in the SU-5416/hypoxia-normoxia experimental model of PAH in vivo
| Variable | Control | SU-5416/hypoxia-normoxia |
P | ||
|---|---|---|---|---|---|
| Untreated | si-Raptor | si-Raptor + Sp | |||
| Weight (g) | 256 (2.1) | 367 (19.7)* | 332 (13.5) | 372 (15.6)* | < 0.01 |
| HR (bpm) | 250 (11.2) | 282 (9.5) | 267 (13.9) | 270 (3.4) | 0.27 |
| RAP (mmHg) | 3.9 (1.2) | 4.9 (0.5) | 5.4 (0.9) | 6.3 (0.3) | 0.31 |
| PASP (mmHg) | 24 (1.3) | 109 (3.9)* | 91 (2.9)*,** | 73.6 (10.9)*,† | < 0.0001 |
| mPAP (mmHg) | 15 (0.6) | 36 (1.3)* | 30 (1.6)** | 21.3 (3.2)*,†,‡ | < 0.0001 |
| MAP (mmHg) | 75 (4.0) | 65 (2.7) | 69 (8.1) | 66 (1.2) | 0.55 |
| LVEDP (mmHg) | 7.8 (0.1) | 7.2 (1.0) | 4.6 (0.7) | 5.2 (1.0) | 0.10 |
| CI (L/min/g) | 210 (9.1) | 100 (15)* | 174 (16)† | 166 (12)‡ | < 0.002 |
| PVRi (mmHg⋅min⋅g/ml) | 36 (2.5) | 329 (60)* | 145 (21)§ | 103 (29)‡ | < 0.01 |
| RV/LV + S | 0.24 (0.03) | 0.46 (0.06)* | 0.34 (0.03) | 0.32 (0.01) | 0.046 |
Male Sprague-Dawley rats (n = 3–6/condition) were injected with SU-5416 (20 mg/kg), exposed to hypoxia (10% fraction of inspired oxygen) for 21 d, and returned to normoxia for 21 d before the experiments were performed. In a disease treatment protocol, rats were treated with Staramine-mPEG (2 mg/kg i.v. injection; 4 total doses/rat) formulated with si-Raptor or si-Raptor plus spironolactone (SP; 25 mg/kg/d) initiated 21 d after injection with SU-5416 and continued until cardiac catheterization was performed on day 42 of the protocol. HR, heart rate; LV + S, left ventricle + septum; LVEDP, LV end-diastolic pressure; MAP, mean arterial pressure; mPAP, mean pulmonary artery pressure; RAP, right atrial pressure. P values indicate results from 1-way ANOVA. Post hoc analyses using the Tukey method indicated the following: *P < 0.05 vs. Control; **P = 0.07 vs. SU-5416/hypoxia-normoxia; †P < 0.0001 vs. SU-5416/hypoxia-normoxia; ‡P ≤ 0.04 vs. SU-5416/hypoxia-normoxia + si-Raptor; §P = 0.058 vs. SU-5416/hypoxia-normoxia. Data are presented as mean (sem). RV/LV+S ratio of weight is given in grams.
To determine whether the favorable changes to cardiopulmonary hemodynamics that were observed in PAH rats could have occurred as a result of off-target effects of therapy on systemic blood vessels, we performed hematoxylin and eosin staining and anti-Raptor immunohistochemistry on renal tissue that was harvested from controls, MCT-PAH rats, and MCT-PAH rats treated with si-Raptor (Supplemental Fig. 3B). We observed no significant differences between these conditions with respect to blood vessel architecture or Raptor expression in small- or medium-sized renal arterioles measuring 20–80 μm in diameter.
