

Abstract
Acetylation of protein lysine residues is a reversible and dynamic process that is controlled by histone acetyltransferases (HATs) and deacetylases (HDACs and SIRTs). Recent studies have revealed that acetylation modulates not only nuclear proteins but also cytoplasmic or mitochondrial proteins, including many metabolic enzymes. In tumors, cellular metabolism is reprogrammed to provide intermediates for biosynthesis such as nucleotides, fatty acids, and amino acids, and thereby favor the rapid proliferation of cancer cells and tumor development. An increasing number of investigations have indicated that acetylation plays an important role in tumor metabolism. Here, we summarize the substrates that are modified by acetylation, especially oncogenes, tumor suppressor genes, and enzymes that are implicated in tumor metabolism.
Keywords: acetylation, tumor metabolism, oncogene, tumor suppressor, metabolic enzyme
Abbreviations
- aces
acetyl-coa synthetase
- ACLY
ATP–citrate lyase
- AMPK
AMP-activated protein kinase
- CMA
chaperone-mediated autophagy
- EHHADH
enoyl coa hydratase/3-hydroxylacyl coa dehydrogenase
- ENO1
enolase 1
- FBP
fructose-1,6-bisphosphate
- FH
fumarate hydratase
- G6PD
glucose-6-phosphate dehydrogenase
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GDH
glutamate dehydrogenase
- GLUT1
glucose transporter isoform 1
- GLS1
glutaminase
- HATs
histone acetyltransferases
- HDACs
histone deacetylases
- HIF
hypoxia-inducible factor
- IDH
isocitrate dehydrogenase
- KATs
lysine acetyltransferases
- LDH-A
lactate dehydrogenase
- LEF/TCF
lymphoid enhancer factor/T-cell factor
- MCT4
monocarboxylate transporter 4
- MDH
malate dehydrogenase
- NADPH
nicotinamide adenine dinucleotide phosphate
- OTC
ornithine transcarbamylase
- PDC
pyruvate dehydrogenase complex
- PDH
pyruvate dehydrogenase
- PEPCK1
phosphoenolpyruvate carboxykinase 1
- PGAM
phosphoglycerate mutase
- PKM2
M2 isoform of pyruvate kinase
- pRB
retinoblastoma protein
- PTEN
phosphatase and tensin homolog
- ROS
reactive oxygen species
- S1P
sphingosine 1-phosphate
- SDH
succinate dehydrogenase
- SIRTs
sirtuins
- SOD
superoxide dismutase
- SPHK
sphingosine kinase
- STAT
signal transducers and activators of transcription
- TPI
triosephosphate isomerase
Protein Acetylation System
Protein lysine acetylation is a dynamic and reversible process that is controlled by protein lysine acetyltransferases (KATs, also called histone acetyltransferases or HATs) and deacetylases (known as histone deacetylases or HDACs).
HATs are a group of enzymes that transfer the acetyl group from acetyl-CoA to lysine residues of substrate proteins. HATs were initially found to acetylate histones and decrease their interaction with negatively charged DNA through neutralization of the positive charge of lysine residues. This process can enhance the binding of transcriptional factors with gene promoters to regulate gene expression during development, cell proliferation, and disease development. Further studies have suggested broader functions for the HAT family in the regulation of lysine acetylation of non-histone proteins, such as transcription factors and nuclear receptors, in order to control gene expression.1
HATs are conserved in evolution from yeast to human. HAT1 was the first reported histone acetyltransferase,2 and to date approximately 19 members of the mammalian HAT family have been reported. On the basis of sequence homology, structural features, and functional roles, HATs are commonly grouped into several families, including p300/CBP, GNATs, MYST HATs, nuclear receptors, and co-activators.
The best-studied HATs belong to the p300/CBP subfamily. CREB-binding protein (CBP or CREBBP) and p300 increase gene expression in various ways, such as by relaxing the chromatin structure at the gene promoters through histone acetyltransferase activity or by recruiting the transcriptional machinery (e.g., RNA polymerase II) to the promoters.3 These proteins bind to many sequence-specific factors involved in cell growth and differentiation, including c-jun and the adenoviral oncoprotein E1A. CBP or p300 knockout mice die at an early embryonic stage, indicating that both proteins are essential for normal embryonic development.4 Loss-of-function mutations in the p300 gene have been identified in several types of cancer, including acute myeloid leukemia (AML), prostate cancer, and breast cancer.
The Gcn5-related N-acetyltransferase (GNAT) family includes GCN5, PCAF, Hat1, Elp3, Hpa2, Hpa3, ATF-2, and Nut1. These HATs are known to acetylate lysine residues on histones H2B, H3, and H4, and share a similar catalytic HAT domain. GCN5 and p300/CBP-associated factor (PCAF) are mammalian GNATs. PCAF can bind together with p300/CBP to directly regulate transcription. The targets of the acetyltransferase activity of PCAF include Fli1, p53, and numerous histone residues. The MYST family of HATs consists of MOZ, Ybf2 (Sas3), Sas2, Tip60, Esa1, MOF, MORF, and HBO1. These HATs acetylate lysine residues on histones H2A, H3, and H4. Tip60 was the first reported MYST family member to exhibit HAT activity in humans. Three important nuclear receptor co-activators that display HAT activity are SRC-1, ACTR, and TIF-2, all of which are known to interact with p300/CBP and PCAF. In addition to these subfamilies, several other proteins exhibit HAT activity, including TAFII250, TFIIIC, Rtt109, and CLOCK.
The HDACs are a group of hydrolases that remove acetyl groups from acetylated lysine residues of histone, allowing the histones to wrap the DNA more tightly to downregulate gene transcription. Although the initial HDAC substrates identified were histones, it is now clear that HDAC substrates extend far beyond these proteins. Therefore, HDACs are now also called lysine deacetylases (KDAC) to reflect their action on non-histone proteins.
HDACs are divided into 4 classes based on sequence homology and function. The first 2 classes are considered “classical” HDACs, with activities that are inhibited by the HDAC inhibitor, trichostatin A (TSA), whereas the third class is a family of NAD+-dependent proteins (Sirtuins or SIRTs) that are affected by nicotinamide (NAM) but not by TSA. The last class has one typical member, HDAC11, which shows only DNA sequence similarity to other HDACs.
Class I HDACs, including HDAC1, HDAC2, HDAC3, and HDAC8, are homologs of the yeast reduced potassium dependency 3 (Rpd3) protein and are the most ubiquitously expressed in tissues. All of these proteins localize in the nucleus, and HDAC8 also partially localizes in the cytoplasm. HDAC1 interacts with Rb protein to control cell proliferation and differentiation, and also deacetylates p53 and modulates its effect on cell growth and apoptosis.5 Class II HDACs, homologs of yeast histone deacetylase 1 (hda1), comprise HDAC4, HDAC5, HDAC6, HDAC7, HDAC9, and HDAC10. HDACs in this class are able to shuttle in and out of the nucleus in response to different signals. Compared with Class I HDACs, class II HDAC proteins have relatively weak deacetylase activity and act primarily as recruiting proteins within large complexes that include class I HDACs and other regulatory elements.6 Mutations in HDAC6 have been associated with Alzheimer's disease, whereas HDAC6 overexpression correlates with tumorigenesis and cancer metastasis.7,8
SIRTs, an evolutionarily conserved multigene family, are structurally distinct from the other classes of HDACs. Members of this family occupy different subcellular compartments—the nucleus (SIRT1, SIRT2, SIRT6, and SIRT7), the cytoplasm (SIRT1 and SIRT2), and the mitochondria (SIRT3, SIRT4, and SIRT5)—and participate in a wide range of cellular processes including transcription, metabolism, aging, apoptosis, inflammation, and stress resistance.9 Some SIRTs are also involved in tumorigenesis, control of circadian clocks, and mitochondrial biogenesis. SIRT4, SIRT5, and SIRT6 have also been found to possess other catalytic activities beyond deacetylation.10
Tumor Metabolism
The dysregulation of cellular metabolism is an important hallmark of cancer that was first described by Otto Warburg, which has a reciprocal causation with tumorigenesis and development. The famous “Warburg phenomenon” is an increased rate of glycolysis in cancer cells even under normal oxygen conditions (aerobic glycolysis), resulting in enhanced lactate production. Cancer cells can benefit from glycolysis through different mechanisms to promote tumor cell growth and biosynthesis of mass. Warburg also proposed that a permanent impairment of oxidative metabolism contributes to increased glycolysis, accounting for the lack of ATP generation.11 However, later studies have demonstrated that tumor cells do not have defects in oxidative metabolism and most tumors retain the capacity for oxidative phosphorylation.12,13 Although glycolysis in tumor cells generates ATP with low efficiency compared with normal cells, tumor cells take up much more glucose to support the high rate of glycolysis flux and produce abundant ATP for tumor survival and growth.14 At the same time, the intermediate metabolites of glycolysis provide precursors for synthesis of biomacromolecules such as nucleotides, fatty acids, and nonessential amino acids to support rapid cancer cell growth (Fig. 1).
Figure 1.
