

Abstract
Tumors need blood vessels for their growth, thus providing the rationale for antiangiogenic therapy in cancer treatment. However, intrinsic and acquired resistance and low response rates have turned out to be major limitations of antiangiogenic therapy. This emphasizes the need to further understand how the vasculature in cancer can be targeted. Although endothelial cells (ECs) rely on multiple growth factors and cytokines to grow, antiangiogenic therapies have mainly centered on targeting vascular endothelial growth factor (VEGF). Phosphoinositide 3-kinases (PI3Ks) form a family of 8 isoenzymes with non-redundant functions in normal biology and cancer. The subgroup of class I PI3Ks are situated at the crossroad of a plethora of proangiogenic signals and control cell growth, survival, motility, and metabolism. These isoenzymes have pleiotropic roles in the tumor microenvironment, including cell-autonomous functions in ECs, underscoring the complexity of targeting this pathway in cancer. Here, we describe how the PI3K axis influences angiogenesis in different cell compartments and summarize the diversity of vascular responses to PI3K inhibition. Targeting PI3K signaling by isoform-selective inhibitors, together with readjusting the current doses below the maximum tolerated dose, may improve clinical responses to class I PI3K anticancer agents.
Keywords: PI3K, VEGF, angiogenesis, tumor-extrinsic and intrinsic roles
Abbreviations
- Ang
Angiopoietin
- Dll4
Delta-like 4
- ECs
Endothelial cells
- EPCs
Endothelial cells precursors
- FGF
Fibroblast growth factor
- GAB
GRB2 associated binding
- GAPs
GTPases-activating proteins
- GEFS
Guanine nucleotide exchange factors
- GPCR
G protein-coupled receptor
- miRs
microRNAs
- mTOR
Mammalian target of rapamycin complex
- MVD
Microvascular density
- PDGFR-b
Platelet-derived growth factor receptor-b
- PI3Ks
Phosphoinositide 3-kinases
- PLG1
Placental growth factor 1
- PtdIns
Phosphatidylinositol
- PtdIns(3,4,5)P3
PtdIns(3,4,5)-trisphosphate
- Rapalogues
Rapamycin analogues
- RBD
Ras-binding domain
- RTKs
Tyrosine kinase receptors
- VEGF
Vascular endothelial growth factor
Introduction
Highly proliferative tumor cells are in continuous need of oxygen and nutrients. To meet these needs, tumor cells produce large amounts of proangiogenic signals that stimulate vessel growth. This observation forms the basis of the idea that blocking angiogenesis would inhibit the capacity of tumor cells to grow and metastasize.1,2 To this end, several antiangiogenic agents have been developed in the last decade for clinical applications. Traditional antiangiogenic therapies have focused on targeting VEGF, which plays a central role in vessel morphogenesis.3-5 Preclinical studies have demonstrated that vessel pruning through inhibition of VEGF delays tumor progression and have provided the rationale for pursuing this strategy in the treatment of human cancers.6-8 However, when used as single agents these drugs have not met therapeutic expectations, and the lack of a widespread antitumor impact, insufficient efficacy, and acquired resistance remain major limitations.8
At least 2 important concepts should be considered in order to improve current antiangiogenic therapies. First, inhibition of vessel growth and pruning results in intratumor hypoxia, which over time promotes invasion and metastasis.9,10 To overcome this aggravation, a novel strategy based on normalizing the vasculature is being tested in preclinical models.7 Although promising data are emerging that suggest a real alternative to vessel pruning, key questions concerning the mode of action of these new drugs still need to be resolved.11 For example, it remains unclear whether vessel normalization will have a negative impact on tumor growth (because of improved delivery of nutrients and oxygen), or how the transient window for normalization will be identified in patients. Second, other proangiogenic factors such as fibroblast growth factor (FGF), ephrins, placental growth factor 1 (PLG1), and angiopoietins4,10 also stimulate tumor angiogenesis; moreover, the contribution of these proangiogenic factors is particularly significant upon resistance to VEGF therapy.4,11 Therefore, multitargeted antiangiogenic drugs that inhibit several different proangiogenic tyrosine kinase receptors are currently being tested in clinical trials.12,13 An important challenge of this strategy is to identify the particular angiokinase receptors to inhibit in different tumor types or individual patients. To overcome this problem, it may be beneficial to target the intracellular signaling hubs that converge in the plethora of angiogenic tyrosine-kinase receptors instead of distinct receptors. In this sense, the class I PI3Ks that are situated at the crossroad of many proangiogenic signals and that control cell growth, survival, motility, and metabolism, might offer a potential target. In this review, we will summarize how PI3K signaling contributes to tumor angiogenesis and the potential therapeutic effects of inhibiting this signaling node.
The PI3K Family as A Key Signaling Hub
PI3Ks are lipid kinases that phosphorylate the 3-hydroxyl group of the inositol ring present in membrane-bound phosphatidylinositol (PtdIns) lipids. By generating different PtdIns species, PI3Ks play a master role in the regulation of many cellular processes including cell cycle progression, cell growth, survival, migration, metabolism, and intracellular vesicle transport.14,15 In vertebrates, the PI3K family consists of 8 catalytic isoenzymes divided into 3 classes (class I, class II, and class III) based on their domain structures and lipid substrate preferences.14,16-19 Class I PI3K consists of 4 catalytic isoforms—p110α (PIK3CA), p110β (PIK3CB), p110γ (PIK3CG), and p110δ (PIK3CD). Class II PI3K, which contains 3 members—PI3K-C2α PIK3C2A), PI3K-C2β (PIK3C2A), and PI3K-C2γ (PIK3C2G)—is the most enigmatic subgroup because the physiological roles of these proteins remain largely unknown. The sole member of class III, vps34 (PIK3C3), is principally involved in vesicle trafficking and autophagy.14 To date, mainly the class I PI3Ks have been implicated in human cancer; hence this review will be principally focused on this subset of isoenzymes. However, class II and III PI3Ks could play a role in tumor angiogenesis, and might have to be considered in the future (see box 1).
