

Abstract
Autosomal dominant polycystic kidney disease (ADPKD) is the most common hereditary kidney disease. Relentless cyst growth substantially enlarges both kidneys and culminates in renal failure. Patients with ADPKD also have vascular abnormalities; intracranial aneurysms (IAs) are found in ~10% of asymptomatic patients during screening and in up to 25% of those with a family history of IA or subarachnoid haemorrhage. As the genes responsible for ADPKD—PKD1 and PKD2—have complex integrative roles in mechanotransduction and intracellular calcium signalling, the molecular basis of IA formation might involve focal haemodynamic conditions exacerbated by hypertension and altered flow sensing. IA rupture results in substantial mortality, morbidity and poor long-term outcomes. In this Review, we focus mainly on strategies for screening, diagnosis and treatment of IAs in patients with ADPKD. Other vascular aneurysms and anomalies—including aneurysms of the aorta and coronary arteries, cervicocephalic and thoracic aortic dissections, aortic root dilatation and cerebral dolichoectasia—are less common in this population, and the available data are insufficient to recommend screening strategies. Treatment decisions should be made with expert consultation and be based on a risk–benefit analysis that takes into account aneurysm location and morphology as well as patient age and comorbidities.
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is the most common inherited kidney disease, affecting 1 in 400 to 1 in 1,000 individuals.1 In 2011, ADPKD accounted for 8% of incident and 4.8% of prevalent patients on dialysis in the USA.2 Mutations in either PKD1 (located on chromosome 16p13.3) or PKD2 (located on chromosome 4q22.1) account for most cases of the disease.3 Expansion of multiple cysts scattered throughout the renal parenchyma is characteristic of ADPKD. Cyst expansion produces a dramatic increase in total kidney volume4 and commonly leads to the development of kidney failure requiring renal replacement therapy by the middle of the 6th decade of life. All of the kidney complications of ADPKD (that is, haematuria, cyst infection, nephrolithiasis, pain, abdominal distension and hypertension) are related to the growth of cysts.5 Mutations in PKD1 and PKD2 trigger a cascade of cellular and molecular events that result in the formation of cysts in the kidney and liver and lead to a multitude of extrarenal pathologies affecting the vasculature, heart valves, seminal vesicles and other tissues. Our understanding of the pathogenesis of the extra-renal manifestations of ADPKD is extremely limited. Vascular abnormalities, particularly those associated with intracranial aneurysm (IA) rupture or arterial dissection are among the most serious complications of ADPKD.
Here, we review the vascular complications of ADPKD, including the clinical manifestations, management, and the relationship to pathophysiological mechanisms. Our discussion focuses primarily on IAs, which are fairly common and have the potential for devastating complications. Aneurysm rupture can result in permanent neurological impairment and death. Other vascular complications are not as common, unpredictable in their onset and less fully studied. Vascular complications are not generally associated with autosomal recessive polycystic kidney disease although isolated case reports exist of IAs in a child and in two young adults with this disease.6–8
Vascular phenotype
Dissections and aneurysms of almost every large artery—including the aorta (Figure 1), coronary arteries, cervico-cephalic arteries, vertebral arteries (Figure 2) and cranial arteries (Figure 3)—have been reported in patients with ADPKD (Tables 1 and 2). The presence of this wide array of vascular abnormalities has led to the hypothesis that polycystins might be required to maintain vascular integrity.9 The expression patterns of polycystin-1 and polycystin-2 are permissive; in mice genetic reporter studies have confirmed high levels of Pkd1 expression throughout the embryonic and adult cardiovascular system, including in the heart, aortic outflow tract and all major vessels.10 On a cellular level both proteins are expressed in the endothelial cells and vascular smooth muscle cells (VSMCs) that make up the vascular wall.11–13
Figure 1.
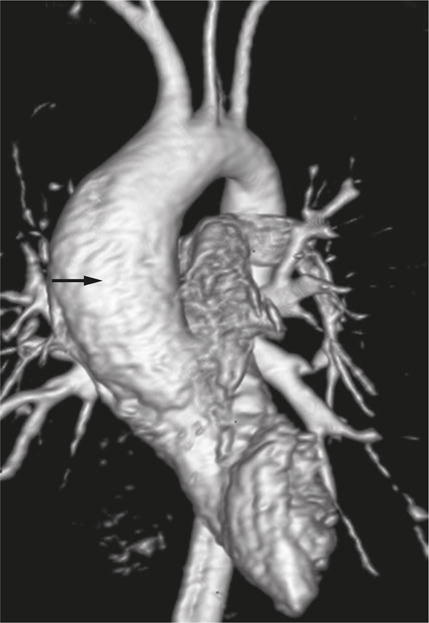
An aortic aneurysm in a 19-year-old patient with ADPKD. 3D magnetic resonance angiogram showing the ascending aortic aneurysm (arrow). Abbreviation: ADPKD, autosomal dominant polycystic kidney disease. Permission obtained from the American Society of Nephrology © Liu, D. et al. J. Am. Soc. Nephrol. 25, 81–91 (2014).
Figure 2.
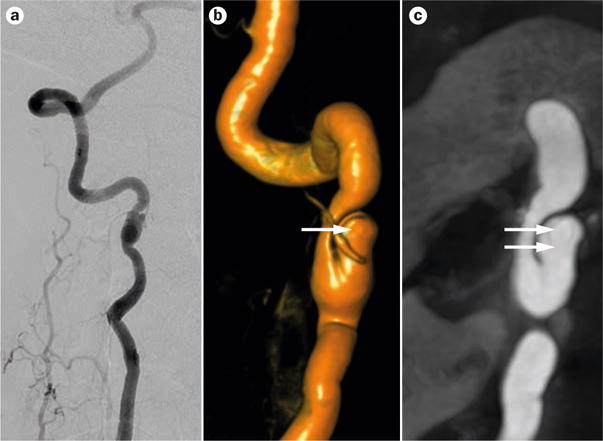
Right vertebral artery dissection in a 47-year-old patient with ADPKD. a | Angiogram b | 3D rotational angiogram and c | 3T magnetic resonance angiogram showing a spontaneous asymptomatic nontraumatic right vertebral artery dissection (dual arrows) and associated pseudoaneurysm (single arrow) with non-haemodynamically significant stenosis. The patient also had an unruptured intracranial aneurysm (shown in Figure 3). Abbreviation: ADPKD, autosomal dominant polycystic kidney disease.
Figure 3.
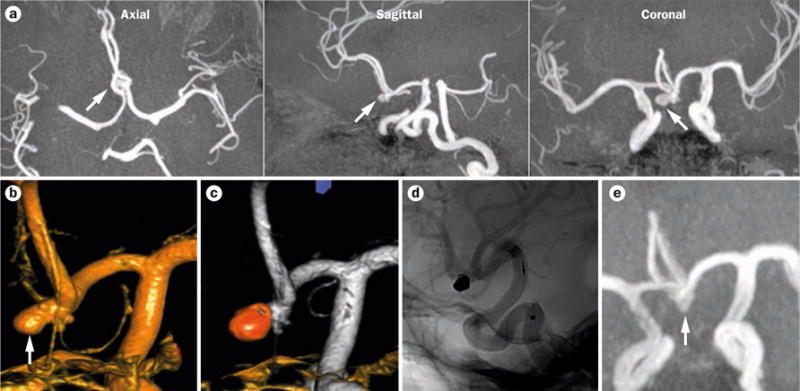
An unruptured intracranial aneurysm in a 47-year-old patient with ADPKD and distant smoking history. The patient underwent screening for assessment of suitability for transplant. a | 3T magnetic resonance angiograms showing an unruptured 5 mm aneurysm in the anterior communicating artery. b–c | 3D rotational angiograms confirming the presence of the aneurysm. d | 3D rotational angiogram showing the aneurysm after treatment using endovascular coiling. e | Magnetic resonance angiogram showing the stable aneurysm at 3-year follow up. Arrows indicate the position of the aneurysm. Abbreviation: ADPKD, autosomal dominant polycystic kidney disease.
