

Abstract
Alzheimer's disease (AD), a fatal progressive neurodegenerative disorder, has no cure to date. One of the causes of AD is the accumulation of amyloid-beta 42 (Aβ42) plaques, which result in the onset of neurodegeneration. It is not known how these plaques trigger the onset of neurodegeneration. There are several animal models developed to (i) study etiology of disease, (ii) look for genetic modifiers, and (iii) identify chemical inhibitors that can block neurodegeneration and help to find cure for this disease. An insect model of Drosophila melanogaster has also provided new insights into the disease. Here we will discuss the utility of the Drosophila eye model to study Alzheimer's disease.
Keywords: Alzheimer's disease, neurodegeneration, amyloid plaques, amyloid hypothesis, animal models, drosophila melanogaster, drosophila eye
Introduction
Alzheimer's disease (AD) represents a complex class of debilitating brain diseases that mostly affects people aged 65 and older. AD is characterized by progressive loss of neurons in the hippocampus and cortex, which results in the shrinkage of brain. Basically, the brain cells required to process, store and retrieve information are killed, which is characteristic of a neurodegenerative process (Singh, 2012). It results in decline in cognitive and behavioral functions like memory, thinking and language skills (O’Brien and Wong, 2010). AD, a common form of dementia, is a progressive neurodegenerative disorder, which killed half a million people in 2010. AD is aptly called as the silver tsunami of the 21st first century. It is the sixth leading cause of death in US. It is estimated that by 2050, the number of people with AD may nearly triple, from 5 million to as many as 16 million, barring the development of medical breakthroughs to prevent, slow or stop the disease (Xu, 2016). To date, there is no effective cure.
AD neuropathology is a proteinopathy, which is an outcome of misfolded proteins. AD is associated with two types of abnormal protein deposition in the human brain: (1) neurofibrillary tangles (NFTs) containing hyper-phosphorylated forms of a microtubule associated protein Tau, and (2) accumulation of the plaques of amyloid-beta (Aβ42) peptide due to aggregation prone amyloidogenic domains (Hardy, 2009; Aguzzi and O’Connor, 2010; Fernandez-Funez et al., 2013; Selkoe and Hardy, 2016).
Generation of Amyloid-beta Plaques in AD
One of the hallmarks of AD is the accumulation of amyloid plaques in the brain. In the healthy brain these plaques are not present. The focus of this review is on the accumulation of Aβ42 polypeptides formed by the improper cleavage of a transmembrane amyloid precursor protein (APP) in the brain (Figure 1) (Hardy, 2009; Aguzzi and O’Connor, 2010; O’Brien and Wong, 2010; Fernandez-Funez et al., 2013). Mutations in the gene encoding for APP have been linked to the familial form of AD. APP is proteolytically processed in the extracellular and intracellular domains by α and then γ-secretase enzymes, which leads to the generation of a forty amino acid long polypeptide (Aβ40), which causes age-dependent learning defects but no neurodegeneration. However, the differential cleavage of APP by the activities of β- and γ-secretase enzymes lead to generation of forty two amino acid long polypeptide, hence Aβ42 (Figure 1) (Hardy, 2009; Aguzzi and O’Connor, 2010; O’Brien and Wong, 2010; Fernandez-Funez et al., 2013; Selkoe and Hardy, 2016). These extra two amino acids cause the amyloid Aβ42 polypeptide to become hydrophobic, resulting in formation of the amyloid plaques. Increased level of the more toxic form of human Aβ42 polypeptide is responsible for the neuropathology seen in AD. These plaques disrupt normal cellular processes through oxidative stress and aberrant signaling, resulting in the loss of synaptic activity and death of neurons.
Figure 1.