Class I PI3Ks are heterodimeric proteins composed of a regulatory subunit and a p110 catalytic subunit. The class IA PI3Ks are composed of a p110α, p110β, or p110δ subunit constitutively bound to one of 5 types of p85 regulatory subunit. p110γ is the sole catalytic subunit of the class IB isoform and binds to either p101 or p84. All class I PI3Ks produce the same lipid but differ in their expression patterns, modes of activation, and physiological functions.14,15,20 Key information on class I catalytic subunits is summarized in Figure 1.
Figure 1.

Class I PI3K isoforms at a glance. Expression patterns, modes of activation, and physiological and pathophysiological roles of class I PI3K isoforms are summarized. p110α and p110β are ubiquitously expressed, in contrast to p110δ and p110γ, which are enriched in leukocytes. p110α and p110δ are preferentially activated by RTKs, whereas p110β and p110γ are activated by GPCRs. Each of the class I catalytic isoforms contain a Ras-binding domain (RBD). p110α, p110δ, and p110γ interact with Ras although a physiological role of this interaction has only been reported for p110α and p110γ. The activation of p110γ by Ras allows RTKs to activate this isoform in some cell types. The RBD of p110δ preferentially interacts with the small G protein TC21. p110β does not bind to Ras but interacts with the Rho subfamily GTPases Rac and Ccd42. p110β also binds to the GTP-bound RAB5 and regulates receptor-mediated endocytosis and autophagy independent of its kinase activity.
The lipid PtdIns(3,4,5)-trisphosphate (PtdIns(3,4,5)P3) is the second messenger made specifically by class I PI3Ks and acts as a membrane tether for multiple proteins, including Ser/Thr and Tyr protein kinases (such as AKT and BTK), adaptor proteins (such as GRB2 associated binding [GAB] 1 and GAB2, TAPP1, and DAPP) and regulators of small GTPases (GTPase-activating proteins [GAPs] and guanine nucleotide exchange factors [GEFs]).14,18,19 The Ser/Thr kinase Akt is responsible for the majority of PI3K-mediated responses as it phosphorylates a number of downstream targets, including the mammalian target of rapamycin complex 1 (mTORC1). Importantly, little attention has been paid to other PI3K downstream targets such as the small GTPases and their regulators, which might very well play a role in endothelial cell biology (Fig. 2).
Figure 2.

General overview of the class I PI3K signaling pathway. The PH domain-containing proteins can be divided into 3 groups: kinases, adaptor proteins, and GAPs and GEFs. Although the role of kinases has been extensively studied, little attention has been paid to how adaptor proteins or GAPs and GEFs drive PI3K cell functions, especially in cancer. The PI3K effector AKT is a master kinase that activates or inhibits a plethora of proteins by phosphorylation; one of its targets is mTORC1. A variety of compounds have been developed to inhibit different nodes of the PI3K/Akt/mTOR signaling axis, including PI3K inhibitors (subdivided based on their selectivity into pan-PI3K-mTOR inhibitors, pan-PI3K inhibitors, and isoform-specific inhibitors), AKT inhibitors (allosteric inhibitors and catalytic inhibitors), and mTOR inhibitors (allosteric inhibitors, commonly known as rapalogues, and mTOR catalytic inhibitors).
Pleiotropic Role of PI3Ks in Angiogenesis
Angiogenesis, meaning vessels sprouting from pre-existing ones, is a process that relies on a variety of different cell types, including endothelial cells (ECs), mural cells, immune cells, astrocytes and, in the context of cancer, tumor cells.3,4,21 Tissue hypoxia triggers vessel morphogenesis through the production of proangiogenic signals, of which VEGF is the most important.22,23 ECs occupy a key position at the inner surface of the tubular vessel network and respond to angiogenic signals including growth factors, guidance cues, and biophysical stimuli.3,4 To acquire full functionality, blood vessels need to be surrounded by perivascular mural cells (pericytes and vascular smooth muscle cells). The vessels with the smallest diameter (venules and capillaries) are associated with solitary pericytes, whereas multiple concentric layers of vascular smooth muscle cells surround larger vessels, in particular the arteries.24 For detailed information about how blood vessels grow, we refer the reader to cited references.3-5
The PI3K signaling pathway is engaged in nearly all cell types involved in vessel growth (Fig. 3). New insights into the biology and the context of activation of PI3K signaling in these different cell types are clarifying the mode of action of PI3K inhibitors in cancer. Although the majority of PI3K-mediated angiogenic functions involve the p110α isoform, the role of other p110 isoforms cannot be ignored. These include the role of p110γ in EC migration and recruitment of inflammatory cells,25,26 p110δ in the proliferation and chemotaxis of macrophages27 and in the immune responses of regulatory T cells 28, and p110β in platelet adhesion,29 all of which probably also contribute to vessel growth. Identifying the specific roles of these isoenzymes in the tumor stroma is particularly important when deciding whether pan-PI3K versus isoform-selective inhibitors should be used in cancer treatment. Summarized below is a detailed description of PI3K signaling in the different cell types that are relevant to angiogenesis.