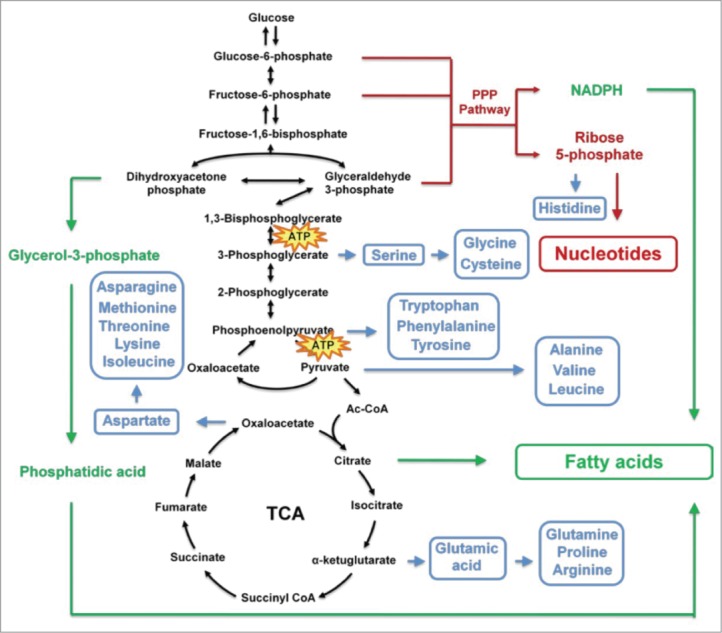
Acetylation controls enzymes involved in tumor metabolism. Enzymes involved in different metabolism pathways, including glucose metabolism, fatty acid metabolism, and glutamine metabolism, are regulated by acetylation modification. Enzymes in the red circle are catalytically activated or exhibit increased protein stability after acetylation whereas those in the blue circle are catalytically inhibited or exhibit increased degradation after acetylation.
Many glycolysis intermediates of tumor cell metabolism, such as glucose-6-phosphate, fructose-6-phosphate, and glyceraldehyde 3-phosphate, can be used to produce ribose 5-phosphate and nicotinamide adenine dinucleotide phosphate (NADPH) through the pentose phosphate pathway (PPP). The generation of ribose 5-phosphate contributes to rapid nucleotide synthesis, thereby supporting DNA duplication and RNA activity during cell proliferation. Furthermore, the production of NADPH can prevent cancer cells from undergoing apoptosis by eliminating intracellular reactive oxygen species (ROS) and also provides reducing power for lipid synthesis to generate membrane structures for newly divided cells. Moreover, 3-phosphoglycerate produced in glycolysis can be used for de novo serine biosynthesis, feeding carbon derived from glucose into the folate cycle. The folate cycle provides purine nucleotides for DNA and RNA synthesis and generates glutathione to maintain the NADPH/ NADP+ ratio for anabolic metabolism. It has been reported that the folate cycle is activated in tumors and correlates with tumorigenesis.15
Several intermediates of glycolysis and the TCA cycle participate in fatty acid synthesis. Dihydroxyacetone phosphate produced during glycolysis can be converted into glycerol-3-phosphate, which is an important substrate for triacylglycerol synthesis by the phosphatidic acid pathway. Synthesis of fatty acids, which are used to produce cellular lipids, requires acetyl-CoA, most of which is generated from citrate transferred from the mitochondria to the cytoplasm (Fig. 1). Oxaloacetate is not only a source of citrate production for lipid synthesis, but is also an important precursor for the synthesis of many types of amino acid. In fact, many intermediates can be used for amino acid synthesis, including 3-phosphoglycerate, phosphoenolpyruvate, pyruvate, and α-ketoglutarate (Fig. 1). In addition, some amino acids, such as glutamine, glycine, and aspartate, are essential for nucleotide synthesis. Table 1.
Table 1.
Acetylation of oncogenes and tumor suppressor genes
| Name | Related tumors | Acetylation site | HATs | HDACs | Acetylation effects |
|---|---|---|---|---|---|
| HIF-1 | Renal cell cancer, breast cancer | K674 | PCAF42 | SIRT143 | Delays HIF-1α degradation to stimulate anaerobic glycolysis |
| Myc | Lymphoma, breast cancer | K323, K417 | P30046; CBP45; GCN5/PCAF/ TIP6047 | — | Stimulates oncogene expression |
| KRAS | Lung carcinoma, pancreatic cancer, colon cancer | K104 | — | HDAC649 SIRT249 | Attenuates transforming activity and suppresses tumor cell growth |
| p53 | Pancreatic cancer, breast cancer, lung cancer | K305, K320, K370, K372, K373, K381, K382 | p300/CBP54; PCAF54 | SIRT1 | Promotes DNA-binding ability; modulates stability |
| pRb | Breast cancer, prostate cancer | K873, K874 | p300/CBP62 | HDAC1 | Blocks its phosphorylation by CDKs; retains pRb in an active state of growth suppression |
| PTEN | Glioblastoma, breast cancer, lung cancer, prostate cancer | K125, K128, K402 | CBP57; PCAF58 | SIRT157 | Promotes interaction with PDZ domain-containing proteins; interferes with catalytic specificity toward PIP3 |
These rapid biosynthesis processes result in depletion of TCA cycle intermediates. These are replenished by the process of anaplerosis, which involves 5 reactions: production of oxaloacetate from pyruvate catalyzed by pyruvate carboxylase; reversible conversion of aspartate to oxaloacetate catalyzed by aspartate transaminase; generation of α-ketoglutarate from glutamate catalyzed by glutamate dehydrogenase; formation of succinyl-CoA by odd-chain fatty acid oxidation; and the generation of fumarate from adenylosuccinate.16 In addition to glycolysis, glutaminolysis is another major source of energy production in cancer cells. Tumor cells use glutamine to generate glutamate and then produce α-ketoglutarate for the remaining TCA cycle steps from α-ketoglutarate to citrate. A high extracellular glutamine concentration not only promotes tumor growth but is also essential for cell transformation. Furthermore, some cancer types display addiction to glutamine, such as pancreatic cancer, glioblastoma, acute myelogenous leukemia, and small-cell lung cancer. The sensitivity of such cancers to glutamine starvation provides a new therapeutic target in cancer treatment.17
During tumorigenesis and development, the cells undergo metabolic reprogramming such that energy production is redirected to balance energy needs and macromolecular biosynthesis, ultimately promoting cell growth. A principal mechanism of metabolic reprogramming in cancer cells is alteration of metabolic enzyme systems. Hypoxia-inducible factor (HIF) is a transcription factor that is primarily activated by hypoxic stress.18 The activation of HIF in tumors can stimulate the conversion of glucose to pyruvate and lactate by upregulation of glucose transporter isoform 1 (GLUT1), hexokinase, monocarboxylate transporter 4 (MCT4), and lactate dehydrogenase A.19 Moreover, HIF-1 also decreases the conversion of pyruvate to acetyl-CoA by upregulating PDH kinase 1 (PDK1) to inhibit pyruvate dehydrogenase (PDH).20,21 Activation of oncogenes (e.g., Myc, Ras) and inactivation of tumor suppressors (e.g. PTEN, P53) also contribute to the regulation of metabolic enzyme transcription during cancer cell reprogramming.22,23 On the other hand, loss-of-function mutations of fumarate hydratase (FH), succinate dehydrogenase (SDH), and isocitrate dehydrogenase (IDH), which have been reported in different tumors, result in accumulation of the TCA cycle intermediates fumarate, succinate, and 2-hydroxyglutarate respectively. These metabolites competitively inhibit the α-ketoglutarate-dependent dioxygenases that regulate epigenetic mechanisms and tumorigenesis.24,25 In this regard, fumarate, succinate, and 2-hydroxyglutarate are also referred to as oncometabolites. This serves as a good example of the notion that metabolic enzymes can be important oncogenic players.
With the development of improved technology for studying tumor biology, the dysregulation of cellular metabolism that occurs before tumorigenesis and progression has gradually been validated by experiments. In 2009, Yun and colleagues found that a lack of glucose promotes KRAS WT cells to gain KRAS and BRAF mutations; this was the first study to demonstrate that dysregulation of cellular metabolism leads to tumorigenesis.26 Subsequently, C-labeled pyruvate molecular imaging technology showed that alterations in glycolysis occur before c-Myc induces tumor formation and shrinkage in vivo.13 In addition, the accumulation of oncometabolites such as fumarate, succinate, and 2-hydroxyglutarate competitively inhibits the α-ketoglutarate-dependent dioxygenases and plays an important role in tumorigenesis.24,25 These studies demonstrate that dysregulation of cellular metabolism plays an initial role in tumorigenesis. These data also suggest a novel approach to antitumor therapy by directly targeting tumor metabolic pathways.
Indirect targets consist of upstream regulators and proteins involved in signaling pathways of metabolism, such as the restoration of apoptosis, interruption of cell-autonomous growth signals, and inhibition of angiogenesis; such proteins include HIF, PI3K, AKT, mTOR, and AMP-activated protein kinase (AMPK).27-29 Studies also proved that some enzymes that are directly involved in cancer metabolism could confer antitumor therapeutic vulnerabilities. In glioblastoma, deletion of the glycolytic gene enolase 1 (ENO1) in tumor cells is tolerated through the expression of ENO2. Muller et al. demonstrated that selective silencing of ENO2 by shRNA inhibits growth, survival, and the tumorigenic potential of ENO1-deleted GBM cells and increases the sensitivity of these cells to enolase inhibitors.30 More common approaches include suppressing the enzymes involved in tumor reprogramming or targeting tumor-specific enzyme isoforms. For example, 3-bromopyruvate inhibits HK2 and results in apoptosis in hepatocellular carcinomas,23 inhibition of PDK1 restores PDH activity and triggers apoptosis in tumor cells,31 LDH-A inhibition induces oxidative stress and inhibits tumor progression,32 and targeted inhibition of mutant IDH1 delays the growth of glioma.33 These therapeutic methods not only limit tumor-specific bioenergetic flow and anabolic reactions, but also reverse the neoplastic phenotype.
Histone Acetylation and Tumor Metabolism
Histone acetylation regulates chromatin modification and affects the expression of numerous genes implicated in oncogenesis. Altered expression and mutations of class I or II HDAC genes have been linked to tumor development through induction of aberrant transcription of key genes regulating cell proliferation, cell cycle transition, differentiation, and apoptosis.34 Moreover, HDAC inhibitors were shown to reverse the transformed cell phenotype and have been considered as promising cancer therapeutics.35,36 These studies indicated the important roles of histone acetylation in tumor metabolism.
