

Keywords: nerve regeneration, brain injury, integrin αvβ3, vascular endothelial growth factor, vascular endothelial growth factor receptor, vascular endothelial growth factor receptor-2, fetal liver kinase 1, ischemic preconditioning; ischemic tolerance, global cerebral ischemia, cerebral ischemia, cerebral infarction, NSFC grant, neural regeneration
Abstract
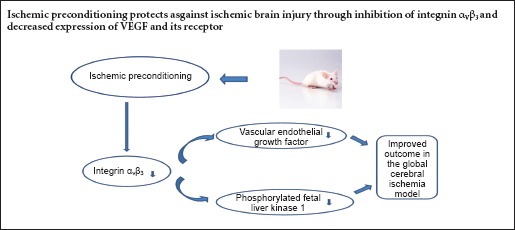
In this study, we hypothesized that an increase in integrin αvβ3 and its co-activator vascular endothelial growth factor play important neuroprotective roles in ischemic injury. We performed ischemic preconditioning with bilateral common carotid artery occlusion for 5 minutes in C57BL/6J mice. This was followed by ischemic injury with bilateral common carotid artery occlusion for 30 minutes. The time interval between ischemic preconditioning and lethal ischemia was 48 hours. Histopathological analysis showed that ischemic preconditioning substantially diminished damage to neurons in the hippocampus 7 days after ischemia. Evans Blue dye assay showed that ischemic preconditioning reduced damage to the blood-brain barrier 24 hours after ischemia. This demonstrates the neuroprotective effect of ischemic preconditioning. Western blot assay revealed a significant reduction in protein levels of integrin αvβ3, vascular endothelial growth factor and its receptor in mice given ischemic preconditioning compared with mice not given ischemic preconditioning 24 hours after ischemia. These findings suggest that the neuroprotective effect of ischemic preconditioning is associated with lower integrin αvβ3 and vascular endothelial growth factor levels in the brain following ischemia.
Introduction
Cerebral ischemia is a leading cause of death and disability globally. Developing new therapeutic strategies for cerebral ischemic injury is a major aim of scientists. Ischemic preconditioning (IP), which induces ischemic tolerance, is a brief, non-lethal, ischemic event one or several days prior to subsequent severe ischemia. Previous studies have found that the neuroprotective effects of IP are mediated by an attenuation of the mechanisms of injury, the activation of innate defense mechanisms, and the enhancement of endogenous repair processes (Gidday et al., 2006). However, the detailed mechanisms remain unclear.
Integrin αVβ3, a member of the integrin family, is involved in angiogenesis and tumor growth, and has been shown to play an important role in animal models of focal brain ischemia (Haring et al., 1996; Okada et al., 1996; Abumiya et al., 1999; Del Zoppo and Mabuchi, 2003). In primates, integrin αVβ3 is not expressed in the non-ischemic basal ganglia, but is expressed exclusively in the microvessels of the ischemic basal ganglia after middle cerebral artery occlusion or middle cerebral artery occlusion/reperfusion (Okada et al., 1996). Furthermore, inhibition of integrin αVβ3 preserves microvascular patency, reduces blood-brain barrier (BBB) breakdown, and ameliorates ischemic damage in animal models of focal brain ischemia (Abumiya et al., 2000; Shimamura et al., 2006a, b; Kiessling et al., 2009).
Vascular endothelial growth factor (VEGF) is one of the most important growth factors involved in vasculogenesis and angiogenesis. Park et al. (2014) found that IP dramatically augments VEGF and phosphorylated fetal liver kinase 1 (pFlk-1) immunoreactivity in the pyramidal cells of the hippocampal CA1 region after transient cerebral ischemia in gerbils. Previous studies demonstrated that the expression levels of VEGF and integrin αVβ3 are closely related (Abumiya et al., 1999), and that integrin αVβ3 plays a role in the activation of the VEGF receptor (Soldi et al., 1999). In animal models of focal brain ischemia, integrin αVβ3 antagonists appear to ameliorate damage by modulating VEGF and its receptor (Shimamura et al., 2006a). This suggests that increased expression of integrin αVβ3 may play a harmful role during early cerebral ischemic injury, and that the neuroprotective effects of integrin αVβ3 inhibition may be mediated through the modulation of VEGF and its receptor.