DISCUSSION
These data demonstrate that activation of Raptor by aldosterone induces PASMC growth and apoptosis resistance in vitro and contributes to the development of pulmonary arteriole remodeling and PAH in vivo. We have shown that MR-dependent Akt-mTORC1 signal transduction in aldosterone-treated PASMCs induced phosphorylation of p70S6K at Thr389, which is known to target S6 ribosomal proteins and to increase cell proliferation and survival (31). Molecular inhibition of Raptor or MR antagonism prevented p70S6K phosphorylation by inhibiting the activation of Raptor-p70S6K, which was associated with attenuated aldosterone-induced PASMC proliferation. The translational relevance of these findings was also assessed: aldosterone levels in pulmonary arterial plasma from patients with PAH were sufficient to up-regulate Raptor in cultured PASMCs, whereas MR antagonism was required for the optimal inhibition of PASMC Raptor to achieve maximal improvements to hypertrophic vascular remodeling and pulmonary hypertension in 2 experimental models of PAH in vivo (this complex mechanism is illustrated in Fig. 9).
Figure 9.

Summary of aldosterone-Raptor signaling in the development of pulmonary arteriole remodeling and pulmonary hypertension in PAH. Elevated circulating levels of aldosterone (ALDO) in PAH stimulate the MR to induce the activation of p70S6K via up-regulation of the Akt/mTOR/Raptor axis in PASMCs. Aldosterone-p70S6K signaling promotes apoptosis resistance, cellular proliferation, and increased cell survival in vitro, which is associated with concentric hypertrophic remodeling of distal pulmonary arterioles and pulmonary hypertension in PAH in vivo. In turn, MR inhibition with spironolactone or eplerenone prevents the adverse effects of aldosterone-Raptor in vitro, whereas molecular inhibition of Raptor, in combination with spironolactone, was more effective than Raptor inhibition alone for reversing or preventing experimental PAH.
Our findings support prior reports that implicate mTOR bioactivity in the pathogenesis of vascular remodeling in PAH (7, 32) and expand this field by identifying hormonal regulation of mTORC1 by aldosterone as a novel mechanism that controls survival and growth patterns in PASMCs. These effects were not contingent on nutrient status in the culture medium of PASMCs: we observed no meaningful difference in aldosterone-stimulated Raptor-p70S6K signaling between serum-starved and serum-replete PASMCs in vitro. Although starvation conditions are known to stimulate mTOR activity (9), our findings are in concert with previous reports that demonstrate that under nutrient-rich conditions, sufficient levels of the mTORC1 substrate remain available for mTORC1 activation in HeLa (33), human embryonic kidney (34), and vascular smooth muscle cells (35).
We have also demonstrated increased mTORC1 in pulmonary blood vessels of MCT-PAH and SU-5416/hypoxia-PAH rats, even though the pathogenesis of PAH in these disease models is characterized by impaired oxygen use and insulin resistance, which, in turn, are implicated in mTORC1 inhibition in pulmonary vascular cells (36–38). This observation raises the possibility that acquired factors in PAH, such as elevated levels of aldosterone, may offset the negative regulatory consequences of metabolic dysfunction on Raptor bioactivity in PASMCs, although this specific mechanism was not tested directly in the current study. Similarly, it is interesting that cross-talk between hypoxic signaling, insulin function, and aldosterone bioactivity is reported in pulmonary and systemic vascular cells (13, 23, 39); however, future studies are required to investigate the extent to which an interaction between these factors may influence mTORC1 in PAH. Moreover, although our data suggest that aldosterone levels from female patients with PAH were sufficient for inducing Raptor activation in PASMCs, the cells used for in vitro experiments were from male donors and in vivo PAH models included only male rats. Thus, the derivative effects on Raptor of female sex hormones, which are implicated increasingly in the pathobiology of pulmonary vascular remodeling and RV function in PAH (40–42), were not tested in this study but could have had important effects on our results; therefore, this requires further analysis.