Metabolism intermediates such as butyrate, acetyl-CoA, and acetate have been reported to influence histone acetylation. Before 1977, n-butyrate had been shown to increase histone acetylation in neuroblastoma, HeLa, and other tumor cells, thereby inhibiting cell proliferation and affecting DNA content and cell morphology.37 Acetyl-CoA is a broad substrate for histone acetylation. Recently, Lee et al. demonstrated that changes in acetyl-CoA availability mediated by oncogenic metabolic reprogramming contribute to histone acetylation levels in tumor cells.38 They also found that oncogenic KRAS or AKT stimulates histone acetylation through the metabolic enzyme ATP-citrate lyase (ACLY). AKT-dependent alterations in nuclear acetyl-CoA level modulate global histone acetylation levels preceding tumor development in gliomas and prostate cancers. These results implicate acetyl-CoA metabolism as a key regulator of histone acetylation in cancer cells.38 Evidence that acetyl-CoA controls HAT activity comes from findings that depletion of acetyl-CoA reduced the activity of p300 and impaired its suppressive effect on autophagy.39 Interestingly, Thompson's lab found that ACLY, which converts glucose-derived citrate into acetyl-CoA, is required for the increase in histone acetylation in response to growth factor stimulation and during differentiation.40 This study also reported that ACLY translocates into the nucleus and generates acetyl-CoA to induce histone acetylation and modulate expression of glycolysis genes in tumor metabolism. In addition to ACLY, pyruvate dehydrogenase complex (PDC) also contributes to acetyl-CoA generation in the nucleus of mammalian cells. A recent elegant investigation showed that knockdown of nuclear PDC decreased acetyl-CoA synthesis and acetylation of core histones41. These authors also found that PDC translocates from mitochondria to the nucleus in a cell cycle-dependent manner and provides a mechanism of nuclear acetyl-CoA synthesis for histone acetylation and epigenetic regulation.41
Regulation of Oncogenes and Tumor Suppressor Genes by Acetylation
Many oncogenes and tumor suppressor genes have been linked to cancer metabolism. Intensive studies have found that this process is heavily regulated by acetylation, indicating that acetylation of these proteins contributes to functions implicated in tumor metabolism. These findings are summarized below.
Oncogenes
As mentioned previously, hypoxia-inducible factor (HIF-1) plays a critical role in the regulation of tumor metabolism by mediating the switch in energy metabolism from oxidative phosphorylation to anaerobic glycolysis during hypoxic adaptation and tumor metabolism. To date, more than 80 genes (including most of the glycolytic enzymes) have been found to be regulated by HIF-1. HIF-1 is a heterodimer composed of HIF-1α and HIF-1β. The stability and activity of HIF-1 are predominantly determined by HIF-1α. It has been reported that PCAF acetylates HIF-1α at K674, which delays its degradation.42 Lim et al. found that SIRT1 deacetylates and inactivates HIF-1α by blocking p300 recruitment, leading to repression of HIF-1 target genes. Under hypoxia, SIRT1 is downregulated to allow the activation of HIF-1α and stimulate anaerobic glycolysis. These authors also showed that SIRT1 deacetylates HIF-1α and suppresses target gene transcription, thereby inhibiting tumor metabolism and tumor growth.43
Myc functions as a transcription factor and binds to 10–15% of all promoter regions. By modifying expression of its target genes, Myc activation plays an important role in numerous biological effects including cell proliferation, apoptosis, differentiation, and stem cell self-renewal. Myc also contributes to the metabolic reprogramming of tumor cells at multiple levels. Myc not only activates expression of the glucose transporter GLUT1, the lactate transporter MCT1, and lactate dehydrogenase (LDH-A) to promote the Warburg effect, but also prevents pyruvate entering the TCA cycle by upregulating PDK. Moreover, Myc stimulates glutamine transporters but inhibits glutaminase (GLS1) to support the glutamine addiction of cancer cells. In addition, Myc activates genes involved in ribosomal and mitochondrial biogenesis, nucleotide synthesis, and lipid synthesis.44 Myc is a strong proto-oncogene and is mutated in many types of cancers. In 2003, Vervoorts et al. reported that Myc is acetylated by CBP.45 Subsequent studies demonstrated that Myc could be also a substrate of the acetyltransferases P300, GCN5, PCAF, and TIP60.46,47 Acetylation was found to inhibit ubiquitin-mediated degradation of Myc and thereby stabilize Myc protein. However, some reports suggested dual roles of p300/CBP in Myc regulation: acting as a coactivator it stabilizes Myc whereas acting as an acetylase it destabilizes Myc.46 Further studies are needed to clarify how acetylation affects Myc, in particular its functions in tumor metabolism.
Normal KRAS performs an essential function in cellular signaling, and activating mutations of KRAS are critical in the development of many cancers, such as lung adenocarcinoma, pancreatic ductal carcinoma, and colorectal carcinoma. Recently, researchers found that KRAS is acetylated at K104. This acetylation attenuates KRAS transforming activity by interfering with GEF-induced nucleotide exchange.48 Later, the same group reported that the deacetylases HDAC6 and SIRT2 could regulate the acetylation of KRAS in cancer cells. Inhibition of these deacetylases suppresses the growth of cancer cells that express activated mutants of KRAS.49 In 2012, Ying et al. revealed that oncogenic KRAS could regulate anabolic glucose metabolism to maintain pancreatic tumors. Transcriptome and metabolome analysis indicated that KRAS (G12D) increases glucose uptake, enhances metabolism of glucose intermediates in the hexosamine biosynthesis and pentose phosphate pathways (PPP), and promotes ribose biogenesis.50 Further study may reveal the effects of KRAS acetylation on tumor metabolism.
Tumor Suppressor Genes
Upon DNA damage, the tumor suppressor p53 can activate DNA repair proteins, arrest the cell cycle at the G1/S checkpoint, and initiate apoptosis. Many studies have investigated a genetic link between TP53 variation and various cancers, such as pancreatic cancer, breast cancer, and lung cancer. P53 controls central carbohydrate metabolism (glycolysis, oxidative phosphorylation, and pentose phosphate pathways), and also regulates lipid metabolism. P53 is tightly associated with cancer metabolism through TIGAR, SCO2, and PGM.51-53 Notably, acetylation plays an important role in p53 function. The acetylation of p53 by p300/CBP at K305, K370, K372, K373, K381, and K382 or by PCAF at K320 increases its DNA-binding ability and activates the transcription of p53 target genes.34 Another study mentioned the role of site-specific p53 acetylation in transcriptional regulation induced by different types of damage. SIRT1 was shown to deacetylate p53 at K382 and inhibit its activity. Later, Chao et al. found that abrogation of acetylation of the K320 homolog in mice enhances p53-mediated apoptosis after DNA damage.54 Acetylation also modulates the stability of p53 because many acetylated lysines are also sites of ubiquitylation. Acetylation can therefore protect p53 from ubiquitin-proteasome degradation.
Phosphatase and tensin homolog (PTEN) is a major tumor suppressor gene that is mutated at high frequency in many cancers, including glioblastoma, breast cancer, lung cancer, and prostate cancer. Up to 70% of patients with prostate cancer have lost a copy of the PTEN gene at the time of diagnosis.55 PTEN promotes oxidative phosphorylation and decreases glycolysis. During tumor development, PTEN mutations lead to metabolic reprogramming in cancer cells and thus increase cell proliferation.56 In 2008, PTEN was reported to be acetylated at K402. This study showed that CBP acetylates PTEN, whereas SIRT1 is mainly responsible for PTEN deacetylation. K402 acetylation modulates PTEN interaction with PDZ domain-containing proteins, indicating its regulatory function.57 Four years later, another group reported that PCAF interacts with and acetylates PTEN at lysines 125/128; this downregulates PTEN activity by interfering with its catalytic specificity toward PIP3.58 The PI3K/AKT/mTOR pathway is a major regulator of aerobic glycolysis and cellular biosynthesis. In contrast, activation of the PI3K system is tightly controlled by the dephosphorylation of phosphatidylinositol species by PTEN. Recently, ENTPD5, a newly discovered component of the PTEN pathway, was found to promote the Warburg effect.59 These studies indicate that acetylation of PTEN may play an important role in tumor metabolism.
The retinoblastoma protein (pRb) is a tumor suppressor that prevents excessive cell growth by inhibiting the cell cycle. Inactivation of pRb can lead to many types of cancer. Recent studies have linked pRb to cell metabolism through regulation of glucose tolerance, mitogenesis, and glutathione synthesis.60 Moreover, pRb controls the expression of genes involved in central carbon metabolism, lipid metabolism, and nucleotide metabolism.61 It has been reported that adenovirus E1A stimulates the acetylation of pRb by p300 and participates in cell cycle control. Acetylation at K873/874 also blocks the phosphorylation of pRb by cyclin-dependent kinases and thereby retains pRb in an active state of growth suppression.62 A subsequent study found that pRb acetylation also plays a role in the DNA damage signaling pathway.63
Control of Metabolic Enzymes by Acetylation
To date, 4 important acetylation proteomic studies have been reported. Together, they identified more than 4,000 potentially acetylated proteins in different cell lines and organ tissues.64-67 These studies have made tremendous contribution to the field of acetylation, especially as most of the potential acetylated proteins identified are non-histone proteins. Notably, nearly all of the enzymes involved in glycolysis, gluconeogenesis, TCA cycle, fatty acid β-oxidation, urea cycle, nitrogen metabolism, and glycogen metabolism are acetylated. More importantly, further studies demonstrate that protein acetylation plays an essential role in metabolism, directing attention away from modification by phosphorylation and ubiquitylation.