Figure 3.
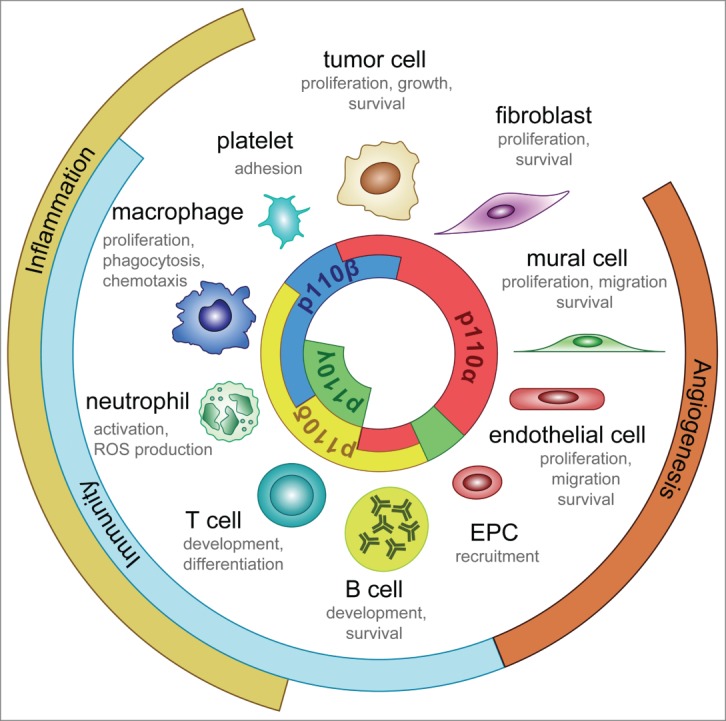
Isoform-specific roles of class (I)PI3Ks. This illustration shows the cell-autonomous roles of class I PI3K isoforms in normal physiology and cancer.
PI3K in endothelial cells: isoforms are key
Class I PI3K signaling in ECs is required in a cell-autonomous manner in order to grow vessels.30 Although ECs express all class I PI3K isoforms, only the catalytic subunit p110α is required for vessel sprouting.30,31 p110α accounts for most of the PtdIns(3,4,5)P3 produced upon activation of tyrosine kinase receptors (RTKs) in ECs.31 It is therefore likely that p110α regulates many functions of the angiogenic process. Cell-based studies have clearly demonstrated that PI3K signaling is activated downstream of VEGF-A, VEGF-C, angiopoietin (Ang)1, Ang2, VE-cadherin, Dll4, and ephrins, among others.30 In contrast, when, by what, and how p110α/PI3K signaling is engaged in vivo remains unknown. Progress in the field has been hampered by the embryonic lethality of constitutive and endothelial-specific kinase-dead p110α mutant mice.31 Further studies using more sophisticated assays and inducible systems are needed to fully elucidate the role of this PI3K isoform in angiogenesis. To date, 2 major in vivo downstream effectors of p110α have been identified in angiogenic ECs, namely Akt132,33 and Arap3.34
To counterbalance class I PI3K signaling, ECs principally use the 3-phosphatase PTEN that converts PtdIns(3,4,5)P3 into PtdIns(4,5)P2. In line with this, PTEN is required in ECs in a cell-autonomous manner for vessel morphogenesis.35 However, to achieve fine-tuned regulation of PtdIns(3,4,5)P3 levels, ECs have evolved an additional layer of regulation by modulating the levels of the p85 regulatory subunits. Loss of both p85α and p85β in ECs results in failure to make vessels.36 Studies in zebrafish, however, have shown that partial inactivation of these regulatory subunits results in improved EC responses. Two specific microRNAs (miRs), miR221 and miR126, have been found to modulate the levels of p85α and p85β respectively.37,38 Upregulation of miR-221 and miR-126 decreases p85α and p85β protein levels but increases PI3K activity, leading to stimulation of angiogenesis.37,38 The most likely explanation for this discrepancy is that during sprouting angiogenesis a pool of “free” p85 regulatory subunits (that lack kinase activity) exists that competes with the functional p85/p110 complex in binding to Tyr-phosphorylated activators.39,40 Interestingly, the boost of PtdIns(3,4,5)P3 generated upon miR221 expression and subsequent p85α downregulation seems to specifically regulate EC migration.37 In contrast, miR126 regulates vascular integrity in a p85β-dependent manner.38 Whether these regulatory mechanisms also occur in mammals still needs to be determined.
Although sprouting angiogenesis is the principal mechanism of vessel formation in health and disease,3,41 other mechanisms, such as the recruitment of endothelial cell precursors (EPCs) from the bone marrow,41,42 also contribute to the growth of vessels. Interestingly, p110γ, rather than p110α, seems to be the PI3K isoenzyme required in EPCs to drive postnatal neovascularization.43 A peculiarity of p110γ in EPCs is that it mediates EPC recruitment in an Akt-dependent manner but independently of the p110γ lipid kinase activity. This favors the hypothesis that, in this context, p110γ acts as a scaffolding protein rather than a lipid kinase.