Table 1.
Prevalence of IA in asymptomatic patients with ADPKD
| Characteristic | Huston et al. (1993)39 | Ruggieri et al. (1994)40 | Irazabal et al. (2011)42 | Xu et al. (2011)41 | Graf et al. (2002)82 |
|---|---|---|---|---|---|
| No. of patients | 85 | 93 | 407 | 355 | 43 |
| Age (years) | Mean 45; range 17–72 | 48 ± 12.8 | Median 50; range 17–82 | Mean 46.5; range 7–87 | Mean 45.7; range 16–73 |
| Type of MRA | 1.5 T | 1.5 T | 1.5 T | 3 T | 0.5 T |
| Overall detection rate (%) | 10.6 | 11.7 | 9.3 | 12.4 | 14* |
| Detection rate in patients with family history of SAH or IA (%) | 22 | 25.8 | 21.1 | 21.6 | 27.3 |
| IA in anterior circulation (%) | 78 | 84.6 | 84 | 100 | 80 |
| Size of IA | All <6.5 mm | All 2–8 mm | Median 3.5 mm; range 2–10 mm | Mean 3.85 mm; one 25 mm, rest <10 mm | Eight <10 mm; two >10 mm |
| Multiple IAs (% of cohort) | 0 | 2.2 | 1.8 | 2.3 | 4.7 |
Patients selected based on either a positive family history of a cerebral event (stroke), cerebral bleeding or known IA. Abbreviations: ADPKD, autosomal dominant polycystic kidney disease; IA, intracranial aneurysm; MRA, magnetic resonance angiography; SAH, subarachnoid haemorrhage.
Table 2.
Frequency of non-IA vascular anomalies in patients with ADPKD
| Anomaly | Frequency | Evidence | Reference(s) |
|---|---|---|---|
| Intracranial dolichoectasia | 4.7% (2 of 43 patients) | MRI screening | 82 |
| 2.2% (7 of 316 patients) | MRI screening, angiography, autopsy | 46 | |
| Coronary artery dissection | Rare | Case reports | 83,84 |
| Cervicocephalic arterial dissection | Rare | Case reports | 83,85 |
| Ascending aortic aneurysm | Rare | Case reports | 16,86 |
| Abdominal aortic aneurysm | Probably not increased | No increase in aortic diameter in 139 patients with ADPKD versus 149 healthy family members | 87 |
Abbreviations: ADPKD, autosomal dominant polycystic kidney disease; IA, intracranial aneurysm.
Several lines of experimental data support the concept of a vascular phenotype in ADPKD. In mice, targeted homozygous mutation of either Pkd1 or Pkd2 results in embryonic lethality with subcutaneous oedema, focal vascular leaks and haemorrhage.9,14 Mice with a hypomorphic Pkd1 allele that results in a significant reduction in polycystin-1 levels are viable but develop extensive aneurysm formation of the descending thoracic and abdominal aorta.15 The Cre-lox system has been used to investigate the role of polycystins in individual cell types. Deletion of either Pkd1 or Pkd2 in endothelial cells partially recapitulates the vascular phenotype observed in knockout mice with increased fetal demise, occasional haemorrhage and defects in branching of placental vessels.13 Deletion of Pkd1 in VSMCs, however, yields a surprisingly mild result with normal survival but a gradual degeneration of elastic fibres in the ascending aorta.16 These partial vascular phenotypes might result from incomplete activity of various Cre recombinases or could reflect a requirement for polycystin inactivation in multiple cell types.
Taken together, the available data suggest that polycystins have an important role in the development and maintenance of the vascular system. The precise cellular pathways that are disrupted by polycystin deficiency are now being investigated. Experimental evidence suggests that the polycystin complex functions as a flow sensor in the primary cilia of renal epithelium.17 Wild-type endothelial cells respond to fluid shear stress by increasing cytosolic calcium and nitric oxide levels, whereas Pkd1−/− and Pkd2−/− fetal aortic endothelial cells lack this response.18,19 Defective release of nitric oxide—a potent vasodilator—in response to fluid shear stress has been hypothesized to contribute to hypertension in ADPKD.18 Although loss of Pkd1 and Pkd2 yield similar endothelial cell phenotypes, a study has suggested that polycystin-1 and polycystin-2 have antagonistic effects in vascular myocytes.20 Deletion of Pkd1 in VSMCs reduced the activity of stretch-activated channels and resulted in decreased arterial myogenic tone, whereas knockdown of Pkd2 in Pkd1−/− arteries rescued the myogenic response. These results are counterintuitive as inactivation of either PKD1 or PKD2 yields identical phenotypes in most other systems. More work is needed to define whether these cellular phenotypes contribute to aneurysm formation in vivo.
Genetics of aneurysm formation
Familial clustering of IAs in patients with ADPKD suggests that features of the germline mutation in PKD1 or PKD2 might predispose to this phenotype. Mutation analyses by two independent groups have identified a recurrent two base pair deletion in PKD1 (5224del2) in unrelated families with ADPKD and IA.21,22 Moreover some, but not all, investigators have reported that the median position of the PKD1 mutation is located closer to the N-terminal in individuals with a vascular phenotype than in those without vascular abnormalities.22,23
The mechanism by which specific PKD mutations predispose to a vascular phenotype remains unclear. Although certain mutations might increase the risk of developing an aneurysm, not all individuals from high-risk families will experience vascular complications. Several models might account for intrafamilial variability with respect to this phenotype. Aneurysm formation could depend on focal, somatic and random loss of the wild-type PKD allele in vascular tissue, as has been demonstrated in the cyst lining epithelium of the kidney and liver.24–26 The finding that endothelial cells that are heterozygous for Pkd2 mutations have a normal response to shear stress is consistent with the requirement for a two-hit mechanism of aneurysm formation.19,27 However, such a mechanism would be challenging to prove in patients because of difficulty in isolating the target cell population from aneurysmal vessels.
Another potential genetic mechanism of aneurysm formation was suggested by reports that haploinsufficiency for Pkd2 results in a variety of cellular and vascular defects in mice.28–31 VSMCs isolated from mice that were haploinsufficient for Pkd2 demonstrated altered intracellular calcium homeostasis with decreased levels of resting calcium, decreased Ca2+ stores in the sarcoplasmic reticulum and a decrease in store-operated Ca2+channel activity.28 These fairly subtle abnormalities in intracellular calcium homeostasis had phenotypic consequences. The haploinsufficient mice had more intracranial vascular abnormalities than their littermate controls when subjected to severe haemodynamic stress including nephrectomy, carotid artery ligation and subcutaneous deoxycorticosterone acetate administration. In addition, aortic rings and mesenteric resistance arteries from these animals exhibited enhanced vascular reactivity in response to phenylephrine. These findings suggest that loss of one Pkd2 allele creates a sensitized state that might be susceptible to genetic modifiers of disease severity.
Genetic analysis of families with ADPKD and features of overlap connective tissue disorders such as Marfan syndrome (MFS) might also provide some clues regarding signalling pathways that could modify the propensity toward aneurysm formation in ADPKD.32 Marfan syndrome is caused by autosomal dominant mutations in the extracelluar matrix protein fibrillin-1 and is characterized by aortic root aneurysm and rupture.33 The pathophysiologic basis of MFS involves up-regulation of TGF-β and downstream pathways.34–36 Data from murine models demonstrate that heterozygosity for Pkd1 can exacerbate the MFS phenotype.16 Both Pkd1+/− VSMCs and Pkd1−/− murine embryonic fibroblasts exhibited a heightened response to TGF-β1, suggesting that this genetic interaction results from further upregulation of TGF-β signalling caused by haploinsufficiency for Pkd1.16 In addition, selective deletion of Pkd1 from VSMCs in the aorta resulted in upregulation of TGF-β signalling and gradual degeneration of elastic fibres (Figure 4). Modifiers that increase the activity of TGF-β signalling might, therefore, increase the risk of vascular complications in ADPKD. Genome-wide association studies (GWAS) in large cohorts of individuals without ADPKD have identified numerous loci that might be risk factors for the development of IA.37 GWAS in the ADPKD population might be similarly successful in identifying those at greatest risk, but will require the enrolment of large numbers of families with ADPKD and aneurysms.38
Figure 4.