Previous studies of integrin αVβ3 focused on ischemic brain damage, but the role of integrin αVβ3 in IP-mediated neuroprotection has rarely been reported. Liu et al. (2010) demonstrated that IP effectively attenuates the upregulation of integrin αVβ3 mRNA expression after ischemia. Therefore, in this study, we investigated the effect of IP on integrin αVβ3, VEGF and its receptor to clarify the relationship between these proteins and neuroprotection.
Materials and Methods
Animals
A total of 78 clean male C57BL/6J mice weighing 22–25 g (certificate No. 0052588) were supplied by the Experimental Animal Center of Sun Yat-sen University in China and housed in separate cages under standard conditions. The animals were fed a standard diet and maintained under a 12-hour light-dark cycle. All surgery was performed under chloral hydrate (350 mg/kg, intraperitoneally) anesthesia, and all efforts were made to minimize pain and stress to the animals. The procedures conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). The animal experiments were performed in accordance with international ethical standards and were approved by the research ethics committee of Sun Yat-sen University in China.
Surgical operation
A total of 78 mice were randomly divided into the following groups: sham operation (Sham; n = 18), IP (n = 20), ischemia (Isch; n = 20), and IP followed by ischemia (IP + Isch; n = 20).
The two-vessel occlusion model of global cerebral ischemia was used (Liu et al., 2010). To induce lethal ischemia, mice were anesthetized with chloral hydrate (350 mg/kg, intraperitoneally) (Weijia Technology, Guangzhou, China) and allowed spontaneous respiration throughout the surgical procedure. Through a midline cervical incision, the bilateral common carotid arteries were exposed and clipped with two vascular clamps simultaneously for 30 minutes. The ischemic preconditioning was produced in a similar manner for a period of 5 minutes (Wu et al., 2001; Cho et al., 2005). In the IP + Isch group, the second ischemic insult (30 minutes) was performed 48 hours following the preconditioning ischemic event. Sham-operated animals received the same surgical procedures except that the carotid arteries were not clipped. The mice were placed on a heating pad after surgery until they recovered from anesthesia.
Evaluation of BBB disruption
The integrity of the BBB was assessed by quantitative measurement of Evans Blue (Sigma-Aldrich, St. Louis, MO, USA) content 24 hours after ischemia or sham surgery in eight animals per group (Kozler et al., 2003). Briefly, sterilized 2% Evans Blue solution was administered intravenously at a dosage of 4 mL/kg. Thirty minutes after injection, mice were perfused with saline to remove intravascular Evans Blue dye. Brains were rapidly removed, and each sample was weighed, homogenized with 2.5 mL phosphate-buffered saline (PBS), and mixed with 2.5 mL 60% trichloroacetic acid to precipitate protein. The samples were centrifuged for 30 minutes at 1,000 × g, and the absorbances of the supernatants were measured at 610 nm using a spectrophotometer (Genesys 10S; Thermo Electron Corporation, Madison, WI, USA). Evans Blue is expressed as µg/g of brain tissue against a standard curve.
Histological evaluation
Mice chosen randomly from the four groups (n = 4 in the Sham group; n = 6 each in the other three groups) were anesthetized with chloral hydrate (350 mg/kg, intraperitoneally) 7 days after cerebral ischemia or sham operation, and then perfused transcardially with normal saline followed by 4% formaldehyde solution. All brains were then postfixed in the same fixative at 4°C, dehydrated, and then embedded in paraffin blocks. Coronal sections of 5 µm thickness were stained with hematoxylin and eosin. The morphology of neurons was observed, and damaged and normal neurons were counted at 200× magnification with a ruled counting plate (Olympus, Tokyo, Japan).