PI3K signaling in mural cells: a hidden gem
Very little is known about the role that PI3K signaling plays in regulating mural cell biology in vivo. The signaling cascade stimulated by platelet-derived growth factor-B/platelet-derived growth factor receptor-β (PDGFR-β) in mural cells is critical for recruiting pericytes to growing vessels and for regulating their proliferation, migration, and survival.24 Broad-spectrum PI3K inhibitors have implicated PI3Ks as key effectors of PDGFR-β signaling in cell-based studies (as described in Refs.44-46 among others). It is therefore tempting to speculate that PI3K signaling also plays a critical role downstream of PDGFR-β in mural cells in vivo; however, roles for the individual class I PI3Ks in mural cells have not yet been identified. The p110α and p110β isoenzymes, which are widely expressed compared to p110γ and p110δ, are likely to generate PtdIns(3,4,5)P3 in mural cells. As few examples of redundancy of p110 isoforms have been found,14 we speculate that p110α is the key isoform engaged by RTKs, with p110β mediating G protein-coupled receptor (GPCR)-mediated signaling47 in mural cells. Of note, inhibition of p110α in the tumor microenvironment of syngeneic tumors alters pericyte marker expression.48 Currently, however, it is not clear whether this is a cell-autonomous effect of p110α in mural cells or simply correlates with the reduced vessel caliber observed upon inhibition of this isoenzyme.48 Low pericyte coverage on tumor vessels facilitates metastasis49,50 and has been associated with the emergence of intratumor hypoxia, epithelial-to-mesenchymal transition, and Met receptor activation,49 all of which are believed to facilitate tumor cell dissemination. This is an important concept to take into account, as it is possible that therapies that aim to reduce pericyte coverage (either by inhibiting their recruitment and proliferation or by inducing their cell death) may have negative therapeutic consequences by promoting cancer progression and metastasis.
PI3K in VEGF producing cells: a proangiogenic link
There are numerous studies documenting a positive role for PI3K signaling in regulating VEGF protein levels (for example,48,51-55). While oncogenic signaling induced by Ras, p110α, or Akt further enhances the levels of VEGF,51,56 inhibition of PI3K signaling reduces intratumor VEGF levels.48,54,55 Mechanistically, activation of the PI3K/mTOR axis induces HIF1α expression and/or stabilization,51-53 which in turn upregulates VEGF protein levels. p110α has been reported to be the principal isoform regulating VEGF levels in cancer cells.51,54 However, inflammatory cells are also an important source of proangiogenic signals.57 With p110γ and p110δ (but not p110α) accounting for most of the class I PI3K signaling in immune cells,58 we hypothesize that these 2 isoenzymes also stimulate the production of proangiogenic factors in health and disease.
Based on these observations, it was thought for many years that the antiangiogenic activity of PI3K inhibitors was mediated by their capacity to reduce VEGF levels. However, inhibition of PI3K signaling does not always mirror the vascular response of anti-VEGF targeted therapies.36,48 Whereas anti-VEGF therapy prunes vessels,21 inhibition of PI3K in some contexts may result in more, but less functional, vessels.48 These observations highlight the fact that the antiangiogenic activity of PI3K inhibitors goes beyond inhibiting VEGF expression.
PI3K as An Antiangiogenic Target
The PI3K pathway is frequently activated in cancer as a consequence of oncogenic RTK and Ras, amplification or mutational activation of PIK3CA, and/or inactivation of the tumor suppressor PTEN. Furthermore, PI3K signaling is required for promotion of tumor stromal functions such as angiogenesis and recruitment of inflammatory cells15,26,48,59 (Fig. 3). This central role of PI3Ks in most hallmarks of cancer has promoted a substantial effort to target this signaling pathway in cancer.15,60 Several strategies aimed to interfere with the PI3K cascade are currently being tested in clinical trials, including pan-class I PI3K inhibitors, isoform-selective PI3K inhibitors, rapamycin analogs (rapalogues), active-site mTOR inhibitors, dual PI3K-mTOR inhibitors, and Akt inhibitors (Fig. 2).15 It is clear, however, that inhibition of the PI3K pathway will not only directly impact tumor cells, but will also affect the tumor stroma, including the vasculature. Indeed, targeting of PI3K, Akt, or mTOR has demonstrated antiangiogenic activity in preclinical tumor models.36,48,59,61-67
Context-dependent vascular responses
Antiangiogenic responses to PI3K inhibitors in preclinical models are not homogenous but can be grouped into 4 different categories depending on their effects on microvascular density (MVD) and vessel function36,48,53,55,59,61,63,64,67-69 (Fig. 4). The different MVD outcomes are likely related to the intrinsic characteristics of the tumor (i.e., mutational status and origin) and therefore the differences in vessel numbers observed upon inhibition of PI3K could simply be explained by the capacity of PI3K inhibitors to target VEGF expression. It is possible that, in those settings where VEGF expression is dependent on PI3K, the final angiogenic outcome upon PI3K inhibition in tumors is reduced MVD. In other cases, where VEGF expression is not dependent on PI3K, stromal-autonomous effects upon PI3K treatment would simply be observed. PI3K signaling regulates both VEGF expression in tumor cells, and Delta-like 4 (Dll4) levels in ECs.48 Whereas anti-VEGF approaches prune vessels, Dll4 blockers result in more, but smaller, vessels.7 An important conclusion to be drawn from the diversity in the MVD responses seen upon treatment with PI3K inhibitors is that the vascular density value is of limited use; instead, a change in the functionality of the vasculature is a more reliable parameter for measuring the antiangiogenic activity of PI3K inhibitors.
