

Abstract
Vascular endothelial growth factor (VEGF) activates unfolded protein response sensors in the endoplasmic reticulum through phospholipase C gamma (PLCγ)-mediated crosstalk with mammalian target of rapamycin complex 1 (mTORC1). Activation of transcription factor 6 (ATF6) and protein kinase RNA-like endoplasmic reticulum kinase (PERK) activate mTORC2, ensuring maximal endothelial cell survival and angiogenic activity through phosphorylation of AKT on Ser473. As mTORC1 is a metabolic sensor, metabolic signals may be integrated with signals from VEGF in the regulation of angiogenesis.
Keywords: AKT, angiogenesis, apoptosis, ATF6, CHOP, IRE1α, PERK, PLCγ, mTORC1, mTORC2, survival, unfolded protein response, VEGF
Abbreviations
- ATF6
activating transcription factor 6
- DAG
diacylglycerol
- EC
endothelial cell
- ER
endoplasmic reticulum
- ERK
extracellular signal-regulated kinase
- FOXO1
forkhead box protein O1
- IP3
inositol 1, 4, 5-trisphosphate
- IRE1
inositol-requiring kinase 1
- mTORC
mammalian target of rapamycin complex
- PDK1
phosphoinositide-dependent kinase-1
- PERK
protein kinase RNA-like endoplasmic reticulum kinase
- PI3K
phosphoinositide 3-kinase
- PIP2
phosphatidylinositol 4, 5-bisphosphate
- PIP3
phosphatidylinositol (3, 4, 5)-trisphosphate
- PKC
protein kinase C
- PLCγ
phospholipase C gamma
- UPR
unfolded protein response
- VEGF
vascular endothelial growth factor
- VEGFR2
vascular endothelial growth factor receptor 2
Author's View
Vascular endothelial growth factor (VEGF) is a key regulator of normal vessel formation and is also involved in the pathogenesis of angiogenic diseases such as cancer.1 Upon VEGF-A binding, phosphorylation of vascular endothelial growth factor receptor 2 (VEGFR2) on Tyr1175 leads to recruitment of phospholipase C gamma (PLCγ), which in turn hydrolyses phosphatidylinositol 4,5-bisphosphate (PIP2) to inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). IP3 opens IP3-gated channels on the endoplasmic reticulum (ER) to release Ca2+, whereas DAG phosphorylates extracellular signal-regulated kinases 1/2 (ERK1/2) via activation of protein kinase C (PKC), leading to proliferation of endothelial cells (ECs). Concomitantly, the active VEGF/VEGFR2 complex recruits GRB2-associated-binding protein 1 (GAB1), which binds to the p85 subunit of phosphoinositide 3-kinase (PI3K), leading to PIP3 generation and phosphorylation of AKT via phosphoinositide-dependent kinase 1 (PDK1). AKT conveys antiapoptotic signals, thus the PI3K/AKT pathway is considered the main survival pathway of VEGF in ECs (reviewed in2 and depicted in Fig. 1, left panel).
Figure 1.