Preparation of tissue extracts
Twenty-four hours after the last surgical operation, six mice per group were killed with an overdose of chloral hydrate and then transcardially perfused with ice-cold PBS (pH 7.4). The brains were removed quickly, and the cerebral cortex and hippocampus were rapidly dissected on a cold plate and frozen immediately in liquid nitrogen. All tissues were stored at −80°C until assay. Brain tissue was homogenized in 1 mL of ice-cold Tris buffer (pH 7.2, 4°C) containing 50 mM Tris, 1 mM ethylenediamine tetraacetic acid, 6 mM MgCl2 and 5% (w/v) protease inhibitor cocktail. After homogenization, samples were sonicated for 10 seconds and then centrifuged at 20,800 × g for 20 minutes at 4°C. Afterwards, supernatants were collected for western blot assay. The protein concentrations were determined in each sample using a commercially available bicinchoninic acid protein assay kit (Key GEN Biotech, Nanjing, China).
Western blot assay
To examine the expression of integrin αVβ3, VEGF and its receptor pFlk-1 in the cortex and hippocampus, western blot assay was performed as described in a previous study (Jiang et al., 2013). Samples from treated mice were resolved using sodium dodecyl sulfate polyacrylamide gradient gels (20 mg protein per lane). Proteins were transferred onto nitrocellulose membranes (Bio-Rad, Hercules, CA, USA). The membranes were blocked in 5% non-fat milk and then incubated with polyclonal mouse anti-integrin αv (1 μg/mL), mouse anti-integrin β3 (1 μg/mL), rabbit anti-VEGF (1 μg/mL) or rabbit anti-pFlk-1 (1 μg/mL) (all from Santa Cruz Biotechnology, Santa Cruz, CA, USA) overnight at 4°C. After three washes with Tris-buffered saline containing Tween-20, the membranes were incubated with anti-mouse-horseradish peroxidase (Santa Cruz Biotechnology) or goat anti-rabbit-horseradish peroxidase (Santa Cruz Biotechnology) for 30 minutes at room temperature. The experiment was performed in triplicate, and β-actin was used as an internal control. The optical density values were calculated with Quantity One image analysis software (Bio-Rad).
Statistical analysis
The data followed a normal distribution. Data were expressed as the mean ± SEM and analyzed with SPSS 13.0 software (SPSS, Chicago, IL, USA). Comparisons were performed using one-way analysis of variance followed by Bonferroni post hoc analysis. P values less than 0.05 were considered statistically significant.
Results
IP protected against ischemic brain injury
Hematoxylin-eosin staining revealed no obvious pathological abnormalities in the hippocampus in the Sham and IP groups. In comparison, neuronal cell loss, dark staining of neurons and nuclear shrinkage were observed in the Isch group. Damaged neurons were fewer in the IP + Isch group (Figure 1).
Figure 1.
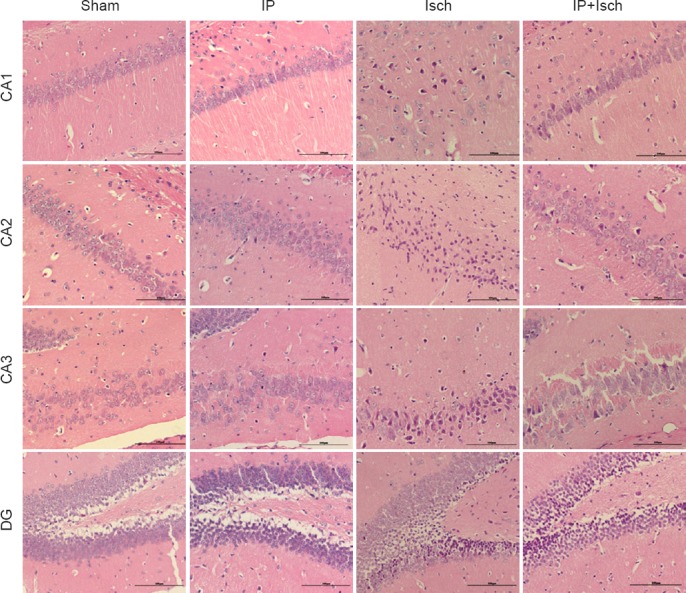
IP improved the histopathology of the hippocampus following global cerebral ischemia in mice (hematoxylin-eosin staining).