Interestingly, acetylation modulates different metabolic enzymes in different manners. Acetylation controls the catalytic activity of enzymes through neutralization of the lysine residue, thus changing the conformation of the active site, antagonizing allosteric activation, blocking substrate interaction, or affecting the structural polymerization state.68 For example, ornithine transcarbamylase (OTC) is acetylated at K88, which is located in the active site for substrate binding. Acetylation most likely inhibits OTC catalytic activity by neutralizing the positive charge of K88 and reducing substrate binding.69
Acetylation could also affect enzyme degradation through the ubiquitin-proteasomal pathway or the lysosomal pathway. Crosstalk between acetylation and ubiquitylation contributes to acetylation-mediated proteasomal degradation of enzymes. In general, as acetyl groups and ubiquitin compete for the same lysine residues, acetylation protects enzymes from ubiquitin-proteasomal degradation; for example, acetylation of ACLY blocks its degradation.70 Interestingly, acetylation of phosphoenolpyruvate carboxykinase 1 (PEPCK1) stimulates its interaction with the E3 ubiquitin ligase UBR5 to promote PEPCK1 ubiquitylation and proteasomal degradation.70 In addition, acetylation regulates lysosomal degradation of enzymes by promoting interaction with chaperones, such as HSC70, that target enzyme transport into the lysosome for degradation. Recently we revealed that acetylation promotes lysosomal degradation of PKM2 and LDHA via chaperone-mediated autophagy.71,72
In addition, acetylation could regulate enzymes in other ways, such as through altered subcellular localization or catalytic reaction. There is emerging evidence that lysine acetylation of metabolic enzymes links signaling pathways to metabolism pathways and plays an important role in cancer cells. Many enzymes that are modulated by acetylation have been reported; in this review we discuss only the acetylated enzymes that play critical roles in tumor metabolism (Fig. 2).
Figure 2.

Tumor metabolism provides precursors for biomacromolecule synthesis. Metabolism in tumor cells is reprogrammed compared to that in normal cells. Upregulation of glycolysis and altered TCA flow produces intermediates that provide precursors for macromolecule biosynthesis, such as nucleotides (pathway in red), fatty acids (green), and amino acids (blue). These alterations in tumor cells support rapid cancer cell growth.
Glucose Metabolism
Glycolysis pathway
In glycolysis and gluconeogenesis, glyceraldehyde 3-phosphate dehydrogenase (GAPDH) catalyzes a reversible reaction between glyceraldehyde 3-phosphate and 1, 3-bisphosphoglycerate. It has been reported that acetylation regulates GAPDH glycolytic activity in Salmonella. Acetylation of GAPDH responds well to different signals such as glucose and acetate, and adjusts the direction of carbon flux according to environmental nutrient changes.73 Recently, we reported a new acetylation site of GAPDH at K254, and showed that a high glucose concentration increases its acetylation and catalytic activity. We also demonstrated the role of GAPDH acetylation at K254 in accelerating tumor growth.74
Mammalian phosphoglycerate mutase (PGAM) catalyzes the reversible conversion of 3phosphoglycerate (3PGA) to 2phosphoglycerate (2PGA) in glycolysis. PGAM contributes to energy production through the generation of reducing power and the biosynthesis of nucleotide precursors and amino acids. Inhibition of PGAM by RNA interference or small molecules induces cell proliferation and tumor growth, and PGAM activity is commonly upregulated in tumor cells. These findings indicate an important role of PGAM in tumor metabolism. Both isoforms of PGAM (PGAM1 and PGAM2) have been reported to be acetylated. Acetylated PGAM1 displays enhanced activity, thereby stimulating glycolysis flux, and is deacetylated by SIRT1. Glucose restriction leads to deacetylation and attenuated activity of PGAM1 by increasing Sirt1 levels.75 Another report found that PGAM2 is acetylated at K100, an active site residue that is invariably conserved from bacteria to mammals. Acetylation of K100 of PGAM2 decreases its activity, and SIRT2 is also the deacetylase for this isoform. Reactive oxygen species (ROS) promote the interaction of PGAM2 with SIRT2, resulting in deacetylation and activation of PGAM2. Moreover, K100 acetylation reduced cellular NADPH production and inhibited cell proliferation and tumor growth.74
In the last 5 years, intensive studies have focused on the M2 isoform of pyruvate kinase (PKM2). PKM2 catalyzes the last step in glycolysis, from phosphoenolpyruvate to pyruvate, and is highly expressed in embryonic tissues and many cancer types. Alternative splicing of the PKM2 gene produces 2 isoforms, M1 and M2. Expression of the inactive PKM2 isoform causes rapidly dividing cells to accumulate glycolytic metabolites that drive macromolecular biosynthesis and cell growth. In 2011, we found that acetylation at K305 decreases PKM2 enzyme activity and promotes its lysosomal-dependent degradation. High glucose concentrations stimulate PKM2 acetylation by increasing its binding to the acetyltransferase PCAF. We also demonstrated that K305 acetylation leads to accumulation of glycolytic intermediates to promote cell proliferation and tumor growth.71
In tumor cells, lactate is generated from pyruvate by lactate dehydrogenase A (LDH-A), which is frequently overexpressed in tumor cells and is important for cell growth. LDH-A regenerates NAD+ for rapid glycolysis in tumor cells in addition to the production of lactate, giving rise to an acidic microenvironment that promotes tumor cell migration and metastasis. Recently, we reported that the acetylation of LDH-A at K5 inhibits its activity and targets LDH-A degradation by chaperone-mediated autophagy. We also showed that LDH-A acetylation decreases cell migration and tumor growth. Moreover, reduced K5 acetylation during the initiation of human pancreatic cancers contributes to accumulation of LDH-A and promotes tumor metabolism and cancer progression.72
TCA cycle pathway
The pyruvate dehydrogenase complex (PDC), which plays an important role in glucose homeostasis, is negatively regulated through phosphorylation of pyruvate dehydrogenase (PDH) by PDH kinase (PDK) and activated by PDH phosphatase (PDP). A recent study reported that PDHA1 and PDP1 are acetylated in EGF-stimulated cells and diverse human cancer cells, and ACAT1 and SIRT3 were identified as the acetyltransferase and deacetylase responsible. K321 acetylation inhibits PDHA1 through recruitment of PDK1, whereas PDP1 acetylation at K202 decreases its interaction with PDHA1, leading to increased cancer cell glycolysis and tumor growth.76
Several enzymes in the TCA cycle have been reported to be acetylated. The activity of mitochondrial isocitrate dehydrogenase 2 (IDH2), which catalyzes the decarboxylation of isocitrate to αketoglutarate, is inhibited by acetylation. Under caloric restriction, IDH2 is deacetylated and activated by SIRT3 to protect against oxidative damage by increasing NAPDH production.77 Malate dehydrogenase (MDH) is also acetylated at K185, K301, K307, and K314 in response to high glucose.64 Succinate dehydrogenase (SDH) is a unique enzyme that participates in both the TCA cycle and in oxidative phosphorylation (as complex II). Deacetylation of one of its subunits, SDHA, by SIRT3 is accompanied by the activation of SDHA complex II activity.78 By mass spectrometry, 4 acetylated lysine residues were found to be located on the hydrophilic surface of SDHA, indicating that acetylation may hinder the entry of substrates into the active site.68 So far, acetylation of these enzymes has not been linked to tumors; however, the products produced by activity of these enzymes, such as αketoglutarate and succinate, have been reported to modulate epigenetics in some cancer types. These findings indicate the potential functions of acetylation of TCA cycle enzymes in tumor metabolism.
Pentose phosphate pathway (PPP)
The PPP is controlled by glucose-6-phosphate dehydrogenase (G6PD), which provides reducing power and ribose phosphate for nucleotide and lipid biosynthesis and redox balance. There is ample clinical evidence that G6PD is frequently overexpressed in multiple myeloma, chronic lymphocytic leukemia, and gastric cancer.79 G6PD has been revealed as a promising target in cancer therapy.80 A recent study reported that G6PD is negatively regulated by acetylation on K403. This acetylation inhibits formation of the active dimer and causes a complete loss of catalytic activity. Knockdown of G6PD sensitizes cells to oxidative stress, and re-expression of a K403 acetylation mimetic mutant impairs the rescue of cells from oxidative injury. Moreover, SIRT2-mediated deacetylation and activation of G6PD stimulates the PPP to supply NADPH in order to resist oxidative damage and protect mouse erythrocytes. KAT9/ELP3 was identified as a potential acetyltransferase of G6PD.74 This study suggested the potential role of G6PD acetylation in tumor metabolism.
Lipid Metabolism
ATP-citrate lyase (ACLY) is an enzyme involved in fatty acid biosynthesis that links carbohydrate metabolism and fatty acid production. ACLY is responsible for the synthesis of cytosolic acetyl-CoA in many tissues. ACLY is upregulated or activated in various types of cancer, and inhibition of ACLY represses cancer cell proliferation. Recently, we found that ACLY is acetylated at K540, K546, and K554. PCAF and SIRT2 are responsible for regulating ACLY acetylation. High glucose stimulates acetylation and increases ACLY stability by blocking its ubiquitylation and degradation. Substitution of these 3 lysine residues stabilizes ACLY and promotes de novo lipid synthesis, cell proliferation, and tumor growth. Moreover, acetylation of ACLY is increased in human lung cancers.81
Microarray-Bioinformatic analysis revealed that many genes involved in fatty acid metabolic pathways are overexpressed in colorectal cancer, including the gene encoding enoyl CoA hydratase/3-hydroxylacyl CoA dehydrogenase (EHHADH).82 EHHADH is the key enzyme in fatty acid β-oxidation and contains 2 domains: enoyl-CoA hydratase (N-terminal) and 3-hydroxyacyl-CoA dehydrogenase (C-terminal). Recently, Zhao et al. reported that EHHADH is modified by acetylation at 4 lysine residues (K165, K171, K346, and K584), which increases its enzymatic activity by up to 80%. Fatty acids in the culture medium stimulate the acetylation and activation of EHHADH.64
Sphingosine kinase (SPHK), an oncogenic lipid kinase, regulates the sphingolipid metabolic pathway. Its 2 isoforms (SPHK1 and SPHK2) catalyze the conversion of D-erythrosphingosine to sphingosine 1-phosphate (S1P), which is a signaling molecule involved in cell proliferation and survival through G protein-coupled receptor signaling. Accumulation of S1P is linked to the progression of various cancers, thus inhibitors of SPHK represent a novel class of compounds that have potential as anticancer agents.83 Some studies have demonstrated that SPHK1 is acetylated by p300 or CBP, and that this acetylation regulates protein stabilization. Acetylation and ubiquitylation compete for the same lysine residues and therefore acetylation inhibits SPHK1 ubiquitylation and degradation.84 These data suggest that SPK1 acetylation plays a key role in cell growth and tumor metabolism.