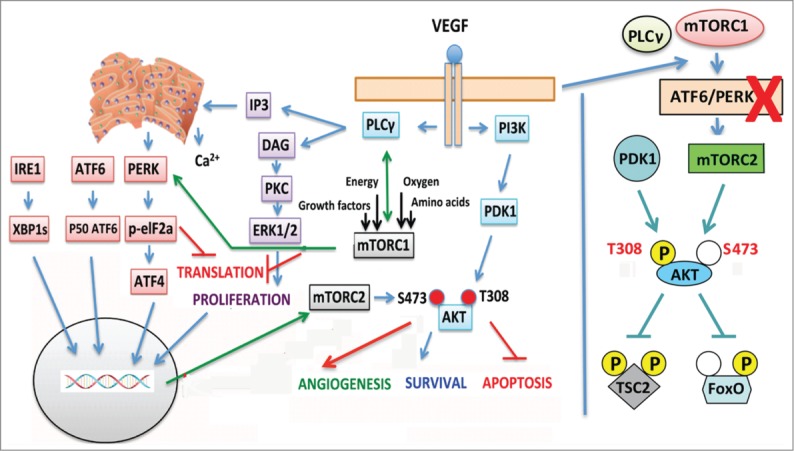
Graphical presentation of VEGF signaling including our findings. Left panel: Upon ligand binding VEGFR2 activates PLCγ, which eventually leads to Ca2+ release from the ER (via IP3) and ERK1/2 MAPK phosphorylation (via DAG) that induces endothelial cell (EC) proliferation. Concomitant activation of the PI3K/AKT axis ensures EC survival via antiapoptotic mechanisms. We have recently shown that VEGF activates IRE1, ATF6, and PERK through PLCγ-mediated crosstalk with the mTORC1 complex in the absence of any ER stress. Thus, even though there are no unfolded proteins in the ER, a UPR transcriptional gene response is initiated by PLCγ–mTORC1 crosstalk, leading to EC survival. Indeed, ATF6 and PERK activation positively regulate mTORC2-mediated phosphorylation of AKT on Ser473 that maintains AKT in a fully active state (see right panel). The crosstalk of mTORC1 with PLCγ provides a link between EC metabolism and angiogenesis. In other words, any decision concerning angiogenesis is probably under the control of the metabolic state of the EC (energy and nutrient levels, oxygen tension) and the growth factor milieu in which the EC resides. Although such a link to metabolism is a rational expectation, the involvement of mTORC1 located on the membranes of the ER provides a starting point to associate EC angiogenic responses, such as survival, with the metabolic state of the EC. Right panel: AKT is fully active when phosphorylated on both Thr308 and Ser473 by PDK1 and mTORC2, respectively. VEGF signals via the ER keep mTORC2 activated and AKT dually phosphorylated. Silencing of ATF6 or eIF2α (downstream of PERK) renders AKT monophosphorylated (only on Thr308). Some substrates of AKT are unaffected but FOXO1 is not phosphorylated sufficiently, allowing expression of genes that should be fully suppressed for initiation of angiogenesis.
We have recently revealed that VEGF rapidly (within 15–30 min) leads to short-term activation of the 3 ER-residing sensors of the unfolded protein response (UPR) (inositol-requiring kinase 1 [IRE1], activating transcription factor 6 [ATF6], and protein kinase RNA-like endoplasmic reticulum kinase [PERK]) via the PLCγ pathway, leading to the transcriptional activation of at least 37 UPR genes of which 24 are upregulated more than 10-fold.3 Interestingly, PLCγ does not activate the ER sensors via its lipid enzymatic activity on PIP2 because neither Ca2+ nor PKC are involved. Instead, this activation appears to be mediated via mammalian target of rapamycin complex 1 (mTORC1) because i) rapamycin inhibits VEGF-induced UPR activation, ii) p-PLCγ and p-mTOR can be co-immunoprecipitated, and iii) VEGF phosphorylates S6K1, a known downstream substrate of mTORC1, within 10 min of activation.
Thus, the ER appears to be critical for VEGF-induced EC survival and angiogenesis. Indeed, silencing of the ATF6 and eukaryotic translation initiation factor 2A (eIF2a) genes reduces VEGF-induced survival of ECs by 50%. Remarkably, VEGF-induced phosphorylation of AKT on Ser473 requires concomitantly active PLCγ-ATF6/PERK, underscoring the importance of mTORC2 activity for this phosphorylation. Ser473-phosphodeficient AKT is only partially active and lacks sufficient activity to phosphorylate all of its substrates4 (Fig. 1, right panel) leading to decreased survival of VEGF-induced ECs in culture and decreased angiogenesis in vivo. Indeed, vessel growth occurs when VEGF, angiopoietin 1 (ANG1), and survival signals suppress the expression of FOXO1-regulated target genes.5,6 Therefore, inhibition of PLCγ or silencing of the ATF6 and PERK genes, which lead to mTORC2 inactivation and deficient phosphorylation on Ser473 of AKT, apparently insufficiently suppresses FOXO1 target genes and results in inhibition of angiogenesis.3
Therefore, VEGF, acting through PLCγ, integrates the ER-residing UPR machinery in its regulatory circuit that controls EC survival and, eventually induces the formation of new vessels. Indeed, the ER consists of an extensive network of membranes that is essentially juxtaposed to all cellular compartments, contributing to the autophagic membranes and actually forming the nuclear envelope. Functionally, the ER is a central modulator of important cellular processes, such as the secretory pathway, calcium storage, lipid synthesis, fatty acid oxidation, gluconeogenesis, and the UPR. As a consequence the bidirectional communication network of the ER is very important.
