

Abstract
Many cellular functions are critically dependent on the folding of complex multimeric proteins, such as p97, a hexameric multi-domain AAA+ chaperone. Given p97’s complex architecture, single-molecule Förster Resonance Energy Transfer (smFRET) would be a powerful tool for studying folding while avoiding ensemble averaging. However, dual site-specific labeling of such a large protein for smFRET is a significant challenge. Here, we address this issue by using bioorthogonal azide-alkyne chemistry to attach an smFRET dye-pair to site-specifically incorporate unnatural amino acids, allowing us to generate p97 variants reporting on inter- or intra-domain structural features. An initial proof-of-principle set of smFRET results demonstrates the strengths of the labeling method. Our results highlight a powerful tool for structural studies of p97 and other large protein machines.
Keywords: p97, FRET, Single-molecule studies, Unnatural amino acid, Protein folding
Graphical Abstract
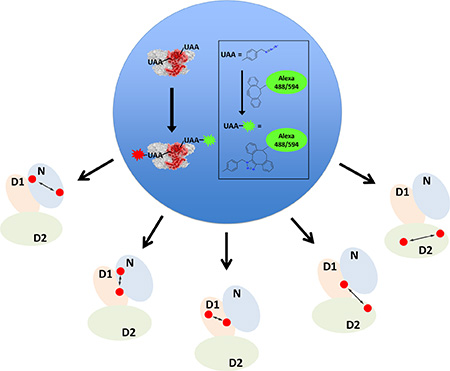
Watching one p97 at a time. The single-molecule biophysical chemistry of p97, a complex, cysteine-rich, essential cellular protein machine, can now be studied using smFRET, thanks to use of advanced site-specific dye labeling via an unnatural amino acid and click chemistry.
p97, also called VCP or Cdc48p (in yeast), is an essential AAA+ chaperone[1] that is involved in numerous critical cellular functions such as proteostasis, cell-cycle progression, membrane fusion, and transcription factor regulation. These diverse biochemical functions also implicate p97 in a variety of pathological states such as neurodegenerative diseases (Huntington’s and Parkinson’s diseases, and amyotrophic lateral sclerosis (ALS)) and cancer.[2] p97 is a type II AAA+ protein with two AAA+ ATPase domains, D1 and D2, and an N-terminal domain. Like other AAA+ chaperones, the active form of p97 is a homohexamer.[3] The precise roles of the D1 and D2 domains remain controversial, however it is well established that the D1 domain is sufficient and necessary to catalyze hexamer assembly, even in the absence of nucleotide.[4] Despite structural and biochemical knowledge of p97 tertiary and quaternary structure, understanding the details of folding and assembly remain opaque. In fact, understanding such details for any large, macromolecular machine remains a challenge.
The development of single-molecule Förster Resonance Energy Transfer (smFRET) has proven to be a powerful tool to reveal structural, stochastic, and kinetic details of events such as protein folding that are critical to biomolecule function and organism survival.[5] However, the application of smFRET to proteins has been generally restricted to smaller proteins, with the complexity of site-specific dye labeling being a major hurdle for studies in larger systems. The most common method for site-specific labeling has been the use of nucleophile-reactive maleimides, by which fluorophores are attached to native or introduced cysteine residues in the target protein sequence. However, many enzymes are large proteins with many, often critical native cysteine residues. In many cases for smFRET, to avoid non-specific labeling, cysteine residues are removed from the native sequence using mutagenesis. However, since the removal of multiple cysteine residues can alter the structural characteristics of the protein under investigation, the cysteine labeling method is limited in scope. p97 falls into this class, as it contains 12 native cysteine residues per monomer (72 total), many that are important for proper tertiary and/or quaternary structure formation.[6] Therefore, alternative labeling methods have been developed, and one such approach, unnatural amino acid mutagenesis, has been used in a few cases, albeit mostly for smaller, test-case proteins.[7] Few studies of large proteins using this method to avoid non-specific cysteine residue labeling have been reported.[8] In this study, we incorporated a bio-orthogonal unnatural amino acid simultaneously at two different sites of the p97 monomer to study its folding characteristics using smFRET. p97 is over 0.5 MDa as a hexamer, which is to our knowledge, the largest protein to have two unnatural amino acids simultaneously and site-specifically incorporated for FRET labeling (Figure 1a). Moreover, since unnatural amino acid incorporation can be applied at any position in the sequence, we were able to prepare multiple labeling variants representing various structural features of p97 for smFRET studies.