Mice were subjected to histopathological assay at 24 hours post ischemia. Hematoxylin-eosin-stained hippocampal tissue from the sham, IP, Isch and IP + Isch groups (× 200). In the sham and IP groups, no obvious pathological abnormalities are observable in the hippocampal CA1–3 and DG region. Marked morphological changes are visible in the hippocampus of the Isch group, especially in the CA1–3 region; neuronal cell loss, nuclear shrinkage and dark staining of neurons are observed. Fewer damaged neurons and more normal neurons are present in these regions in the IP + Isch group, compared with the Isch group. Scale bars: 100 μm. IP: Ischemic preconditioning; Isch: ischemia; CA: cornu ammonis; DG: dentate gyrus.
IP reduced BBB dysfunction in cerebral ischemic mice
Evans Blue assay showed that the levels of Evans Blue in the brain were higher in the Isch group than in the Sham group (P < 0.001). IP decreased Evans Blue content in the brain (P < 0.01; Figure 2).
Figure 2.

IP reduced blood-brain barrier dysfunction in mice with global cerebral ischemia.
(A–D) The whole brains in the sham, IP, Isch and IP + Isch groups. (E) The permeability of the blood-brain barrier was determined by Evans Blue extravasation test 24 hours after ischemia or sham surgery. Evans Blue content is expressed as µg/g brain tissue. Data are presented as the mean ± SEM, with eight samples in each group. One-way analysis of variance followed by Bonferroni post hoc analysis was used. **P < 0.01, vs. sham group; ##P < 0.01, vs. Isch group. IP: Ischemic preconditioning; Isch: ischemia.
IP suppressed the increase in integrin αV and β3 expression in the cerebral cortex and hippocampus following cerebral ischemia
Twenty-four hours after the ischemic insult, integrin αv protein levels in the cortex and hippocampus of mice were determined. As shown in Figure 3A, B, integrin αv protein levels in the cortex were not significantly different among Sham, IP and IP + Isch groups (~1.14-fold increase over Sham in both the IP and IP + Isch groups, with no significant difference among them). However, integrin αv levels were significantly increased in the Isch group compared with the Sham group (~2.01-fold increase; P < 0.05). Integrin αv levels were lower in the IP + Isch group compared with the Isch group. In the hippocampus, integrin αv levels in the IP group were increased slightly compared with the Sham group (~1.50-fold increase over Sham; no significant difference). Integrin αv levels were significantly elevated in the Isch group (~4.13-fold increase over Sham; P < 0.01). Levels in the IP + Isch group (~1.80-fold increase over Sham) were lower than those in the Isch group (P < 0.05).
Figure 3.

IP prevented the increase in integrin αV and β3. expression in the cerebral cortex and hippocampus of mice with global cerebral ischemia 24 hours after the ischemic event.
(A, C) Representative bands of the western blot assay for integrin αV and β3 protein expression, respectively. (B, D) The relative intensities of integrin αV and β3, respectively. β-Actin was used as an internal control. *P < 0.05, **P < 0.01, vs. sham group; #P < 0.05, vs. Isch group. Data are expressed as the mean ± SEM from six independent animals (one-way analysis of variance followed by Bonferroni post hoc analysis). The experiment was performed in triplicate. IP: Ischemic preconditioning; Isch: ischemia.
Changes in integrin β3 expression among the four groups were similar to those observed for integrin αv (Figure 3C, D). The integrin β3 levels in the IP group were 1.31-fold (cortex) and 0.96-fold (hippocampus) those in the Sham group (no significant differences). In the Isch group, the levels were elevated 3.92-fold in the cortex and 2.38-fold in the hippocampus (both P < 0.05, compared with the Sham group). In the IP + Isch group, integrin β3 levels were 1.32-fold (cortex) and 1.11-fold (hippocampus) the levels in the Sham group (both P < 0.05). Therefore, IP seems to attenuate the increase in integrin αVβ3 induced by ischemic insult.