Pkd1 deletion in VSMCs results in abnormal proximal ascending aortic architecture. Ascending aortic sections from 6-month-old mice with (Pkd1VSMC−) and without (Pkd1VSMC+) selective deletion of Pkd1 in VSMCs were stained with Verhoeff–Van Gieson. a | Intact elastic fibre architecture in the aorta of a Pkd1VSMC+ mouse. b–d | Aortic sections from Pkd1VSMC− mice demonstrate diffuse fragmentation of elastic fibres and increased extracellular matrix deposition. e | Blinded quantitative histologic evaluation of aorta architecture scored by three observers (n = 8). *P = 0.001. Abbreviation: VSMC, vascular smooth muscle cell. Permission obtained from the American Society of Nephrology © Liu, D. et al. J. Am. Soc. Nephrol. 25, 81–91 (2014).
Aysmptomatic intracranial aneurysms
Natural history
Limited information on the natural history of IAs in patients with ADPKD is available, and is primarily from observational studies. IAs are more frequent in patients with ADPKD (~9–12%)39–42 than in the general population (~2–3%; Table 1)43 and are found in families with PKD1 or PKD2 mutations.22,42,44 The difference in rates of IA rupture in patients with ADPKD and the general population is consistent with the difference in prevalence, suggesting a similar rupture risk in these populations. For example, in Rochester, MN, USA, the rupture rate for patients with ADPKD is ~1 in 2,000 person years,45 compared with 1 in 10,000 person years in the general population.46 The average age of IA rupture in patients with ADPKD is 41 years, approximately 10 years earlier than in the general population.45,47–49
A study by Belz and colleagues that included 20 patients with ADPKD and IAs—11 of whom had ruptured IAs at the time of initial diagnosis—reported that the earliest change in the size of an existing aneurysm or appearance of a new aneurysm occurred after 10.4 years of follow-up.50 During a mean follow-up period of 15.2 years, five patients developed a new IA in a different location from their initial IA and two of these patients also experienced an increase in the size of their existing IA. 10 patients underwent further IA surgery at a mean of 8.1 years (range 1.3–17 years) after their initial diagnosis, one patient experienced recurrent rupture, and no patients died during follow-up. Risk factors for the development of a new IA or an increase in the size of an existing IA were not identified.
The risk of IA expansion and rupture in patients with ADPKD was also assessed in a study of 38 patients with unruptured saccular IAs detected during pre-symptomatic screening.42 No aneurysm ruptures were reported during a cumulative clinical follow-up period of 316 years (mean 7.9 ± 6.2 years). At diagnosis, the median diameter of the 45 detected IAs was 3 mm (range 2–10 mm); 84% of IAs were located in the anterior circulation. Two existing IAs expanded in size during follow-up (4.5–5.9 mm over 69 months and 4.7–6.2 mm over 184 months) and one patient developed a new IA, which expanded from 2 mm to 4.4 mm over 144 months. Thus the risk of rapid expansion or rupture of small unruptured IAs detected by presymptomatic screening in patients with ADPKD is quite low, although rapid expansion might not equate to interval growth.
Screening
Imaging techniques
Screening for IA can be performed using high-resolution CT angiography (CTA) or time-of-flight magnetic resonance angiography (MRA) (Figures 3 and 5). A direct comparison of 3T time-of-flight MRA with 64 channel multi-detector CTA showed that these techniques have comparable sensitivity and specificity for detection of IA and can detect IAs as small as 2–3 mm.51–53 MRA is, however, the screening method of choice as gadolinium-based contrast agents are not required.
Figure 5.
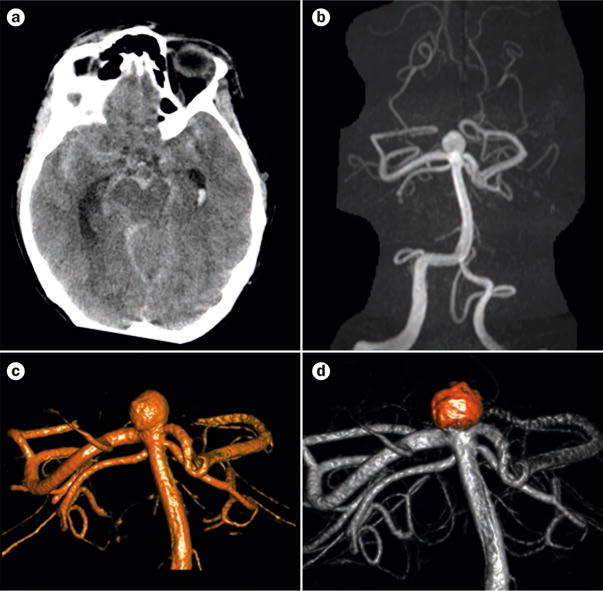
A ruptured intracranial aneurysm in a 46-year-old patient with ADPKD. a | CT scan showing diffuse subarachnoid haemorrhage and hydrocephalus. b | 3 T magnetic resonance angiogram showing the ruptured basilar bifurcation. c | 3D rotational angiography confirmed the presence of a wide-necked aneurysm. d | The aneurysm following treatment using endovascular coiling, which had an excellent result. Abbreviation: ADPKD, autosomal dominant polycystic kidney disease.
Strategy
IA rupture causing subarachnoid or intracerebral haemorrhage is the most serious complication of ADPKD, resulting in grave neurological morbidity or mortality in more than 50% of cases.54,55 As IA rupture is frequently catastrophic, early detection and intervention in appropriate individuals is highly desirable. The only factor that predicts an increased likelihood of IA detection in patients with ADPKD, however, is a positive family history of IA or subarachnoid haemorrhage (SAH). Among patients with asymptomatic ADPKD, the prevalence of unruptured IAs in those with such a family history ranges from 21–25% (Table 1), compared with 5–6% in those without a positive family history.39,42
As the prevalence of IAs and the risk of rupture of small IAs is low, screening of patients with ADPKD has generally been advised only for those with a family history of ruptured IA or SAH, a previous IA rupture or SAH, a high risk occupation where rupture would place the individual or others at high risk, those who require surgery that is associated with haemodynamic instability and hypertension, and those who need the reassurance of a negative result.56 Presymptomatic screening in these individuals should not be initiated before the age of 18 years because rupture of IA in children is very rare. Initial screening should, however, be performed by the age of 30 years, unless the family history of IA is particularly strong, in which case screening should be initiated earlier. If no IA is found during screening in a patient aged >30 years, rescreening at 5–10 year intervals has been suggested based on the findings of Belz et al.50 Those with known IA, previous rupture or SAH should be screened at more frequent intervals.
Several decision analyses have been performed to evaluate the benefits of screening for IAs in patients with ADPKD and in other populations.57,58 Decision analyses performed for the general population are applicable to patients with ADPKD if assumptions regarding the parameters used in the analyses are selected appropriately. Parameters that have been used in these analyses include age, size and location of IA, prior probability of IA, prior detection of IA or SAH, and surgical or endovascular morbidity and mortality. Butler and colleagues predicted that their strategy of screening all patients with ADPKD using MRA would provide on average one additional year of life without neurological disability for a patient with ADPKD aged 20 years, but an additional 10.8 years of life if an IA was discovered and treated.58 The benefit of screening decreased with increasing age. Their screening decision model was most sensitive to the prevalence of IA, the annual rupture incidence, and the morbidity and mortality rates associated with rupture.