Figure 1.
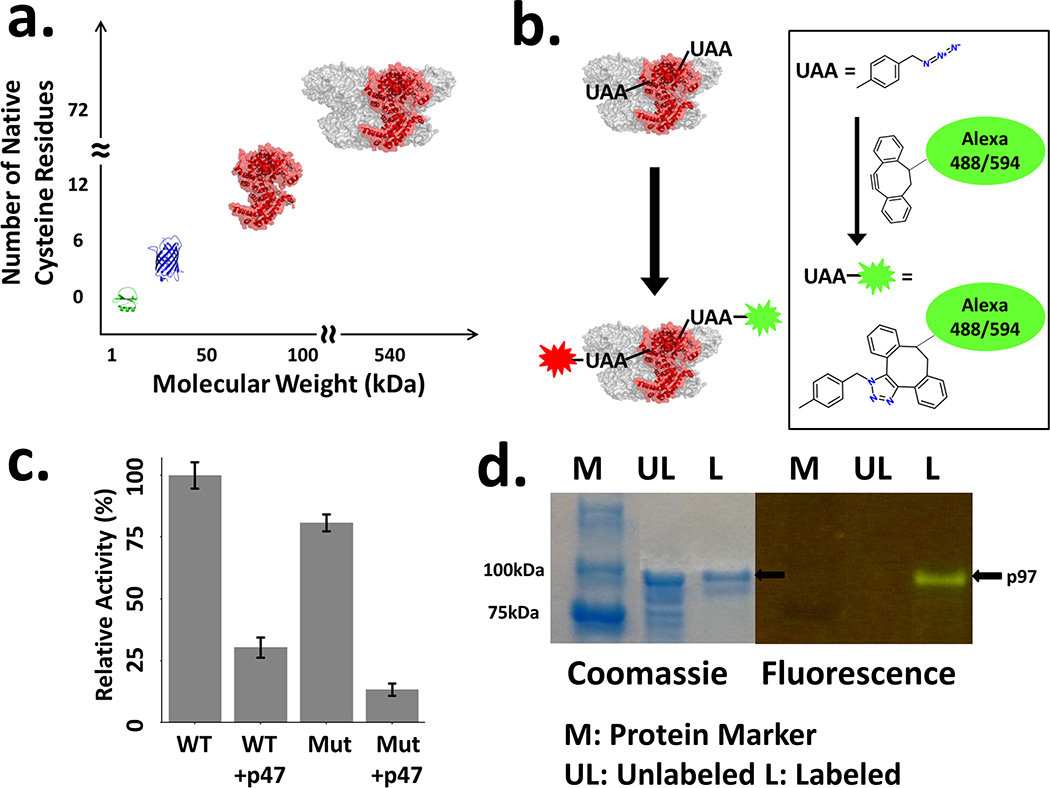
(a) Size comparison of p97 and other proteins studied by site specific labeling and smFRET. CI2 (Green, PDB ID 1YPC), the first 2-state protein studied by smFRET. GFP (Blue, PDB ID 1EMA), commonly used model protein for unnatural amino acid incorporation. p97 monomer and hexamer (Red/Gray, PDB ID 3CF1), the monomer is colored red. (b) Schematic of fluorophore labeling of a multi-unnatural amino acid p97 mutant with copper-free ‘click’ chemistry. (c) ATPase activity assay of p97 wild-type (with C-terminal His-6 tag) and mutant (F131Q382) in the absence or presence of p47. Error bars indicate twice the standard deviation of the measured activities. (d) SDS-PAGE for Alexa Fluor 488 labeled p97 (F131Q382). Coommassie stain (left) and fluorescence (right, excitation wavelength 450 nm). Multiple bands in unlabeled protein lane indicate impurities, which are removed during further purification of labeled protein. Only the relevant portion of the gel is shown. For original images, see Supporting Information, Figure S1.