IP prevented the increase in VEGF and pFlk-1 expression in the cerebral cortex and hippocampus 24 hours after ischemia
As shown in Figure 4A, B, VEGF expression levels in the cortex and hippocampus in the IP group were slightly increased in comparison with the Sham group (1.45-fold and 1.44-fold increases over Sham, respectively [both P > 0.05]). Expression levels in the cortex and hippocampus were increased significantly in the Isch group (~9.08-fold and ~4.20-fold increases over Sham, respectively [both P < 0.05]). The expression levels were lower in the IP + Isch group than in the Isch group (~2.37-fold and ~2.86-fold lower in the cortex and hippocampus, respectively). The difference between the Isch and IP + Isch groups was significant in the cortex (P < 0.05), but no significant difference was observed for the hippocampus. Similar trends were observed in pFlk-1 expression (Figure 4C, D). The levels of pFlk-1 in the IP group were 0.90-fold (cortex) and 1.31-fold (hippocampus) those in the Sham group (P > 0.05). In the Isch group, pFlk-1 levels were elevated 3.80-fold and 2.02-fold in the cortex and hippocampus, respectively (both P < 0.05, compared with Sham group). Expression levels in the IP + Isch group were significantly lower than those in the Isch group (1.38-fold in the cortex and 1.39-fold in the hippocampus [P < 0.05, only for the cortex]).
Figure 4.

IP prevented the increase in VEGF and pFlk-1 expression in the cerebral cortex and hippocampus in mice with global cerebral ischemia 24 hours after ischemia.
(A, C) Representative bands of the western blot assay for VEGF and pFlk-1 protein expression. B and D show the relative intensities of VEGF and pFlk-1. β-Actin was used as an internal control. *P < 0.05, vs. sham group; #P < 0.05, vs. Isch group. Data are expressed as the mean ± SEM from six independent animals (one-way analysis of variance followed by Bonferroni post hoc analysis). The experiment was performed in triplicate. IP: Ischemic preconditioning; Isch: ischemia; VEGF: vascular endothelial growth factor; pFlk-1: phosphorylated fetal liver kinase 1.
Discussion
Clinical studies suggest that IP is beneficial to the human brain. In a retrospective study, patients with a previous ipsilateral transient ischemic attack (TIA) had a more favorable outcome after cerebral infarction than patients without a prior TIA (Moncayo et al., 2000). This suggests that ischemic tolerance induced by the TIA results in a better neurological outcome after a more severe subsequent ischemic event. Numerous animal studies on the neuroprotective mechanisms of IP suggest that ischemic tolerance is produced by multiple mechanisms, including vascular changes (Gidday et al., 2006).
In this study, the expression levels of integrin αVβ3 were substantially elevated after global cerebral ischemia, consistent with other studies (Okada et al., 1996; Shimamura et al., 2006a; Kang et al., 2008). We found that IP inhibited this increase in expression of integrin αVβ3 after global cerebral ischemia. Furthermore, this effect of IP was associated with reduced ischemic injury to the brain.
To examine how IP affects integrin αVβ3 expression, two proteins, VEGF and its receptor pFlk-1, which are linked to integrin αVβ3 expression following ischemic injury, were assessed. We observed that VEGF and pFlk-1 expression levels were reduced by IP, suggesting that this reduction in VEGF levels after ischemic injury may be beneficial.
Integrin αVβ3, a member of the integrin family, plays a major role in angiogenesis and tumor growth, and has been shown to play a critical role in animal models of focal brain ischemia (Haring et al., 1996; Okada et al., 1996; Abumiya et al., 1999; Del Zoppo and Mabuchi, 2003). Previous studies have shown that inhibition of integrin αVβ3 helps preserve microvascular patency (Okada et al., 1996), reduces BBB breakdown, and ameliorates damage resulting from focal brain ischemia (Abumiya et al., 2000; Shimamura et al., 2006a, b; Kiessling et al., 2009). It has been conjectured that increased expression of integrin αVβ3 may play a harmful role during early cerebral ischemic injury, and that inhibition of integrin αVβ3 expression may reduce ischemic damage.
VEGF is an endothelial cell mitogen that enhances vascular permeability during angiogenesis. Flk-1, also known as vascular endothelial growth factor receptor-2, is a receptor for VEGF (Rosenstein et al., 1998). Flk-1 is active in its phosphorylated form. Previous studies reported that VEGF was elaborated during ischemic stroke (Hayashi et al., 1997). Although VEGF has been shown to induce angiogenesis in the penumbra and to contribute to the recovery of neuronal function after an ischemic event (Zhang et al., 2000; Manoonkitiwongsa et al., 2004; Yano et al., 2005; Udo et al., 2008), it also has myriad deleterious effects in early ischemic stroke, including increasing BBB leakage, elevating the risk of hemorrhagic transformation, widening the infarction zone (Zhang et al., 2000; Kaya et al., 2005), and increasing platelet adhesion (Verheul et al., 2004). In this study, we found that VEGF levels were lowered by IP, and that the brain was protected by IP, consistent with a deleterious role of VEGF in stroke. Therefore, the beneficial effects of IP may involve integrin αVβ3, VEGF and its receptor. Although these results are preliminary, our findings provide potential new therapeutic targets for ischemic injury.
It is known that integrin αVβ3 can activate VEGF receptors, and that the inhibition of integrin αVβ3 expression reduces phosphorylation of VEGF receptors, thereby limiting the biological effects of VEGF (Soldi et al., 1999). Furthermore, VEGF was reported to induce integrin αVβ3 expression in vitro, and expression was highly correlated with integrin αVβ3 in vivo (Abumiya et al., 1999). Hence, integrin αVβ3 expression may also be suppressed in a VEGF-dependent manner by IP. The relationship between integrin αVβ3 and VEGF is very complex, and their roles in ischemic injury remain unclear.
One of the limitations of the present study is that all animals were killed at 24 hours, whereas ischemic stroke and ischemic tolerance may occur over several days. This study focused on early brain injury, and the long-term effects of IP on integrin αVβ3 were not studied. Future studies will need to evaluate the long-term impact of IP on integrin αVβ3 and more time points should be analyzed. In addition, inhibitors of integrin αVβ3 and VEGF should be used before and after ischemia to determine the role of these proteins in the neuroprotective effects of IP.
In summary, IP improved outcome in the global cerebral ischemia model, and its effects were associated with inhibition of integrin αVβ3 through decreased expression of VEGF and its receptor. Although the mechanisms of ischemic tolerance remain unclear, this study provides insight into the mechanisms of endogenous neuroprotection, and may help in the development of novel therapeutic strategies for stroke.
Footnotes
Conflicts of interest: None declared.
Funding: This work was supported by grants from the National Natural Science Foundation of China, No. 81071068, the Israel Science Foundation-the National Natural Science Foundation of China (Joint Program), No. 813111290; and the Natural Science Foundation of Guangdong Province in China, No. 2014A030313172.
Plagiarism check: This paper was screened twice using Cross-Check to verify originality before publication.
Peer review: This paper was double-blinded and stringently reviewed by international expert reviewers.