The question of whether patients with ADPKD who are not deemed to be at high risk of IA rupture should undergo screening is controversial. Rozenfeld and colleagues recommend screening for IA using 3 T time-of-flight MRA in all patients with ADPKD at initial diagnosis, and follow-up MRA imaging at intervals of 2–10 years, depending on patient-specific risk factors including family history of IA or SAH, hypertension, smoking, alcohol abuse, high risk professions or need for major elective surgery with potential haemodynamic instability.53 Other experts recommend initial screening only in patients with increased risk of IA rupture defined as those with a family history of IA or SAH, a high-risk profession or those undergoing major elective surgery with potential haemodynamic instability.42 The rationale for the latter, less-stringent screening strategy is that most detected IAs are small with low risk of rupture. In view of the current uncertainty regarding the benefits of screening and the lack of data from randomized prospective clinical trials, it is reasonable to offer, but not necessarily recommend, screening for IA to all adult patients with ADPKD, including those without identified risk factors.
Screening for IAs prior to listing for kidney transplantation is performed in some centres, but the impact of this screening has not yet been systematically addressed. Limited data are available and do not demonstrate a high likelihood of IA rupture after transplantation,59 although an increased likelihood of haemorrhagic stroke after kidney transplantation has been reported in patients with ADPKD.60 As the average age of IA rupture (41 years)47 is well below the average age of progression to end-stage renal disease (55 years),61 little benefit might be obtained from screening individuals aged ≥50 years before transplantation. We do not recommend an absolute upper age limit for screening of patients with ADPKD in those considered to be at high risk, but it seems reasonable to defer screening after the age of 60 years, particularly if multiple previous screenings have been negative.
Some of the screening recommendations listed above are not strictly evidence based. In certain instances a consensus between experts might, therefore, be lacking. A reason able approach is to perform screening if risk–benefit considerations including the vital status, age and overall health of the patient are favourable and would dictate intervention if an IA is identified. For example, individuals aged >60 years on dialysis would be predicted to have increased risks of complications associated with IA repair, which might outweigh the benefits of intervention.
Management
Decisions regarding the management of IA should involve multidisciplinary consultation with neurosurgeons and neurointerventional radiologists. In general, both the decision to intervene and the choice of intervention is determined by the size and location of the IA, the general health and age of the affected individual, and the risk of rupture. No specific data on outcomes of IA interventions in patients with ADPKD are available. In the general population reported complications of IA interventions include in-hospital mortality, requirement for discharge to a long-term facility, headache, aphasia, hemiplegia, hemiparesis, hydrocephalus, cerebral artery occlusion, postoperative cardiac complications, surgical complications, postoperative infections, neurologic complications, performance of tracheostomy, placement of an endotracheal tube, performance of ventriculostomy and ventriculoperitoneal shunt surgery.62 In the USA, periprocedural morbidity and mortality is lower after endovascular coiling than after clipping,62 and centres that preferentially perform coiling have the best outcomes.63
Assessing rupture risk
The risk of IA rupture has been extensively evaluated in non-ADPKD populations. The prospective ISUIA study, which followed 1,692 patients with unruptured IAs of ≥2 mm for a mean of 4.1 years, reported that increasing aneurysm size, location in the posterior versus the anterior circulation and prior SAH were associated with an increased rate of rupture.64 The 5-year cumulative rupture rate for IAs <7 mm in the anterior circulation was 0% versus a rupture rate of 2.5% for IA <7 mm located in the posterior circulation.64 By contrast the SUAVe prospective study from Japan, which monitored 448 aneurysms of <5 mm over 41 months, reported an overall yearly rupture risk of 0.54%. Age <50 years, IA size >4 mm, the presence of multiple IAs and comorbid hypertension were associated with increased rupture risk.65,66
The UCAS study from Japan followed 5,720 patients aged >20 years with a total of 6,697 aneurysms measuring >3 mm (mean 5.7 ± 3.6 mm) over a 3-year period.67 91% of the aneurysms were detected incidentally and most were located in the middle cerebral (36%) or internal carotid arteries (34%). The overall rate of IA rupture was 0.95% per year. Aneurysms in anterior communicating arteries (ACOM) or posterior communicating arteries (PCOM) were nearly twice as likely to rupture as lesions in the middle cerebral artery. This study confirmed an important role of aneurysm size in rupture risk and also identified shape as a possible risk factor; the presence of an irregular surface protrusion or daughter sac on the aneurysm conferred a 1.63-fold increased risk of rupture.
In contrast to the ISUIA study, which suggested that IAs of <7 mm located in the anterior circulation were unlikely to rupture,64 UCAS reported that aneurysms in the ACOM have non-negligible yearly rupture rates that increase with size: 0.9% at 3–4 mm, 0.75% at 5–6 mm, 1.97% at 7–9 mm, 5.24% at 10–24 mm and 39.77 at >25 mm.67 Similarly aneurysms in the internal carotid or PCOM had yearly rupture rates of 0.41% at 3–4 mm, 1.00% at 5–6 mm, 3.19% at 7–9 mm, 6.12% at 10–24 mm and >100% (reflecting multiple ruptures) at >25 mm. Although the prevalence of ADPKD in the UCAS study was low (0.3%), the findings confirmed an increased risk of IA rupture in women and in patients with hypertension, and a nonsignificant trend towards protection by hypercholesterolaemia. Unexpectedly, UCAS also showed that smoking, family history or presence of other aneurysms did not increase rupture risk.
The results of the UCAS study suggest that the perception that aneurysms with a dome size <7 mm do not require intervention might be unjustified, and point towards a shape and location-dependent individualized care plan for IA.67 To develop a personalized risk score for IA rupture, the PHASES study pooled data from six international prospective cohort studies on the natural history of unruptured aneurysms.68 The study reported 3.6-fold and 2.8-fold higher rates of IA rupture in Finnish and Japanese patients, respectively, than in other European and North American participants. The overall mean rupture rate was 1.4% with a 5-year cumulative risk of 3.4%; age, hypertension, previous SAH and the size and location of the IA were significant predictors of rupture. The resulting PHASES risk score assigns one point each for hypertension, age ≥70 years and prior SAH; three points for Japanese and five points for Finnish nationality; up to four points for aneurysm location and up to 10 points for size. The score enables estimation of a 5-year risk of rupture from 0.4% for a score of two or less, 4.3% for a score of nine and 17.8 for a score ≥12.
Follow-up monitoring
No standardized criteria exist for the frequency of follow-up imaging after IA detection in patients with ADPKD.53 Wiebers and colleagues propose that aneurysms develop rapidly and either rupture soon after or are stabilized by the elastic component of the aneurysm wall and further strengthened by deposition of collagen.69 When the size of the IA exceeds 10 mm, however, the resulting high wall stress might promote further growth. Thus, small aneurysms might rupture soon after formation and newly detected IAs should be monitored frequently, at least yearly on initial presentation, to assure stability. Follow-up can be less frequent once stability has been determined. Similarly, Rozenfeld and colleagues recommend biannual screening for the first 2 years after detection and every 2–5 years thereafter if the IA is stable.53 Lifestyle modifications should also be implemented, including smoking cessation, blood pressure control and avoidance of heavy alcohol consumption, stimulant medications, illicit drugs, excessive straining and Valsalva maneuvers.53,70 Anticoagulant and antiplatelet medications do not seem to increase the risk of aneurysm rupture but might worsen the severity of SAH.70
Although aspirin use has traditionally been avoided in patients with IA, some evidence suggests that its anti-inflammatory effects might be beneficial in this population. Inflammation is thought to be a critical contributor to the pathogenesis of aneurysm rupture71 and inflammatory changes (increased uptake of ferumoxytol by macrophages and upregulation of inflammatory mediators) have been demonstrated in the walls of unruptured aneurysms.72,73 In five patients with IAs, daily treatment with 81 mg of aspirin for 3 months resulted in a decrease in inflammation in aneurysm tissues.72 Moreover, a case-controlled study in which 58 patients with ruptured IAs were matched to 213 patients with unruptured IAs reported that those who used aspirin (3–7 times per week) had a significantly lower odds of haemorrhage than those who did not take aspirin.74 These findings suggest that aspirin avoidance in patients with IA might need to be re-examined, but additional prospective data on the prophylactic use of aspirin in patients with IA is required.