Figure 4.
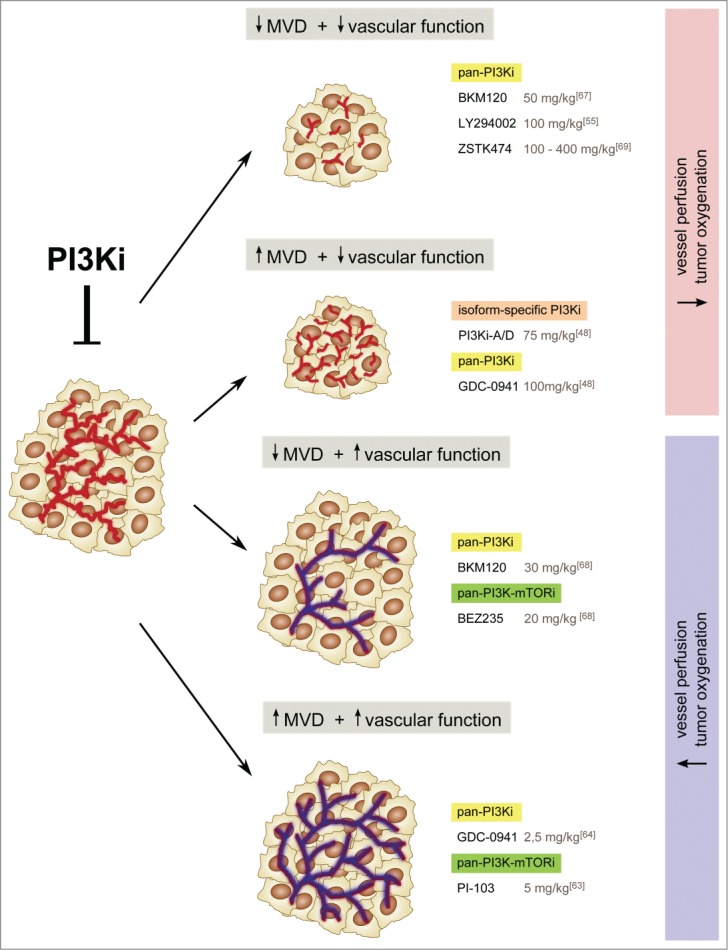
Four modes of vascular responses to PI3K inhibitors in tumors. Antiangiogenic responses to PI3K inhibitors are not homogenous; in fact, these agents can have opposite effects on vessel number and vessel function. Current data seem to indicate that vessel function, rather than number, determines the antitumor efficacy of PI3K inhibitors. Thus, reduced vessel function leads to a decrease in tumor growth, whereas enhanced blood flow and perfusion does not. The doses of the PI3K inhibitors used in preclinical models are shown for comparison.
Inhibition of PI3K in cancer also results in different vessel function responses. Frequently, PI3K inhibitors (irrespective of their impact on vessel number) result in reduced vessel function, as shown by reduced vascular flow and perfusion, which in turn enhances intratumor hypoxia.36,48,53,55,59,61,62,67,69 High hypoxic levels have been associated with increased tumor dissemination and metastasis.9,10 It is therefore possible that resistance to PI3K inhibitors60 may also be attributed to their antiangiogenic activity. An important question to consider, though, is whether there is a threshold of hypoxia that determines the switch toward a more malignant phenotype. It is possible that the long-term beneficial effects of the antiangiogenic therapy may depend on the amount of hypoxia generated. Therefore, it would be interesting to compare the strength and the durability of inhibiting PI3K versus traditional anti-VEGF agents.
In sharp contrast, recent studies have shown that PI3K inhibitors can also improve tumor vessel function. In this context, PI3K inhibitors enhance tumor vascular flow and perfusion, and reduce tumor hypoxia.63,64,68 Although at first glance this seems paradoxical, several possible explanations can be put forward: First, the tumor models tested in Refs.63,64,68 exhibit oncogenic activation of the PI3K pathway, re-addressing the issue that vascular response may depend on tumor-intrinsic mutations; Second, the PI3K inhibitors used have several off-target effects, suggesting a possible PI3K-independent vascular response; Third, low doses (below the maximum-tolerated doses and even below the EC50) of PI3K inhibitors were used by Qayum et al. and Fokas et al. (Ref 63,64,68). These authors claim that low doses of PI3K inhibitors target only some tumor functions, with no impact on stromal cells. However, 2 important issues remain unclear: First, what is the threshold at which PI3K inhibitors selectively impair only some tumor functions? Second, how are these effects related to changes in the tumor vasculature? A possible explanation is that low doses of PI3K inhibitors principally interfere with the production of cytokines by tumor cells, and therefore reduce immune cell infiltration. Immune cells, such as tumor-infiltrating macrophages and T cells, produce proangiogenic signals that may in turn impact tumor angiogenesis. Although blocking all class PI3K activity may result in immune suppression, low doses of these agents may bypass intolerable side effects by leaving residual immune functions.