Targeting of Akt by MR signal transduction is recognized increasingly in the pathogenesis of cardiovascular disease. In vascular endothelial cells, pharmacologic inhibition of the MR by spironolactone prevents the effects of acute aldosterone treatment on Akt-dependent uncoupling of endothelial nitric oxide synthase and subsequent cytoskeletal rearrangement (43), whereas intimal hyperplasia in abdominal aortas of transgenic mice overproducing angiotensin II (16) is prevented via the inhibition of Akt by MR blockade. To our knowledge, in the current study we have demonstrated for the first time the significance of Akt targeting by MR signal transduction to the pathobiology of pulmonary vascular disease. Specifically, these data illustrate a novel mechanism downstream of MR stimulation by which to account for maladaptive changes to vascular structure and function: the activation of Raptor to promote a PASMC pathophenotype that is characterized by apoptosis resistance and dysregulated cell growth.
Raptor or mTOR as a potential treatment target in PAH has been tested previously in various experimental models of PAH (44) and, recently, in a small clinical study (45). However, the therapeutic efficacy of these interventions in PAH varies across reports, and off-target drug effects have emerged as an important consideration due to the functionality of mTOR in numerous tissue types, in particular in systemic blood vessels (45). To minimize off-target side effects of Raptor inhibition in this study, we used a cationic lipopolyamine nanoparticle delivery system reported previously to selectively target lung vascular cells (20), and we observed no significant effect on central hemodynamics or Raptor inhibition in resistance vessels by using this strategy. However, in MCT-PAH and SU-5416/hypoxia-normoxia-PAH rats, maximal inhibition of Raptor in pulmonary arterioles and the attendant beneficial effects on pulmonary hypertension severity was achieved by the addition of MR antagonism with spironolactone to si-Raptor, which was observed in both disease prevention and treatment protocols. Taken together, these observations identify elevated levels of aldosterone as one acquired factor that mediates Raptor functionality and provides evidence to support the targeting of converging pathways to achieve maximal Raptor inhibition in PAH.
A number of limitations warrant consideration when interpreting the results of this study. First, Akt/Raptor/p70S6K post-translational modifications that were selected for analysis were based on prior reports that indicated activation of the corresponding protein in similar cell types and experimental systems (27); however, the effect of aldosterone on other phosphorylation sites implicated in (counter)regulation of these intermediaries was not tested, and, thus, the ramifications of MR-dependent signaling on Raptor in PASMCs is not fully characterized by this work. Second, our findings implicate up-regulation of pulmonary arteriole Rictor by aldosterone in PAH in vivo, although acute aldosterone treatment did not activate Rictor in PASMCs in vitro. These findings suggest that Rictor expression is affected by chronic exposure to aldosterone and/or by other as yet undetermined in vivo factors; however, further analysis is required to characterize the differential effects of Akt activation by mTORC2 (46) and/or aldosterone on PASMC proliferation in PAH (7, 8). Along these lines, our findings on the expression pattern of p70S6K in remodeled pulmonary arterioles in experimental PAH in vivo are consistent with prior reports (47, 48) that suggest that cell types other than PASMCs known to mediate the vasculopathy of PAH may also express increased Raptor. Therefore, future studies are required to determine whether mTORC1 and mTORC2 are involved in p70S6K-mediated cell growth, proliferation, and apoptosis resistance in aldosterone-treated PAMSCs as well as in other cell types implicated in PAH pathobiology. Third, although elevated levels of aldosterone were confirmed in patients with PAH and the studied animal models, we did not measure circulating glucocorticoid hormone levels, and, thus, the extent to which up-regulation of Raptor occurred in our experiments in vivo through glucocorticoid-MR cross reactivity was not assessed.
In summary, our findings demonstrate hormonal regulation of mTORC1 by aldosterone that induces viability, proliferation, and apoptosis resistance in PASMCs to promote vascular remodeling and pulmonary hypertension in PAH. These data identify a novel mechanism that controls PASMC survival patterns that is activated by MR stimulation and requires Raptor. Collectively, our findings suggest that optimal strategies to abrogate vascular proliferation in PAH may require the inhibition of converging pathways that influence p70S6K, including involvement of aldosterone, and lend support to future studies analyzing the effect of therapeutic strategies that aim to achieve maximal inhibition of Raptor and other critical PAH disease-promoting targets.