The p97 constructs used in the present studies were ex-pressed in BL21 (DE3) E. coli cells from a T7 RNA polymerase driven promoter. The wild type full-length human p97 sequence was used as the template for unnatural amino acid incorporation with a HisTag incorporated at the C-terminus for rapid purification using immobilized metal affinity chromatography. The C-terminus was chosen to facilitate purification of full-length p97 from truncated products resulting from inefficient suppression of the engineered amber mutations (TAG). As the initial test case for double UAA incorporation two amber mutations were introduced, F131 (N domain) and Q382 (D1 domain), so the corresponding smFRET experiment would report on N-D1 interdomain structure. We chose surface residues because we reasoned that mutations at the surface were less likely to perturb the structure and function of p97, and the crystal structure indicated that the distance between the N and D1 domains are appropriate for FRET investigations (~30Å). Moreover, these residues showed low conservation scores (PDBsum), again arguing against perturbations. Based on SDS PAGE analysis, over 50% of the p97 successfully incorporated the unnatural amino acid at both sites. After optimization, large-scale expression resulted in ~10 mg of full-length, pure protein per 1 L of LB media. (Supporting Information Table S1) Site-specific incorporation of p-azidomethylphenylalanine (Figure 1b), which is more photostable due to the one carbon homologation, and not reduced in the E. coli cytosol like the more commonly used p-azidophenylalanine,[9] was carried out using a previously reported amber suppression procedure.[10] SDS-PAGE analysis of the protein sample was used to confirm protein size, and a standard ATPase activity assay was performed using malachite green[11] (Figure 1c, d). Moreover, subjecting the wild-type protein without unnatural amino acids to the labeling conditions did not show detectable labeling, which confirmed that the labeling was specific to the unnatural amino acid. It has been shown in the literature that only hexameric p97 has ATPase activity, which together with a similarly observed level of activity of the F131Q382 p97 (Figure 1c), led us to conclude that the variant p97 assembles into the correct quaternary structure. To further probe biochemical function, we measured binding of the cofactor, p47. p47 is known to inhibit p97 ATPase activity,[12] which was observed when p47 was added to p97 (Figure 1c). Finally, the site-specificity of unnatural amino acid incorporation was confirmed by mass spectrometry (Supporting Information, Figure S2).
We next used the copper-free, bio-orthogonal click reaction with a strained and electronically activated alkyne (DIBO, Figure 1b) to couple fluorescent dyes to the site-specifically incorporated unnatural amino acids. Thus, DIBO substituted Alexa Fluor 488 (donor) and 594 (acceptor) dyes were used to label p97 (Figure 1b). The concentration of the protein and dyes in the labeled species was measured using UV-vis absorption spectroscopy, revealing an ~1.2 to 1.4 ratio of total dyes to proteins. This again indicated successful incorporation of multiple unnatural amino acids on the protein. Moreover, the labeled protein was analyzed on SDS-PAGE gel, which confirmed covalent labeling of full-length p97 (Figure 1d).
Next, a home-built confocal microscope system was used to test this N-D1 labeled variant by smFRET, following procedures described previously.[15] Briefly, 488 nm laser light was used to excite donor dyes in a solution of ~250 pM freely diffusing labeled p97 molecules. Unlabeled wildtype protein was added in excess (100 nM monomer) to reduce sticking of labeled protein to the walls of the smFRET cuvette. Donor and acceptor signals were simultaneously recorded, and used to measure FRET efficiencies (EFRET) from individual molecules. EFRET for a number of single molecules, one at a time, were collected in this way, and then plotted in the form of an EFRET histogram (Figure 2a), which provides information about populations of molecules as a function of EFRET. Previous literature indicated that the p97 hexamer disassembles into monomer in a predominantly two-state fashion, by non-denaturing gel analysis.[14] The same trend was also observed here by smFRET with F131Q382 labeling, which represented the folding between N domain and D1 domains (Figure 2a). The histograms showed loss of the native peak and growth of a denatured state peak as a function of increasing urea concentration. Together, the above results showed that this method of p97 labeling was effective for dual-labeling for smFRET experiments.