Improved vascular responses associated with low doses of PI3K inhibitors may offer an alternative to vascular normalization therapy.7 However, these agents should be used in combination with cytotoxic agents to avoid the risk of enhancing perfusion of the tumor vasculature. Interestingly, treatment with a low dose of GDC-0941, a pan-class I PI3K inhibitor, resulted in improved doxorubicin delivery64 and a dramatic reduction in tumor growth. Although promising, these findings warrant further mechanistic experiments to elucidate the target of low doses of PI3K inhibitors in tumor cells.
Tumor-extrinsic effects
The importance of PI3K signalling in the tumour stroma has recently been addressed by inoculation of syngeneic tumour cell lines in mice with genetic inhibition of class I PI3K isoforms.36,48,59 An important conclusion from these studies is that inhibition of PI3K in the tumor stroma is sufficient to reduce tumor growth.36,48,59 These data suggest that inhibition of PI3K may offer therapeutic benefits even in those scenarios in which tumors do not rely on PI3K to grow. Given that clinical responses to PI3K inhibitors do not always correlate with PIK3CA mutations15,59,70, an important point to consider is whether the effectiveness of PI3K inhibitors is related to their capacity to impact stromal or malignant cells. It is tempting to speculate that the tumor stroma, and in particular the vasculature, could be a predictive factor for the therapeutic response to PI3K inhibitors. More experiments are needed to further elucidate how the tumor microenvironment determines the effectiveness of PI3K inhibitors.
Isoform-specific PI3K functions call for the use of PI3K isoform-selective inhibitors
A current clinical debate is whether the PI3K members should be targeted at the isoform-specific level. Given that tumor cells express all 4 class I PI3K isoforms, pan-class I PI3K inhibitors are expected to offer higher effectiveness. However, long-term inhibition at the doses needed to fully block all class I PI3K isoforms is believed to be not tolerable.15,60,71 It is also worth mentioning that the first generation of PI3K inhibitors showed several off-target effects, which have hampered efforts to understand the effects of these agents. Isoform-selective PI3K inhibitors may have the ability to block the relevant target with fewer side effects.60,72
Current data seem to indicate that there is no obvious difference in the antiangiogenic properties of p110α inhibitors and pan-class I PI3Ks inhibitors. This suggests that p110α-selective agents may be a better antiangiogenic option due to their reduced toxicity. However, the possible positive impact of inhibiting other members of the family in the tumor microenvironment cannot be ignored. Recent studies in preclinical models have shown that blocking p110γ activity restricts tumor growth by inhibiting recruitment of inflammatory cells.25 Additionally, inhibiting p110δ suppresses the function of regulatory T cells, allowing cytotoxic T-cell responses to tumors,28 which in turn decreases tumor growth and metastasis. One could imagine that using either a pan-PI3K inhibitor or a dual p110α/p110δ inhibitor would be the best strategy by providing both an antiangiogenic effect and a procytotoxic T cell response. Unfortunately, though, this strategy may result in immune suppression as, upon p110δ inhibition, p110α compensates for B cell development in the bone marrow and B cell survival in the spleen.73,74 An interesting alternative could be to use p110α-selective and p110δ-selective inhibitors sequentially, which would offer the advantage of inhibiting each isoform individually (antiangiogenic activity upon inhibition of p110α and procytotoxic T cell responses upon inhibition of p110δ) without inducing immune suppression. However, given the exciting vascular phenotype observed upon administration of low doses of pan-PI3K inhibitors (described above), the idea of using low doses of a dual p110α/p110δ inhibitor should not be neglected in the attempt to redefine the landscape of clinical trials with PI3K inhibitors.
Contrasting effects of PI3K versus mTOR inhibition
Inhibition of mTOR using rapalogues reduces tumor vascular density,75 but it is still unclear whether this is an endothelial intrinsic effect or simply due to reduced intratumor VEGF levels.75 Although cell-based studies have shown than rapalogues reduce EC proliferation,61,76,77 such thorough analyses have not been performed in vivo. Rapalogues, though, often show limited efficacy due to the activation of mTORC2 signaling via a p70S6K-IRS1-mediated negative feedback loop.78 Indeed, treatment with an ATP-competitive inhibitor that targets both mTORC1 and mTORC2 signaling shows a higher antiangiogenic response than rapamycin.79 BEZ235 and PI-103, dual pan-PI3K/mTOR inhibitors, also show stronger antiangiogenic activity than rapalogues, suggesting that PI3K regulates vessel growth through additional downstream effectors. This additive effect could also be explained by tumor and stromal effects, which result from PI3K inhibition with only tumor intrinsic effects upon mTOR inhibition. These observations suggest that PI3K blockers are a better choice, especially in those settings where a link between VEGF levels and PI3K/mTOR signaling is not clear, as at least stromal therapeutic benefits could be obtained.
Conclusions and Perspectives
Current limitations of antivascular therapy call for the identification of common signaling hubs that target multiple angiogenic signals in a broader spectrum of tumor types. PI3K is a key hub that is engaged by multiple, if not all, proangiogenic signals. The identification of specific and non-redundant roles of class I PI3K isoforms in the tumor microenvironment is revealing the way in which this hub dictates cancer progression. Although promising results of targeting PI3K as an antiangiogenic strategy are emerging, the pleiotropic role of PI3Ks in different cell types (including ECs) have highlighted the complexity of targeting this pathway in cancer. Furthermore, PI3K inhibitors show context-dependent responses depending on their ability to block VEGF levels, which cannot be ignored.