Intervention
Aneurysm treatment must balance the risk of continued observation with the anticipated risk of medical, surgical or endovascular therapy. The method of treatment should be determined using an individualized approach made by an expert team with expertise in the various treatment modalities (Figure 6). Treatment decisions must take into account patient age, medical risk factors, aneurysm location and shape as well as accessibility by either surgical or endovascular routes.
Figure 6.
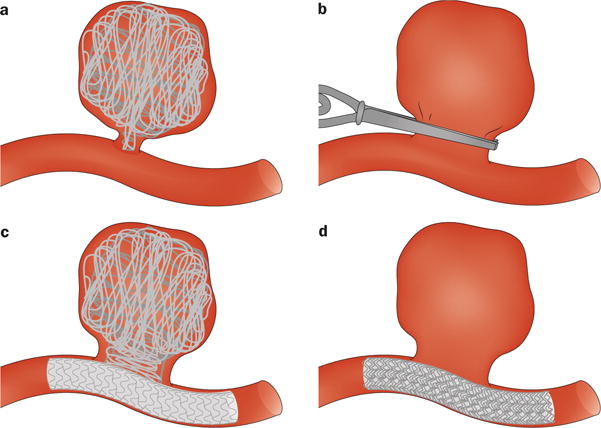
Surgical and angiographic techniques for the treatment of intracranial aneurysms. a | Microsurgical clipping. b | Endovascular coiling. c | Stent-supported coiling. d | Flow-diverter therapy.
Until the development of endovascular coiling in 1991,75,76 treatment of IA involved a craniotomy and use of microsurgical techniques under high-power magnification to enter the subarachnoid space and carefully dissect the aneurysm after obtaining proximal control of the parent vessel. The aneurysm would then be secured by applying a specially shaped stainless steel or titanium clip to permanently close the neck of the aneurysm whilst maintaining flow in the daughter branches of the parent vessel.
Improvements in microcatheter design and delivery methods as well as radiographic and angiographic techniques enabled the use of size-matched, complex-shaped embolic coils to fill the aneurysm from a percutaneous transfemoral endovascular route. These coils dampen the forces in the artery wall, mitigate rupture and promote progressive clotting and hopefully healing at the aneurysm neck via re-endothelialization.76,77 Initially limited to narrow-necked aneurysms, the use of adjunctive balloon-remodelling and stent-supported coiling has further expanded the spectrum of aneurysms that are amenable to endovascular treatment.
Finely-braided intracranial stent constructs known as flow-diverters are also now available for the treatment of IA. Flow-diverters are deployed across side-wall aneurysms and promote involution by slowing blood flow velocity in the dome, and progressive healing by providing a scaffold for re-endothelialization at the aneurysm neck.78
Symptomatic or ruptured intracranial aneurysm
The sudden onset of an extremely severe headache or new neurological symptoms in a patient with ADPKD should prompt further evaluation for IA. Typically a non-contrast CT scan is used to look for evidence of haemorrhage (Figure 5a). A full discussion of the management of symptomatic or ruptured IA is beyond the scope of this Review, but expert guidelines are available.79 Patients should be cared for in an intensive care setting with continuous haemodynamic and neurological monitoring. Common complications include re-bleeding, vasospasm, cerebral ischaemia, hydrocephalus, increased intracranial pressure, seizures and cardiac complications. A good outcome is achieved in approximately one-third of patients.79–81
Other vascular abnormalities
Unfortunately, very few systematic studies of the incidence and prevalence of vascular abnormalities other than IA in patients with ADPKD exist and validated recommendations for screening and follow up cannot be made. However, in patients who have a strong family history of a particular vascular complication (such as aortic root dilatation), intensive evaluation and follow-up monitoring seems reasonable.
Conclusions
The molecular basis of aneurysm formation in ADPKD is complex and remains to be fully delineated. PKD1 or PKD2 haploinsufficiency, alterations in intracellular calcium signalling, modifier genes and alterations in TGF-β signalling could contribute to aneurysm formation. Upregulation of TGF-β signalling in the setting of Pkd1 haploinsufficiency suggests a potential mechanism that will require further exploration.
As IA rupture results in substantial morbidity and mortality, patients at increased risk of rupture should undergo screening. If an asymptomatic IA is detected, decisions regarding intervention should involve multidisciplinary consultations. Small aneurysms in the anterior circulation have a low risk of rupture and can be monitored with repeated imaging. Lifestyle interventions to decrease risk of rupture should also be implemented. The development of acute onset, severe headache mandates urgent evaluation for intracranial haemorrhage with a CT scan and neurosurgical consultation.
Other vascular complications of ADPKD are rare and cannot currently be predicted based on clinical screening or genetic analysis. Thus, definitive recommendations for screening cannot be provided. Association of particular mutations with a vascular phenotype or familial clustering of vascular anomalies could, however, justify screening in affected families.
Although substantial progress has been made in the diagnosis and treatment of vascular anomalies—particularly IAs—in patients with ADPKD, important questions remain. More research is required to define which patients are at risk of vascular anomalies and their complications and to better understand the mechanisms of aneurysm formation and arterial dissection, including the role of inflammation and the interactions of ADPKD genetic mutations with TGF-β and other signalling pathways.
Key points.
-
■
Intracranial aneurysms (IAs) are the most common vascular manifestation of autosomal dominant polycystic kidney disease (ADPKD)
-
■
Individuals with ADPKD and increased risk of IA—including those with a family or personal history of IA or subarachnoid haemorrhage—should undergo screening
-
■
Other vascular abnormalities in ADPKD include aneurysms and dissections of the thoracic aorta, coronary arteries and cervicocephalic arteries, aortic root dilatation and cerebral dolichoectasia; screening is not usually indicated
-
■
Asymptomatic IAs detected by screening are frequently small and have a low risk of rupture
-
■
Intervention, either surgical or endovascular, is indicated based on the size and location of the aneurysm
-
■
The relationship between PKD1 and PKD2 mutations and the development of vascular abnormalities is undefined; modifier genes that increase TGF-β signalling might increase the risk of vascular complications in ADPKD
Acknowledgments
T.W.’s work is supported in part by grants from the National Institute of Diabetes and Digestive and Kidney Diseases (P30DK090868, DK076017 and DK095036). R.D.P.’s work is supported in part by grants from the National Institute of Diabetes and Digestive and Kidney Diseases (DK62411), and from the National Center for Advancing Translational Sciences Clinical and Translational Science Awards (UL1TR001064 awarded to Tufts University).
R.D.P. has received research funding from Sanofi–Genzyme and is a consultant for Otsuka and Vertex.
Biographies
Ronald D. Perrone is Associate Chief of the Division of Nephrology, Medical Director of Kidney Transplantation and Scientific Director of the Clinical and Translational Research Center at Tufts Medical Center as well as Professor of Medicine at Tufts University School of Medicine, Boston, MA, USA. He focuses on clinical research in poly-cystic kidney disease (PKD) and the role of total kidney volume as a biomarker in response to clinical therapies. Dr Perrone served as site Principal Investigator and was a member of the steering committee for the TEMPO 251 and 210 studies of tolvaptan in autosomal dominant PKD (ADPKD). He is currently the site Principal Investigator and a member of the steering committee for the NIDDK-sponsored HALT PKD study and the clinical lead for the PKD Outcomes Consortium, which is seeking to validate total kidney volume as a clinical trial and regulatory end point in ADPKD.