Figure 2.
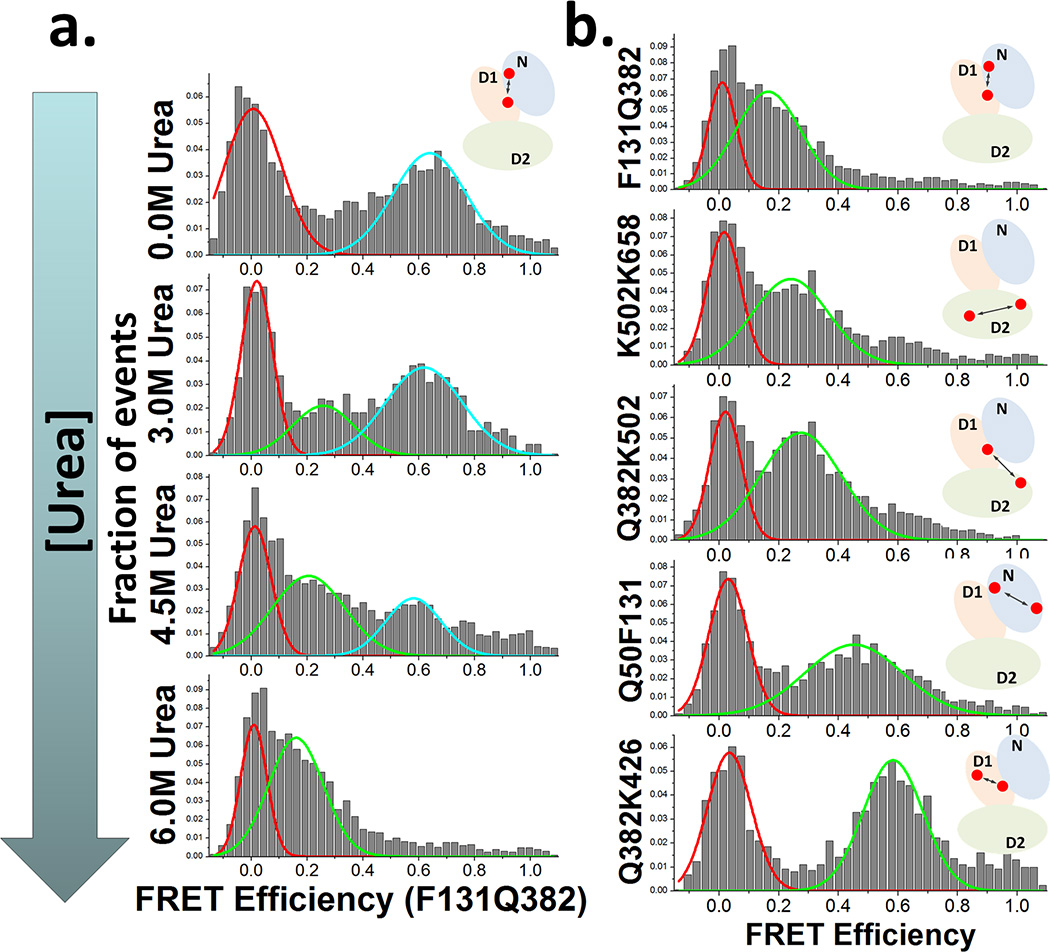
(a) EFRET histograms of p97F131Q382 as a function of increasing amounts of urea. The y-axis for each histogram is the relative fraction of events per histogram column. Solid lines indicate Gaussian fits to the data. The peaks centered at EFRET ~ 0 indicate proteins labelled with only the donor dye or otherwise missing fluorescent acceptor. (b) EFRET histograms of several labelling variants at 6M urea.
