

Abstract
The metabolic function of p53 is important for its oncosuppressive function. Mutant p53 (mutp53) with gain of oncogenic function can regulate cell metabolism. Our recent study revealed a novel transcription-independent mechanism for a gain-of-function mutp53 that directly inhibits activation of adenosine monophosphate-activated protein kinase (AMPK) to promote cancer cell metabolism.
Keywords: mutant p53, p53 mutation, AMPK, metabolism, gain of function, p53
As one of the most frequent genetic alterations in all cancer types, mutation of TP53 can result in loss of the oncosuppressive functions of wild-type p53 (wtp53). Some p53 mutants also have a dominant-negative effect and can bind and inhibit the function of remaining wtp53. Moreover, p53 mutations can invoke activities different from those resulting from a simple loss of wtp53 oncosuppressive function. Many of these mutants actually acquire oncogenic properties that enable them to promote invasion, metastasis, genomic instability, cancer inflammation, cancer cell proliferation, and survival. However, how gain-of-function (GOF) p53 mutants acquire these properties remains a mystery that has attracted much attention from researchers in recent years.
Role of p53 in Tumor Suppression
Once activated, wtp53 mostly functions as a transcription factor that targets many downstream genes involved in various processes including cell cycle arrest, apoptosis, senescence, DNA repair, and metabolism. However, it is unclear which of these functions is the most important in the oncosuppressive role of p53 under physiological conditions. Regulation of cell cycle arrest, apoptosis, and senescence have been widely accepted as the major functions contributing to the oncosuppressive role of p53, but recent studies with animal models have challenged this paradigm. For example, p53 efficiently suppresses tumor development in the complete absence of its cell cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa. Moreover, mice expressing p53 with mutations at 3 acetylation sites (K117, K161, and K162) that abolish p53-mediated cell cycle arrest, apoptosis, and senescence are not prone to early onset spontaneous tumorigenesis, suggesting that these functions are dispensable for the p53 oncosuppressive activity in mice. More importantly, the acetylation site-mutated p53 retained the ability to inhibit glucose uptake, glycolysis, and reactive oxygen species production by regulating the metabolic p53 target genes GLS2 and TIGAR, strongly suggesting that p53-mediated cell metabolism plays an important role in p53 oncosuppressive function (for a review see ref.1).
As a tumor suppressor, p53 can regulate several aspects of cell metabolism and thereby counteract many of the metabolic alterations associated with cancer development and progression. For example, p53 generally dampens aerobic glycolysis and the pentose phosphate pathway and promotes mitochondrial respiration through multiple mechanisms. p53 also functions as a negative regulator of lipid synthesis by enhancing fatty acid oxidation and inhibiting fatty acid synthesis.2
Mutp53 Gain of Function and Cancer Metabolism
In contrast to wtp53, mutp53 that has gained oncogenic function promotes glycolysis by targeting RhoA/ROCK/GLUT-1, and lipid synthesis by targeting SREBPs and Pla2g16.1,3 Thus, GOF mutp53 may be actively involved in the regulation of cancer metabolism, which is important for the oncogenic function of GOF mutp53.
As the master regulator of cellular energy homeostasis, adenosine monophosphate-activated protein kinase (AMPK) senses the cellular energy status and regulates the balance between anabolism and catabolism. Once activated, AMPK targets many downstream metabolic enzymes and transcription factors or cofactors to regulate cell metabolism and gene expression.4 Although stimulation of AMPK often leads to the phosphorylation and activation of p53, it is unclear whether p53 is a direct target of AMPK.4 AMPK was, however, recently shown to activate p53 indirectly through the phosphorylation and inactivation of MDMX 5 and the p53 deacetylase SIRT1.6 Activated p53 can, in turn, increase AMPK activity through transcriptional activation of the gene encoding the β subunit of AMPK 7 and sestrin,8 providing a positive feedback effect on AMPK function (Fig. 1). In contrast, we recently showed that GOF mutp53 directly inhibits AMPK activation to promote glycolysis and lipid synthesis in head and neck squamous cell carcinoma cells,9 providing the latest evidence that GOF mutp53 regulates cancer cell metabolism in an opposing manner to wtp53.
Figure 1.
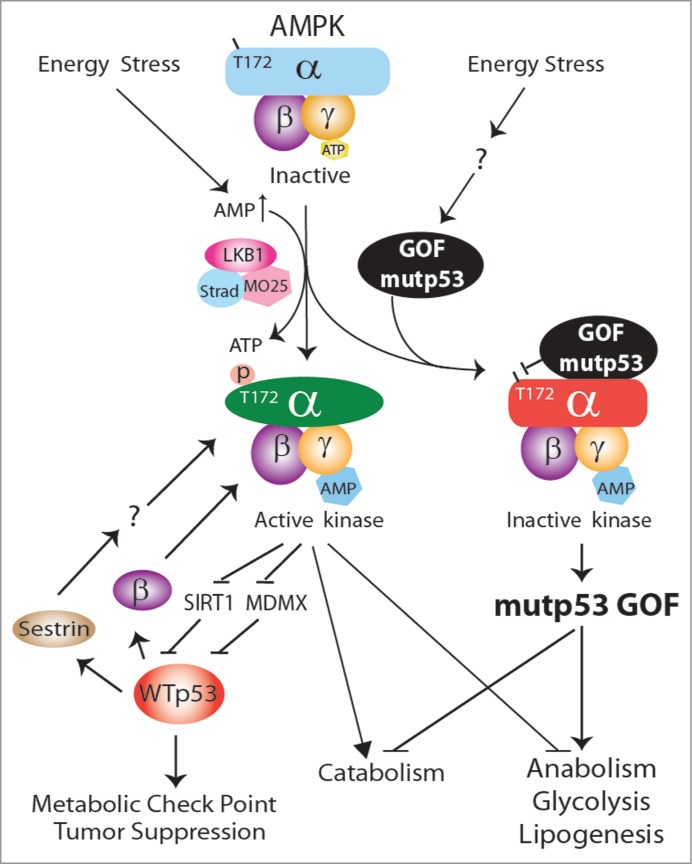
Gain-of-function mutant p53 inhibits adenosine monophosphate-activated protein kinase (AMPK) activation in head and neck squamous cell carcinoma. The wild-type p53 protein functions indirectly both upstream and downstream of AMPK. The gain-of-function mutant p53 functions directly upstream of AMPK and inhibits its activation, impairing the metabolic checkpoint and increasing anabolic tumor growth and progression.