Glutamine Metabolism
In order to replenish oxaloacetate in the TCA cycle, many cancer cells require elevated levels of glutamine. Glutamate dehydrogenase (GDH) catalyzes the reversible conversion of glutamate to α-ketoglutarate and ammonium in the truncated TCA. Indeed, inhibition of GDH activity sensitizes glioblastoma cells to glucose deprivation. GDH has been reported to be acetylated, and calorie restriction stimulates its acetylation in response to different nutrient conditions. In vitro deacetylation of GDH by SIRT3 activates the enzyme, and studies in Sirt3 knockout mice indicated that GDH is a prominent acetylated protein in the liver.85,86 SIRT4, which is localized in the mitochondria, has NAD+-dependent ADP-ribosylase activity but not NAD+-dependent deacetylase activity.87 GDH is ADP-ribosylated by SIRT4 in vitro and this modification appears to inhibit GDH activity. Moreover, GDH activity is significantly decreased in Sirt4-deficient cells. These studies have established the complex regulation of GDH through different post-translational modifications by SIRT3 and SIRT4.
Acetate Metabolism
Acetyl-CoA synthetase (AceS) was the first enzyme shown to be acetylated. AceS catalyzes the transformation of acetate into acetyl-CoA, which enters the TCA cycle for energy production or the glycolysis pathway for intermediate biosynthesis. AceS is essential for growth of bacteria on acetate or propionate as carbon sources. Acetylation of AceS of S. entericais at K609 is essential for AceS catalysis.88 Acetylation strongly inhibits AceS activity both in vitro and in vivo. AceS purified from a CobB (SIRT homolog in S. enterica) mutant strain shows significantly reduced activity compared to that purified from wild-type cells, leading to poor growth of CobB mutant cells on propionate or acetate medium. Several studies have also reported that the 2 mammalian AceS isoforms (AceS1 in cytoplasm and AceS2 in mitochondria) are similarly regulated by acetylation. PAT, a bacterial protein acetyltransferase, acetylates AceS1 in vitro primarily at K661, which is conserved with K609 in S. enterica, thus inactivating the enzyme. In vivo experiments have found that acetylated AceS1 can be deacetylated by SIRT1, resulting in activation.89 Another investigation demonstrated that mitochondrial AceS2 is also acetylated by PAT in vitro and deacetylated by SIRT3. Acetylation similarly inhibits AceS2 catalytic activity.90 In mammalian cells, the cytosolic AceS1 produces acetyl-CoA to provide a critical substrate for biosynthesis, such as fatty acid synthesis, whereas mitochondrial AceS2 generates acetyl-CoA for cellular energy production through the TCA cycle. Studies have found that acetate is used as a carbon source in tumor metabolism to support rapid tumor growth, indicating that regulation of AceS1 and AceS2 by acetylation may play an important role in tumor metabolism.91
ROS Metabolism
An early hypothesis suggested that the tumor suppressive role of SIRT3 comes from protection against cellular ROS levels, and is supported by a series of studies reporting that superoxide dismutase (SOD) is a substrate of SIRT3. SOD catalyzes the dismutation of superoxide (O2−) into oxygen and hydrogen peroxide, and is therefore an important antioxidant defense for cells that are exposed to oxygen. Superoxide is quite toxic in vivo, and knockout of SOD not only shortens individual lifespan, but also accelerates certain features of aging. Deficiency of SOD is linked to various human diseases including cancers. Three investigations found that acetylation inhibits SOD2 catalytic activity, whereas oxidative stress-induced deacetylation by SIRT3 results in SOD2 activation and ROS reduction; however, these studies identified different acetylated lysine residues in SOD2.92-94
Future Prospects
A number of studies have recently established the critical role of acetylation in the regulation of metabolism, especially cancer metabolism. However, despite the rapid progress in this field many important questions remain largely unanswered.
Non-Metabolic Functions of Metabolic Enzymes
Although the enzymes discussed above are commonly responsible for tumor metabolism, recent studies have revealed that some of them have non-metabolic functions in cancer.
Recent studies have proven that PKM2 has non-metabolic functions in the nucleus as a protein kinase and a transcriptional cofactor. Luo et al. reported that PKM2 interacts with HIF-1α in the nucleus to promote the transactivation of HIF-1 target genes for the reprogramming of glucose metabolism in cancer cells.95 However, Yang et al. reported that EGFR induces nuclear localization of PKM2 to promote its interaction with β-catenin and thereby stimulate cyclin D1 expression.96 Gao et al. reported that homodimers of PKM2 could phosphorylate STAT3.97 More recently, PKM2 was found to directly phosphorylate histone H3 upon EGFR activation, leading to the activation of oncogenes.98 Interestingly, we found that PKM2 is acetylated by p300 at K433, which is unique to PKM2 and directly contacts its allosteric activator, fructose-1,6-bisphosphate (FBP). Acetylation prevents PKM2 activation by interfering with FBP binding and promotes PKM2 nuclear accumulation and its protein kinase activity, leading to cell proliferation and tumorigenesis.99
Several studies have found that GAPDH is not only a classic metabolic protein, but also plays important roles in a variety of critical nuclear pathways, including apoptosis, transcriptional regulation, nuclear membrane fusion, and the maintenance of telomere structure.100 The translocation of GAPDH into the nucleus can be influenced by cellular growth conditions and is regulated by acetylation. Mammalian GAPDH is acetylated by PCAF, leading to GAPDH nuclear translocation, and treatment of cells with the HDAC inhibitor TSA promotes GAPDH nuclear accumulation.101
Another example is triosephosphate isomerase (TPI), which converts dihydroxyacetone phosphate into glyceraldehyde-3-phosphate in glycolysis. TPI deficiency leads to serious neurological symptoms. Roland et al. found that the Drosophila TPI sugarkill mutant, which closely models TPI deficiency, can be genetically complemented by catalytically inactive TPI; this indicates a non-metabolic function of TPI, loss of which contributes to neurological dysfunction.102
These studies suggest that the regulatory roles of acetylation on metabolic enzymes go beyond their classic catalytic functions in human diseases. We expect that more of these enzymes will be shown to have non-metabolic functions in the future.
Multiple Signaling Pathways Modulated by Acetylation
Acetylation is an important regulator of histones, transcriptional factors, and metabolic enzymes. In addition, many other processes are modulated by acetylation, such as mRNA splicing, mRNA transport, mRNA integrity, and protein translation. Multiple signaling pathways in cancer are regulated by acetylation, but due to space limitations we will discuss only the NF-κB pathway, Wnt pathway, and EGFR signaling pathway.
As a regulator of gene transcription, NF-κB controls cell proliferation and cell survival. Many types of tumor have been shown to have dysregulated NF-κB. The activity of NF-κB is tightly regulated by acetylation. NF-κB is a heterodimer of RelA/p65 and p50/p52 subunits. In resting cells, NF-κB is associated with its repressor and localized in the cytoplasm. Upon stimulation, the repressor is inhibited and the released NF-κB translocates to the nucleus. The p65 subunit is then acetylated by p300/CBP or PCAF at K218, K221, and K310 to enhance NF-κB binding with DNA and promote its transcriptional activation ability. Moreover, deacetylation by HDAC3 promotes NF-κB nuclear export through binding with its repressor.103 Therefore, the antitumor effect of HDACs inhibitors also partially depends on the deacetylation of NF-κB to promote transcription of apoptosis genes.
The signal transducers and activators of transcription (STAT) protein family regulate cell survival, growth, and differentiation. Dysregulation of STATs is frequently observed in primary tumors and leads to increased tumor growth and angiogenesis. Acetylation at K685 by CBP/p300 modulates the dimerization of STAT3, which enhances DNA binding and gene transcription (such as cyclin D1, Bcl-xL), and HDAC3 is primarily responsible for its deacetylation.104 In melanoma cells, CBP acetylates STAT1 at K410 and K413 and induces cell apoptosis.105 Acetylated STAT1 also preferentially binds to p65 and enhances the transcription of NF-κB target genes.
The Wnt signaling pathway has a crucial role during embryonic development, whereas its dysregulation causes cellular transformation and a variety of human cancers. Lymphoid enhancer factor/T-cell factor (LEF/TCF) mediates Wnt signaling in the nucleus by recruiting β-catenin and its co-activators to target genes. In C. elegans, acetylation enhances the nuclear retention of POP-1, the LEF/TCF homolog, and activates its biological function during embryogenesis.106 Another central component of the Wnt pathway, β-catenin, is also acetylated by CBP at K49, which is located in an important region that is frequently mutated in cancers. Mutation of K49 increases the ability of β-catenin to activate the c-Myc gene. Many investigations have indicated that Wnt signaling is also responsible for the metabolic programming of glycolysis in some cancers.107 These results suggest that acetylation of the Wnt/β-catenin pathway is involved in tumor metabolism.108
Alteration of EGFR is involved in many cancers and has been intensively investigated. There is evidence that acetylation of EGFR plays a pivotal role in controlling its function and metabolism. EGFR can be acetylated by CBP. This acetylation influences the tyrosine phosphorylation of EGFR, which may contribute to resistance of cancer cells to HDAC inhibitors. This study indicated that a combination of HDAC inhibitors and EGFR inhibitors enhances therapeutic outcomes.109
So far, many investigations have shown that acetylation plays an important role in the development and progression of human cancers, and several HDAC inhibitors such as SAHA have been approved by the FDA for cancer therapy in clinical practice. Recent studies found that several SIRT family members act as both oncogenes and tumor suppressor genes in a cell context-dependent manner. The identification of an increasing number of acetylation-modified substrates suggests that the acetylation network responsible for the regulation of tumor metabolism is more complicated than previously thought. Efforts to understand how acetylation coordinately modulates different metabolic and related pathways globally and contributes to tumorigenesis and tumor development are ongoing, but it is hoped that the results of these studies will provide a number of potential targets for cancer therapy.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This review was supported by MOST 973 (Grant No. 2009CB918401, 2011CB910600), NSFC (Grant No. 31271454, 81225016), Shanghai Key basic research program (12JC1401100), “100 Talents” Program of Shanghai Health (Grant No. XBR2011041), Scholar of “Dawn” Program of Shanghai Education Commission, and Shanghai Outstanding Academic Leader (Grant No.13XD1400600) to QYL.