Supplementary Material
Acknowledgments
This work was supported by the British Heart Foundation and a C.R.T Fellowship (FS/10/42/28372) (to R.A.); the U.S. National Institutes of Health (NIH), National Heart, Lung, and Blood Institute (NHLBI) Grant 1K08HL11207-01A1, American Heart Association Grant AHA 15GRNT25080016, the Pulmonary Hypertension Association, the Cardiovascular Medical Research and Education Fund (CMREF), the Klarman Foundation at Brigham and Women’s Hospital, and the Gilead Young Scholars Foundation (to B.A.M.); by the Dunlevie Family Fund (to A.R.O.); by NIH NHLBI Grant 1U01HL125215-01 (to J.A.L.); and by NIH NHLBI Grants R37 HL061795, HL108630 (MAPGEN), U54HL119145, and PPGHL048743 (to J.L.). B.A.M. is involved with investigator-initiated research that is supported by Gilead Sciences. A.R.O. is involved with investigator-initiated research that is supported by Actelion Pharmaceuticals.
Glossary
- a.u.
arbitrary units
- BrdU
bromodeoxyuridine
- CI
cardiac index
- CM
conditioned medium
- FBS
fetal bovine serum
- LV
left ventricle
- MCT
monocrotaline
- mPEG
monomethoxy polyethylene glycol
- MR
mineralocorticoid receptor
- mTOR
mammalian target of rapamycin
- mTORC1
mTOR complex 1
- PAH
pulmonary arterial hypertension
- PASMC
pulmonary artery smooth muscle cell
- PASP
pulmonary artery systolic pressure
- PVRi
indexed pulmonary vascular resistance
- RV
right ventricle
- si-Raptor
Raptor siRNA
- siRNA
small interfering RNA
- si-Scc
small inhibitory RNA scrambled (negative) control
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Marsboom G., Toth P. T., Ryan J. J., Hong Z., Wu X., Fang Y. H., Thenappan T., Piao L., Zhang H. J., Pogoriler J., Chen Y., Morrow E., Weir E. K., Rehman J., Archer S. L. (2012) Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ. Res. 110, 1484–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taraseviciene-Stewart L., Kasahara Y., Alger L., Hirth P., Mc Mahon G., Waltenberger J., Voelkel N. F., Tuder R. M. (2001) Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 15, 427–438 [DOI] [PubMed] [Google Scholar]
- 3.Galiè N., Olschewski H., Oudiz R. J., Torres F., Frost A., Ghofrani H. A., Badesch D. B., McGoon M. D., McLaughlin V. V., Roecker E. B., Gerber M. J., Dufton C., Wiens B. L., Rubin L. J.; Ambrisentan in Pulmonary Arterial Hypertension, Randomized, Double-Blind, Placebo-Controlled, Multicenter, Efficacy Studies (ARIES) Group (2008) Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 117, 3010–3019 [DOI] [PubMed] [Google Scholar]
- 4.Humbert M., Sitbon O., Chaouat A., Bertocchi M., Habib G., Gressin V., Yaici A., Weitzenblum E., Cordier J. F., Chabot F., Dromer C., Pison C., Reynaud-Gaubert M., Haloun A., Laurent M., Hachulla E., Simonneau G. (2006) Pulmonary arterial hypertension in France: results from a national registry. Am. J. Respir. Crit. Care Med. 173, 1023–1030 [DOI] [PubMed] [Google Scholar]
- 5.Bockmeyer C. L., Maegel L., Janciauskiene S., Rische J., Lehmann U., Maus U. A., Nickel N., Haverich A., Hoeper M. M., Golpon H. A., Kreipe H., Laenger F., Jonigk D. (2012) Plexiform vasculopathy of severe pulmonary arterial hypertension and microRNA expression. J. Heart Lung Transplant. 31, 764–772 [DOI] [PubMed] [Google Scholar]