Adel M. Malek is Director of Cerebrovascular and Endovascular Neurosurgery at Tufts Medical Center and Professor of Neurosurgery, Radiology and Neurology at Tufts University School of Medicine, Boston, MA, USA. He studied electrical engineering at Massachusetts Institute of Technology (MIT), Cambridge, USA, and graduated in medicine from Harvard–MIT before completing doctoral work on haemodynamic shear-stress-induced endothelial gene expression. Dr Malek trained in Neurosurgery at Brigham and Women’s Hospital and Boston Children’s Hospital and in Neurointerventional Radiology at University of California in San Francisco, USA. His clinical practice centers on treatment of cerebral aneurysms and arteriovenous malformations. His research is focused on understanding the link between mechanical forces and aneurysm development, imaging-based rupture risk stratification, and clinical outcomes.
Terry Watnick is Associate Professor of Medicine in the Division of Nephrology at the University of Maryland School of Medicine, Baltimore, MD, USA. She directs the Baltimore Polycystic Kidney Disease Research and Clinical Core Center. Dr Watnick completed medical school at Yale School of Medicine, New Haven, CT, USA, and postgraduate training in Medicine and Nephrology at Yale New Haven Hospital and Johns Hopkins Hospital, respectively. Her research focuses on understanding the vascular phenotype that is associated with autosomal dominant polycystic kidney disease.
Footnotes
Competing interests
The other authors declare no competing interests.
Author contributions
All authors researched the data, made substantial contributions to discussion of content, wrote the article and reviewed/edited the manuscript before submission
References
- 1.Iglesias C, et al. Epidemiology of adult polycystic kidney disease, Olmsted County, Minnesota: 1935–1980. Am J Kidney Dis. 1983;2:630–639. doi: 10.1016/s0272-6386(83)80044-4. [DOI] [PubMed] [Google Scholar]
- 2.Collins AJ, et al. United States Renal Data System 2011 Annual Data Report: Atlas of chronic kidney disease & end-stage renal disease in the United States. Am J Kidney Dis. 2012;59(Suppl. 1):e1–e420. doi: 10.1053/j.ajkd.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 3.Ong A, Harris P. Molecular pathogenesis of ADPKD: the polycystin complex gets complex. Kidney Int. 2005;67:1234–1247. doi: 10.1111/j.1523-1755.2005.00201.x. [DOI] [PubMed] [Google Scholar]
- 4.Grantham J, et al. Volume progression in polycystic kidney disease. N Engl J Med. 2006;354:2122–2130. doi: 10.1056/NEJMoa054341. [DOI] [PubMed] [Google Scholar]
- 5.Grantham J, Chapman A, Torres V. Volume progression in autosomal dominant polycystic kidney disease: the major factor determining clinical outcomes. Clin J Am Soc Nephrol. 2006;1:148–157. doi: 10.2215/CJN.00330705. [DOI] [PubMed] [Google Scholar]
- 6.Lilova M, Petkov D. Intracranial aneurysms in a child with autosomal recessive polycystic kidney disease. Pediatr Nephrol. 2001;16:1030–1032. doi: 10.1007/s004670100019. [DOI] [PubMed] [Google Scholar]
- 7.Neumann H, Krumme B, van Velthoven V, Orszagh M, Zerres K. Multiple intracranial aneurysms in a patient with autosomal recessive polycystic kidney disease. Nephrol Dial Transplant. 1999;14:936–939. doi: 10.1093/ndt/14.4.936. [DOI] [PubMed] [Google Scholar]
- 8.Chalhoub V, Abi-Rafeh L, Hachem K, Ayoub E, Yazbeck P. Intracranial aneurysm and recessive polycystic kidney disease: the third reported case. JAMA Neurol. 2013;70:114–116. doi: 10.1001/jamaneurol.2013.584. [DOI] [PubMed] [Google Scholar]
- 9.Kim K, Drummond I, Ibraghimov-Beskrovnaya O, Klinger K, Arnaout M. Polycystin 1 is required for the structural integrity of blood vessels. PNAS. 2000;97:1731–1736. doi: 10.1073/pnas.040550097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boulter C, et al. Cardiovascular, skeletal, and renal defects in mice with a targeted disruption of the Pkd1 gene. Proc Natl Acad Sci USA. 2001;98:12174–12179. doi: 10.1073/pnas.211191098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qian Q, Ming L, Somlo S, Harris H, Torres V. Analysis of polycystins in vascular smooth muscle cells. J Am Soc Nephrol. 2003;14:2280–2287. doi: 10.1097/01.asn.0000080185.38113.a3. [DOI] [PubMed] [Google Scholar]
- 12.Torres V, et al. Vascular expression of polycystin-2. J Am Soc Nephrol. 2001;12:1–9. doi: 10.1681/ASN.V1211. [DOI] [PubMed] [Google Scholar]
- 13.Garcia-Gonzalez M, et al. Pkd1 and Pkd2 are required for normal placental development. PLoS ONE. 2010;5:e12821. doi: 10.1371/journal.pone.0012821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu G, et al. Cardiac defects and renal failure in mice with targeted mutations in Pkd2. Nat Genet. 2000;24:75–78. doi: 10.1038/71724. [DOI] [PubMed] [Google Scholar]
- 15.Hassane S, et al. Pathogenic sequence for dissecting aneurysm formation in a hypomorphic polycystic kidney disease 1 mouse model. Arterioscler Thromb Vasc Biol. 2007;27:2177–2183. doi: 10.1161/ATVBAHA.107.149252. [DOI] [PubMed] [Google Scholar]
- 16.Liu D, et al. A Pkd1–Fbn1 genetic interaction implicates TGF-β signaling in the pathogenesis of vascular complications in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2014;25:81–91. doi: 10.1681/ASN.2012050486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nauli S, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33:129–137. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 18.Nauli S, et al. Endothelial cilia are fluid shear sensors that regulate calcium signaling and nitric oxide production through polycystin-1. Circulation. 2008;117:1161–1171. doi: 10.1161/CIRCULATIONAHA.107.710111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.AbouAlaiwi W, et al. Ciliary polycystin-2 is a mechanosensitive calcium channel involved in nitric oxide signaling cascades. Circ Res. 2009;104:860–869. doi: 10.1161/CIRCRESAHA.108.192765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharif-Naeini R, et al. Polycystin-1 and -2 dosage regulates pressure sensing. Cell. 2009;139:587–596. doi: 10.1016/j.cell.2009.08.045. [DOI] [PubMed] [Google Scholar]
- 21.Watnick T, et al. Mutation detection of PKD1 identifies a novel mutation common to three families with aneurysms and/or very-early-onset disease. Am J Hum Genet. 1999;65:1561–1571. doi: 10.1086/302657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rossetti S, et al. Association of mutation position in polycystic kidney disease 1 (PKD1) gene and development of a vascular phenotype. Lancet. 2003;361:2196–2201. doi: 10.1016/S0140-6736(03)13773-7. [DOI] [PubMed] [Google Scholar]
- 23.Neumann H, et al. Characteristics of intracranial aneurysms in the else kröner-fresenius registry of autosomal dominant polycystic kidney disease. Cerebrovasc Dis Extra. 2012;2:71–79. doi: 10.1159/000342620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qian F, Watnick T, Onuchic L, Germino G. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell. 1996;87:979–987. doi: 10.1016/s0092-8674(00)81793-6. [DOI] [PubMed] [Google Scholar]