References
- 1. Allis CD, Berger SL, Cote J, Dent S, Jenuwien T, Kouzarides T, Pillus L, Reinberg D, Shi Y, Shiekhattar R, et al. New nomenclature for chromatin-modifying enzymes. Cell 2007; 131:633-6; PMID:18022353; http://dx.doi.org/ 10.1016/j.cell.2007.10.039 [DOI] [PubMed] [Google Scholar]
- 2. Kleff S, Andrulis ED, Anderson CW, Sternglanz R. Identification of a gene encoding a yeast histone H4 acetyltransferase. J Biol Chem 1995; 270:24674-7; PMID:7559580; http://dx.doi.org/ 10.1074/jbc.270.42.24674 [DOI] [PubMed] [Google Scholar]
- 3. Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev 2000; 14:1553-77; PMID:10887150 [PubMed] [Google Scholar]
- 4. Yao TP, Oh SP, Fuchs M, Zhou ND, Ch'ng LE, Newsome D, Bronson RT, Li E, Livingston DM, Eckner R. Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell 1998; 93:361-72; PMID:9590171; http://dx.doi.org/ 10.1016/S0092-8674(00)81165-4 [DOI] [PubMed] [Google Scholar]
- 5. Ito A, Kawaguchi Y, Lai CH, Kovacs JJ, Higashimoto Y, Appella E, Yao TP. MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J 2002; 21:6236-45; PMID:12426395; http://dx.doi.org/ 10.1093/emboj/cdf616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Akimova T, Beier UH, Liu Y, Wang L, Hancock WW. Histone/protein deacetylases and T-cell immune responses. Blood 2012; 119:2443-51; PMID:22246031; http://dx.doi.org/ 10.1182/blood-2011-10-292003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cook C, Gendron TF, Scheffel K, Carlomagno Y, Dunmore J, DeTure M, Petrucelli L. Loss of HDAC6, a novel CHIP substrate, alleviates abnormal tau accumulation. Hum Mol Genet 2012; 21:2936-45; PMID:22492994; http://dx.doi.org/ 10.1093/hmg/dds125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee YS, Lim KH, Guo X, Kawaguchi Y, Gao Y, Barrientos T, Ordentlich P, Wang XF, Counter CM, Yao TP. The cytoplasmic deacetylase HDAC6 is required for efficient oncogenic tumorigenesis. Cancer Res 2008; 68:7561-9; PMID:18794144; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-0188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Preyat N, Leo O. Sirtuin deacylases: a molecular link between metabolism and immunity. J Leukoc Biol 2013; 93:669-80; PMID:23325925; http://dx.doi.org/ 10.1189/jlb.1112557 [DOI] [PubMed] [Google Scholar]
- 10. Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH, et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 2011; 334:806-9; PMID:22076378; http://dx.doi.org/ 10.1126/science.1207861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Semenza GL. Regulation of cancer cell metabolism by hypoxia-inducible factor 1. Semin Cancer Biol 2009; 19:12-6; PMID:19114105; http://dx.doi.org/ 10.1016/j.semcancer.2008.11.009 [DOI] [PubMed] [Google Scholar]
- 12. Brand K. Glutamine and glucose metabolism during thymocyte proliferation. Pathways of glutamine and glutamate metabolism. Biochem J 1985; 228:353-61; PMID:2861809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hedeskov CJ. Early effects of phytohaemagglutinin on glucose metabolism of normal human lymphocytes. Biochem J 1968; 110:373-80; PMID:5726214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guppy M, Greiner E, Brand K. The role of the Crabtree effect and an endogenous fuel in the energy metabolism of resting and proliferating thymocytes. Eur J Biochem 1993; 212:95-9; PMID:8444168; http://dx.doi.org/ 10.1111/j.1432-1033.1993.tb17637.x [DOI] [PubMed] [Google Scholar]
- 15. Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer 2013; 13:572-83; PMID:23822983; http://dx.doi.org/ 10.1038/nrc3557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Owen OE, Kalhan SC, Hanson RW. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem 2002; 277:30409-12; PMID:12087111; http://dx.doi.org/ 10.1074/jbc.R200006200 [DOI] [PubMed] [Google Scholar]
- 17. Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci 2010; 35:427-33; PMID:20570523; http://dx.doi.org/ 10.1016/j.tibs.2010.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Taylor CT, Pouyssegur J. Oxygen, hypoxia, and stress. Ann N Y Acad Sci 2007; 1113:87-94; PMID:17483207; http://dx.doi.org/ 10.1196/annals.1391.004 [DOI] [PubMed] [Google Scholar]
- 19. Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006; 441:437-43; PMID:16724055; http://dx.doi.org/ 10.1038/nature04871 [DOI] [PubMed] [Google Scholar]
- 20. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 2006; 3:177-85; PMID:16517405; http://dx.doi.org/ 10.1016/j.cmet.2006.02.002 [DOI] [PubMed] [Google Scholar]
- 21. Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 2006; 3:187-97; PMID:16517406; http://dx.doi.org/ 10.1016/j.cmet.2006.01.012 [DOI] [PubMed] [Google Scholar]
- 22. Wang HQ, Altomare DA, Skele KL, Poulikakos PI, Kuhajda FP, Di Cristofano A, Testa JR. Positive feedback regulation between AKT activation and fatty acid synthase expression in ovarian carcinoma cells. Oncogene 2005; 24:3574-82; PMID:15806173; http://dx.doi.org/ 10.1038/sj.onc.1208463 [DOI] [PubMed] [Google Scholar]
- 23. Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006; 441:424-30; PMID:16724053; http://dx.doi.org/ 10.1038/nature04869 [DOI] [PubMed] [Google Scholar]
- 24. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Xiao MT, Liu LX, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011; 19:17-30; PMID:21251613; http://dx.doi.org/ 10.1016/j.ccr.2010.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, Liu L, Liu Y, Yang C, Xu Y, et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev 2012; 26:1326-38; PMID:22677546; http://dx.doi.org/ 10.1101/gad.191056.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, Schmidt K, Willson JK, Markowitz S, Zhou S, et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009; 325:1555-9; PMID:19661383; http://dx.doi.org/ 10.1126/science.1174229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. MacKenzie ED, Selak MA, Tennant DA, Payne LJ, Crosby S, Frederiksen CM, Watson DG, Gottlieb E. Cell-permeating alpha-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol Cell Biol 2007; 27:3282-9; PMID:17325041; http://dx.doi.org/ 10.1128/MCB.01927-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Deberardinis RJ, Lum JJ, Thompson CB. Phosphatidylinositol 3-kinase-dependent modulation of carnitine palmitoyltransferase 1A expression regulates lipid metabolism during hematopoietic cell growth. J Biol Chem 2006; 281:37372-80; PMID:17030509; http://dx.doi.org/ 10.1074/jbc.M608372200 [DOI] [PubMed] [Google Scholar]
- 29. Evans JM, Ogston SA, Emslie-Smith A, Morris AD. Risk of mortality and adverse cardiovascular outcomes in type 2 diabetes: a comparison of patients treated with sulfonylureas and metformin. Diabetologia 2006; 49:930-6; PMID:16525843; http://dx.doi.org/ 10.1007/s00125-006-0176-9 [DOI] [PubMed] [Google Scholar]
- 30. Muller FL, Colla S, Aquilanti E, Manzo VE, Genovese G, Lee J, Eisenson D, Narurkar R, Deng P, Nezi L, et al. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature 2012; 488:337-42; PMID:22895339; http://dx.doi.org/ 10.1038/nature11331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Harry G, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007; 11:37-51; PMID:17222789; http://dx.doi.org/ 10.1016/j.ccr.2006.10.020 [DOI] [PubMed] [Google Scholar]
- 32. Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A 2010; 107:2037-42; PMID:20133848; http://dx.doi.org/ 10.1073/pnas.0914433107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013; 340:626-30; PMID:23558169; http://dx.doi.org/ 10.1126/science.1236062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene 2007; 26:5420-32; PMID:17694083; http://dx.doi.org/ 10.1038/sj.onc.1210610 [DOI] [PubMed] [Google Scholar]
- 35. Spiegel S, Milstien S, Grant S. Endogenous modulators and pharmacological inhibitors of histone deacetylases in cancer therapy. Oncogene 2012; 31:537-51; PMID:21725353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rius M, Lyko F. Epigenetic cancer therapy: rationales, targets and drugs. Oncogene 2012; 31:4257-65; PMID:22179827; http://dx.doi.org/ 10.1038/onc.2011.601 [DOI] [PubMed] [Google Scholar]
- 37. Riggs MG, Whittaker RG, Neumann JR, Ingram VM. n-Butyrate causes histone modification in HeLa and Friend erythroleukaemia cells. Nature 1977; 268:462-4; PMID:268489; http://dx.doi.org/ 10.1038/268462a0 [DOI] [PubMed] [Google Scholar]
- 38. Lee JV, Carrer A, Shah S, Snyder NW, Wei S, Venneti S, Worth AJ, Yuan ZF, Lim HW, Liu S, et al. Akt-Dependent Metabolic Reprogramming Regulates Tumor Cell Histone Acetylation. Cell Metabolism 2014; 20:306-19; PMID:24998913; http://dx.doi.org/ 10.1016/j.cmet.2014.06.00 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Marino G, Pietrocola F, Eisenberg T, Kong YL, Malik SA, Andryushkova A, Schroeder S, Pendl T, Harger A, Niso-Santano M, et al. Regulation of autophagy by cytosolic acetyl-coenzyme A. Molecular Cell 2014; 53:710-25; PMID:24560926; http://dx.doi.org/ 10.1016/j.molcel.2014.01.016 [DOI] [PubMed] [Google Scholar]