As the cellular energy sensor, AMPK acts as a tumor suppressor or an oncogene depending on the context.10 Earlier work with a variety of cell lines with different genetic backgrounds (e.g., wtp53 versus mutp53) yielded seemingly inconsistent results. Given that p53 functions both upstream and downstream of AMPK and that GOF mutp53 inhibits AMPK (Fig. 1), we believe that p53 status plays an important role in determining the outcome of AMPK function as an oncogene or a tumor suppressor in different contexts.
Most of the proposed mechanisms for GOF mutp53 suggest that it functions as a transcriptional coactivator or a corepressor (or both) that interacts with many transcription factors or cofactors (e.g., p63, p73, Sp1, NF-κB, NF-Y, VDR, ETS, SREBP) to modulate gene expression.11 Our results, however, suggest that cytoplasmic inhibition of AMPK under metabolic stress is also important in the tumor-promoting function of GOF mutp53,9 indicating a novel transcription-independent mechanism for GOF mutp53 function. Consistent with this proposal, a recent study showed that a predominantly cytoplasmic localized p53 mutant (p53ψ) gains oncogenic function to induce a metastatic program in a transcription-independent manner.12 Finally, given that AMPK can target many transcription factors and cofactors,4 we expect that GOF mutp53 can also regulate gene expression indirectly via AMPK.
Conclusion
Although our results suggest that AMPK inhibition is an important mechanism in the tumor-promoting function of GOF mutp53, several questions—such as which signals control the mutp53–AMPK interaction and mutp53 cytoplasmic localization—remain to be answered.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This research was supported by a grant from the University Cancer Foundation via the Institutional Research Grant program at The University of Texas MD Anderson Cancer Center (IRG Grant Fred 35608) (to GZ) and by NIH grants RO1 DE14613, P50CA097007, and CA016672 (to JNM).
References
- 1. Liu J, Zhang C, Hu W, Feng Z. Tumor suppressor p53 and its mutants in cancer metabolism. Cancer Lett 2013; pii: S0304-3835(13)00863-X; http://dx.doi.org/ 10.1016/j.canlet.2013.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Berkers CR, Maddocks OD, Cheung EC, Mor I, Vousden KH. Metabolic regulation by p53 family members. Cell Metab 2013; 18:617-33; PMID:23954639; http://dx.doi.org/ 10.1016/j.cmet.2013.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Xiong S, Tu H, Kollareddy M, Pant V, Li Q, Zhang Y, Jackson JG, Suh YA, Elizondo-Fraire AC, Yang P, et al. . Pla2g16 phospholipase mediates gain-of-function activities of mutant p53. Proc Natl Acad Sci U S A 2014; 111:11145-50; PMID:25024203; http://dx.doi.org/ 10.1073/pnas.1404139111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 2012; 13:251-62; PMID:22436748; http://dx.doi.org/ 10.1038/nrm3311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. He G, Zhang YW, Lee JH, Zeng SX, Wang YV, Luo Z, Dong XC, Viollet B, Wahl GM, Lu H. AMP-activated protein kinase induces p53 by phosphorylating MDMX and inhibiting its activity. Mol Cell Biol 2014; 34:148-57; PMID:24190973; http://dx.doi.org/ 10.1128/MCB.00670-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee CW, Wong LL, Tse EY, Liu HF, Leong VY, Lee JM, Hardie DG, Ng IO, Ching YP. AMPK promotes p53 acetylation via phosphorylation and inactivation of SIRT1 in liver cancer cells. Cancer Res 2012; 72:4394-404; PMID:22728651; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-0429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S, Levine AJ. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res 2007; 67:3043-53; PMID:17409411; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-4149 [DOI] [PubMed] [Google Scholar]
- 8. Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008; 134:451-60; PMID:18692468; http://dx.doi.org/ 10.1016/j.cell.2008.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhou G, Wang J, Zhao M, Xie TX, Tanaka N, Sano D, Patel AA, Ward AM, Sandulache VC, Jasser SA, et al. . Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Mol Cell 2014; 54:960-74; PMID:24857548; http://dx.doi.org/ 10.1016/j.molcel.2014.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liang J, Mills GB. AMPK: a contextual oncogene or tumor suppressor? Cancer Res 2013; 73:2929-35; PMID:23644529; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-3876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Muller PA, Vousden KH. p53 mutations in cancer. Nat Cell Biol 2013; 15:2-8; PMID:23263379; http://dx.doi.org/ 10.1038/ncb2641 [DOI] [PubMed] [Google Scholar]
- 12. Senturk S, Yao Z, Camiolo M, Stiles B, Rathod T, Walsh AM, Nemajerova A, Lazzara MJ, Altorki NK, Krainer A, et al. . p53Psi is a transcriptionally inactive p53 isoform able to reprogram cells toward a metastatic-like state. Proc Natl Acad Sci U S A 2014; 111:E3287-96; PMID:25074920; http://www.pnas.org/cgi/doi/10.1073/pnas.1321640111 [DOI] [PMC free article] [PubMed] [Google Scholar]