- 25.Watnick T, et al. Somatic mutation in individual liver cysts supports a two-hit model of cystogenesis in autosomal dominant polycystic kidney disease. Mol Cell. 1998;2:247–251. doi: 10.1016/s1097-2765(00)80135-5. [DOI] [PubMed] [Google Scholar]
- 26.Pei Y, et al. Somatic PKD2 mutations in individual kidney and liver cysts support a “two-hit” model of cystogenesis in type 2 autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1999;10:1524–1529. doi: 10.1681/ASN.V1071524. [DOI] [PubMed] [Google Scholar]
- 27.Patel A, Honoré E. Polycystins and renovascular mechanosensory transduction. Nat Rev Nephrol. 2010;6:530–538. doi: 10.1038/nrneph.2010.97. [DOI] [PubMed] [Google Scholar]
- 28.Qian Q, et al. Pkd2 haploinsufficiency alters intracellular calcium regulation in vascular smooth muscle cells. Hum Mol Genet. 2003;12:1875–1880. doi: 10.1093/hmg/ddg190. [DOI] [PubMed] [Google Scholar]
- 29.Morel N, et al. PKD1 haploinsufficiency is associated with altered vascular reactivity and abnormal calcium signaling in the mouse aorta. Pflugers Arch. 2009;457:845–856. doi: 10.1007/s00424-008-0561-y. [DOI] [PubMed] [Google Scholar]
- 30.Qian Q, et al. Pkd2± vascular smooth muscles develop exaggerated vasocontraction in response to phenylephrine stimulation. J Am Soc Nephrol. 2007;18:485–493. doi: 10.1681/ASN.2006050501. [DOI] [PubMed] [Google Scholar]
- 31.Du H, Wang X, Wu J, Qian Q. Phenylephrine induces elevated RhoA activation and smooth muscle α-actin expression in Pkd2± vascular smooth muscle cells. Hypertens Res. 2010;33:37–42. doi: 10.1038/hr.2009.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Somlo S, et al. A kindred exhibiting cosegregation of an overlap connective tissue disorder and the chromosome 16 linked form of autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1993;4:1371–1378. doi: 10.1681/ASN.V461371. [DOI] [PubMed] [Google Scholar]
- 33.Ramirez F, Dietz H. Marfan syndrome: from molecular pathogenesis to clinical treatment. Curr Opin Genet Dev. 2007;17:252–258. doi: 10.1016/j.gde.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 34.Neptune E, et al. Dysregulation of TGF-β activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33:407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- 35.Habashi J, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holm T, et al. Noncanonical TGFβ signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science. 2011;332:358–361. doi: 10.1126/science.1192149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hussain I, Duffis E, Gandhi C, Prestigiacomo C. Genome-wide association studies of intracranial aneurysms: an update. Stroke. 2013;44:2670–2675. doi: 10.1161/STROKEAHA.113.001753. [DOI] [PubMed] [Google Scholar]
- 38.Rossetti S, Harris P. The genetics of vascular complications in autosomal dominant polycystic kidney disease (ADPKD) Curr Hypertens Rev. 2013;9:37–43. doi: 10.2174/1573402111309010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huston J, III, Torres V, Sulivan P, Offord K, Wiebers D. Value of magnetic resonance angiography for the detection of intracranial aneurysms in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1993;3:1871–1877. doi: 10.1681/ASN.V3121871. [DOI] [PubMed] [Google Scholar]
- 40.Ruggieri P, et al. Occult intracranial aneurysms in polycystic kidney disease: screening with MR angiography. Radiology. 1994;191:33–39. doi: 10.1148/radiology.191.1.8134594. [DOI] [PubMed] [Google Scholar]
- 41.Xu H, Yu S, Mei C, Li M. Screening for intracranial aneurysm in 355 patients with autosomal-dominant polycystic kidney disease. Stroke. 2011;42:204. doi: 10.1161/STROKEAHA.110.578740. [DOI] [PubMed] [Google Scholar]
- 42.Irazabal M, et al. Extended follow-up of unruptured intracranial aneurysms detected by presymptomatic screening in patients with autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2011;6:1274–1285. doi: 10.2215/CJN.09731110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vlak M, Algra A, Brandenburg R, Rinkel G. Prevalence of unruptured intracranial aneurysms, with emphasis on sex, age, comorbidity, country, and time period: a systematic review and meta-analysis. Lancet Neurol. 2011;10:626–636. doi: 10.1016/S1474-4422(11)70109-0. [DOI] [PubMed] [Google Scholar]
- 44.van Dijk M, Chang P, Peters D, Breuning M. Intracranial aneurysms in polycystic kidney disease linked to chromosome 4. J Am Soc Nephrol. 1995;6:1670–1673. doi: 10.1681/ASN.V661670. [DOI] [PubMed] [Google Scholar]
- 45.Schievink W, Torres V, Piepgras D, Wiebers D. Saccular intracranial aneurysms in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1992;3:88–95. doi: 10.1681/ASN.V3188. [DOI] [PubMed] [Google Scholar]
- 46.Schievink W, Torres V, Wiebers D, Huston J., 3rd Intracranial arterial dolichoectasia in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1997;8:1283–1291. doi: 10.1681/ASN.V881298. [DOI] [PubMed] [Google Scholar]
- 47.Chauveau D, et al. Intracranial aneurysms in autosomal dominant polycystic kidney disease. Kidney Int. 1994;45:1140–1146. doi: 10.1038/ki.1994.151. [DOI] [PubMed] [Google Scholar]
- 48.Lozano A, Leblanc R. Familial intracranial aneurysms. J Neurosurg. 1987;66:522–528. doi: 10.3171/jns.1987.66.4.0522. [DOI] [PubMed] [Google Scholar]
- 49.Chauveau D, et al. Extrarenal manifestations in autosomal dominant polycystic kidney disease. Adv Nephrol. 1997;26:265–289. [PubMed] [Google Scholar]
- 50.Belz M, et al. Recurrence of intracranial aneurysms in autosomal-dominant polycystic kidney disease. Kidney Int. 2003;63:1824. doi: 10.1046/j.1523-1755.2003.00918.x. [DOI] [PubMed] [Google Scholar]
- 51.Hiratsuka Y, et al. Diagnosis of unruptured intracranial aneurysms: 3T MR angiography versus 64-channel multi-detector row CT angiography. Magn Reson Med Sci. 2008;7:169–178. doi: 10.2463/mrms.7.169. [DOI] [PubMed] [Google Scholar]
- 52.Sailer A, Wagemans B, Nelemans P, de Graaf R, van Zwam W. Diagnosing intracranial aneurysms with MR angiography. Systematic review and meta-analysis. Stroke. 2014;45:119–126. doi: 10.1161/STROKEAHA.113.003133. [DOI] [PubMed] [Google Scholar]
- 53.Rozenfeld M, et al. Should patients with autosomal dominant polycystic kidney disease be screened for cerebral aneurysms? Am J Neuroradiol. 2014;35:3–9. doi: 10.3174/ajnr.A3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Broderick J, Brott T, Duldner J, Tomsick T, Leach A. Initial and recurrent bleeding are the major causes of death following subarachnoid hemorrhage. Stroke. 1994;25:1342–1347. doi: 10.1161/01.str.25.7.1342. [DOI] [PubMed] [Google Scholar]
- 55.Phillips L, 2nd, Whisnant J, O’Fallon W, Sundt T., Jr The unchanging pattern of subarachnoid hemorrhage in a community. Neurology. 1980;30:1034–1040. doi: 10.1212/wnl.30.10.1034. [DOI] [PubMed] [Google Scholar]
- 56.Grantham J. Clinical practice. Autosomal dominant polycystic kidney disease. N Engl J Med. 2008;359:1477–1485. doi: 10.1056/NEJMcp0804458. [DOI] [PubMed] [Google Scholar]
- 57.Takao H, Nojo T. Treatment of unruptured intracranial aneurysms: decision and cost-effectiveness analysis. Radiology. 2007;244:755–766. doi: 10.1148/radiol.2443061278. [DOI] [PubMed] [Google Scholar]