How can we design novel strategies to maximize the effect of PI3K inhibitors as antiangiogenics? Based on the current data, 3 options could be considered: First, sequential treatment of tumors with p110α- and p110δ-selective inhibitors might provide the benefits of inhibiting each isoform individually but avoid complete immune suppression. Second, drugs could be tested below their maximum-tolerated dose to study whether stromal responses are better modulated, especially when targeting of more than one stromal compartment is expected. In such settings, both pan-PI3K inhibitors and isoform selective inhibitors may prove to be efficient as low doses of PI3K inhibitors are not expected to target all tumor functions. This could also minimize the risk of developing tumor-intrinsic resistance mechanisms. It would also be pertinent to compare agents with even subtle differences in potency against different isoforms, as their degree of efficacy, tolerability, and beneficial stromal responses may differ. Finally, it is currently believed that selecting patients based on their cancer mutations will be critical to achieve better responses to PI3K inhibitors. We propose that this selection ought to include analysis of the nature of the tumor stroma as these non-malignant cells may be an even better predictor of response to these agents.
BOX 1 – Role of class II PI3Ks in angiogenesis
Class II PI3Ks are monomers of high molecular weight. They principally generate PtdIns(3)P and are constitutively bound to intracellular membranes. The three members of class II PI3Ks—PI3K-C2α, PI3K-C2β and PI3K-C2γ—represent the least-studied members of the PI3K family. According to in vitro data, both PI3K-C2α and PI3K-C2β regulate EC functions.80 However, work to date on vascular morphogenesis in vivo has included only PI3K-C2α. Both global and endothelial-specific deletion of the PI3K-C2α isoform lead to embryonic lethality due to severe vascular defects associated with impaired vesicle trafficking, junction assembly, and VEGF receptor internalization.81,82 Inoculation of tumor cells upon endothelial-specific deletion of PI3K-C2α reduces tumor growth by reducing vascular density,81 suggesting that inhibition of PI3K-C2α could be a promising antiangiogenic agent. More experiments are needed to further elucidate the potential of PI3K-C2α inhibitors in tumor angiogenesis.
Acknowledgments
We apologize to authors whose work could not be cited because of the limit on the number of references. We thank Maria Whitehead, UCL Cancer Institute, UCL, and Oriol Casanovas, Research Laboratory, Catalan Institute of Oncology, IDIBELL for critical review of the manuscript.
Funding
The research in the laboratory of M.G. is supported by the Spanish Government (Ramon y Cajal program and SAF2010-15661 and SAF2013-46542-P from MICINN (Spain)), 2014-SGR-725 from the Catalan Government, Ajuts Joves Investigators from IDIBELL, and from the People Program (Marie Curie Actions) of the European Union's Seventh Framework Program FP7/2007–2013/ (REA grant agreement 317250).
References
- 1. Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med 285, 1182-6 1971; PMID:4938153; http://dx.doi.org/ 10.1056/NEJM197108122850711 [DOI] [PubMed] [Google Scholar]
- 2. Folkman J. Tumor angiogenesis. Adv Cancer Res 1974; 19:331-58; PMID:4605404; http://dx.doi.org/ 10.1016/S0065-230X(08)60058-5 [DOI] [PubMed] [Google Scholar]
- 3. Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell 2011; 146:873-87; PMID:21925313; http://dx.doi.org/ 10.1016/j.cell.2011.08.039 [DOI] [PubMed] [Google Scholar]
- 4. Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol 2007; 8:464-78; PMID:17522591 [DOI] [PubMed] [Google Scholar]
- 5. Geudens I, Gerhardt H. Coordinating cell behaviour during blood vessel formation. Development 2011; 138:4569-83; PMID:21965610; http://dx.doi.org/ 10.1242/dev.062323 [DOI] [PubMed] [Google Scholar]
- 6. Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer 2008; 8:592-603; PMID:18650835; http://dx.doi.org/ 10.1038/nrc2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov 2011; 10:417-27; PMID:21629292; http://dx.doi.org/ 10.1038/nrd3455 [DOI] [PubMed] [Google Scholar]
- 8. Moserle L, Jimenez-Valerio G, Casanovas O. Antiangiogenic therapies: going beyond their limits. Cancer Discov 2014; 4:31-41; PMID:24356098; http://dx.doi.org/ 10.1158/2159-8290.CD-13-0199 [DOI] [PubMed] [Google Scholar]
- 9. Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Viñals F, Inoue M, Bergers G, Hanahan D, Casanovas O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009; 15:220-31; PMID:19249680; http://dx.doi.org/ 10.1016/j.ccr.2009.01.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ebos JM., Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 2009; 15, 232-9; PMID:19249681; http://dx.doi.org/ 10.1016/j.ccr.2009.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 2005; 307:58-62; PMID:15637262; http://dx.doi.org/ 10.1126/science.1104819 [DOI] [PubMed] [Google Scholar]