Copyedited by Patel B, Wysong S, Wang J, Qiu Y, Li CH, Song LP, Zhao M
References
- Abumiya T, Lucero J, Heo JH, Tagaya M, Koziol JA, Copeland BR, del Zoppo GJ. Activated microvessels express vascular endothelial growth factor and integrin alpha(v)beta3 during focal cerebral ischemia. J Cereb Blood Flow Metab. 1999;19:1038–1050. doi: 10.1097/00004647-199909000-00012. [DOI] [PubMed] [Google Scholar]
- Abumiya T, Fitridge R, Mazur C, Copeland BR, Koziol JA, Tschopp JF, Pierschbacher MD, del Zoppo GJ. Integrin alpha(IIb)beta(3) inhibitor preserves microvascular patency in experimental acute focal cerebral ischemia. Stroke. 2000;31:1402–1409. doi: 10.1161/01.str.31.6.1402. [DOI] [PubMed] [Google Scholar]
- Cho S, Park EM, Zhou P, Frys K, Ross ME, Iadecola C. Obligatory role of inducible nitric oxide synthase in ischemic preconditioning. J Cereb Blood Flow Metab. 2005;25:493–501. doi: 10.1038/sj.jcbfm.9600058. [DOI] [PubMed] [Google Scholar]
- del Zoppo GJ, Mabuchi T. Cerebral microvessel responses to focal ischemia. J Cereb Blood Flow Metab. 2003;23:879–894. doi: 10.1097/01.WCB.0000078322.96027.78. [DOI] [PubMed] [Google Scholar]
- Gidday JM. Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci. 2006;7:437–448. doi: 10.1038/nrn1927. [DOI] [PubMed] [Google Scholar]
- Haring HP, Akamine BS, Habermann R, Koziol JA, Del Zoppo GJ. Distribution of integrin-like immunoreactivity on primate brain microvasculature. J Neuropathol Exp Neurol. 1996;55:236–245. doi: 10.1097/00005072-199602000-00012. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Abe K, Suzuki H, Itoyama Y. Rapid induction of vascular endothelial growth factor gene expression after transient middle cerebral artery occlusion in rats. Stroke. 1997;28:2039–2044. doi: 10.1161/01.str.28.10.2039. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Zou Y, Chen S, Zhu C, Wu A, Liu Y, Ma L, Zhu D, Ma X, Liu M, Kang Z, Pi R, Peng F, Wang Q, Chen X. The anti-inflammatory effect of donepezil on experimental autoimmune encephalomyelitis in C57BL/6 mice. Neuropharmacology. 2013;73:415–424. doi: 10.1016/j.neuropharm.2013.06.023. [DOI] [PubMed] [Google Scholar]
- Kang WS, Choi JS, Shin YJ, Kim HY, Cha JH, Lee JY, Chun MH, Lee MY. Differential regulation of osteopontin receptors, CD44 and the alpha(v) and beta(3) integrin subunits, in the rat hippocampus following transient forebrain ischemia. Brain Res. 2008;1228:208–216. doi: 10.1016/j.brainres.2008.06.106. [DOI] [PubMed] [Google Scholar]
- Kaya D, Gursoy-Ozdemir Y, Yemisci M, Tuncer N, Aktan S, Dalkara T. VEGF protects brain against focal ischemia without increasing blood--brain permeability when administered intracerebroventricularly. J Cereb Blood Flow Metab. 2005;25:1111–1118. doi: 10.1038/sj.jcbfm.9600109. [DOI] [PubMed] [Google Scholar]
- Kiessling JW, Cines DB, Higazi AA, Armstead WM. Inhibition of integrin alphavbeta3 prevents urokinase plasminogen activator-mediated impairment of cerebrovasodilation after cerebral hypoxia/ischemia. Am J Physiol Heart Circ Physiol. 2009;296:H862–867. doi: 10.1152/ajpheart.01141.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozler P, Pokorný J. Altered blood-brain barrier permeability and its effect on the distribution of Evans blue and sodium fluorescein in the rat brain applied by intracarotid injection. Physiol Res. 2003;52:607–614. [PubMed] [Google Scholar]
- Liu M, Ma X, Chen X, Jiang Y, Wu A, Peng F, Liu Y, Pi R. Ischemic preconditioning partially suppresses and postpones integrin αV β 3 mRNA expression following transient global cerebral ischemia in C57BL/6 mice. Neural Regen Res. 2010;5:1782–1786. [Google Scholar]
- Manoonkitiwongsa PS, Schultz RL, McCreery DB, Whitter EF, Lyden PD. Neuroprotection of ischemic brain by vascular endothelial growth factor is critically dependent on proper dosage and may be compromised by angiogenesis. J Cereb Blood Flow Metab. 2004;24:693–702. doi: 10.1097/01.WCB.0000126236.54306.21. [DOI] [PubMed] [Google Scholar]
- Moncayo J, de Freitas GR, Bogousslavsky J, Altieri M, van Melle G. Do transient ischemic attacks have a neuroprotective effect? Neurology. 2000;54:2089–2094. doi: 10.1212/wnl.54.11.2089. [DOI] [PubMed] [Google Scholar]
- Okada Y, Copeland BR, Hamann GF, Koziol JA, Cheresh DA, del Zoppo GJ. Integrin alphavbeta3 is expressed in selected microvessels after focal cerebral ischemia. Am J Pathol. 1996;149:37–44. [PMC free article] [PubMed] [Google Scholar]
- Park YS, Cho JH, Kim IH, Cho GS, Park JH, Ahn JH, Chen BH, Shin BN, Shin MC, Tae HJ, Cho YS, Lee YL, Kim YM, Won MH, Lee JC. Effects of ischemic preconditioning on VEGF and pFlk-1 immunoreactivities in the gerbil ischemic hippocampus after transient cerebral ischemia. J Neurol Sci. 2014;347:179–187. doi: 10.1016/j.jns.2014.09.044. [DOI] [PubMed] [Google Scholar]
- Rosenstein JM, Mani N, Silverman WF, Krum JM. Patterns of brain angiogenesis after vascular endothelial growth factor administration in vitro and in vivo. Proc Natl Acad Sci U S A. 1998;95:7086–7091. doi: 10.1073/pnas.95.12.7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura N, Matchett G, Solaroglu I, Tsubokawa T, Ohkuma H, Zhang J. Inhibition of integrin alphavbeta3 reduces blood-brain barrier breakdown in focal ischemia in rats. J Neurosci Res. 2006a;84:1837–1847. doi: 10.1002/jnr.21073. [DOI] [PubMed] [Google Scholar]
- Shimamura N, Matchett G, Yatsushige H, Calvert JW, Ohkuma H, Zhang J. Inhibition of integrin alphavbeta3 ameliorates focal cerebral ischemic damage in the rat middle cerebral artery occlusion model. Stroke. 2006b;37:1902–1909. doi: 10.1161/01.STR.0000226991.27540.f2. [DOI] [PubMed] [Google Scholar]
- Soldi R, Mitola S, Strasly M, Defilippi P, Tarone G, Bussolino F. Role of alphavbeta3 integrin in the activation of vascular endothelial growth factor receptor-2. EMBO J. 1999;18:882–892. doi: 10.1093/emboj/18.4.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udo H, Yoshida Y, Kino T, Ohnuki K, Mizunoya W, Mukuda T, Sugiyama H. Enhanced adult neurogenesis and angiogenesis and altered affective behaviors in mice overexpressing vascular endothelial growth factor 120. J Neurosci. 2008;28:14522–14536. doi: 10.1523/JNEUROSCI.3673-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheul HM, Jorna AS, Hoekman K, Broxterman HJ, Gebbink MF, Pinedo HM. Vascular endothelial growth factor-stimulated endothelial cells promote adhesion and activation of platelets. Blood. 2000;96:4216–4221. [PubMed] [Google Scholar]
- Wu C, Zhan RZ, Qi S, Fujihara H, Taga K, Shimoji K. A forebrain ischemic preconditioning model established in C57Black/Crj6 mice. J Neurosci Methods. 2001;107:101–106. doi: 10.1016/s0165-0270(01)00356-9. [DOI] [PubMed] [Google Scholar]
- Yano A, Shingo T, Takeuchi A, Yasuhara T, Kobayashi K, Takahashi K, Muraoka K, Matsui T, Miyoshi Y, Hamada H, Date I. Encapsulated vascular endothelial growth factor-secreting cell grafts have neuroprotective and angiogenic effects on focal cerebral ischemia. J Neurosurg. 2005;103:104–114. doi: 10.3171/jns.2005.103.1.0104. [DOI] [PubMed] [Google Scholar]
- Zhang ZG, Zhang L, Jiang Q, Zhang R, Davies K, Powers C, Bruggen N, Chopp M. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J Clin Invest. 2000;106:829–838. doi: 10.1172/JCI9369. [DOI] [PMC free article] [PubMed] [Google Scholar]