- 58.Butler W, Barker FG, Crowell R. Patients with polycystic kidney disease would benefit from routine magnetic resonance angiographic screening for intracerebral aneurysms: a decision analysis. Neurosurgery. 1996;38:506–515. doi: 10.1097/00006123-199603000-00018. [DOI] [PubMed] [Google Scholar]
- 59.Alam A, Perrone R. Management of ESRD in patients with autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis. 2010;17:164–172. doi: 10.1053/j.ackd.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 60.Abedini S, et al. Cerebrovascular events in renal transplant recipients. Transplantation. 2009;87:112–117. doi: 10.1097/TP.0b013e31818bfce8. [DOI] [PubMed] [Google Scholar]
- 61.Perrone R, Ruthazer R, Terrin N. Survival after end-stage renal disease in autosomal dominant polycystic kidney disease: contribution of extrarenal complications to mortality. Am J Kidney Dis. 2001;38:777–784. doi: 10.1053/ajkd.2001.27720. [DOI] [PubMed] [Google Scholar]
- 62.Brinjikji W, et al. Better outcomes with treatment by coiling relative to clipping of unruptured intracranial aneurysms in the United States, 2001–2008. Am J Neuroradiol. 2011;32:1071–1075. doi: 10.3174/ajnr.A2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brinjikji W, Rabinstein A, Lanzino G, Kallmes D, Cloft H. Patient outcomes are better for unruptured cerebral aneurysms treated at centers that preferentially treat with endovascular coiling: a study of the National Inpatient Sample 2001–2007. Am J Neuroradiol. 2011;32:1065–1070. doi: 10.3174/ajnr.A2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wiebers D, et al. Unruptured intracranial aneurysms: natural history, clinical outcome, and risks of surgical and endovascular treatment. Lancet. 2003;362:103–110. doi: 10.1016/s0140-6736(03)13860-3. [DOI] [PubMed] [Google Scholar]
- 65.UCAS Japan Investigators et al. The natural course of unruptured cerebral aneurysms in a Japanese cohort. N Engl J Med. 2012;366:2474–2482. doi: 10.1056/NEJMoa1113260. [DOI] [PubMed] [Google Scholar]
- 66.Sonobe M, Yamazaki T, Yonekura M, Kikuchi H. Small unruptured intracranial aneurysm verification study: SUAVe study, Japan. Stroke. 2010;41:1969–1977. doi: 10.1161/STROKEAHA.110.585059. [DOI] [PubMed] [Google Scholar]
- 67.Investigators, U. J. The natural course of unruptured cerebral aneurysms in a Japanese cohort. N Engl J Med. 2012;366:2474–2482. doi: 10.1056/NEJMoa1113260. [DOI] [PubMed] [Google Scholar]
- 68.Greving J, et al. Development of the PHASES score for prediction of risk of rupture of intracranial aneurysms: a pooled analysis of six prospective cohort studies. Lancet Neurol. 2014;13:59–66. doi: 10.1016/S1474-4422(13)70263-1. [DOI] [PubMed] [Google Scholar]
- 69.Wiebers D, et al. Reprint of Symposium on Cerebrovascular Diseases. Pathogenesis, natural history, and treatment of unruptured intracranial aneurysms. Neuroradiol J. 2006;19:504–515. doi: 10.1177/197140090601900409. [DOI] [PubMed] [Google Scholar]
- 70.Williams L, Brown R., Jr Management of unruptured intracranial aneurysms. Neurol Clin Pract. 2013;3:99–108. doi: 10.1212/CPJ.0b013e31828d9f6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chalouhi N, et al. Biology of intracranial aneurysms: role of inflammation. J Cereb Blood Flow Metab. 2012;32:1659–1676. doi: 10.1038/jcbfm.2012.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hasan DM, et al. Imaging aspirin effect on macrophages in the wall of human cerebral aneurysms using ferumoxytol-enhanced MRI: preliminary results. J Neuroradiol. 2013;40:187–191. doi: 10.1016/j.neurad.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hasan DM, et al. Evidence that acetylsalicylic acid attenuates inflammation in the walls of human cerebral aneurysms: preliminary results. J Am Heart Assoc. 2013;2:e000019. doi: 10.1161/JAHA.112.000019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hasan DM, et al. Aspirin as a promising agent for decreasing incidence of cerebral aneurysm rupture. Stroke. 2011;42:3156–3162. doi: 10.1161/STROKEAHA.111.619411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guglielmi G, Vinuela F, Dion J, Duckwiler G. Electrothrombosis of saccular aneurysms via endovascular approach. Part 2: Preliminary clinical experience. J Neurosurg. 1991;75:8–14. doi: 10.3171/jns.1991.75.1.0008. [DOI] [PubMed] [Google Scholar]
- 76.Guglielmi G, Vinuela F, Sepetka I, Macellari V. Electrothrombosis of saccular aneurysms via endovascular approach. Part 1: Electrochemical basis, technique, and experimental results. J Neurosurg. 1991;75:1–7. doi: 10.3171/jns.1991.75.1.0001. [DOI] [PubMed] [Google Scholar]
- 77.Brinjikji W, Kallmes D, Kadirvel R. Mechanisms of healing in coiled intracranial aneurysms: a review of the literature. Am J Neuroradiol. doi: 10.3174/ajnr.A4175. http://dx.doi.org/10.3174/ajnr.A4175. [DOI] [PMC free article] [PubMed]
- 78.Kadirvel R, et al. Cellular mechanisms of aneurysm occlusion after treatment with a flow diverter. Radiology. 2014;270:394–399. doi: 10.1148/radiol.13130796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Connolly E, Jr, et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2012;43:1711–1737. doi: 10.1161/STR.0b013e3182587839. [DOI] [PubMed] [Google Scholar]
- 80.Tidswell P, Dias P, Sagar H, Mayes A, Battersby R. Cognitive outcome after aneurysm rupture: relationship to aneurysm site and perioperative complications. Neurology. 1995;45:875–882. doi: 10.1212/wnl.45.5.876. [DOI] [PubMed] [Google Scholar]
- 81.Broderick J, Brott T, Duldner J, Tomsick T, Leach A. Initial and recurrent bleeding are the major causes of death following subarachnoid hemorrhage. Stroke. 1994;25:1342–1347. doi: 10.1161/01.str.25.7.1342. [DOI] [PubMed] [Google Scholar]
- 82.Graf S, et al. Intracranial aneurysms and dolichoectasia in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant. 2002;17:819–823. doi: 10.1093/ndt/17.5.819. [DOI] [PubMed] [Google Scholar]
- 83.Afari M, Quddus A, Bhattarai M, John A, Broderick R. Spontaneous coronary dissection in polycystic kidney disease. R I Med J. 2013;96:44–45. [PubMed] [Google Scholar]
- 84.Lee C, et al. Computed tomography angiographic demonstration of an unexpected left main coronary artery dissection in a patient with polycystic kidney disease. J Thorac Imaging. 2011;26:W4–W6. doi: 10.1097/RTI.0b013e3181dc2a53. [DOI] [PubMed] [Google Scholar]
- 85.Larrangaga J, Rutecki G, Whittier F. Spontaneous vertebral artery dissection as a complication of autosomal polycystic kidney disease. Am J Kidney Dis. 1995;25:70–74. doi: 10.1016/0272-6386(95)90629-0. [DOI] [PubMed] [Google Scholar]
- 86.Kim J, et al. A case of severe aortic valve regurgitation caused by an ascending aortic aneurysm in a young patient with autosomal dominant polycystic kidney disease and normal renal function. Korean Circ J. 2012;42:136–139. doi: 10.4070/kcj.2012.42.2.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Torra R, et al. Abdominal aortic aneurysms and autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1996;7:2483–2486. doi: 10.1681/ASN.V7112483. [DOI] [PubMed] [Google Scholar]