

Abstract
Unrestrained oncogene activity triggers DNA damage. Cancer cells exploit various stratagems to deal with this potentially lethal event. We found that hematologic cancer cells inactivate the Hippo co-activator Yes-associated protein 1 (YAP1). A synthetic lethal approach is proposed whereby inhibition of the serine/threonine kinase 4 (STK4) could be exploited to restore YAP1 levels in hematologic cancers.
Keywords: multiple myeloma, STK4, tumor suppressor, YAP-1
In epithelial cancers, the unbridled activity of oncogenes that leads to rampant proliferation has clear advantages for the tumor cell. However, such activity also has potentially lethal consequences for cancer cells: impetuous proliferation is associated with replicative stress, which in turn leads to DNA damage.1 In healthy cells, the DNA damage repair checkpoint would react first in the attempt to mend the damaged DNA and, if overwhelmed, channel the cell toward an apoptotic fate. The main (and highly effective) strategy employed by epithelial cancers to prevent this deadly outcome is early inactivation during tumor evolution of the p53 (TP53, best known as p53) or ATM genes, crucial mediators of this surveillance mechanism. As a result, these cells thrive despite intense and continuous DNA damage, in fact turning a potential drawback to their advantage by increasing their evolutionary pool through enhanced genomic instability.
The presence of DNA damage as one of the main outcomes resulting from disrupted oncogene activation was only recently recognized in hematologic cancers. In our recent publication,2 we confirmed and extended previous observations suggesting that ongoing DNA damage is also a pervasive event in lymphomas, leukemia, and multiple myeloma cells. How do hematologic cancer cells cope with this time bomb? Disabling p53 is probably not the main strategy employed by these cells to prevent death. In fact, unlike epithelial cancers, hematologic tumors do not inactivate p53 until the late stages, and even then only in a small subset of patients. We thus sought to identify alternative pathways to p53 that are activated by DNA damage in hematologic cancers and that cancer cells may attempt to curb. At the end of the 90s, several groups described a second pathway that is triggered by DNA damage and leads to apoptosis, centered upon the non-receptor tyrosine kinase ABL1, the same kinase that is translocated in chronic myeloid leukemia (CML) and has been effectively targeted with imatinib. These studies demonstrated that after pharmacologically induced DNA damage, ABL1 in its wild type form shuttles from the cytoplasm to the nucleus, thereby inducing apoptosis and behaving as a powerful tumor suppressor.3,4 We tested whether hematologic cancer cells show ABL1 nuclear localization in basal conditions as a result of ongoing DNA damage and found strong evidence of nuclear ABL1 in all of the hematologic cancers examined. Notably, DNA damage was present at comparable levels in both p53 mutated and p53 wild type cancer cells. Thus, we could conclude that ABL1 re-localization is not only the result of drug-induced DNA damage, but represents a more comprehensive mechanism that is present in more physiological conditions as a response to oncogene-induced DNA damage.
The pressing question then was how do hematologic cancer tumor cells manage to inactivate this potentially deadly pathway, which combines rampant DNA damage and nuclear ABL1? To induce apoptosis, ABL1 forms a complex with the Hippo coactivator Yes-associated protein 1 (YAP1) (5). The Hippo pathway exerts a prominent role in controlling organ size. YAP1 has been implicated as an oncogene in several epithelial cancers, including liver and breast carcinomas.6 However, analysis of gene expression profiles derived from different cancer types revealed a striking pattern: while cancer cells of epithelial origin showed increased levels of expression of YAP1 compared with their normal counterparts, in hematologic cancers YAP1 was instead consistently downregulated. Moreover, by reassessing previous array comparative genomic hybridization (aCGH) data we were able to demonstrate that YAP1 is homozygously deleted in more than 10% of myeloma patients. In addition to this subset of patients, it seemed that most patients with hematologic cancers present low levels of YAP1 levels, which is associated with poor prognosis. These results suggested that YAP1 behaves as a tumor suppressor in hematologic cancers, unlike its oncogenic role in epithelial tumors. We proved this hypothesis in myeloma cell lines deleted for YAP1 and showed that re-expression of YAP1 triggered intense cell death, mediated by the presence of active ABL1. Together, these data suggested that YAP1 is a powerful tumor suppressor in hematologic cancer cells, which is kept at bay to prevent DNA damage and ABL1-mediated apoptosis. Notably, similar results were obtained in the much larger group of tumor cells that do not present deletion of the YAP1 locus but nevertheless show low expression levels for this protein. In fact, we found that low expression of YAP1 in most hematologic cancer cells is not due to genomic ablation, but rather a result of poorly understood transcriptional and post-transcriptional events.