- 40. Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009; 324:1076-80; PMID:19461003; http://dx.doi.org/ 10.1126/science.1164097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sutendra G KA, Dromparis P, Paulin R, Stenson TH, Haromy A, Hashimoto K, Zhang N, Flaim E, Michelakis ED. A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell 2014; 158:84-97; PMID:24995980; http://dx.doi.org/ 10.1016/j.cell.2014.04.046 [DOI] [PubMed] [Google Scholar]
- 42. Xenaki G, Ontikatze T, Rajendran R, Stratford IJ, Dive C, Krstic-Demonacos M, Demonacos C. PCAF is an HIF-1alpha cofactor that regulates p53 transcriptional activity in hypoxia. Oncogene 2008; 27:5785-96; PMID:18574470; http://dx.doi.org/ 10.1038/onc.2008.192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lim JH, Lee YM, Chun YS, Chen J, Kim JE, Park JW. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol Cell 2010; 38:864-78; PMID:20620956; http://dx.doi.org/ 10.1016/j.molcel.2010.05.023 [DOI] [PubMed] [Google Scholar]
- 44. Wahlstrom T, Henriksson MA. Impact of MYC in regulation of tumor cell metabolism. Biochim Biophys Acta 2014; S1874-9399:00192-8; PMID:25038584 [DOI] [PubMed] [Google Scholar]
- 45. Vervoorts J, Luscher-Firzlaff JM, Rottmann S, Lilischkis R, Walsemann G, Dohmann K, Austen M, Luscher B. Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP. EMBO Rep 2003; 4:484-90; PMID:12776737; http://dx.doi.org/ 10.1038/sj.embor.embor821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Faiola F, Liu X, Lo S, Pan S, Zhang K, Lymar E, Farina A, Martinez E. Dual regulation of c-Myc by p300 via acetylation-dependent control of Myc protein turnover and coactivation of Myc-induced transcription. Mol Cell Biol 2005; 25:10220-34; PMID:16287840; http://dx.doi.org/ 10.1128/MCB.25.23.10220-10234.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Patel JH, Du Y, Ard PG, Phillips C, Carella B, Chen CJ, Rakowski C, Chatterjee C, Lieberman PM, Lane WS, et al. The c-MYC oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Mol Cell Biol 2004; 24:10826-34; PMID:15572685; http://dx.doi.org/ 10.1128/MCB.24.24.10826-10834.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yang MH, Nickerson S, Kim ET, Liot C, Laurent G, Spang R, Philips MR, Shan Y, Shaw DE, Bar-Sagi D, et al. Regulation of RAS oncogenicity by acetylation. Proc Natl Acad Sci U S A 2012; 109:10843-8; PMID:22711838; http://dx.doi.org/ 10.1073/pnas.1201487109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yang MH, Laurent G, Bause AS, Spang R, German N, Haigis MC, Haigis KM. HDAC6 and SIRT2 regulate the acetylation state and oncogenic activity of mutant K-RAS. Mol Cancer Res 2013; 11:1072-7; PMID:23723075; http://dx.doi.org/ 10.1158/1541-7786.MCR-13-0040-T [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012; 149:656-70; PMID:22541435; http://dx.doi.org/ 10.1016/j.cell.2012.01.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006; 126:107-20; PMID:16839880; http://dx.doi.org/ 10.1016/j.cell.2006.05.036 [DOI] [PubMed] [Google Scholar]
- 52. Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM. p53 regulates mitochondrial respiration. Science 2006; 312:1650-3; PMID:16728594; http://dx.doi.org/ 10.1126/science.1126863 [DOI] [PubMed] [Google Scholar]
- 53. Corcoran CA, Huang Y, Sheikh MS. The regulation of energy generating metabolic pathways by p53. Cancer Biol Ther 2006; 5:1610-3; PMID:17204863; http://dx.doi.org/ 10.4161/cbt.5.12.3617 [DOI] [PubMed] [Google Scholar]
- 54. Chao C, Wu Z, Mazur SJ, Borges H, Rossi M, Lin T, Wang JY, Anderson CW, Appella E, Xu Y. Acetylation of mouse p53 at lysine 317 negatively regulates p53 apoptotic activities after DNA damage. Mol Cell Biol 2006; 26:6859-69; PMID:16943427; http://dx.doi.org/ 10.1128/MCB.00062-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005; 436:725-30; PMID:16079851; http://dx.doi.org/ 10.1038/nature03918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ortega-Molina A SM. PTEN in cancer, metabolism, and aging. Trends Endocrinol Metab 2013; 24:184-9; PMID:23245767; http://dx.doi.org/ 10.1016/j.tem.2012.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ikenoue T, Inoki K, Zhao B, Guan KL. PTEN acetylation modulates its interaction with PDZ domain. Cancer Res 2008; 68:6908-12; PMID:18757404; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-1107 [DOI] [PubMed] [Google Scholar]
- 58. Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol 2012; 13:283-96; PMID:22473468 [DOI] [PubMed] [Google Scholar]
- 59. Fang M, Shen Z, Huang S, Zhao L, Chen S, Mak TW, Wang X. The ER UDPase ENTPD5 promotes protein N-glycosylation, the Warburg effect, and proliferation in the PTEN pathway. Cell 2010; 143:711-24; PMID:21074248; http://dx.doi.org/ 10.1016/j.cell.2010.10.010 [DOI] [PubMed] [Google Scholar]
- 60. Nicolay BN, Dyson NJ. The multiple connections between pRB and cell metabolism. Curr Opin Cell Biol 2013; 25:735-40; PMID:23916769; http://dx.doi.org/ 10.1016/j.ceb.2013.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Takahashi C, Sasaki N, Kitajima S. Twists in views on RB functions in cellular signaling, metabolism and stem cells. Cancer Sci 2012; 103:1182-8; PMID:22448711; http://dx.doi.org/ 10.1111/j.1349-7006.2012.02284.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chan HM, Krstic-Demonacos M, Smith L, Demonacos C, La Thangue NB. Acetylation control of the retinoblastoma tumour-suppressor protein. Nat Cell Biol 2001; 3:667-74; PMID:11433299; http://dx.doi.org/ 10.1038/35083062 [DOI] [PubMed] [Google Scholar]
- 63. Markham D, Munro S, Soloway J, O'Connor DP, La Thangue NB. DNA-damage-responsive acetylation of pRb regulates binding to E2F-1. EMBO Rep 2006; 7:192-8; PMID:16374512; http://dx.doi.org/ 10.1038/sj.embor.7400591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, et al. Regulation of cellular metabolism by protein lysine acetylation. Science 2010; 327:1000-4; PMID:20167786; http://dx.doi.org/ 10.1126/science.1179689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009; 325:834-40; PMID:19608861; http://dx.doi.org/ 10.1126/science.1175371 [DOI] [PubMed] [Google Scholar]
- 66. Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell 2006; 23:607-18; PMID:16916647; http://dx.doi.org/ 10.1016/j.molc-el.2006.06.026 [DOI] [PubMed] [Google Scholar]
- 67. Lundby A LK, Weinert BT, Bekker-Jensen DB, Secher A, Skovgaard T, Kelstrup CD, Dmytriyev A, Choudhary C, Lundby C, Olsen JV. Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Reports 2012; 2:419-31; PMID:22902405; http://dx.doi.org/ 10.1016/j.celrep.2012.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Xiong Y, Guan KL. Mechanistic insights into the regulation of metabolic enzymes by acetylation. J Cell Biol 2012; 198:155-64; PMID:22826120; http://dx.doi.org/ 10.1083/jcb.201202056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yu W, Lin Y, Yao J, Huang W, Lei QY, Xiong Y, Zhao SM, Guan KL. Lysine 88 Acetylation Negatively Regulates Ornithine Carbamoyltransferase Activity in Response to Nutrient Signals. J Biol Chem 2009; 284:13669-75; PMID:19318352; http://dx.doi.org/ 10.1074/jbc.M901921200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Jiang WQ, Wang SW, Xiao MT, Lin Y, Zhou LS, Lei QY, Xiong Y, Guan KL, Zhao SM. Acetylation Regulates Gluconeogenesis by Promoting PEPCK1 Degradation via Recruiting the UBR5 Ubiquitin Ligase. Mol Cell 2011; 43:33-44; PMID:21726808; http://dx.doi.org/ 10.1016/j.molcel.2011.04.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, Zha Z, Liu Y, Li Z, Xu Y, et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell 2011; 42:719-30; PMID:21700219; http://dx.doi.org/ 10.1016/j.molcel.2011.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhao D, Zou SW, Liu Y, Zhou X, Mo Y, Wang P, Xu YH, Dong B, Xiong Y, Lei QY, et al. Lysine-5 acetylation negatively regulates lactate dehydrogenase A and is decreased in pancreatic cancer. Cancer Cell 2013; 23:464-76; PMID:23523103; http://dx.doi.org/ 10.1016/j.ccr.2013.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Zhao W, Yao Y, et al. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 2010; 327:1004-7; PMID:20167787; http://dx.doi.org/ 10.1126/science.1179687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Li T, Liu M, Feng X, Wang Z, Das I, Xu Y, Zhou X, Sun Y, Guan KL, Xiong Y, et al. Glyceraldehyde-3-phosphate dehydrogenase is activated by lysine 254 acetylation in response to glucose signal. J Biol Chem 2014; 289:3775-85; PMID:24362262; http://dx.doi.org/ 10.1074/jbc.M113.531640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hallows WC, Yu W, Denu JM. Regulation of glycolytic enzyme phosphoglycerate mutase-1 by Sirt1 protein-mediated deacetylation. J Biol Chem 2012; 287:3850-8; PMID:22157007; http://dx.doi.org/ 10.1074/jbc.M111.317404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Fan J, Shan C, Kang HB, Elf S, Xie J, Tucker M, Gu TL, Aguiar M, Lonning S, Chen H, et al. Tyr phosphorylation of PDP1 toggles recruitment between ACAT1 and SIRT3 to regulate the pyruvate dehydrogenase complex. Mol Cell 2014; 53:534-48; PMID:24486017; http://dx.doi.org/ 10.1016/j.molcel.2013.