Next, we leveraged the flexibility of the labeling scheme to demonstrate labeling at other strategically chosen pairs of positions in p97. We again labeled surface residues, but now at positions that reported on other domain characteristics of p97. The Q50F131, Q382K426, and K502K658 intradomain labeling variants are designed to report on the N, D1, and D2 domains, respectively. We also prepared a Q382K502 variant, which would provide information about D1-D2 interdomain conformational properties. To demonstrate proof-of-principle, we carried out single molecule experiments as described above for these variants and present smFRET histograms for the denatured states of these proteins in 6M urea in Figure 2b. The denatured states of the five different variants show increasing FRET efficiencies in the order F131Q382 (N-D1), K502K658 (D2), Q382K502 (D1-D2), Q50F131 (N) and Q382K426 (D1), as anticipated from the decreasing sequence separation of the two dyes along the protein. The data are therefore consistent with covalent and site-specific attachment of the donor and acceptor dyes.
In summary, we demonstrate site-specific FRET labeling of a large, multimeric, multidomain enzyme p97 by use of unnatural amino acid mutagenesis and click chemistry in a background of a large number of native cysteine residues. The flexibility of the method was demonstrated by producing p97 variants site-specifically labeled at multiple different pairs of sites that were designed to allow smFRET studies of different folding features of the protein. An initial set of smFRET data is consistent with the different sequence separations of the labeling variants. Given the complexity of this protein system, the reaction pathway from unfolded monomer to folded hexamer most likely consists of a number of steps, including folding within domains, and interactions between domains in a monomer and between monomers in an oligomer. Thus, the current work now opens the door to more detailed equilibrium and kinetic studies (see discussion in Supporting Information) of these and other p97 constructs to better understand folding, assembly and function of this complex protein system. Finally, our study also highlights the power of the unnatural amino acid labeling technique for potentiating single-molecule studies on a broad range of large protein machines and complexes that are critical in cellular function.
Experimental Section
More detailed experimental procedures can be found in the Supporting Information.
Supplementary Material
Acknowledgments
This work was supported by grants MCB 1121959 (National Science Foundation, to A.A.D.), RO1 GM066833 (NIGMS, Na-tional Institutes of Health, to A.A.D), start-up funds provided by the University of Arizona and National Institute of Environmental Health Sciences RO1 ES023758 (E.C.). T.C.L. was supported by scholarship from Kwanjeong Educational Foundation for his work.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Contributor Information
Prof. Dr. Eli Chapman, Email: chapman@pharmacy.arizona.edu.
Prof. Dr. Ashok A. Deniz, Email: deniz@scripps.edu.
References
- 1.a) Davies JM, Brunger AT, Weis WI. Structure. 2008;16:715–726. doi: 10.1016/j.str.2008.02.010. [DOI] [PubMed] [Google Scholar]; b) Huang C, Li G, Lennarz WJ. Proc Natl Acad Sci U S A. 2012;109:9792–9797. doi: 10.1073/pnas.1205853109. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Li G, Huang C, Zhao G, Lennarz WJ. Proc Natl Acad Sci U S A. 2012;109:3737–3741. doi: 10.1073/pnas.1200255109. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Noi K, Yamamoto D, Nishikori S, Arita-Morioka KI, Kato T, Ando T, Ogura T. Structure. 2013 doi: 10.1016/j.str.2013.08.017. [DOI] [PubMed] [Google Scholar]; e) Pye VE, Dreveny I, Briggs LC, Sands C, Beuron F, Zhang X, Freemont PS. J Struct Biol. 2006;156:12–28. doi: 10.1016/j.jsb.2006.03.003. [DOI] [PubMed] [Google Scholar]; f) Song C, Wang Q, Li CC. J Biol Chem. 2003;278:3648–3655. doi: 10.1074/jbc.M208422200. [DOI] [PubMed] [Google Scholar]
- 2.a) Chapman E, Fry AN, Kang M. Mol Biosyst. 2011;7:700–710. doi: 10.1039/c0mb00176g. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Meyer H, Bug M, Bremer S. Nat Cell Biol. 2012;14:117–123. doi: 10.1038/ncb2407. [DOI] [PubMed] [Google Scholar]; c) Tang WK, Xia D. J Struct Biol. 2012;179:83–92. doi: 10.1016/j.jsb.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Yamanaka K, Sasagawa Y, Ogura T. Biochim Biophys Acta. 2012;1823:130–137. doi: 10.1016/j.bbamcr.2011.07.001. [DOI] [PubMed] [Google Scholar]; e) Chapman E, Maksim N, de la Cruz F, La Clair JJ. Molecules. 2015;20:3027–3049. doi: 10.3390/molecules20023027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Erzberger JP, Berger JM. Annu Rev Biophys Biomol Struct. 2006;35:93–114. doi: 10.1146/annurev.biophys.35.040405.101933. [DOI] [PubMed] [Google Scholar]
- 4.Wang Q, Song C, Yang X, Li CC. J Biol Chem. 2003;278:32784–32793. doi: 10.1074/jbc.M303869200. [DOI] [PubMed] [Google Scholar]
- 5.a) Kim H, Ha T. Rep Prog Phys. 2013;76:016601. doi: 10.1088/0034-4885/76/1/016601. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Banerjee PR, Deniz AA. Chem Soc Rev. 2014;43:1172–1188. doi: 10.1039/c3cs60311c. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Schuler B, Eaton WA. Curr Opin Struct Biol. 2008;18:16–26. doi: 10.1016/j.sbi.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Tao S, Tillotson J, Wijeratne EM, Xu YM, Kang M, Wu T, Lau EC, Mesa C, Mason DJ, Brown RV, La Clair JJ, Gunatilaka AA, Zhang DD, Chapman E. ACS Chem Biol. 2015 doi: 10.1021/acschembio.5b00367. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kang MJ, Wu T, Wijeratne EM, Lau EC, Mason DJ, Mesa C, Tillotson J, Zhang DD, Gunatilaka AA, La Clair JJ, Chapman E. Chembiochem. 2014;15:2125–2131. doi: 10.1002/cbic.201402258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Brustad EM, Lemke EA, Schultz PG, Deniz AA. J Am Chem Soc. 2008;130:17664–17665. doi: 10.1021/ja807430h. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chatterjee A, Sun SB, Furman JL, Xiao H, Schultz PG. Biochemistry. 2013;52:1828–1837. doi: 10.1021/bi4000244. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kim J, Seo MH, Lee S, Cho K, Yang A, Woo K, Kim HS, Park HS. Anal Chem. 2013;85:1468–1474. doi: 10.1021/ac303089v. [DOI] [PubMed] [Google Scholar]; d) McLoughlin SY, Kastantin M, Schwartz DK, Kaar JL. Proc Natl Acad Sci U S A. 2013;110:19396–19401. doi: 10.1073/pnas.1311761110. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Tyagi S, Lemke EA. Curr Opin Struct Biol. 2015;32:66–73. doi: 10.1016/j.sbi.2015.02.009. [DOI] [PubMed] [Google Scholar]
- 8.a) Cooper DR, Dolino DM, Jaurich H, Shuang B, Ramaswamy S, Nurik CE, Chen J, Jayaraman V, Landes CF. Biophys J. 2015;109:66–75. doi: 10.1016/j.bpj.2015.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dolino DM, Cooper D, Ramaswamy S, Jaurich H, Landes CF, Jayaraman V. J Biol Chem. 2015;290:797–804. doi: 10.1074/jbc.M114.605436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bazewicz CG, Liskov MT, Hines KJ, Brewer SH. J Phys Chem B. 2013;117:8987–8993. doi: 10.1021/jp4052598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Young TS, Ahmad I, Yin JA, Schultz PG. J Mol Biol. 2010;395:361–374. doi: 10.1016/j.jmb.2009.10.030. [DOI] [PubMed] [Google Scholar]
- 11.Geladopoulos TP, Sotiroudis TG, Evangelopoulos AE. Anal Biochem. 1991;192:112–116. doi: 10.1016/0003-2697(91)90194-x. [DOI] [PubMed] [Google Scholar]
- 12.Meyer HH, Kondo H, Warren G. FEBS Lett. 1998;437:255–257. doi: 10.1016/s0014-5793(98)01232-0. [DOI] [PubMed] [Google Scholar]
- 13.Ferreon AC, Gambin Y, Lemke EA, Deniz AA. Proc Natl Acad Sci USA. 2009;106:5645–5650. doi: 10.1073/pnas.0809232106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Q, Song C, Li CC. Biochem Biophys Res Commun. 2003;300:253–260. doi: 10.1016/s0006-291x(02)02840-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.