We were then faced by the final, most challenging, question: are there any ways to restore YAP1 levels in hematologic cancers in order to revitalize a healthy tumor suppressive response in these cancers? It has been reported that 2 serine-threonine kinases, serine/threonine kinase 3 (STK3) and serine/threonine kinase 4 (STK4), appear to control YAP1 levels.7,8 We therefore tested whether downregulation of these kinases could restore YAP1 levels, thus inducing apoptosis. Indeed, a panel of shRNAs targeting different portions of STK4 succeeded in lifting the suppression of YAP1 both in vitro and in vivo, driving cell death. We therefore propose a model whereby myeloma, lymphoma, and leukemia cells present pervasive DNA damage with nuclear localization of ABL1 (Fig. 1). However, apoptosis does not ensue because of genetic inactivation or reduced levels of the Hippo co-transcription factor YAP1. Inhibition of the kinase STK4 reactivates YAP1 and triggers apoptosis, providing the rationale for the development of novel STK4 inhibitors for clinical application in the setting of hematologic malignancies. Since this pathway does not seem to rely on p53, we envisage obtaining clinical benefit also in the patient subset presenting with p53 inactivation, which is often associated with a dismal prognosis. In conclusion, restoring YAP1 levels could represent a novel treatment strategy even in cancers with p53 inactivation and poor prognosis that are often refractory to even the most aggressive chemotherapeutic regimens.
Figure 1.
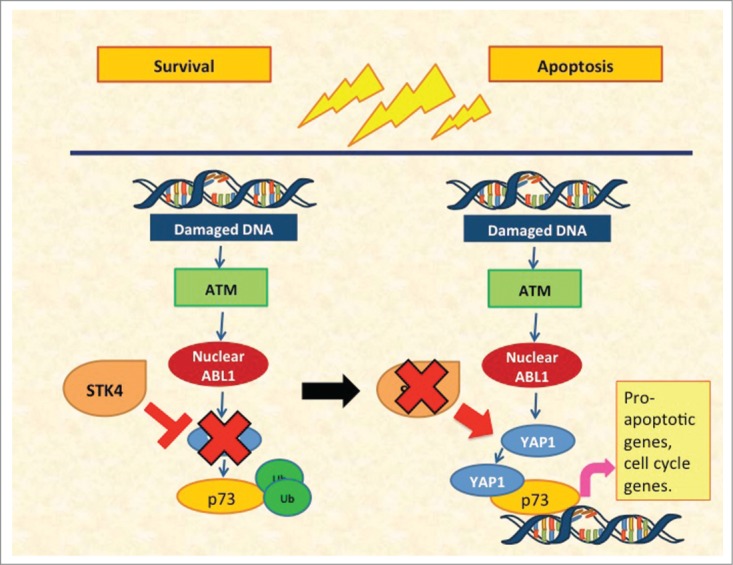
Yes-associated protein 1 (YAP1)-mediated apoptosis in hematologic cancers. Left panel: ongoing DNA damage induces shuttling of ABL1 into the nucleus. Serine/threonine kinase 4 (STK4)-mediated suppression of YAP1 prevents the occurrence of apoptosis. Right panel: STK4 downregulation relieves the inhibition on YAP1, which in turn interacts with ABL1 and p73, inducing apoptosis.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank members of the Anderson and Tonon lab for the scientific exchanges and discussions that helped shaped this commentary.
Funding
This work was supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC Investigator Grants and Special Program Molecular Clinical Oncology, 5 per mille no. 9965 to GT). KCA is an American Cancer Society Clinical Research Professor and is supported by NIH grants NIH SPORE P50 100707, PO-1 78378 and RO-1 50947.
References
- 1. Halazonetis T.D, et al. An oncogene-induced DNA damage model for cancer development. Science 2008; 319(5868):1352-5; PMID:18323444; http://dx.doi.org/ 10.1126/science.1140735 [DOI] [PubMed] [Google Scholar]
- 2. Cottini F, et al. Rescue of Hippo coactivator YAP1 triggers DNA damage-induced apoptosis in hematological cancers. Nat Med 2014; 20(6):599-606; PMID:24813251; http://dx.doi.org/ 10.1038/nm.3562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kharbanda S, et al, Functional role for the c-Abl protein tyrosine kinase in the cellular response to genotoxic stress. Biochim Biophys Acta, 1997; 1333(2):O1-7; PMID:9395286 [DOI] [PubMed] [Google Scholar]
- 4. Yuan Z M, et al. , Regulation of DNA damage-induced apoptosis by the c-Abl tyrosine kinase. Proc Natl Acad Sci U S A 1997; 94(4):1437-1440; PMID:9037071; http://dx.doi.org/ 10.1073/pnas.94.4.1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Levy D, et al. Yap1 phosphorylation by c-Abl is a critical step in selective activation of proapoptotic genes in response to DNA damage. Mol Cell 2008; 29(3):350-61; PMID:18280240; http://dx.doi.org/ 10.1016/j.molcel.2007.12.022 [DOI] [PubMed] [Google Scholar]
- 6. Zender L, et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 2006; 125(7):1253-67; PMID:16814713; http://dx.doi.org/ 10.1016/j.cell.2006.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhou D, et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 2009; 16(5):425-38; PMID:19878874; http://dx.doi.org/ 10.1016/j.ccr.2009.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou D, et al. Mst1 and Mst2 protein kinases restrain intestinal stem cell proliferation and colonic tumorigenesis by inhibition of Yes-associated protein (Yap) overabundance. Proc Natl Acad Sci U S A 2011; 108(49):E1312-1320; PMID:22042863; http://dx.doi.org/ 10.1073/pnas.1110428108 [DOI] [PMC free article] [PubMed] [Google Scholar]