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, Tanokura M, Denu JM, Prolla TA. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 2010; 143:802-12; PMID:21094524; http://dx.doi.org/ 10.1016/j.cell.2010.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Cimen H, Han MJ, Yang Y, Tong Q, Koc H, Koc EC. Regulation of succinate dehydrogenase activity by SIRT3 in mammalian mitochondria. Biochemistry 2010; 49:304-11; PMID:20000467; http://dx.doi.org/ 10.1021/bi901627u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. McBrayer SK, Yarrington M, Qian J, Feng G, Shanmugam M, Gandhi V, Krett NL, Rosen ST. Integrative gene expression profiling reveals G6PD-mediated resistance to RNA-directed nucleoside analogues in B-cell neoplasms. PLoS One 2012; 7:e41455; PMID:22848499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zhang C, Zhang Z, Zhu Y, Qin S. Glucose-6-phosphate dehydrogenase: a biomarker and potential therapeutic target for cancer. Anticancer Agents Med Chem 2014; 14:280-9; PMID:24066844; http://dx.doi.org/ 10.2174/18715206113136660337 [DOI] [PubMed] [Google Scholar]
- 81. Lin R, Tao R, Gao X, Li T, Zhou X, Guan KL, Xiong Y, Lei QY. Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth. Mol Cell 2013; 51:506-18; PMID:23932781; http://dx.doi.org/ 10.1016/j.molcel.2013.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yeh CS, Wang JY, Cheng TL, Juan CH, Wu CH, Lin SR. Fatty acid metabolism pathway play an important role in carcinogenesis of human colorectal cancers by Microarray-Bioinformatics analysis. Cancer Lett 2006; 233:297-308; PMID:15885896; http://dx.doi.org/ 10.1016/j.canlet.2005.03.050 [DOI] [PubMed] [Google Scholar]
- 83. Pyne S, Bittman R, Pyne NJ. Sphingosine kinase inhibitors and cancer: seeking the golden sword of Hercules. Cancer Res 2011; 71:6576-82; PMID:21940750; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-2364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Yu H, Shao Y, Gao L, Zhang L, Guo K, Wu C, Hu X, Duan H. Acetylation of sphingosine kinase 1 regulates cell growth and cell-cycle progression. Biochem Biophys Res Commun 2012; 417:1242-7; PMID:22227192; http://dx.doi.org/ 10.1016/j.bbrc.2011.12.117 [DOI] [PubMed] [Google Scholar]
- 85. Schlicker C, Gertz M, Papatheodorou P, Kachholz B, Becker CF, Steegborn C. Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J Mol Biol 2008; 382:790-801; PMID:18680753; http://dx.doi.org/ 10.1016/j.jmb.2008.07.048 [DOI] [PubMed] [Google Scholar]
- 86. Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol 2007; 27:8807-14; PMID:17923681; http://dx.doi.org/ 10.1128/MCB.01636-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ, Valenzuela DM, Yancopoulos GD, Karow M, Blander G, et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 2006; 126:941-54; PMID:16959573; http://dx.doi.org/ 10.1016/j.cell.2006.06.057 [DOI] [PubMed] [Google Scholar]
- 88. Starai VJ, Celic I, Cole RN, Boeke JD, Escalante-Semerena JC. Sirt2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science 2002; 298:2390-2; PMID:12493915; http://dx.doi.org/ 10.1126/science.1077650 [DOI] [PubMed] [Google Scholar]
- 89. Hallows WC, Lee S, Denu JM. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc Natl Acad Sci U S A 2006; 103:10230-5; PMID:16790548; http://dx.doi.org/ 10.1073/pnas.0604392103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc Natl Acad Sci U S A 2006; 103:10224-9; PMID:16788062; http://dx.doi.org/ 10.1073/pnas.0603968103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Yoshimoto M, Waki A, Yonekura Y, Sadato N, Murata T, Omata N, Takahashi N, Welch MJ, Fujibayashi Y. Characterization of acetate metabolism in tumor cells in relation to cell proliferation: acetate metabolism in tumor cells. Nucl Med Biol 2001; 28:117-22; PMID:11295421; http://dx.doi.org/ 10.1016/S0969-8051(00)00195-5 [DOI] [PubMed] [Google Scholar]
- 92. Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab 2010; 12:662-7; PMID:21109198; http://dx.doi.org/ 10.1016/j.cmet.2010.11.015 [DOI] [PubMed] [Google Scholar]
- 93. Chen Y, Zhang J, Lin Y, Lei Q, Guan KL, Zhao S, Xiong Y. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep 2011; 12:534-41; PMID:21566644; http://dx.doi.org/ 10.1038/embor.2011.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Tao R, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang H, Kim HS, Flynn CR, Hill S, Hayes McDonald W, et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell 2010; 40:893-904; PMID:21172655; http://dx.doi.org/ 10.1016/j.molcel.2010.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Luo W, Hu H, Chang R, Zhong J, Knabel M, O'Meally R, Cole RN, Pandey A, Semenza GL. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 2011; 145:732-44; PMID:21620138; http://dx.doi.org/ 10.1016/j.cell.011.03.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, Gao X, Aldape K, Lu Z. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature 2011; 480:118-22; PMID:22056988; http://dx.doi.org/ 10.1038/nature10598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Gao X, Wang H, Yang JJ, Liu X, Liu ZR. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol Cell 2012; 45:598-609; PMID:22306293; http://dx.doi.org/ 10.1016/j.molc-el.2012.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, Aldape K, Hunter T, Alfred Yung WK, Lu Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 2012; 150:685-96; PMID:22901803; http://dx.doi.org/ 10.1016/j.cell.2012.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lv L, Xu YP, Zhao D, Li FL, Wang W, Sasaki N, Jiang Y, Zhou X, Li TT, Guan KL, et al. Mitogenic and oncogenic stimulation of K433 acetylation promotes PKM2 protein kinase activity and nuclear localization. Mol Cell 2013; 52:340-52; PMID:24120661; http://dx.doi.org/ 10.1016/j.molcel.2013.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Sirover MA. New nuclear functions of the glycolytic protein, glyceraldehyde-3-phosphate dehydrogenase, in mammalian cells. J Cell Biochem 2005; 95:45-52; PMID:15770658; http://dx.doi.org/ 10.1002/jcb.20399 [DOI] [PubMed] [Google Scholar]
- 101. Ventura M, Mateo F, Serratosa J, Salaet I, Carujo S, Bachs O, Pujol MJ. Nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase is regulated by acetylation. Int J Biochem Cell Biol 2010; 42:1672-80; PMID:20601085; http://dx.doi.org/ 10.1016/j.biocel.2010.06.014 [DOI] [PubMed] [Google Scholar]
- 102. Roland BP, Stuchul KA, Larsen SB, Amrich CG, Vandemark AP, Celotto AM, Palladino MJ. Evidence of a triosephosphate isomerase non-catalytic function crucial to behavior and longevity. J Cell Sci 2013; 126:3151-8; PMID:23641070; http://dx.doi.org/ 10.1242/jcs.124586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Chen L, Fischle W, Verdin E, Greene WC. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science 2001; 293:1653-7; PMID:11533489; http://dx.doi.org/ 10.1126/science.1062374 [DOI] [PubMed] [Google Scholar]
- 104. Yuan ZL, Guan YJ, Chatterjee D, Chin YE. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 2005; 307:269-73; PMID:15653507; http://dx.doi.org/ 10.1126/science.1105166 [DOI] [PubMed] [Google Scholar]
- 105. Kramer OH, Baus D, Knauer SK, Stein S, Jager E, Stauber RH, Grez M, Pfitzner E, Heinzel T. Acetylation of Stat1 modulates NF-kappaB activity. Genes Dev 2006; 20:473-85; PMID:16481475; http://dx.doi.org/ 10.1101/gad.364306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Gay F, Calvo D, Lo MC, Ceron J, Maduro M, Lin R, Shi Y. Acetylation regulates subcellular localization of the Wnt signaling nuclear effector POP-1. Genes Dev 2003; 17:717-22; PMID:12651889; http://dx.doi.org/ 10.1101/gad.1042403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Pate KT, Stringari C, Sprowl-Tanio S, Wang K, TeSlaa T, Hoverter NP, McQuade MM, Garner C, Digman MA, Teitell MA, et al. Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J 2014; 33:1454-73; PMID:24825347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Wolf D, Rodova M, Miska EA, Calvet JP, Kouzarides T. Acetylation of beta-catenin by CREB-binding protein (CBP). J Biol Chem 2002; 277:25562-7; PMID:11973335; http://dx.doi.org/ 10.1074/jbc.M201196200 [DOI] [PubMed] [Google Scholar]
- 109. Song H, Li CW, Labaff AM, Lim SO, Li LY, Kan SF, Chen Y, Zhang K, Lang J, Xie X, et al. Acetylation of EGF receptor contributes to tumor cell resistance to histone deacetylase inhibitors. Biochem Biophys Res Commun 2011; 404:68-73; PMID:21094134; http://dx.doi.org/ 10.1016/j.bbrc.2010.11.064 [DOI] [PMC free article] [PubMed] [Google Scholar]