- 6.Jia G., Aroor A. R., Martinez-Lemus L. A., Sowers J. R. (2014) Overnutrition, mTOR signaling, and cardiovascular diseases. Am. J. Physiol. Regul. Integr. Comp. Physiol. 307, R1198–R1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goncharov D. A., Kudryashova T. V., Ziai H., Ihida-Stansbury K., DeLisser H., Krymskaya V. P., Tuder R. M., Kawut S. M., Goncharova E. A. (2014) Mammalian target of rapamycin complex 2 (mTORC2) coordinates pulmonary artery smooth muscle cell metabolism, proliferation, and survival in pulmonary arterial hypertension. Circulation 129, 864–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krymskaya V.P., Snow J., Ceasarone G., Khavin I., Goncharov D.A., Lim P.N., Veasey S.C., Ihidia-Stansbury K., Jones P.L., Goncharova E.A. (2011) mTOR is required for pulmonary artery vascular smooth muscle cell proliferation under chronic hypoxia. FASEB J. 25, 1922–1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sengupta S., Peterson T. R., Sabatini D. M. (2010) Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol. Cell 40, 310–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maron B. A., Opotowsky A. R., Landzberg M. J., Loscalzo J., Waxman A. B., Leopold J. A. (2013) Plasma aldosterone levels are elevated in patients with pulmonary arterial hypertension in the absence of left ventricular heart failure: a pilot study. Eur. J. Heart Fail. 15, 277–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maron B. A., Zhang Y.-Y., White K., Chan S. Y., Handy D. E., Mahoney B. A., Loscalzo J., Leopold J. A. (2012) Aldosterone inactivates the endothelin-B receptor via a cysteinyl thiol redox switch to decrease pulmonary endothelial nitric oxide levels and modulate pulmonary arterial hypertension. Circulation 126, 963–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Preston I. R., Sagliani K. D., Warburton R. R., Hill N. S., Fanburg B. L., Jaffe I. Z. (2013) Mineralocorticoid receptor antagonism attenuates experimental pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 304, L678–L688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maron B. A., Leopold J. A. (2015) Emerging concepts in the molecular basis of pulmonary arterial hypertension: Part II: neurohormonal signaling contributes to the pulmonary vascular and right ventricular pathophenotype of pulmonary arterial hypertension. Circulation 131, 2079–2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Su H., Gu Y., Li F., Wang Q., Huang B., Jin X., Ning G., Sun F. (2013) The PI3K/AKT/mTOR signaling pathway is overactivated in primary aldosteronism. PLoS One 8, e62399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang B., Ding W., Zhang M., Li H., Gu Y. (2015) Rapamycin attenuates aldosterone-induced tubulointerstitial inflammation and fibrosis. Cell. Physiol. Biochem. 35, 116–125 [DOI] [PubMed] [Google Scholar]
- 16.Sakurabayashi-Kitade S., Aoka Y., Nagashima H., Kasanuki H., Hagiwara N., Kawana M. (2009) Aldosterone blockade by Spironolactone improves the hypertensive vascular hypertrophy and remodeling in angiotensin II overproducing transgenic mice. Atherosclerosis 206, 54–60 [DOI] [PubMed] [Google Scholar]
- 17.Lee C.-C., Wang C.-N., Lee Y.-L., Tsai Y.-R., Liu J.-J. (2015) High mobility group box 1 induced human lung myofibroblasts differentiation and enhanced migration by activation of MMP-9. PLoS One 10, e0116393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang L., Guo L. J., Liu J., Wang W., Yuan J. X., Zhao L., Wang J., Wang C. (2013) MicroRNA expression profile of pulmonary artery smooth muscle cells and the effect of let-7d in chronic thromboembolic pulmonary hypertension. Pulm. Circ. 3, 654–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mill C., Monk B. A., Williams H., Simmonds S. J., Jeremy J. Y., Johnson J. L., George S. J. (2014) Wnt5a-induced Wnt1-inducible secreted protein-1 suppresses vascular smooth muscle cell apoptosis induced by oxidative stress. Arterioscler. Thromb. Vasc. Biol. 34, 2449–2456 [DOI] [PubMed] [Google Scholar]
- 20.McLendon J. M., Joshi S. R., Sparks J., Matar M., Fewell J. G., Abe K., Oka M., McMurtry I. F., Gerthoffer W. T. (2015) Lipid nanoparticle delivery of a microRNA-145 inhibitor improves experimental pulmonary hypertension. J. Control. Release 210, 67–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.White K., Lu Y., Annis S., Hale A. E., Chau B. N., Dahlman J. E., Hemann C., Opotowsky A. R., Vargas S. O., Rosas I., Perrella M. A., Osorio J. C., Haley K. J., Graham B. B., Kumar R., Saggar R., Saggar R., Wallace W. D., Ross D. J., Khan O. F., Bader A., Gochuico B. R., Matar M., Polach K., Johannessen N. M., Prosser H. M., Anderson D. G., Langer R., Zweier J. L., Bindoff L. A., Systrom D., Waxman A. B., Jin R. C., Chan S. Y. (2015) Genetic and hypoxic alterations of the microRNA-210-ISCU1/2 axis promote iron-sulfur deficiency and pulmonary hypertension. EMBO Mol. Med. 7, 695–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abe K., Toba M., Alzoubi A., Ito M., Fagan K. A., Cool C. D., Voelkel N. F., McMurtry I. F., Oka M. (2010) Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation 121, 2747–2754 [DOI] [PubMed] [Google Scholar]
- 23.Maron B. A., Oldham W. M., Chan S. Y., Vargas S. O., Arons E., Zhang Y.-Y., Loscalzo J., Leopold J. A. (2014) Upregulation of steroidogenic acute regulatory protein by hypoxia stimulates aldosterone synthesis in pulmonary artery endothelial cells to promote pulmonary vascular fibrosis. Circulation 130, 168–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.French C. J., Spees J. L., Zaman A. K. M. T., Taatjes D. J., Sobel B. E. (2009) The magnitude and temporal dependence of apoptosis early after myocardial ischemia with or without reperfusion. FASEB J. 23, 1177–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McMurtry M. S., Bonnet S., Wu X., Dyck J. R. B., Haromy A., Hashimoto K., Michelakis E. D. (2004) Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ. Res. 95, 830–840 [DOI] [PubMed] [Google Scholar]