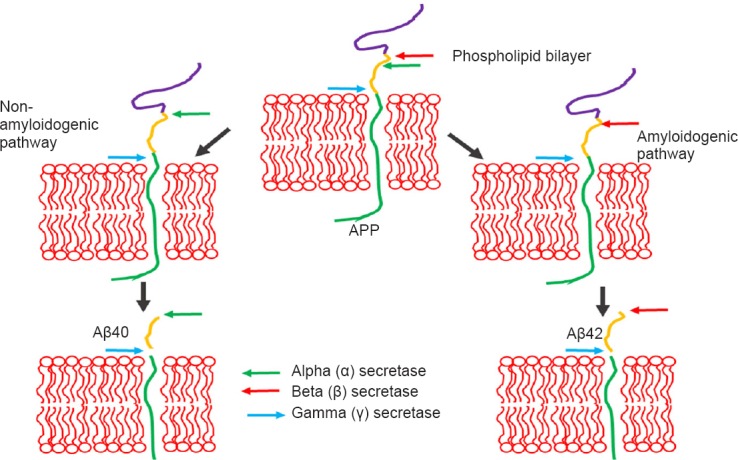
Schematic presentation of generation of amyloid-beta 42 (Aβ42) plaques.
Aβ42 are generated by improper cleavage of amyloid precursor proteins (APP). APP a transmembrane protein, is cleaved by alpha (α)-sectretase and gamma (γ)-secretase to generate forty amino-acid (Aβ40) long polypeptide. However, when APP is cleaved by beta (β)-secretase and gamma (γ)-secretase it generates forty two amino-acid (Aβ42) long polypeptide. Aβ42 polypeptide oligomerize to form a plaque.
Even though many gene mutations responsible for AD have been identified, the detailed genetic signaling mechanism(s) responsible for this neurodegeneration and progression of AD remain indefinable. A great deal of efforts are channeled towards discerning the underlying mechanisms that induce Aβ42 metabolism and the pathways (e.g., apoptosis and autophagy) that trigger neuronal cell death. The rationale is to identify the molecular genetic mechanism of generation of amyloid (Aβ42) accumulation, which will allow (a) development of biomarkers for early detection of AD, and (b) development of strategies to prevent accumulation of amyloid plaques, thereby leading to identification of drug targets for AD. These insights can lead to a cure or better therapeutic strategies for this disease.
Animal Models for AD
Clinical trials largely aimed at removing these Aβ42 plaques have not yet been efficient. Aβ42 mediated neurodegeneration is a dominant gain-of-function phenotype. Misexpression of human Aβ42 polypeptides trigger neurodegeneration in traditional mammalian models like the Mouse (Mus musculus) as well as in the genetically tractable alternative models like zebrafish (Danio rerio), roundworm (Caenorhabditis elegans), and the fruit fly (Drosophila melanogaster) (Iijima and Iijima-Ando, 2008; Moloney et al., 2010; Harvey et al., 2011; Singh and Irvine, 2012). It has been seen that the primary structures of the Aβ peptide in rats and mice differ from their human counterpart at three amino acid residues. Furthermore, the complete Aβ-peptide profile produced by the processing of APP in these rodents exhibits major differences in comparison to that of humans. In guinea pigs, the rabbit Aβ peptide sequence is identical to human but does not present AD pathology spontaneously. Therefore, alternative animal models for AD have been developed to understand the mechanism(s) of regulation of human Aβ42 protein accumulation, and identification of the signaling pathways (e.g., cell death) involved to generate insights into the etiology of the disease (Bier, 2005; Singh and Irvine, 2012; Moloney et al., 2010; Pandey and Nichols, 2011; Tare et al., 2011).