- 12. Mesange P, Poindessous V, Sabbah M, Escargueil AE, de Gramont A, Larsen AK. Intrinsic bevacizumab resistance is associated with prolonged activation of autocrine VEGF signaling and hypoxia tolerance in colorectal cancer cells and can be overcome by nintedanib, a small molecule angiokinase inhibitor. Oncotarget 2014; 5:4709-21; PMID:25015210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Reck M, Kaiser R, Mellemgaard A, Douillard JY, Orlov S, Krzakowski M, von Pawel J, Gottfried M, Bondarenko I, Liao M, et al. Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer LUME-Lung 1): a phase 3, double-blind, randomised controlled trial. Lancet Oncol 2014; 15:143-55; PMID:24411639; http://dx.doi.org/ 10.1016/S1470-2045(13)70586-2 [DOI] [PubMed] [Google Scholar]
- 14. Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol 2010; 11:329-41; PMID:20379207; http://dx.doi.org/ 10.1038/nrm2882 [DOI] [PubMed] [Google Scholar]
- 15. Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov 2014; 13:140-56; PMID:24481312; http://dx.doi.org/ 10.1038/nrd4204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vanhaesebroeck B, Leevers SJ, Panayotou G, Waterfield MD. Phosphoinositide 3-kinases: a conserved family of signal transducers. Trends Biochem Sci 1997; 22:267-72; PMID:9255069; http://dx.doi.org/ 10.1016/S0968-0004(97)01061-X [DOI] [PubMed] [Google Scholar]
- 17. Hawkins PT, Anderson KE, Davidson K, Stephens LR. Signalling through Class I PI3Ks in mammalian cells. Biochem Soc Trans 2006; 34:647-62; PMID:17052169 [DOI] [PubMed] [Google Scholar]
- 18. Cantley LC. The phosphoinositide 3-kinase pathway. Science 2002; 296:1655-7; PMID:12040186; http://dx.doi.org/ 10.1126/science.296.5573.1655 [DOI] [PubMed] [Google Scholar]
- 19. Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol 2012; 13:195-203; PMID:22358332; http://dx.doi.org/ 10.1038/nrm3290 [DOI] [PubMed] [Google Scholar]
- 20. Fritsch R, Downward J. SnapShot: class I PI3K isoform signaling. Cell 2013; 154:940-940 e1; PMID:23953121; http://dx.doi.org/ 10.1016/j.cell.2013.07.045 [DOI] [PubMed] [Google Scholar]
- 21. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011; 473:298-307; PMID:21593862; http://dx.doi.org/ 10.1038/nature10144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Carmeliet P. Angiogenesis in health and disease. Nat Med 2003; 9:653-60; PMID:12778163; http://dx.doi.org/ 10.1038/nm0603-653 [DOI] [PubMed] [Google Scholar]
- 23. Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 1992; 359:843-5; PMID:1279431; http://dx.doi.org/ 10.1038/359843a0 [DOI] [PubMed] [Google Scholar]
- 24. Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell 2011; 21:193-215; PMID:21839917; http://dx.doi.org/ 10.1016/j.devcel.2011.07.001 [DOI] [PubMed] [Google Scholar]
- 25. Schmid MC, Avraamides CJ, Dippold HC, Franco I, Foubert P, Ellies LG, Acevedo LM, Manglicmot JR, Song X, Wrasidlo W, et al. Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kgamma, a single convergent point promoting tumor inflammation and progression. Cancer Cell 2011; 19:715-27; PMID:21665146; http://dx.doi.org/ 10.1016/j.ccr.2011.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hirsch E, Ciraolo E, Franco I, Ghigo A, Martini M. PI3K in cancer-stroma interactions: bad in seed and ugly in soil. Oncogene. 2014 Jun 12;33(24):3083-90. http://dx.doi.org/10.1038/onc.2013.265 [DOI] [PubMed] [Google Scholar]
- 27. Papakonstanti EA, Zwaenepoel O, Bilancio A, Burns E, Nock GE, Houseman B, Shokat K, Ridley AJ, Vanhaesebroeck B. Distinct roles of class IA PI3K isoforms in primary and immortalised macrophages. J Cell Sci 2008; 121:4124-33; PMID:19033389 [DOI] [PubMed] [Google Scholar]
- 28. Ali K., Soond DR, Piñeiro R, Hagemann T, Pearce W, Lim EL, Bouabe H, Scudamore CL, Hancox T, Maecker H, et al. Inactivation of PI(3)K p110delta breaks regulatory T-cell-mediated immune tolerance to cancer. Nature 2014; 510:407-11; PMID:24919154; http://dx.doi.org/ 10.1038/nature13444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Martin V, et al. Deletion of the p110beta isoform of phosphoinositide 3-kinase in platelets reveals its central role in Akt activation and thrombus formation in vitro and in vivo. Blood 2010; 115:2008-13; PMID:20065293; http://dx.doi.org/ 10.1182/blood-2009-04-217224 [DOI] [PubMed] [Google Scholar]
- 30. Graupera M, Potente M. Regulation of angiogenesis by PI3K signaling networks. Exp Cell Res 2013; 319:1348-55; PMID:23500680; http://dx.doi.org/ 10.1016/j.yexcr.2013.02.021 [DOI] [PubMed] [Google Scholar]
- 31. Graupera M, Guillermet-Guibert J, Foukas LC, Phng LK, Cain RJ, Salpekar A, Pearce W, Meek S, Millan J, Cutillas PR, et al. Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature 2008; 453:662-6; PMID:18449193; http://dx.doi.org/ 10.1038/nature06892 [DOI] [PubMed] [Google Scholar]
- 32. Ackah E, Yu J, Zoellner S, Iwakiri Y, Skurk C, Shibata R, Ouchi N, Easton RM, Galasso G, Birnbaum MJ, et al. Akt1/protein kinase Balpha is critical for ischemic and VEGF-mediated angiogenesis. J Clin Invest 2005; 115:2119-27; PMID:16075056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lee MY, Luciano AK, Ackah E, