

Summary
Turkey connects the Middle East, Europe, and Asia and has experienced major population movements. We examined the population structure and genetic relatedness of samples from three regions of Turkey using over 500,000 SNP genotypes. The data were analyzed together with Human Genome Diversity Panel data. To obtain a more representative sampling from Central Asia, Kyrgyz samples (Bishkek, Kyrgyzstan) were genotyped and analyzed. Principal component (PC) analysis reveals a significant overlap between Turks and Middle Easterners and a relationship with Europeans and South and Central Asians; however, the Turkish genetic structure is unique. FRAPPE, STRUCTURE, and phylogenetic analyses support the PC analysis depending upon the number of parental ancestry components chosen. For example, supervised STRUCTURE (K = 3) illustrates a genetic ancestry for the Turks of 45% Middle Eastern (95% CI, 42–49), 40% European (95% CI, 36–44), and 15% Central Asian (95% CI, 13–16), whereas at K = 4 the genetic ancestry of the Turks was 38% European (95% CI, 35–42), 35% Middle Eastern (95% CI, 33–38), 18% South Asian (95% CI, 16–19), and 9% Central Asian (95% CI, 7–11). PC analysis and FRAPPE/STRUCTURE results from three regions in Turkey (Aydin, Istanbul, and Kayseri) were superimposed, without clear subpopulation structure, suggesting the selected samples were rather homogeneous. Thus, this study demonstrates admixture of Turkish people reflecting the population migration patterns.
Introduction
Analysis of population genetic substructure has been improved by using high-density single nucleotide polymorphism (SNP) arrays. Knowledge of the patterns of variation within continental populations is useful for several reasons, such as understanding the origin and migration of population groups and providing information on allele frequency for genetic association studies. Recent genome-wide association studies have shown that discovering and accounting for differences (e.g., controlling for population structure even at a fine level within a seemingly homogeneous population) in substructure can reduce error rates in association studies (McClellan & King, 2010; Price et al., 2010; Rosenberg et al., 2010; Tian et al., 2008).
The Human Genome Diversity Panel (HGDP) (Cavalli-Sforza, 2005) has facilitated the discovery of the origin of human genetic diversity, genetic relatedness, and population structure among world populations by providing samples of genomic DNA and genotype data (Li et al., 2008; Rosenberg et al., 2002) (Cann et al., 2002). In addition, several non-HGDP populations have been analyzed (Hunter-Zinck et al., 2010; Teo et al., 2009; Xing et al., 2010; Xu & Jin, 2008) together with HGDP samples. However, the structure of the Turkish population has not been analyzed using high-density SNP genotypes. The Anatolian peninsula (present-day Turkey) connects the Middle East, Europe, and Asia, and thus has been subject to major population movements (Findley, 2005b; Grousset, 1970; Güvenç, 1993). Previous studies of genetic variations in the Turkish population examined mitochondrial DNA sequence variation (Calafell et al., 1996; Mergen et al., 2004; Quintana-Murci et al., 2004), polymorphic markers on the Y chromosome (Cinnioğlu et al., 2004; Regueiro et al., 2006), and some polymorphic loci in autosomal chromosomes (Berkman et al., 2008; Di Benedetto et al., 2001) with relatively few genetic markers.
Previously we have studied the risk factors for coronary artery disease in the Turkish population (Bersot et al., 1999; Mahley et al., 1995; Mahley et al., 2000; Mahley et al., 2001), a population known to have a high prevalence of heart disease (Onat, 2001; Onat et al., 2003). One of the major risk factors is low levels of high density lipoprotein-cholesterol (Bersot et al., 2003). Association studies of candidate genes of lipid metabolism (Hodoğlugil et al., 2005a; Hodoğlugil et al., 2005b; Hodoğlugil et al., 2006; Hodoğlugil et al., 2010) and a genome-wide scan (Ling et al., 2009; Yu et al., 2005) have identified multiple genes that contribute to the Turkish lipid phenotype. Recently, a unique gene—glucuronic acid epimerase—was shown to be associated with both high density lipoprotein-cholesterol and triglyceride levels in the Turkish population (Hodoğlugil et al., 2011). Interestingly the SNP frequency pattern across the locus for this gene more resembled an Asian pattern, whereas the SNP frequency surrounding this locus on chromosome 15q21-23 was more similar to a European pattern, suggesting the importance of recombination events explaining unique population-specific phenotypes. Thus, in the present study, we sought to analyze the genetic ancestry of the Turkish population with respect to publicly available HGDP samples (http://hagsc.org/hgdp/files.html) (Li et al., 2008).
To achieve a more representative sampling from Central Asia relevant to Turkish history (Findley, 2005b; Grousset, 1970; Güvenç, 1993), we also genotyped samples from another Central Asian population, Kyrgyz from Bishkek, Kyrgyzstan. The Central Asian populations in the HGDP are represented by the Uygur and Hazara populations. In addition, to determine whether subpopulations exist among our study subjects, we analyzed Turkish samples from three regions in Turkey (Istanbul, Aydin and Kayseri) (Fig. 1). Thus, we genotyped 64 Turkish and 16 Kyrgyz samples, and then combined the data sets with the HGDP data set to examine genetic relatedness and population substructure among Eurasian populations.
Figure 1.
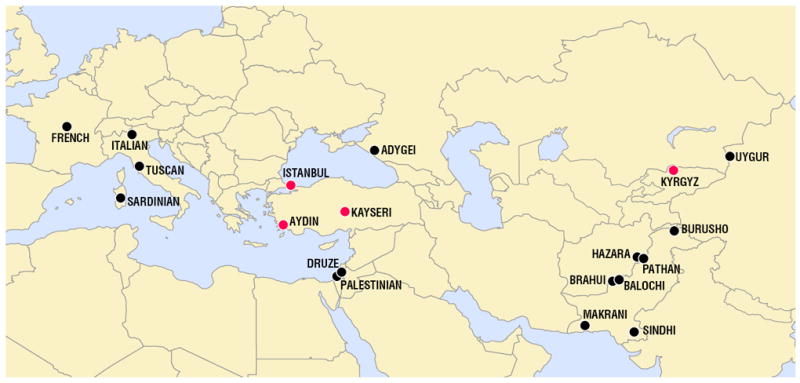
Geographical locations of samples used in this study. Turkish (Istanbul, Aydin, and Kayseri) and Kyrgyz samples are shown in red; populations from the HGDP are shown in black.
Materials and Methods
Study Population, Genotyping, and SNP Quality Control
Sixty-four unrelated Turkish samples (including one duplicate pair) from three locations in Turkey (Istanbul, Aydin, and Kayseri) were selected from participants in the Turkish Heart Study (Mahley et al., 1995). Istanbul is a cosmopolitan city of over 12 million and a major hub for other parts of Turkey. All Istanbul samples were selected from the city itself. Aydin is a mid-size city (population: 188,000) near the Aegean coast, and Kayseri is a relatively large city (population: 1,200,000) in central Turkey. Samples from the Aydin and Kayseri regions were selected from city centers and from several nearby towns and villages. All samples were obtained from individuals who were born and lived in these regions at the time the samples were collected (Fig. 1). In addition, 16 Kyrgyz samples were randomly selected from a Kyrgyz cohort obtained at the Kyrgyz National Center of Cardiology and Internal Medicine in Bishkek, Kyrgyzstan. All participants were queried about their ethnicity, and only participants indicating Turkish or Kyrgyz ethnicity were included in the study. Equal numbers of males and females were included, and all samples were obtained from healthy individuals under controlled conditions as described (Mahley et al., 1995). The protocols were approved by the Committee on Human Research of the University of California, San Francisco, and were in accordance with the Helsinki Declaration.
DNA was extracted from blood with a Qiagen blood kit. DNA samples with an A260/A280 ratio >1.8 quantified with a Nanodrop spectrophotometer were utilized for genotyping with Infinium Human 610-quad BeadChip assays (Illumina, San Diego, CA), according to the manufacturer’s specifications. All samples had call rates >98%. The rate of concordance between a pair of duplicate samples was >99.99%. SNPs were filtered out if they differed between duplicate samples or if their call rates across the 80 samples were <95%. Hardy-Weinberg equilibrium was tested separately in Turkish and Kyrgyz populations. SNPs that deviated from Hardy-Weinberg equilibrium (p < 0.001, n = 590 for Turks and n = 1781 for Kyrgyz) were also excluded. Only autosomal chromosomes were utilized. These filtering and exclusion criteria resulted in 571,852 high-quality SNPs. The genotype data set is available upon request from the authors.
Recently, HGDP samples were genotyped (n = 1043) with Illumina HumanHap650K BeadChips (Illumina), and the genotype data were made publicly available (Li et al., 2008). HGDP genotype data from unrelated HGDP subjects (n = 938) (Rosenberg, 2006) were combined with our filtered 79-sample set (excluding one individual from a duplicate pair) and resulted in high-quality genotypes for 533,261 SNPs.
Three different SNP sets were used in the analysis—all SNPs (533,261), linkage disequilibrium (LD)–pruned SNPs (105,382), and a further trimmed smaller set of SNPs (6,408). To prune SNPs for pairwise LD threshold r2 > 0.2, we used PLINK (Purcell et al., 2007) and the --indep-pairwise command, which removes one of a pair of SNPs if r2 > 0.2 in 50-SNP windows, repeats this process for every pair, and then shifts the window 5 SNPs forward and repeats the entire procedure again. This resulted in 105,382 SNPs in the Turkish samples. The LD-pruned (r2 < 0.2) SNP set was further trimmed by using high Fst SNPs between HapMap (phase II) European and East Asian samples. First, high Fst SNPs (CEU vs. CHB + JPT > 0.250) were selected and thinned if adjacent SNPs were <0.1 cM apart and were filled with SNPs 0.25 > Fst > 0.20 if they were >1 cM apart. This resulted in 6,408 SNPs. Pairwise HapMap Fst and mapping (cM) data for individual SNPs were provided by Stephen Schaffner (Broad Institute of MIT and Harvard) and Tara Matise (Rutgers University), respectively.
Principal Component Analysis for Inference of Population Affinities
Autosomal SNP genotypes were used to examine the relationship between individuals by principal component (PC) analysis with the smartpca program distributed with EIGENSTRAT (Patterson et al., 2006). The LD-pruned (r2 < 0.2, n = 105,382) SNP set was used, and no genetic outliers were removed. PC analysis was conducted on all samples and on selected samples from Eurasia separately without using population labels. The pairwise combinations of up to four components were plotted to illustrate the genetic relatedness among individuals/populations. Turkish and Kyrgyz samples were combined with the HGDP samples and analyzed with smartpca.
To confirm the validity of results, we computed the identity-by-state (IBS) matrix among the 1017 individuals (Turkish, Kyrgyz, and HGDP samples) with PLINK, producing a 1017-by-1017 matrix utilizing all SNPs. We then performed multi-dimensional scaling plots on this IBS matrix and used the top two components to illustrate the genetic relatedness among individuals.
Inference of Population Clustering with FRAPPE, STRUCTURE, and CLUMMP
To assess population substructure from the high-density genetic marker data, we used FRAPPE 1.1 (EM algorithm) (Tang et al., 2005) and STRUCTURE v2.2 (Bayesian clustering algorithm) (Falush et al., 2003). For FRAPPE analysis, owing to computer time constraints, the LD-pruned (r2 < 0.2, n = 105,382) SNP set was utilized with 20 populations selected from all continental/geographical regions representing 339 individuals. This FRAPPE analysis considers each person’s genome as having originated from K parental populations (K = 2–7), whose contributions are described by coefficients that add up to 100% for each individual. For STRUCTURE analysis, default parameter settings of 30,000 replicates and 30,000 burn-in cycles were used. Because STRUCTURE has a large memory demand, the set of 6,408 SNPs was used. Ancestry coefficient estimates from 10 individual STRUCTURE runs for each parental population (K = 2–7) were conducted with a lab computer or computer clusters at the Computational Biology Service Unit, Cornell University (http://cbsuapps.tc.cornell.edu/index.aspx) utilizing all samples or a subset of samples. The estimated ln probability of data [ln Pr(X∣K)] was consistent across independent runs, and the appropriate number of clusters is six or seven for this data set (Falush et al., 2003). STRUCTURE results were analyzed with CLUMMP (Jakobsson & Rosenberg, 2007), which permutes the cluster output by independent runs of clustering programs such as STRUCTURE, so that they match up as closely as possible. Supervised STRUCTURE analysis was performed using selected parental populations as described in the text.
Fst Calculations and Phylogenetic Tree Building
By including population labels in the parameter file while running the program, we calculated an Fst matrix with the smartpca function of EIGENSTRAT simultaneously with the PC analysis. The phylogenetic tree was built with the Fst matrix in MEGA4 using the neighbor-joining method (Saitou & Nei, 1987; Tamura et al., 2007).
Allele Frequency Spectrum Comparison
Genome-wide allele frequency comparisons between population pairs were completed utilizing all SNPs, and heat maps were used to visualize the allele frequency distributions across pairs of populations (R, hexbin package, http://cran.r-project.org/). Frequencies of reference forward allele are reported. All allele frequency values were used, and no cut-off values were applied. Pearson’s correlation was calculated for population pairs.
Patterns of Decay of LD and Haplotype Diversity
For each chromosome, we randomly selected a 1-Mb region, avoiding centromeres, genomic regions with low SNP density, and known segmental duplications. Genotype data were phased with fastPhase (Scheet & Stephens, 2006) software separately for each population, using default parameters. LD (r2 and D’) was measured by pairwise comparison between SNP markers that had a minor allele frequency ≥15% using phased genotype data in Haploview (Barrett et al., 2005). The LD between a focal SNP and any SNP within a 250-kb upstream or downstream region of the focal SNP was calculated. Haplotype blocks were calculated with the Gabriel method (Barrett et al., 2005) in Haploview using phased genotype data, and haplotypes (frequency >5%) were counted in each selected genomic region for each population separately.
The number of subjects in the HGDP populations varies greatly. To avoid the effects of different sample sizes on comparisons of LD decay and haplotype diversity, populations from similar geographic regions were combined, and 48 subjects were selected for each group: Turkish (48), European (14 French, 12 Italian, 8 Tuscan, and 14 Sardinian), Middle Eastern (24 Druze and 24 Palestinian), Central Asian (22 Hazara, 10 Uygur, and 16 Kyrgyz), South Asian (8 Balochi, 8 Brahui, 8 Burusho, 8 Makrani, 8 Pathan, and 8 Sindhi), Northeast Asian (8 Mongola, 8 Tu, 8 Oroqen, 8 Xibo, 8 Daur, and 8 Hezhen), native American (7 Colombian, 8 Surui, 11 Karitiana, 11 Maya, and 11 Pima), and African (11 Bantu, 8 Biaka Pygmy, 8 Mbuti Pygmy, 8 Mandenka, 8 Yoruba, and 5 San).
Relatedness, Identity-by-Descent, IBS, and Runs-of-Homozygosity
Whole-genome genotype data were used to calculate identity-by-descent (IBD) and IBS values in PLINK (Purcell et al., 2007) utilizing all individuals. PI_HAT values (proportion of IBD) were evaluated for cryptic relatedness for Turkish and Kyrgyz samples. Pairwise IBS sharing within a subpopulation was used to evaluate genetic similarity in a given population.
To calculate runs of homozygosity (ROH) in our samples, we used the default parameters in PLINK. To avoid the effects of different sample sizes on calculations, we used the same 48-subject groups (Turkish, European, Middle Eastern, Central Asian, South Asian, and Northeast Asian). To eliminate the effect of LD on detection of ROHs, SNPs were LD pruned (r2 < 0.2, n = 105,382) for each population group separately.
Results
Population Structure, Relatedness, and Admixture
PC analysis is useful for revealing relationships among individuals and exploring the extent of differentiation among populations. We used data from the unrelated subjects in the HGDP, a collection of 52 populations across the globe, and included data from our Turkish and Kyrgyz samples utilizing the LD-pruned SNP set (r2 < 0.2, n = 105,382). Figure 2A shows the first two components of this analysis by smartpca. Population groupings (major geographical regions) were assigned only after the analysis. Subjects from the same geographical region clustered among themselves. Turkish samples clustered tightly among themselves and together with Europeans, Middle Easterners, and South Asians (Pakistani). Krygyz samples also clustered tightly among themselves and between Central Asians (Uygur and Hazara) and East Asians.
Figure 2.
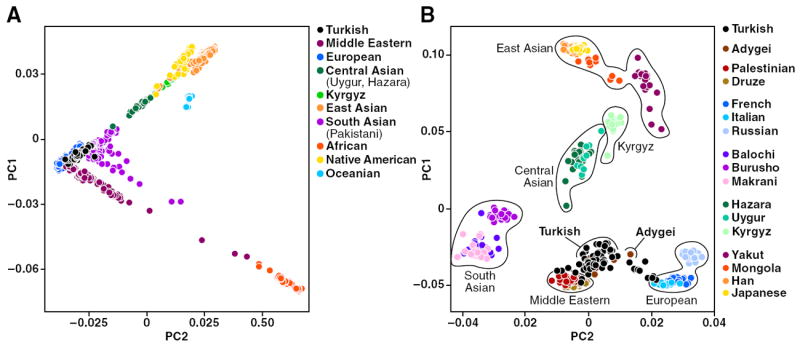
PC analysis demonstrating genetic relatedness across major geographic regions, including HGDP, Turkish, and Kyrgyz samples. Each symbol represents one individual. (A) PC analysis of 52 populations from the HGDP (n = 938), Turkish (n = 63), and Kyrgyz (n = 16) samples. (B) PC analysis focusing on selected Eurasian populations (including Turkish and Kyrgyz populations) (n = 451).
To examine fine-scale population structure and relatedness, we removed African, Oceanian, and native American populations. Representative populations from Eurasia were selected, and the analysis was repeated (Fig. 2B). Turkish samples clustered with Middle Eastern and European populations, particularly with the Adygei population from the Caucasus. South Asian populations clustered separately and did not overlap with Turkish samples. Kyrgyz samples clustered with other Central Asian populations, but they were relatively closer to East Asian populations (Fig. 2B). These results demonstrate that the PC analysis for the Eurasian region clearly delineates fine-scale population structure.
To examine finer-scale population clustering among populations and to identify any subpopulation structure among our subjects from different regions of Turkey, we analyzed Turkish samples together with European and Middle Eastern populations (Fig. 3A) or with South Asian and Central Asian populations (including Kyrgyz) (Fig. 3B) after examining the pattern of clustering of populations in Fig. 2A and B. The Turkish samples were easily separated from the Middle Eastern and European populations, and to some extent from the Adygei population. Importantly, samples from the three regions in Turkey (Aydin, Istanbul, and Kayseri) overlapped, suggesting no clear subpopulation structure in our samples (Fig. 3A). Additional pairwise PCs were plotted using Turkish, European, and Middle Eastern populations. The third PC clearly distinguished Middle Eastern populations of Palestinians and Druze (Fig. S1A and C), while the Turkish samples from different regions overlapped (Fig. S1A–D). Similarly, Turkish samples were clearly separated from South Asian and Central Asian populations as shown in the first two PCs (Fig. 3B). In addition, adding the third and fourth PCs showed that the Turkish samples from the different regions overlapped (Fig. S2A–D) as we observed with the first two PCs (Fig. 3B).
Figure 3.
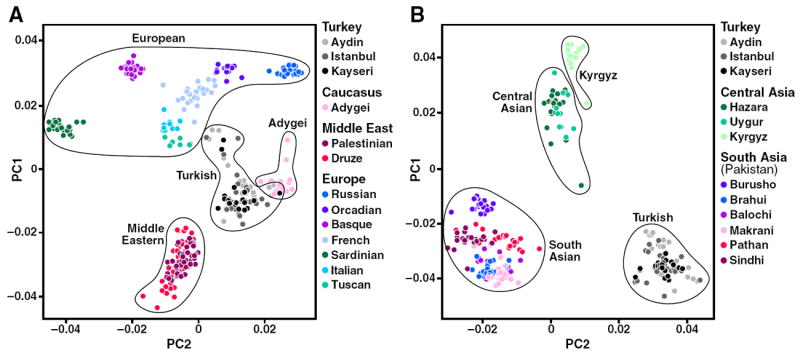
PC analysis demonstrating genetic relatedness in selected HGDP, Turkish, and Kyrgyz samples. Each symbol represents one individual. (A) PC analysis of Turks vs. European and Middle Eastern populations. Turkish samples were from three regions of Turkey (Aydin, Istanbul, and Kayseri). (B) PC analysis of Turks vs. Central Asian (including Kyrgyz) and South Asian (Pakistani) populations.
We repeated the PC analysis using only the 63 Turkish samples and observed that the samples from the different regions overlapped (Fig. S3A–D). In addition, PC analysis of the Turkish and Adygei populations together clearly separated the Adygei population from the Turkish population at the first PC, and again our samples from the different regions overlapped (data not shown). These results demonstrate that our Turkish samples are rather homogeneous and clustered away from other Eurasian populations (Figs. 3 and S1–3). We analyzed up to six PCs and the results did not suggest there were any differences between our samples from the three regions. Including additional population(s) or different groupings (e.g., eliminating a few) did not change the overall interpretation of the PC analysis results for Turkish or Kyrgyz populations (data not shown). To check the validity of the PC analysis results, the IBS matrix was used to create a multidimensional scaling plot for all samples (HGDP, Turkish, and Kyrgyz) including all SNPs (Fig. S4). First and second dimensions were plotted with similar labeling of major geographical regions (Fig. 2A) to illustrate the genetic relatedness among individuals or populations. The results were very similar to those obtained with smartpca. Using multidimensional scaling analysis with the LD-pruned SNP set (r2 < 0.2, n = 105,382), we also obtained similar results (data not shown). Furthermore, removing some populations as we did previously (Figs. 2B and 3A and B) also gave very similar results (data not shown). This demonstrates the validity of population clustering results obtained by two different statistical methods.
The population structure of the Turkish and Kyrgyz samples was further examined with FRAPPE (K = 2–7, Fig. S5) and STRUCTURE (K = 2–7, Fig. S6) along with the HGDP samples. Previous analysis of HGDP samples with FRAPPE and STRUCTURE revealed that individuals from the same geographic region or predefined population nearly always shared similar parental ancestry components (Li et al., 2008; Rosenberg et al., 2002). In FRAPPE, the genetic structure of the Turkish samples revealed four parental ancestries (>1%) at K = 7 (Fig. 4). The largest portion (light blue), about 53% averaged across the Turkish samples, was present as the major ancestry in European populations, and this ancestry was also present in the Middle Eastern and Central Asian populations. About 26% of ancestry (dark blue) in the Turkish population represented the major ancestry in South Asians and present to a lesser extent in Central Asian and Middle Eastern populations but not present in European populations. About 14% of ancestry (green) in Turks was present in Middle Eastern populations, and about 6% of Turkish ancestry (red) was present to a significant extent in Central Asian and to a major extent in East Asian populations. Samples from different regions of Turkey had similar mean parental ancestry estimates (Table S1). Results from the Caucasus region (Adygei population) were similar to the Turks.
Figure 4.
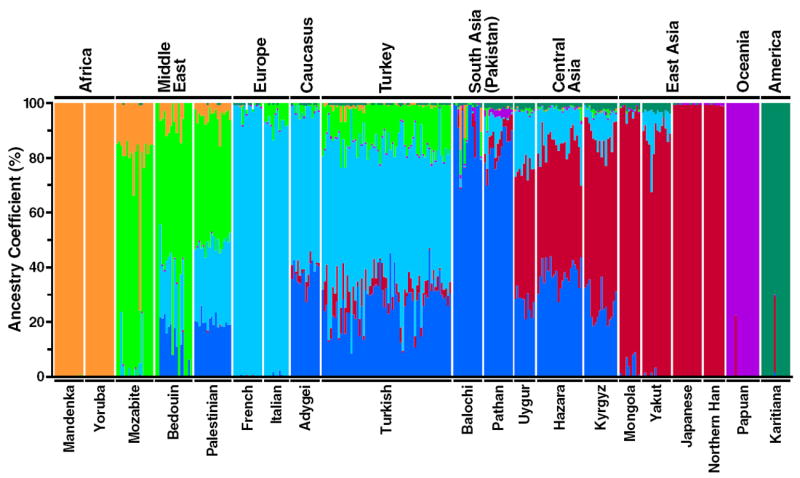
Estimated individual ancestry and population structure in 339 individuals by FRAPPE analysis. Representative HGDP populations selected from all continental/geographical regions and combined with Turkish and Kyrgyz samples (n = 339). Populations are labeled above the figure, with their geographic affiliations below. Each individual is represented by a thin vertical line, which is partitioned into K colored segments (K = 7). Colors represent the inferred ancestry from parental populations. White lines separate individuals of different populations.
Parental ancestry estimates for our Kyrgyz samples were similar to other Central Asian samples (Uygur and Hazara) except that the ‘red’ ancestry coefficient (major ancestry in East Asian populations) was slightly higher in Kyrgyz than other Central Asians (Fig. 4). This finding is consistent with the PC analysis results (Fig. 2A and B).
The population structure of the Turkish and Kyrgyz samples was also examined with STRUCTURE (Fig. S6). At K = 7, parental ancestry estimates for Turkish subjects were higher for ancestry coefficients in which the major ancestry component was European (77%, ‘light blue’) and lower in South Asian (12%, ‘dark blue’) and Middle Eastern (4%, ‘light green’) populations and similar to Central Asian population (6%, ‘red’). FRAPPE distinguished South Asian populations from Middle Eastern and European populations and Middle Eastern populations from European populations as seen in the original HGDP analysis (Li et al., 2008). To determine whether SNP selection affects the results, random SNPs were selected (1st, 84th, 167th, etc. up to 6407 SNPs) and run on STRUCTURE at K = 7. Random selection gave results similar to those of the selection process described previously (data not shown).
Supervised clustering with STRUCTURE (Falush et al., 2003) was also used to analyze the Turkish genetic ancestry by forcing separate clustering of HGDP populations. Supervised analysis was performed using individuals from the Middle East (Druze and Palestinian), Europe (French, Italian, Tuscan, and Sardinian), and Central Asia (Uygur, Hazara, and Kyrgyz) at K = 3 (Fig. 5A). The contributions were 45%, 40%, and 15% for the Middle Eastern, European and Central Asian populations, respectively. Supervised analysis was also performed using Middle Eastern, European, Central Asian, and South Asian (Pakistani) populations (K = 4) (Fig. 5B). Parental ancestry coefficients for our Turkish samples were found to be 38% European, 35% Middle Eastern, 18% South Asian, and 9% Central Asian.
Figure 5.
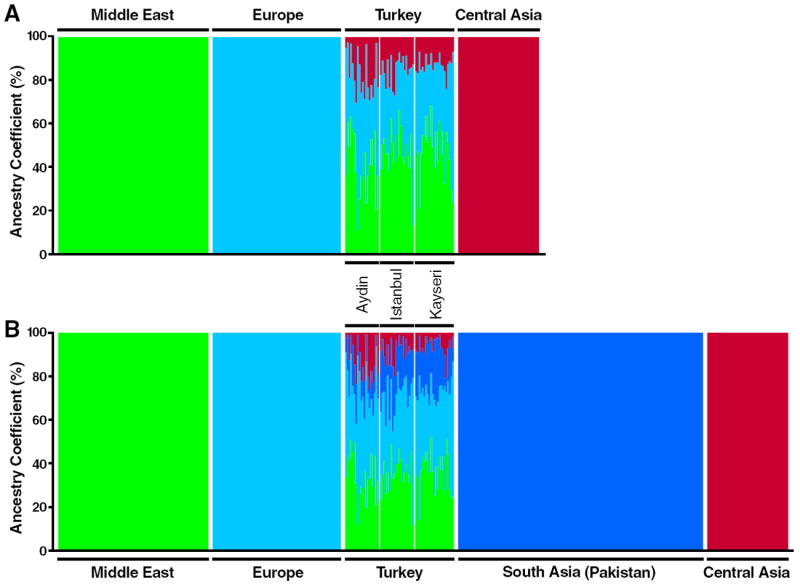
Supervised population structure analysis. Parental ancestry contributions were calculated for Turkish samples using supervised STRUCTURE analysis. Each individual is represented by a thin vertical line. White lines separate individuals of different populations. (A) Three clusters were forced to correspond to Middle Eastern (Druze and Palestinian), European (French, Italian, Tuscan, and Sardinian), and Central Asian (Uygur, Hazara, and Kyrgyz) populations at K = 3. (B) Four clusters were forced to correspond to Middle Eastern, European, South Asian (Balochi, Brahui, Burusho, Makrani, Pathan, and Sindhi), and Central Asian populations at K = 4.
Fst Calculations and Phylogenetic Tree Building
To measure genetic distances between HGDP, Turkish, and Kyrgyz populations, we calculated pairwise Fst values between populations. Results for selected Eurasian populations (Table 1) and all populations in this study (Table S2) are shown. Turks had the lowest pairwise Fst with Adygei, Middle Eastern, and European populations, followed by South Asian and Central Asian populations. Kyrgyz had the lowest pairwise Fst with Uygur and Hazara populations followed by East Asian populations. These pairwise Fst distances are in concordance with the results from the PCA and STRUCTURE analyses. The phylogenetic tree for selected Eurasian populations (Fig. 6) supported the aforementioned relationship that Turks are closer to Adygei and Middle Eastern populations and to some degree to European and South Asian populations.
Table 1.
Fst matrix among selected Eurasian populations
| Turkish | Druze | Palestinian | French | Italian | Adygei | Balochi | Burusho | Uygur | Hazara | Kyrgyz | Han | Mongola | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Turkish | 0.008 | 0.007 | 0.006 | 0.005 | 0.004 | 0.010 | 0.016 | 0.023 | 0.025 | 0.041 | 0.094 | 0.077 | |
|
| |||||||||||||
| Druze | 0.008 | 0.009 | 0.014 | 0.012 | 0.012 | 0.019 | 0.029 | 0.039 | 0.040 | 0.058 | 0.114 | 0.097 | |
| Palestinian | 0.007 | 0.009 | 0.014 | 0.011 | 0.012 | 0.017 | 0.026 | 0.036 | 0.037 | 0.055 | 0.108 | 0.092 | |
|
| |||||||||||||
| French | 0.006 | 0.014 | 0.014 | 0.002 | 0.009 | 0.020 | 0.026 | 0.035 | 0.036 | 0.054 | 0.111 | 0.094 | |
| Italian | 0.005 | 0.012 | 0.011 | 0.002 | 0.009 | 0.020 | 0.028 | 0.037 | 0.038 | 0.056 | 0.113 | 0.096 | |
| Adygei | 0.004 | 0.012 | 0.012 | 0.009 | 0.009 | 0.012 | 0.018 | 0.028 | 0.028 | 0.046 | 0.100 | 0.083 | |
|
| |||||||||||||
| Balochi | 0.010 | 0.019 | 0.017 | 0.020 | 0.020 | 0.012 | 0.011 | 0.023 | 0.023 | 0.040 | 0.090 | 0.074 | |
| Burusho | 0.016 | 0.029 | 0.026 | 0.026 | 0.028 | 0.018 | 0.011 | 0.016 | 0.017 | 0.031 | 0.073 | 0.058 | |
|
| |||||||||||||
| Uygur | 0.023 | 0.039 | 0.036 | 0.035 | 0.037 | 0.028 | 0.023 | 0.016 | 0.003 | 0.009 | 0.032 | 0.019 | |
| Hazara | 0.025 | 0.040 | 0.037 | 0.036 | 0.038 | 0.028 | 0.023 | 0.017 | 0.003 | 0.012 | 0.037 | 0.023 | |
| Kyrgyz | 0.041 | 0.058 | 0.055 | 0.054 | 0.056 | 0.046 | 0.040 | 0.031 | 0.009 | 0.012 | 0.032 | 0.018 | |
|
| |||||||||||||
| Han | 0.094 | 0.114 | 0.108 | 0.111 | 0.113 | 0.100 | 0.090 | 0.073 | 0.032 | 0.037 | 0.032 | 0.007 | |
| Mongola | 0.077 | 0.097 | 0.092 | 0.094 | 0.096 | 0.083 | 0.074 | 0.058 | 0.019 | 0.023 | 0.018 | 0.007 | |
Fst matrix was calculated using the smartpca function of the EIGENSTRAT program utilizing the LD-pruned (r2 < 0.2) SNP data set.
Populations from major geographic regions are grouped and separated by thin black lines.
Figure 6.
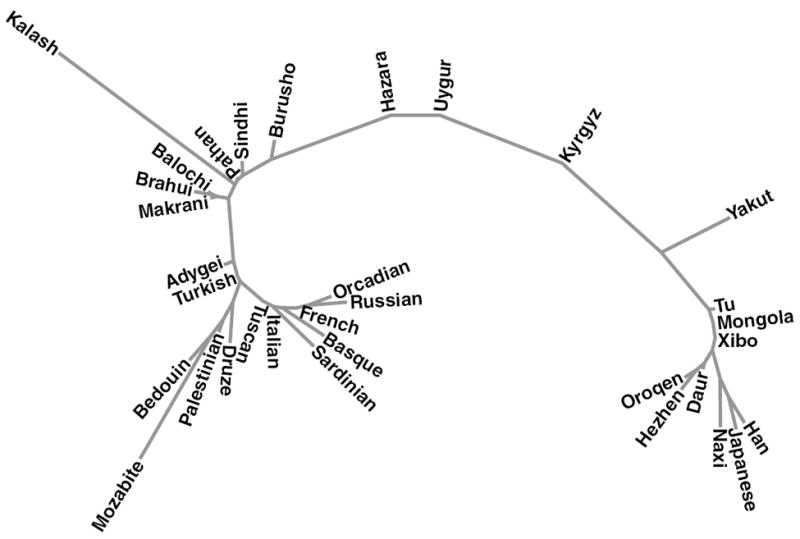
Phylogenetic tree of Eurasian populations. Neighbor-joining tree of 33 Eurasian populations (selected from HGDP, Turkish, and Kyrgyz populations) based on pairwise Fst matrix calculated with smartpca. The phylogenetic tree was constructed with MEGA4 software.
Allele Frequency Comparison Among Populations
Forward reference allele frequencies in Turkish vs. other HGDP populations were compared and visualized (Fig. S7). The highest correlations were between Turks and Middle Easterners (r = 0.923, Druze and Palestinian), Europeans (r = 0.914, French, Italian, Tuscan, and Sardinian), and South Asian populations (r = 0.894, Pakistani). There was some degree of correlation with Central Asian populations (r = 0.747, Hazara and Uygur) (Fig. S6). These results are in line with results of the PC analysis, FRAPPE, and STRUCTURE analyses. Allele frequency correlations between Kyrgyz and HGDP populations were also calculated. The highest correlations were with other Central Asian (r = 0.834), Northeast Asian (r = 0.854), and Chinese populations (r = 0.808).
Patterns of Decay of LD and Haplotype Diversity
To investigate haplotype diversity in our Turkish samples and other population groups, randomly selected 1-Mb regions from each chromosome were analyzed. Population groups contained equal numbers of subjects to avoid the effects of different sample sizes. The number and average size of haplotype blocks and the number of common haplotypes (>5%) were rather similar among Turkish, European, Middle Eastern, Central Asian, and South Asian groups (Table 2). Haplotype block counts were lower, the average size was shorter, and the number of common haplotypes was lower in Africans, whereas the haplotype blocks were much larger in native Americans than in other populations (including Turks).
Table 2.
Haplotype diversity in different population groups
| Population Groups | Blocka (n) | Mean ± SEMb (bp) | Median (bp) | Range (bp) | Distinct haplotypec (n) |
|---|---|---|---|---|---|
| Turkish | 266 | 14,871 ± 2,086 | 4,869 | 352,664 | 801 |
| Middle Eastern | 258 | 15,227 ± 2,092 | 5,765 | 419,903 | 786 |
| Central Asian (including Kyrgyz) | 256 | 15,565 ± 2,143 | 5,276 | 350,723 | 748 |
| European | 267 | 15,623 ± 1,979 | 5,261 | 337,316 | 819 |
| Northeast Asian | 247 | 17,092 ± 2,361 | 5,902 | 417,962 | 724 |
| South Asian (Pakistani) | 251 | 17,171 ± 2,808 | 5,285 | 465,263 | 746 |
| African | 144 | 7,301 ± 1,044 | 3,427 | 89,641 | 417 |
| Native American | 248 | 24,269 ± 2,800 | 7,856 | 350,723 | 718 |
Haplotype blocks were calculated with phased genotype data using Haploview (Gabriel method). Population group size, n = 48 individuals.
Total number of haplotype blocks in 22 different 1-Mb regions.
Average size (bp) of haplotype blocks.
Total number of distinct, common haplotypes (>5%).
Turkish, European, Middle Eastern, Central Asian, and South Asian groups exhibited similar rates of LD decay with increasing distance (Fig. 7). The half-life of LD decay with genomic distances was substantially shorter in African samples and longer in native American samples. The difference among populations started to disappear over 100-kb distances.
Figure 7.
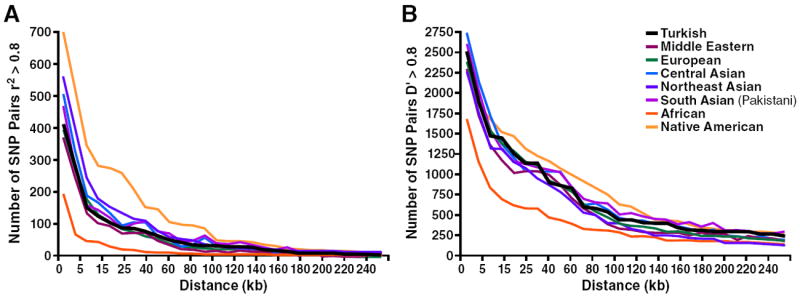
Decay of LD over distance. SNP pairs were partitioned into bins at 5-kb intervals; for each bin number, SNP pairs with r2 > 0.8 (A) and D′ > 0.8 (B) were plotted. Each group has 48 individuals to eliminate possible effects of sample size. The populations shown are European (French, Italian, Tuscan, and Sardinian), Middle Eastern (Druze and Palestinian), Central Asian (Hazara, Uygur, and Kyrgyz), South Asian (Balochi, Brahui, Burusho, Makrani, Pathan, and Sindhi), Northeast Asian (Mongola, Tu, Oroqen, Xibo, Daur, and Hezhen), native American (Colombian, Surui, Karitiana, Maya, and Pima), and African (Bantu, Biaka Pygmy, Mbuti Pygmy, Mandenka, Yoruba, and San).
Relatedness, IBD, IBS, and ROH
Cryptic relatedness was determined by estimating IBD across the genome for all possible pairwise sample combinations for Turkish and Kyrgyz samples separately. All pairwise PI_HAT values (proportion of IBD) were <0.05 for Turkish samples and <0.07 for Kyrgyz samples, suggesting that relatedness was not an issue for our samples. Pairwise IBS sharing values were used to evaluate genetic similarity in a given population and are shown for selected populations (Table S3). Average IBS sharing within populations were quite similar except for Papuan and Piman populations, which were slightly elevated. Samples from different regions of Turkey have also similar IBS sharing values (Table S3).
ROH (extended homozygosity in a locus with two identical alleles) was examined in several population groups containing equal numbers of subjects from Eurasia after SNPs were LD pruned separately in each group. Middle Eastern and South Asian populations showed significantly more ROH as seen by higher count and longer segments in the histogram (Fig. S8). Turkish, Central Asian (including Kyrgyz), European, and Northeast Asian populations showed similar degrees of ROH.
Discussion
The Anatolian peninsula (present-day Turkey), located on the Silk Road, served as a bridge between the West and East and was subject to migration from different regions throughout history. The most recent migration was by Turkic-speaking nomadic groups, mainly Oghuz groups. Starting in the 10th century, they spread away from their homeland in Central Asia (Findley, 2005b; Grousset, 1970; Güvenç, 1993), began to admix with local inhabitants, and established the Anatolian Seljuk Empire (10th–13th centuries). After the collapse of this Turkish Empire by Mongol invasion, another Turkish empire, the Ottomans, ruled (13th–20th centuries) the Anatolian peninsula, the Middle East, and stretching to southeastern Europe and southwestern Asia (Faroqhi, 2007; Findley, 2005a). These major historical events are reflected in the genetic structure of present-day Turkish people, as described in this study.
We analyzed the population structure and genetic relatedness of Turkish and Kyrgyz populations and compared them to other Eurasian populations utilizing HGDP data. PC and FRAPPE/STRUCTURE analyses indicated that the Turkish population has a close genetic similarity to Middle Eastern and European populations and some degree of similarity to South Asian and Central Asian populations. Kyrgyz samples showed genetic relatedness (clustered together) with other Central Asian populations (Uygur and Hazara) in the HGDP set. The PC and FRAPPE results are generally consistent with the phylogenetic tree and the relative paired Fst values with respect to the distance separation among the different population groups. Results from our samples, collected from three regions in Turkey (Aydin, Istanbul, and Kayseri), overlapped without a clear subpopulation structure, suggesting a rather homogeneous and distinct genetic ancestry. The potential weakness of our sampling strategy is that we do not have the parental/grandparental ancestry of our samples, which may cause difficulties in the interpretation of genetic ancestry inference. The complex origins, unrecorded/unknown immigrations, and recent intermarriages with other population/ancestry groups preclude the possibility of unambiguously identifying the ancestry of our samples. However, clear overlapping of our samples from three different regions of Turkey, including samples from a cosmopolitan city such as Istanbul (which may reflect the more general picture of present-day Turkey), and data from about samples that were obtained from individuals who were born and lived in their designated regions give us confidence in our interpretation of the results, at least for the regions and samples included in this study.
Genetic distance also depends on the markers used; the panel of more than 500,000 SNPs we used is biased toward common polymorphisms discovered in European and East Asian (mainly Japanese) populations. Nevertheless, fine population structure has been documented in several studies even when subsets of these high-density markers were selected (Auton et al., 2009; Bonnen et al., 2006; Bryc et al., 2010; Hunter-Zinck et al., 2010; Silva-Zolezzi et al., 2009; Xing et al., 2010; Xu et al., 2008). Importantly, the ancestry proportions inferred from this analysis are affected by the populations used in the study. The HGDP has extensive coverage of the world’s major geographic regions, although some are not well represented (e.g., Central Asia). However, extensive and rigorous analyses have demonstrated that the estimated genetic clusters are not artifacts of noncontinuous sampling of people (Li et al., 2008; Rosenberg et al., 2002).
To obtain better estimates of some calculations in this study, geographic populations in close proximity were grouped together. Populations of Mongola, Tu, Xibo, Oroqen, Hezhen, and Daur were grouped together as Northeast Asians, since these groups reside at high latitudes and speak languages of the Altaic family (Cavalli-Sforza, 2005; Li et al., 2008), of which Turkic is a subdivision (Georg et al., 1998). Uygur and Kyrgyz populations also speak a Turkic language (Georg et al., 1998). Although Hazaran samples were collected from Pakistan (Cann et al., 2002), they are genetically more similar to Central Asian populations than to Pakistani populations as seen in this and other studies (Li et al., 2008; Quintana-Murci et al., 2004; Rosenberg et al., 2002; Xing et al., 2010); therefore, we grouped Hazarans together with Uygur and Kyrgyz populations as Central Asians. The Middle Eastern group consists of Druze and Palestinian populations, since Mozabites have a large African component, and Bedouins are an admixed population (Li et al., 2008). European populations on the Mediterranean Sea (French, Italian, Tuscan, and Sardinian) were grouped as Europeans for supervised STRUCTURE, allele frequency spectrum comparison, patterns of decay of LD, and haplotype diversity analyses, whereas all or representative European populations were used for PC, FRAPPE, and STRUCTURE analyses as described.
Our population substructure analyses are consistent with historic admixture events (Figs. 4, 5, S5, and S6). In Turks, the largest parental ancestry estimates (light blue) were also present as a major ancestry component in European and, to a lesser extent, in Middle Eastern and Central Asian populations. However, the second largest parental ancestry estimates (dark blue) were present in South Asian, Central Asian, and Middle Eastern populations but not in European populations. The third largest ancestry estimates (light green) in Turks have a major component in Middle Easterners. The fourth largest ancestry estimates (red) in Turks were major ancestry estimates in East Asian and Central Asian populations, possibly demonstrating admixture events in Central Asian (Comas et al., 1998; Frye, 1996; Nasidze et al., 2004; Wells et al., 2001; Zerjal et al., 2002) and Turkish populations, but these estimates (red) were absent in European and Middle Eastern populations. PC and phylogenetic tree analyses also supported these conclusions.
The Adygei population from the Caucasus showed a closer genetic affinity to our Turkish samples; however, when the Turkish and Adygei populations were analyzed together, the first PC clearly separated these two populations. Although the Adygei sample set was small (n = 17), it clustered tightly with other populations from the Caucasus (Nasidze et al., 2004; Xing et al., 2010), suggesting that it is a valid finding, not an artifact of low sample size. The Caucasus region, close to present-day Turkey, was also subjected to major population movements and the Caucasus Mountains did not seem to act as a barrier to gene flow (Nasidze et al., 2004). Studies of Y chromosome (Wells et al., 2001) and mitochondrial markers (Quintana-Murci et al., 2004) showed closer affinities of Turkish and Caucasus samples.
Many contemporary Central Asian populations speak a Turkic language (Georg et al., 1998) as do the majority of people in Turkey. Several studies have attempted to quantify the Central Asian contribution to the Turkish gene pool utilizing mitochondrial DNA, Y chromosome, and autosomal markers (Alu insertion polymorphism). Mean estimates varied widely; analysis of mitochondrial markers found that the admixture percent of Central Asian was 22% (Berkman, 2006) to 30% (Di Benedetto et al., 2001); for Y chromosome markers, the percent was <9% (Cinnioğlu et al., 2004), 13% (Berkman, 2006), and 30% (Di Benedetto et al., 2001); and for the Alu insertion polymorphism, it was 13% (Berkman et al., 2008) and 15% (Berkman, 2006) in the Turkish gene pool. Although these markers provide some insights about the relative contributions of different sexes, their haploid nature (mitochondrial and Y chromosome markers) makes them more vulnerable to genetic drift than autosomal markers. However, in the present study we used autosomal high-density SNP genotypes across the genome to more accurately reflect the Central Asian admixture with Turks. To compare our samples with published reports (Berkman, 2006; Berkman et al., 2008; Cinnioğlu et al., 2004; Di Benedetto et al., 2001), we used supervised clustering with STRUCTURE (Falush et al., 2003). Individuals from the Middle East (Druze and Palestinian), Europe (French, Italian, Tuscan, and Sardinian), and Central Asia (Uygur, Hazara, and Kyrgyz) were forced into separate clusters, and supervised analysis of Turkish samples was performed at K = 3. The Central Asian contribution was found to be about 15% (with 45% Middle Eastern and 40% European) (Fig. 5A). We inferred parental populations from contemporary populations living in these locations, although these populations may have experienced population movement (e.g., migration, admixture) or genetic drift. Having different populations than the available ones used in this analysis (e.g., populations closer to Turkey or more populations from Central Asia) may also affect the calculated contributions. Nevertheless, our results compare favorably with published results of the Central Asian contribution to today’s Turkish genome (Berkman, 2006; Berkman et al., 2008; Cinnioğlu et al., 2004; Di Benedetto et al., 2001).
Although separated by large geographic distances, Europe and South Asia (e.g., Pakistan) have some genetic relatedness (Fig. 2A) (Li et al., 2008; Rosenberg et al., 2002) that may reflect the documented gene flow from Central Asia, the Middle East, and Iran to Pakistan (Quintana-Murci et al., 2004; Regueiro et al., 2006; Wells et al., 2001) and the common ancestry of these population groups (Auton et al., 2009; Quintana-Murci et al., 2004). Similarities between our samples and South Asian (Pakistani) samples may reflect those earlier migratory and admixture events. Nevertheless, we did similar supervised clustering in which individuals from South Asia, the Middle East, Europe, and Central Asia were forced into separate clusters. The parental ancestry coefficient for our Turkish samples was 38% European, 35% Middle Eastern, 18% South Asian, and 9% Central Asian at K = 4 (Fig. 5B).
ROH may arise from consanguinity, reduced population size, or prolonged isolation of a population. Middle Eastern and South Asian populations, where consanguinity is relatively common (Hunter-Zinck et al., 2010; Hussain, 1999), have clearly more ROH (in terms of number and size) than Turkish, Central Asian, European, and Northeast Asian populations (Fig. S8). ROH might also result from hemizygosity (copy number variations, such as deletions). Copy number changes were not taken into account in our study. However, no significant differences in mean total length of ROHs were observed when deletions were considered (McQuillan et al., 2008).
The approaches used in our study allowed us to investigate the genetic ancestry of our Turkish samples with respect to HGDP samples and to assess the extent of admixture in our samples. Although the complex origins, historical immigrations, and intermarriages among populations make it hard to be precise, we found that individual parental ancestries can be estimated from the high-density SNP genotype data. A more thorough knowledge of between-population genetic variation is important in improving the design and interpretation of the genetics of complex diseases. Furthermore, since genetic studies are currently aiming at identifying smaller and smaller effects, recognizing and controlling for population structure, even at a fine level within a seemingly homogeneous population, is important to avoid confounding and spurious associations.
Supplementary Material
Acknowledgments
Turkish samples were provided through the Turkish Heart Study. Kyrgyz samples were obtained at the Kyrgyz National Center of Cardiology and Internal Medicine, Bishkek, Kyrgystan with the support of Drs. M. M. Mirrakhimov and E. M. Mirrakhimov. The authors thank Dr. Vivian G. Cheung (University of Pennsylvania) and Dr. Katherine Pollard (Gladstone Institute of Cardiovascular Disease) for valuable input and critical reading of the manuscript, Sylvia Richmond for manuscript preparation, and Gary Howard and Stephen Ordway for editorial assistance. The authors also thank Dr. Stephen Schaffner (BROAD Institute of MIT and Harvard) and Dr. Tara Matise (Rutgers University) for providing pairwise HapMap Fst and mapping (cM) data for individual SNPs, respectively. In addition, the authors are indebted to their associates at the American Hospital, Istanbul, especially Drs. K. Erhan Palaoğlu, Oryal Gökdemir, Sinan Özbayrakcı, Kerem Özer, Guy Pépin, Sibel Tanir, Judy Dawson-Pépin, and Linda L. Mahley. The authors acknowledge the generous support of the American Hospital, especially Mr. George Rountree, and the J. David Gladstone Institutes. This work was supported in part by grant R01 HL71027 from the National Institutes of Health.
Abbreviations
- HGDP
Human Genome Diversity Panel
- LD
linkage disequilibrium
- IBD
identity-by-descent
- IBS
identity-by-state
- PC analysis
principal component analysis
- ROH
runs-of-homozygosity
- SNP
single nucleotide polymorphism
Footnotes
Conflict of Interest
The Authors have no conflict of interest to disclose.
References
- Auton A, Bryc K, Boyko AR, Lohmueller KE, Novembre J, Reynolds A, Indap A, Wright MH, Degenhardt JD, Gutenkunst RN, King KS, Nelson MR, Bustamante CD. Global distribution of genomic diversity underscores rich complex history of continental human populations. Genome Res. 2009;19:795–803. doi: 10.1101/gr.088898.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Berkman CC. Ph D Thesis. Middle East Technical University; Ankara, Turkey: 2006. Comparative analyses from the Central Asian contribution to Anatolian gene pool with reference to Balkans. http://etd.lib.metu.edu.tr/upload/12607764/index.pdf. [Google Scholar]
- Berkman CC, Dinc H, Sekeryapan C, Togan I. Alu insertion polymorphisms and an assessment of the genetic contribution of Central Asia to Anatolia with respect to the Balkans. Am J Phys Anthropol. 2008;136:11–18. doi: 10.1002/ajpa.20772. [DOI] [PubMed] [Google Scholar]
- Bersot TP, Vega GL, Grundy SM, Palaoğlu KE, Atagündüz P, Özbayrakçi S, Gökdemir O, Mahley RW. Elevated hepatic lipase activity and low levels of high density lipoprotein in a normotriglyceridemic, nonobese Turkish population. J Lipid Res. 1999;40:432–438. [PubMed] [Google Scholar]
- Bersot TP, Pépin GM, Mahley RW. Risk determination of dyslipidemia in populations characterized by low levels of high-density lipoprotein cholesterol. Am Heart J. 2003;146:1052–1060. doi: 10.1016/S0002-8703(03)00516-7. [DOI] [PubMed] [Google Scholar]
- Bonnen PE, Pe’er I, Plenge RM, Salit J, Lowe JK, Shapero MH, Lifton RP, Breslow JL, Daly MJ, Reich DE, Jones KW, Stoffel M, Altshuler D, Friedman JM. Evaluating potential for whole-genome studies in Kosrae, an isolated population in Micronesia. Nat Genet. 2006;38:214–217. doi: 10.1038/ng1712. [DOI] [PubMed] [Google Scholar]
- Bryc K, Auton A, Nelson MR, Oksenberg JR, Hauser SL, Williams S, Froment A, Bodo J-M, Wambebe C, Tishkoff SA, Bustamante CD. Genome-wide patterns of population structure and admixture in West Africans and African Americans. Proc Natl Acad Sci USA. 2010;107:786–791. doi: 10.1073/pnas.0909559107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calafell F, Underhill P, Tolun A, Angelicheva D, Kalaydjieva L. From Asia to Europe: Mitochondrial DNA sequence variability in Bulgarians and Turks. Ann Hum Genet. 1996;60:35–49. doi: 10.1111/j.1469-1809.1996.tb01170.x. [DOI] [PubMed] [Google Scholar]
- Cann HM, de Toma C, Cazes L, Legrand M-F, Morel V, Piouffre L, Bodmer J, Bodmer WF, Bonne-Tamir B, Cambon-Thomsen A, Chen Z, Chu J, Carcassi C, Contu L, Du R, Excoffier L, Ferrara GB, Friedlaender JS, Groot H, Gurwitz D, Jenkins T, Herrera RJ, Huang X, Kidd J, Kidd KK, Langaney A, Lin AA, Mehdi SQ, Parham P, Piazza A, Pistillo MP, Qian Y, Shu Q, Xu J, Zhu S, Weber JL, Greely HT, Feldman MW, Thomas G, Dausset J, Cavalli-Sforza LL. A human genome diversity cell line panel. Science. 2002;296:261–262. doi: 10.1126/science.296.5566.261b. [DOI] [PubMed] [Google Scholar]
- Cavalli-Sforza LL. The Human Genome Diversity Project: Past, present and future. Nat Rev Genet. 2005;6:333–340. doi: 10.1038/nrg1596. [DOI] [PubMed] [Google Scholar]
- Cinnioğlu C, King R, Kivisild T, Kalfoğlu E, Atasoy S, Cavalleri GL, Lillie AS, Roseman CC, Lin AA, Prince K, Oefner PJ, Shen P, Semino O, Cavalli-Sforza LL, Underhill PA. Excavating Y-chromosome haplotype strata in Anatolia. Hum Genet. 2004;114:127–148. doi: 10.1007/s00439-003-1031-4. [DOI] [PubMed] [Google Scholar]
- Comas D, Calafell F, Mateu E, Pérez-Lezaun A, Bosch E, Martínez-Arias R, Clarimon J, Facchini F, Fiori G, Luiselli D, Pettener D, Bertranpetit J. Trading genes along the Silk Road: mtDNA sequences and the origin of Central Asian populations. Am J Hum Genet. 1998;63:1824–1838. doi: 10.1086/302133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Benedetto G, Ergüven A, Stenico M, Castrì L, Bertorelle G, Togan I, Barbujani G. DNA diversity and population admixture in Anatolia. Am J Phys Anthropol. 2001;115:144–156. doi: 10.1002/ajpa.1064. [DOI] [PubMed] [Google Scholar]
- Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faroqhi S. The Ottoman Empire and the World Around It. London: I.B.Tauris & Co; 2007. On the margins of empire: Clients and dependants; pp. 75–97. [Google Scholar]
- Findley CV. The Turks in World History. New York: Oxford University Press; 2005a. Islamic empires from Temur to the “gunpowder era”; pp. 93–132. [Google Scholar]
- Findley CV. The Turks in World History. New York: Oxford University Press; 2005b. Islam and empire from the Seljuks through the Mongols; pp. 56–92. [Google Scholar]
- Frye RN. The Heritage of Central Asia: From Antiquity to the Turkish Expansion. Princeton: Marcus Wiener Publishers; 1996. The present is born; pp. 233–239. [Google Scholar]
- Georg S, Michalove PA, Ramer AM, Sidwell PJ. Telling general linguists about Altaic. J Linguistics. 1998;35:65–98. [Google Scholar]
- Grousset R. The Empire of the Steppes: A History of Central Asia. New Brunswick: Rutgers University Press; 1970. The Turks and Islam to the thirteenth century; pp. 141–170. [Google Scholar]
- Güvenç B. Türk Kimliği Kültür Tarihinin Kaynaklari (in Turkish) (Turkish Identity Sources of Cultural History) Ankara: Kültür Bakanlığı; 1993. Türklerin Kimliği: Kim Bu Türkler? (Identity of Turks: Who are the Turks?) pp. 19–52. [Google Scholar]
- Hodoğlugil U, Williamson DW, Huang Y, Mahley RW. An interaction between the TaqIB polymorphism of cholesterol ester transfer protein and smoking is associated with changes in plasma high-density lipoprotein cholesterol levels in Turks. Clin Genet. 2005a;68:118–127. doi: 10.1111/j.1399-0004.2005.00467.x. [DOI] [PubMed] [Google Scholar]
- Hodoğlugil U, Williamson DW, Huang Y, Mahley RW. Common polymorphisms of ATP binding cassette transporter A1, including a functional promoter polymorphism, associated with plasma high density lipoprotein cholesterol levels in Turks. Atherosclerosis. 2005b;183:199–212. doi: 10.1016/j.atherosclerosis.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Hodoğlugil U, Tanyolaç S, Williamson DW, Huang Y, Mahley RW. Apolipoprotein A-V: A potential modulator of plasma triglyceride levels in Turks. J Lipid Res. 2006;47:144–153. doi: 10.1194/jlr.M500343-JLR200. [DOI] [PubMed] [Google Scholar]
- Hodoğlugil U, Williamson DW, Mahley RW. Polymorphisms in the hepatic lipase gene affect plasma HDL-cholesterol levels in a Turkish population. J Lipid Res. 2010;51:422–430. doi: 10.1194/jlr.P001578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodoğlugil U, Williamson DW, Yu Y, Farrer LA, Mahley RW. Glucuronic acid epimerase is associated with plasma triglyceride and high-density lipoprotein cholesterol levels in Turks. Ann Hum Genet. 2011;75:398–417. doi: 10.1111/j.1469-1809.2011.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter-Zinck H, Musharoff S, Salit J, Al-Ali KA, Chouchane L, Gohar A, Matthews R, Butler MW, Fuller J, Hackett NR, Crystal RG, Clark AG. Population genetic structure of the people of Qatar. Am J Hum Genet. 2010;87:17–25. doi: 10.1016/j.ajhg.2010.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain R. Community perceptions of reasons for preference for consanguineous marriages in Pakistan. J Biosoc Sci. 1999;31:449–461. doi: 10.1017/s0021932099004496. [DOI] [PubMed] [Google Scholar]
- Jakobsson M, Rosenberg NA. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007;23:1801–1806. doi: 10.1093/bioinformatics/btm233. [DOI] [PubMed] [Google Scholar]
- Li JZ, Absher DM, Tang H, Southwick AM, Casto AM, Ramachandran S, Cann HM, Barsh GS, Feldman M, Cavalli-Sforza LL, Myers RM. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- Ling H, Waterworth DM, Stirnadel HA, Pollin TI, Barter PJ, Kesäniemi YA, Mahley RW, McPherson R, Waeber G, Bersot TP, Cohen JC, Grundy SM, Mooser VE, Mitchell BD. Genome-wide linkage and association analyses to identify genes influencing adiponectin levels: The GEMS study. Obesity. 2009;17:737–744. doi: 10.1038/oby.2008.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley RW, Palaoğlu KE, Atak Z, Dawson-Pepin J, Langlois A-M, Cheung V, Onat H, Fulks P, Mahley LL, Vakar F, Özbayrakçı S, Gökdemir O, Winkler W. Turkish Heart Study: Lipids, lipoproteins, and apolipoproteins. J Lipid Res. 1995;36:839–859. [PubMed] [Google Scholar]
- Mahley RW, Pépin J, Palaoğlu KE, Malloy MJ, Kane JP, Bersot TP. Low levels of high density lipoproteins in Turks, a population with elevated hepatic lipase: High density lipoprotein characterization and gender-specific effects of apolipoprotein E genotype. J Lipid Res. 2000;41:1290–1301. [PubMed] [Google Scholar]
- Mahley RW, Arslan P, Pekcan G, Pépin GM, Ağaçdiken A, Karaağaoğlu N, Rakicioğlu N, Nursal B, Dayaniklı P, Palaoğlu KE, Bersot TP. Plasma lipids in Turkish children: Impact of puberty, socioeconomic status, and nutrition on plasma cholesterol and HDL. J Lipid Res. 2001;42:1996–2006. [PubMed] [Google Scholar]
- McClellan J, King M-C. Genetic heterogeneity in human disease. Cell. 2010;141:210–217. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
- McQuillan R, Leutenegger A-L, Abdel-Rahman R, Franklin CS, Pericic M, Barac-Lauc L, Smolej-Narancic N, Janicijevic B, Polasek O, Tenesa A, MacLeod AK, Farrington SM, Rudan P, Hayward C, Vitart V, Rudan I, Wild SH, Dunlop MG, Wright AF, Campbell H, Wilson JF. Runs of homozygosity in European populations. Am J Hum Genet. 2008;83:359–372. doi: 10.1016/j.ajhg.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mergen H, Öner R, Öner C. Mitochondrial DNA sequence variation in the Anatolian peninsula (Turkey) J Genet. 2004;83:39–47. doi: 10.1007/BF02715828. [DOI] [PubMed] [Google Scholar]
- Nasidze I, Ling EYS, Quinque D, Dupanloup I, Cordaux R, Rychkov S, Naumova O, Zhukova O, Sarraf-Zadegan N, Naderi GA, Asgary S, Sardas S, Farhud DD, Sarkisian T, Asadov C, Kerimov A, Stoneking M. Mitochondrial DNA and Y-chromosome variation in the Caucasus. Ann Hum Genet. 2004;68:205–221. doi: 10.1046/j.1529-8817.2004.00092.x. [DOI] [PubMed] [Google Scholar]
- Onat A. Risk factors and cardiovascular disease in Turkey. Atherosclerosis. 2001;156:1–10. doi: 10.1016/s0021-9150(01)00500-7. [DOI] [PubMed] [Google Scholar]
- Onat A, Hergenç G, Uzunlar B, Ceyhan K, Uyarel H, Yazıcı M, Dogan Y, Özmay M, Toprak S, Sansoy V. Determinants of HDL-cholesterol and its prediction of coronary disease among Turks (in Turkish) Arch Turk Soc Cardiol. 2003;31:5–13. [Google Scholar]
- Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Zaitlen NA, Reich D, Patterson N. New approaches to population stratification in genome-wide association studies. Nat Rev Genet. 2010;11:459–463. doi: 10.1038/nrg2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, Sham PC. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana-Murci L, Chaix R, Wells RS, Behar DM, Sayar H, Scozzari R, Rengo C, Al-Zahery N, Semino O, Santachiara-Benerecetti AS, Coppa A, Ayub Q, Mohyuddin A, Tyler-Smith C, Qasim Mehdi S, Torroni A, McElreavey K. Where West meets East: The complex mtDNA landscape of the southwest and Central Asian corridor. Am J Hum Genet. 2004;74:827–845. doi: 10.1086/383236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regueiro M, Cadenas AM, Gayden T, Underhill PA, Herrera RJ. Iran: Tricontinental nexus for Y-chromosome driven migration. Hum Hered. 2006;61:132–143. doi: 10.1159/000093774. [DOI] [PubMed] [Google Scholar]
- Rosenberg NA, Pritchard JK, Weber JL, Cann HM, Kidd KK, Zhivotovsky LA, Feldman MW. Genetic structure of human populations. Science. 2002;298:2381–2385. doi: 10.1126/science.1078311. [DOI] [PubMed] [Google Scholar]
- Rosenberg NA. Standardized subsets of the HGDP-CEPH Human Genome Diversity Cell Line Panel, accounting for atypical and duplicated samples and pairs of close relatives. Ann Hum Genet. 2006;70:841–847. doi: 10.1111/j.1469-1809.2006.00285.x. [DOI] [PubMed] [Google Scholar]
- Rosenberg NA, Huang L, Jewett EM, Szpiech ZA, Jankovic I, Boehnke M. Genome-wide association studies in diverse populations. Nat Rev Genet. 2010;11:356–366. doi: 10.1038/nrg2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Scheet P, Stephens M. A fast and flexible statistical model for large-scale population genotype data: Applications to inferring missing genotypes and haplotypic phase. Am J Hum Genet. 2006;78:629–644. doi: 10.1086/502802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva-Zolezzi I, Hidalgo-Miranda A, Estrada-Gil J, Fernandez-Lopez JC, Uribe-Figueroa L, Contreras A, Balam-Ortiz E, del Bosque-Plata L, Velazquez-Fernandez D, Lara C, Goya R, Hernandez-Lemus E, Davila C, Barrientos E, March S, Jimenez-Sanchez G. Analysis of genomic diversity in Mexican Mestizo populations to develop genomic medicine in Mexico. Proc Natl Acad Sci USA. 2009;106:8611–8616. doi: 10.1073/pnas.0903045106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Tang H, Peng J, Wang P, Risch NJ. Estimation of individual admixture: Analytical and study design considerations. Genet Epidemiol. 2005;28:289–301. doi: 10.1002/gepi.20064. [DOI] [PubMed] [Google Scholar]
- Teo Y-Y, Sim X, Ong RTH, Tan AKS, Chen J, Tantoso E, Small KS, Ku C-S, Lee EJD, Seielstad M, Chia K-S. Singapore Genome Variation Project: A haplotype map of three Southeast Asian populations. Genome Res. 2009;19:2154–2162. doi: 10.1101/gr.095000.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian C, Kosoy R, Lee A, Ransom M, Belmont JW, Gregersen PK, Seldin MF. Analysis of East Asia genetic substructure using genome-wide SNP arrays. PLoS One. 2008;3:e3862. doi: 10.1371/journal.pone.0003862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells RS, Yuldasheva N, Ruzibakiev R, Underhill PA, Evseeva I, Blue-Smith J, Jin L, Su B, Pitchappan R, Shanmugalakshmi S, Balakrishnan K, Read M, Pearson NM, Zerjal T, Webster MT, Zholoshvili I, Jamarjashvili E, Gambarov S, Nikbin B, Dostiev A, Aknazarov O, Zalloua P, Tsoy I, Kitaev M, Mirrakhimov M, Chariev A, Bodmer WF. The Eurasian heartland: A continental perspective on Y-chromosome diversity. Proc Natl Acad Sci USA. 2001;98:10244–10249. doi: 10.1073/pnas.171305098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing J, Watkins WS, Shlien A, Walker E, Huff CD, Witherspoon DJ, Zhang Y, Simonson TS, Weiss RB, Schiffman JD, Malkin D, Woodward SR, Jorde LB. Toward a more uniform sampling of human genetic diversity: A survey of worldwide populations by high-density genotyping. Genomics. 2010;96:199–210. doi: 10.1016/j.ygeno.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Huang W, Qian J, Jin L. Analysis of genomic admixture in Uyghur and its implication in mapping strategy. Am J Hum Genet. 2008;82:883–894. doi: 10.1016/j.ajhg.2008.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Jin L. A genome-wide analysis of admixture in Uyghurs and a high-density admixture map for disease-gene discovery. Am J Hum Genet. 2008;83:322–336. doi: 10.1016/j.ajhg.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Wyszynski DF, Waterworth DM, Wilton SD, Barter PJ, Kesäniemi YA, Mahley RW, McPherson R, Waeber G, Bersot TP, Ma Q, Sharma SS, Montgomery DS, Middleton LT, Sundseth SS, Mooser V, Grundy SM, Farrer LA. Multiple QTLs influencing triglyceride and HDL and total cholesterol levels identified in families with atherogenic dyslipidemia. J Lipid Res. 2005;46:2202–2213. doi: 10.1194/jlr.M500137-JLR200. [DOI] [PubMed] [Google Scholar]
- Zerjal T, Wells RS, Yuldasheva N, Ruzibakiev R, Tyler-Smith C. A genetic landscape reshaped by recent events: Y-chromosomal insights into Central Asia. Am J Hum Genet. 2002;71:466–482. doi: 10.1086/342096. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.