The mTORC1 and mTORC2 complexes control cellular processes related to nutrient and energy sensing through a multitude of extracellular signals thus allowing efficient transitioning between anabolism and catabolism, a critical prerequisite for survival and growth when the supply of nutrients is not assured. In this context, mTORC1 senses the presence of certain growth factors and the level of energy, oxygen, and amino acids and accordingly adapts the cell (reviewed in7). It is, therefore, not surprising that the mTOR protein is tightly associated with the outer ER membrane via amino acids 1362–14438 or that the mTORC2 complex resides on ER membranes9 and also functionally interacts with ribosomes,10 thus constituting a powerful mTOR/ER metabolic control unit.
Integration of the mTOR/ER unit into VEGF signaling via PLCγ to control phosphorylation of AKT on Ser473, survival of ECs, and in vivo angiogenesis may have important consequences. VEGF signals are filtered through a metabolic hub that probably influences their final output depending on the metabolic status of the EC. Further understanding of the mechanisms by which VEGF utilizes the UPR machinery to achieve cell survival by integrating context-dependent mTORC1 checkpoints that sense the metabolic state of the cell might help elucidate the underlying mechanisms of the different cell fates supported by the UPR machinery – apoptosis, survival, and differentiation.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1. Kerbel R, Folkman J. Clinical translation of angiogenesis inhibitors. Nat Rev Cancer 2002; 2:727-39; PMID:12360276; http://dx.doi.org/ 10.1038/nrc905. [DOI] [PubMed] [Google Scholar]
- 2. Koch S, Tugues S, Li X, Gualandi L, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Biochem J 2011; 437:169-83; PMID:21711246; http://dx.doi.org/ 10.1042/BJ20110301. [DOI] [PubMed] [Google Scholar]
- 3. Karali E, Bellou S, Stellas D, Klinakis A, Murphy C, Fotsis T. VEGF Signals through ATF6 and PERK to promote endothelial cell survival and angiogenesis in the absence of ER stress. Mol Cell 2014; 54:559-72; PMID:24746698; http://dx.doi.org/ 10.1016/j.molcel.2014.03.022. [DOI] [PubMed] [Google Scholar]
- 4. Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006; 127:125-37; PMID:16962653; http://dx.doi.org/ 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 5. Potente M, Urbich C, Sasaki K, Hofmann WK, Heeschen C, Aicher A, Kollipara R, DePinho RA, Zeiher AM, Dimmeler S. Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J Clin Invest 2005; 115:2382-92; PMID:16100571; http://dx.doi.org/ 10.1172/JCI23126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Daly C, Wong V, Burova E, Wei Y, Zabski S, Griffiths J, Lai KM, Lin HC, Ioffe E, Yancopoulos GD, et al. Angiopoietin-1 modulates endothelial cell function and gene expression via the transcription factor FKHR (FOXO1). Genes Dev 2004; 18:1060-71; PMID:15132996; http://dx.doi.org/ 10.1101/gad.1189704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149:274-93; PMID:22500797; http://dx.doi.org/ 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Drenan RM, Liu X, Bertram PG, Zheng XF. FKBP12-rapamycin-associated protein or mammalian target of rapamycin (FRAP/mTOR) localization in the endoplasmic reticulum and the Golgi apparatus. J Biol Chem 2004; 279:772-8; PMID:14578359; http://dx.doi.org/ 10.1074/jbc.M305912200. [DOI] [PubMed] [Google Scholar]
- 9. Boulbes DR, Shaiken T, Sarbassov dos D. Endoplasmic reticulum is a main localization site of mTORC2. Biochemical and biophysical research communications 2011; 413:46-52; PMID:21867682; http://dx.doi.org/ 10.1016/j.bbrc.2011.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Oh WJ, Wu CC, Kim SJ, Facchinetti V, Julien LA, Finlan M, Roux PP, Su B, Jacinto E. mTORC2 can associate with ribosomes to promote cotranslational phosphorylation and stability of nascent Akt polypeptide. EMBO J 2010; 29:3939-51; PMID:21045808; http://dx.doi.org/ 10.1038/emboj.2010.271. [DOI] [PMC free article] [PubMed] [Google Scholar]