Using an insect Drosophila melanogaster (a.k.a. fruit fly), to model human disease is beneficial for many reasons (Bier, 2005). Most importantly, their entire genome is sequenced, and is highly conserved with humans and is significantly less redundant (Lenz et al., 2013). Furthermore, they are smaller in size and thus can be stored efficiently, are cost effective, and can produce two generations in a month (Bier, 2005; Singh and Irvine, 2012; Lenz et al., 2013). It makes Drosophila an ideal model for studying age related progressive diseases like AD. Several models of AD have been developed in Drosophila. These include transgenic flies misexpressing human Aβ42, human Aβ40, human APP, BACE, β-secretase and Drosophila Psn, FAD (Cauchi and van den Heuvel, 2006; Cao et al., 2008; Iijima and Iijima-Ando, 2008; Tare et al., 2011; Fernandez-Funez et al., 2013). These flies display several aspects of clinical AD neuropathology and symptoms including the generation of amyloid aggregates, external morphological abnormalities, dramatic neuroanatomical changes and defects in motor reflex behavior and memory. As a proof of concept, treatment with a γ-secretase inhibitor was shown to suppress these phenotypes. Drosophila has also proved informative as a model of other neurodegenerative diseases like Parkinson's disease, Huntington's disease, fronto-temporal lobar degeneration (FTLD), and amyotrophic lateral sclerosis (Bier, 2005; Pandey and Nichols, 2011; Singh and Irvine, 2012; Fernandez-Funez et al., 2013).
Drosophila Eye as Disease Model System
Drosophila has a fully functional nervous system with a structural design that separates specialized functions such as vision, olfaction, learning and memory. The Drosophila eye develops from a simple monolayer epithelium called the eye-antennal imaginal disc which grows during larval stages to form a highly organized compound eye comprised of photoreceptor cells, cone cells, and pigment cells (Ready et al., 1976; Kumar, 2011; Singh et al., 2012; Kumar, 2013; Tare et al., 2013). The precise structure of Drosophila eye makes it very sensitive to genetic disruptions/manipulations and it can be easily screened using a stereo microscope for phenotypes generated in the eye field. Thus, Drosophila eye allows quick screening of large sample size. The Drosophila model has a repertoire of tools that make it indispensable, including the ability to express foreign genes along spatio-temporal axes that can mimic several important neurodegenerative disorders in the compound eye (Bier, 2005; Singh and Irvine, 2012). Adding to this, the eye is not essential for the viability or fertility of the fly. Therefore, the Drosophila eye is an ideal organ system to assay the effect of neurodegeneration. Furthermore, the genes involved in eye development are structurally and functionally similar between insects and humans. The main advantage of the Drosophila eye is the ability to directly visualize the cellular and developmental defects induced by neurodegeneration causing agents.
We have utilized Drosophila eye to model Aβ42 mediated neurodegeneration. Using Gal4/UAS transgenic systems (Brand and Perrimon, 1993), we misexpressed human Aβ42 in the differentiating photoreceptor neurons of the developing eye (Tare et al., 2011; Moran et al., 2013; Steffensmeier et al., 2013; Cutler et al., 2015). It resulted in progressive neurodegenerative phenotype; Cutler et al., 2015 in the Drosophila eye, which is similar to AD like neuropathology (Figure 2). However, Drosophila model has some challenges too. In comparison to the vertebrates, Drosophila has less complex and adaptive immune system. Furthermore, effects of drugs on the organism might differ strongly (Prussing et al., 2013).
Figure 2.
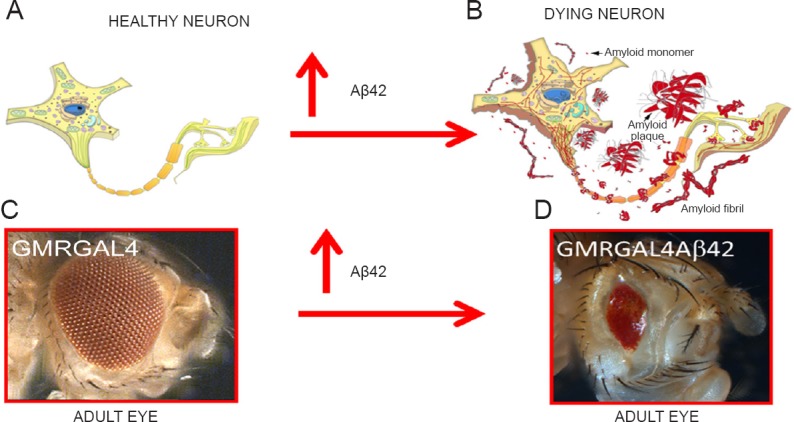
Drosophila melanogaster (fruit fly) eye model exhibits neurodegeneration due to accumulation of amyloid-beta 42 (Aβ42) plaques.
Accumulation of Aβ42 plaques in (A) healthy neuron triggers (B) death of a meuron. (C, D) Misexpression of human Aβ42 in healthy photoreceptor neurons of Drosophila eye using transgenic approach causes a (C) wild-type adult compound eye to change into a (D) highly reduced adult eye due to induction of neuronal cell death (Tare et al., 2011).
Drosophila Model for Genetic Screens
Drosophila model has been extensively used to screen (i) for genetic modifiers responsible for onset and manifestation of disease and (ii) chemical libraries to look for therapeutic targets to find cure for the disease. Drosophila model with the repertoire of genetic tools allows several different approaches like a forward genetic screens, which involve a random mutagenesis via chemicals or X-ray radiation and then to identify the genes responsible for that phenotype, or reverse genetic screen where you mutate a gene and then look for the phenotype caused by loss-of-function of that gene (Bier, 2005; Singh and Irvine, 2012). Forward genetic screens are decidedly useful as evidenced by the Nobel prize awarded to C. Nüsslein-Volhard, E. Wieschaus and E.B. Lewis in 1995 for their work in identifying genes involved in early fruit fly development (Nusslein-Volhard and Wieschaus, 1980; St Johnston, 2002; Lenz et al., 2013). However, mapping of mutations using a classical genetic approach can be time consuming. The advent of new genetic tools, like single nucleotide polymorphisms (SNPs), has alleviated some of this problem in forward genetic screens (Lenz et al., 2013). Despite this, few strictly forward genetic screens have been undertaken in an AD background using a Drosophila model. In reverse genetic screens, a known gene is disrupted, typically utilizing the Gal4/UAS system (Brand and Perrimon, 1993) to misexpress a gene or silence a gene using RNA interference (RNAi) (Lenz et al., 2013).
Modifier (also known as enhancer or suppressor) screens are highly valuable because they combine the advantages of both forward and reverse genetics and carried out using easily assayable phenotypes (Lenz et al., 2013; Prussing et al., 2013). In AD, the Aβ42 neurodegenerative phenotype and the rough eye phenotype are easily assayable phenotypes (Tare et al., 2011). A Drosophila eye model was established where Aβ42 is misexpressed in the developing fly eye results in highly reduced neurodegenerative eye (Cao et al., 2008; Iijima and Iijima-Ando, 2008; Tare et al., 2011). We employed this model for a small scale modifier screen to look for the genetic modifiers which can either enhance/suppress the AD eye phenotype by increasing levels of highly conserved signaling pathways (one at a time) (Tare et al., 2011; Moran et al., 2013). Using this screen, we have identified the homeotic gene teashirt (tsh), highly conserved apical basal polarity marker Crumbs (Crb) (Steffensmeier et al., 2013) as modifiers of Aβ42 mediated neurodegeneration, making these genes as possible target for AD therapies.
The similar logic was utilized for several high throughput screens (HTS) as well as smaller screens to identify genetic modifiers of AD. Though many genetic screens for modifiers of Tau induced neurodegeneration have been carried out, few large-scale screens have investigated modifiers of the Aβ42 neurodegenerative phenotype (Lenz et al., 2013; Prussing et al., 2013). Several other earlier genetic screens to identify modifiers of the Aβ42 phenotype have resulted in a list of potential downstream genetic targets (Cao et al., 2008). Tan et al. used a screen to identify loss of function mutations that either suppressed or enhanced the Aβ42 neurodegenerative phenotype (Tan et al., 2008). Using a IInd and IIIrd chromosome deficiency kit they identified a deficiency that uncovers Toll (Tl) suppresses the Aβ42 mediated neurodegeneration. Binding of the Toll extracellular domain initiates a signaling cascade that eventually leads to activation the NFκB signaling, a pathway which plays a role in innate immunity and inflammatory responses. Tan et al. (2008) identified this inflammatory signaling as playing a key role in promoting neurodegenerative processes.
Ultimately, the goal of these types of genetic screens is to identify a particular protein or signaling pathway member, which is involved in AD mediated neuropathology that can finally be targeted with a chemical compound (Pandey and Nichols, 2011). Identification of such a chemical compound is typically accomplished by in vitro HTS of a chemical library. Once a compound is identified, it is optimized for human usage through a series of tests in animal models. This process can be inefficient when utilizing traditional in vitro cell cultures or biochemical assays. Furthermore, the paradigm in therapeutic treatments has shifted from “one disease-one target” to an understanding that many diseases result from a variety of factors and interactions. Based on these two problems with traditional methods in drug development, Drosophila has emerged as a valuable model (Pandey and Nichols, 2011).
It has already been noted that Drosophila as a model is space, time and resource efficient (Lenz et al., 2013). In terms of drug development, flies present an advantageous supplement or alternative to traditional methods because they allow chemical compounds to be tested in vivo in a whole organism as opposed to an isolated culture (Pandey and Nichols, 2011). Though considerations regarding the discrepancies between flies and humans in pharmacokinetics, pharmacodynamics and the toxicity of particular drug administered in Drosophila are necessary, overall the model has emerged as highly useful in secondary post screening validation. Even though other disease systems have utilized Drosophila as primary screening models, there are currently no studies which have utilized Drosophila as a primary screening model for novel drug discovery in AD (Pandey and Nichols, 2011).
Drug administration typically takes the form of solid media to which drugs are added and larvae are allowed to grow on. Though Drosophila are useful for HTS, the amount of chemicals screened is significantly lower, typically 500 to 1,000 small molecules per month, than traditional cell culture assays, which can screen as many as 10,000 small molecules per month. Despite this, screens utilizing flies typically result in a higher proportion of hits with therapeutic value. In AD models like Drosophila, traditional HTS are frequently utilized first to take advantage of their efficiency and identified small molecules are then tested in flies (Pandey and Nichols, 2011).
One such screen to utilize Drosophila as secondary post screening validation identified a particular small molecule that inhibited Aβ peptide aggregation. Using a traditional HTS based on Aβ42-GFP fusion they identified an inhibitor of Aβ aggregation. The most effective inhibitor, D737, was administered to transgenic flies with Aβ42 misexpressed in neural tissues. They found that not only did the compound reduce the toxicity of Aβ42 oligomers, but that it also increased the lifespan of the Aβ42 transgenic flies and their locomotive activity (McKoy et al., 2012).
A recent study examined the neuroprotective effect of a flavonoid derivative, 2-(4′ Benzloxyphenyl)-3-hydroxychromen-4-one, on the rough eye phenotype of transgenic Aβ42 flies (Singh et al., 2014). The compound was identified using an in silico docking approach, then administered to transgenic Aβ42 flies to test in vivo. Approximately 70% of Aβ42 transgenic flies showed a rescued eye phenotype and there was a significant reduction in plaques in Aβ42 transgenic larvae (Singh et al., 2014). While this type of post screening validation is certainly valuable, other disease models, including epilepsy and Fragile X syndrome (Stilwell et al., 2006), have taken advantage of Drosophila as a primary screening tool.
Conclusion
Here the vast contribution and potential of the Drosophila eye as a model for understanding the cellular and molecular mechanisms of neurodegeneration was explored. The eye model can be exploited for initial screening of the genetic modifiers or chemical inhibitor targets, as well as candidate validation. The Drosophila eye as a validation tool in drug discovery has gained momentum, which allows identification of compounds that would likely be effective in mammalian models and humans. Genetic and pharmacological screens in the fly eye will serve as a strong screening tool for therapeutic approaches in the future.
References
- Aguzzi A, O’Connor T. Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nat Rev Drug Discov. 2010;9:237–248. doi: 10.1038/nrd3050. [DOI] [PubMed] [Google Scholar]
- Bier E. Drosophila, the golden bug, emerges as a tool for human genetics. Nat Rev Genet. 2005;6:9–23. doi: 10.1038/nrg1503. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Cao W, Song HJ, Gangi T, Kelkar A, Antani I, Garza D, Konsolaki M. Identification of novel genes that modify phenotypes induced by Alzheimer's beta-amyloid overexpression in Drosophila. Genetics. 2008;178:1457–1471. doi: 10.1534/genetics.107.078394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauchi RJ, van den Heuvel M. The fly as a model for neurodegenerative diseases: is it worth the jump? Neurodegener Dis. 2006;3:338–356. doi: 10.1159/000097303. [DOI] [PubMed] [Google Scholar]
- Cutler T, Sarkar A, Moran M, Steffensmeier A, Puli OR, Mancini G, Tare M, Gogia N, Singh A. Drosophila eye model to study neuroprotective role of CREB binding protein (CBP) in Alzheimer's disease. PLoS One. 2015;10:e0137691. doi: 10.1371/journal.pone.0137691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Funez P, Sanchez-Garcia J, Ribncon-Limas DE. Unraveling the basis of neurodegeneration using the developing eye. New York: Springer; 2013. [Google Scholar]
- Hardy J. The amyloid hypothesis for Alzheimer's disease: a critical reappraisal. J Neurochem. 2009;110:1129–1134. doi: 10.1111/j.1471-4159.2009.06181.x. [DOI] [PubMed] [Google Scholar]
- Harvey BK, Richie CT, Hoffer BJ, Airavaara M. Transgenic animal models of neurodegeneration based on human genetic studies. J Neural Transm. 2011;118:27–45. doi: 10.1007/s00702-010-0476-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima K, Iijima-Ando K. Drosophila models of Alzheimer's amyloidosis: the challenge of dissecting the complex mechanisms of toxicity of amyloid-beta 42. J Alzheimers Dis. 2008;15:523–540. doi: 10.3233/jad-2008-15402. [DOI] [PubMed] [Google Scholar]
- Kumar J. Catching the next wave: patterning of the Drosophila eye by the morphogenetic furrow. In: Singh A, Kango-Singh M, editors. Molecular Genetics of Axial Patterning, Growth and Disease in the Drosophila Eye. New York: Springer; 2013. pp. 75–97. [Google Scholar]
- Kumar JP. My what big eyes you have: How the Drosophila retina grows. Dev Neurobiol. 2011;71:1133–1152. doi: 10.1002/dneu.20921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz S, Karsten P, Schulz JB, Voigt A. Drosophila as a screening tool to study human neurodegenerative diseases. J Neurochem. 2013;127:453–460. doi: 10.1111/jnc.12446. [DOI] [PubMed] [Google Scholar]
- McKoy AF, Chen J, Schupbach T, Hecht MH. A novel inhibitor of amyloid beta (Abeta) peptide aggregation: from high throughput screening to efficacy in an animal model of Alzheimer disease. J Biol Chem. 2012;287:38992–39000. doi: 10.1074/jbc.M112.348037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moloney A, Sattelle DB, Lomas DA, Crowther DC. Alzheimer's disease: insights from Drosophila melanogaster models. Trends Biochem Sci. 2010;35:228–235. doi: 10.1016/j.tibs.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran MT, Tare M, Kango-Singh M, Singh A. Homeotic Gene teashirt (tsh) has a neuroprotective function in amyloid-beta 42 mediated neurodegeneration. PLoS One. 2013;8:e80829. doi: 10.1371/journal.pone.0080829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature. 1980;287:795–801. doi: 10.1038/287795a0. [DOI] [PubMed] [Google Scholar]
- O’Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer's disease. Annu Rev Neurosci. 2011;34:185–204. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey UB, Nichols CD. Human disease models in Drosophila melanogaster and the role of the fly in therapeutic drug discovery. Pharmacol Rev. 2011;63:411–436. doi: 10.1124/pr.110.003293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prussing K, Voigt A, Schulz JB. Drosophila melanogaster as a model organism for Alzheimer's disease. Mol Neurodegener. 2013;8:35. doi: 10.1186/1750-1326-8-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ready DF, Hanson TE, Benzer S. Development of the Drosophila retina, a neurocrystalline lattice. Dev Biol. 1976;53:217–240. doi: 10.1016/0012-1606(76)90225-6. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med. 2016 doi: 10.15252/emmm.201606210. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A. Neurodegeneration a means to an end. J Cell Sci Ther. 2012;3:e107. [Google Scholar]
- Singh A, Irvine KD. Drosophila as a model for understanding development and disease. Dev Dyn. 2012;241:1–2. doi: 10.1002/dvdy.23712. [DOI] [PubMed] [Google Scholar]
- Singh A, Tare M, Puli OR, Kango-Singh M. A glimpse into dorso-ventral patterning of the Drosophila eye. Dev Dyn. 2012;241:69–84. doi: 10.1002/dvdy.22764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Gaur R, Kumar A, Fatima R, Mishra L, Srikrishna S. The flavonoid derivative 2-(4′ Benzyloxyphenyl)-3-hydroxy-chromen-4-one protects against Abeta42-induced neurodegeneration in transgenic Drosophila: insights from in silico and in vivo studies. Neurotox Res. 2014;26:331–350. doi: 10.1007/s12640-014-9466-z. [DOI] [PubMed] [Google Scholar]
- St Johnston D. The art and design of genetic screens: Drosophila melanogaster. Nat Rev Genet. 2002;3:176–188. doi: 10.1038/nrg751. [DOI] [PubMed] [Google Scholar]
- Steffensmeier AM, Tare M, Puli OR, Modi R, Nainaparampil J, Kango-Singh M, Singh A. Novel neuroprotective function of apical-basal polarity gene crumbs in amyloid beta 42 (abeta42) mediated neurodegeneration. PLoS One. 2013;8:e78717. doi: 10.1371/journal.pone.0078717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stilwell GE, Saraswati S, Littleton JT, Chouinard SW. Development of a Drosophila seizure model for in vivo high-throughput drug screening. Eur J Neurosci. 2006;24:2211–2222. doi: 10.1111/j.1460-9568.2006.05075.x. [DOI] [PubMed] [Google Scholar]
- Tan L, Schedl P, Song HJ, Garza D, Konsolaki M. The Toll-->NFkappaB signaling pathway mediates the neuropathological effects of the human Alzheimer's Abeta42 polypeptide in Drosophila. PLoS One. 2008;3:e3966. doi: 10.1371/journal.pone.0003966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tare M, Puli OR, Singh A. Molecular genetic mechanisms of axial patterning: mechanistic insights into generation of axes in the developing eye. In: Singh A, Kango-Singh M, editors. Molecular Genetics of Axial Patterning, Growth and Disease in the Drosophila Eye. New York: Springer; 2013. pp. 37–75. [Google Scholar]
- Tare M, Modi RM, Nainaparampil JJ, Puli OR, Bedi S, Fernandez-Funez P, Kango-Singh M, Singh A. Activation of JNK signaling mediates amyloid-ss-dependent cell death. PLoS One. 2011;6:e24361. doi: 10.1371/journal.pone.0024361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J. Mortality among centenarians in the United States, 2000-2014. NCHS Data Brief. 2016;233:1–8. [PubMed] [Google Scholar]