- 26.Wang W., Liu J., Ma A., Miao R., Jin Y., Zhang H., Xu K., Wang C., Wang J. (2014) mTORC1 is involved in hypoxia-induced pulmonary hypertension through the activation of Notch3. J. Cell Physiol. 229, 2117–2125 [DOI] [PubMed] [Google Scholar]
- 27.Lee J. W., Park S., Takahashi Y., Wang H. G. (2010) The association of AMPK with ULK1 regulates autophagy. PLoS One 5, e15394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gwinn D. M., Shackelford D. B., Egan D. F., Mihaylova M. M., Mery A., Vasquez D. S., Turk B. E., Shaw R. J. (2008) AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Foster K. G., Fingar D. C. (2010) Mammalian target of rapamycin (mTOR): conducting the cellular signaling symphony. J. Biol. Chem. 285, 14071–14077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hei Y. J., Pelech S. L., Chen X., Diamond J., McNeill J. H. (1994) Purification and characterization of a novel ribosomal S6 kinase from skeletal muscle of insulin-treated rats. J. Biol. Chem. 269, 7816–7823 [PubMed] [Google Scholar]
- 31.Pearson R. B., Dennis P. B., Han J. W., Williamson N. A., Kozma S. C., Wettenhall R. E., Thomas G. (1995) The principal target of rapamycin-induced p70s6k inactivation is a novel phosphorylation site within a conserved hydrophobic domain. EMBO J. 14, 5279–5287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ranchoux B., Antigny F., Rucker-Martin C., Hautefort A., Péchoux C., Bogaard H. J., Dorfmüller P., Remy S., Lecerf F., Planté S., Chat S., Fadel E., Houssaini A., Anegon I., Adnot S., Simonneau G., Humbert M., Cohen-Kaminsky S., Perros F. (2015) Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 131, 1006–1018 [DOI] [PubMed] [Google Scholar]
- 33.Oshiro N., Rapley J., Avruch J. (2014) Amino acids activate mammalian target of rapamycin (mTOR) complex 1 without changing Rag GTPase guanyl nucleotide charging. J. Biol. Chem. 289, 2658–2674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sancak Y., Peterson T. R., Shaul Y. D., Lindquist R. A., Thoreen C. C., Bar-Peled L., Sabatini D. M. (2008) The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thompson A. M., Martin K. A., Rzucidlo E. M. (2014) Resveratrol induces vascular smooth muscle cell differentiation through stimulation of SirT1 and AMPK. PLoS One 9, e85495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McMurtry M. S., Archer S. L., Altieri D. C., Bonnet S., Haromy A., Harry G., Bonnet S., Puttagunta L., Michelakis E. D. (2005) Gene therapy targeting survivin selectivity induces pulmonary vascular apoptosis and reverses pulmonary arterial hypertension. J. Clin. Invest. 115, 1479–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hansmann G., Wagner R. A., Schellong S., Perez V. A., Urashima T., Wang L., Sheikh A. Y., Suen R. S., Stewart D. J., Rabinovitch M. (2007) Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator-activated receptor-gamma activation. Circulation 115, 1275–1284 [DOI] [PubMed] [Google Scholar]
- 38.Duan Y., Li F., Tan K., Liu H., Li Y., Liu Y., Kong X., Tang Y., Wu G., Yin Y. (2015) Key mediators of intracellular amino acids signaling to mTORC1 activation. Amino Acids 47, 857–867 [DOI] [PubMed] [Google Scholar]
- 39.Sherajee S. J., Fujita Y., Rafiq K., Nakano D., Mori H., Masaki T., Hara T., Kohno M., Nishiyama A., Hitomi H. (2012) Aldosterone induces vascular insulin resistance by increasing insulin-like growth factor-1 receptor and hybrid receptor. Arterioscler. Thromb. Vasc. Biol. 32, 257–263 [DOI] [PubMed] [Google Scholar]
- 40.Chen X., Talati M., Fessel J.P., Hemnes A.R., Gladson S., Fench J., Shay S., Trammell A., Phillips J.A., Hamid R., Cogan J.D., Dawson E.P., Womble K.E., Hedges L.K., Martinez E.G., Wheeler L.A., Majka S.J., Lloyd J.E., West J., Austin E.D. (2016) Estrogen metabolite 16α-hydroxyestrone exacerbates bone morphogenetic protein receptor type II-associated pulmonary arterial hypertension through microRNA-29-mediated modulation of cellular metabolism. Circulation 133, 82–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frump A. L., Goss K. N., Vayl A., Albrecht M., Fisher A., Tursunova R., Fierst J., Whitson J., Cucci A. R., Brown M. B., Lahm T. (2015) Estradiol improves right ventricular function in rats with severe angioproliferative pulmonary hypertension: effects of endogenous and exogenous sex hormones. Am. J. Physiol. Lung Cell. Mol. Physiol. 308, L873–L890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wallace E., Morrell N. W., Yang X. D., Long L., Stevens H., Nilsen M., Loughlin L., Mair K. M., Baker A. H., MacLean M. R. (2015) A sex-specific MicroRNA-96/5-hydroxytryptamine 1B axis influences development of pulmonary hypertension. Am. J. Respir. Crit. Care Med. 191, 1432–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kirsch T., Beese M., Wyss K., Klinge U., Haller H., Haubitz M., Fiebeler A. (2013) Aldosterone modulates endothelial permeability and endothelial nitric oxide synthase activity by rearrangement of the actin cytoskeleton. Hypertension 61, 501–508 [DOI] [PubMed] [Google Scholar]
- 44.Li L., Wang X., Wang L., Qu L., Zhu X., Li M., Dang X., Li P., Gao Y., Peng Z., Pan L., Wan L. (2015) Mammalian target of rapamycin overexpression antagonizes chronic hypoxia-triggered pulmonary arterial hypertension via the autophagic pathway. Int. J. Mol. Med. 36, 316–322 [DOI] [PubMed] [Google Scholar]
- 45.Seyfarth H.-J., Hammerschmidt S., Halank M., Neuhaus P., Wirtz H. R. (2013) Everolimus in patients with severe pulmonary hypertension: a safety and efficacy pilot trial. Pulm. Circ. 3, 632–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Manning B. D., Cantley L. C. (2007) AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gerasimovskaya E. V., Tucker D. A., Weiser-Evans M., Wenzlau J. M., Klemm D. J., Banks M., Stenmark K. R. (2005) Extracellular ATP-induced proliferation of adventitial fibroblasts requires phosphoinositide 3-kinase, Akt, mammalian target of rapamycin, and p70 S6 kinase signaling pathways. J. Biol. Chem. 280, 1838–1848 [DOI] [PubMed] [Google Scholar]
- 48.Fattori R., Piva T. (2003) Drug-eluting stents in vascular intervention. Lancet 361, 247–249 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
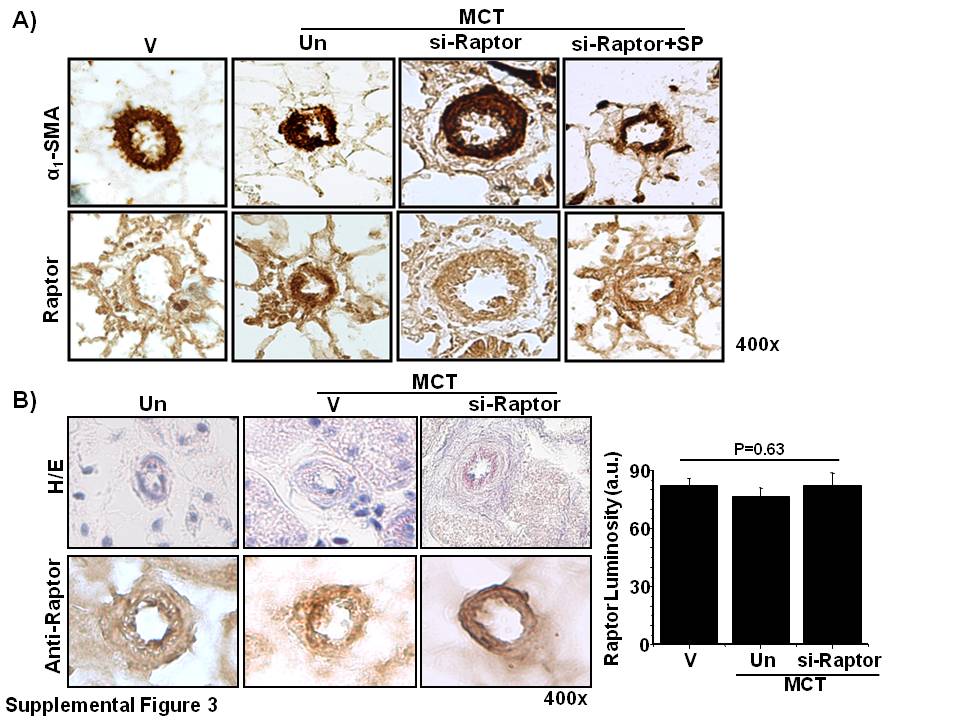{kind=link}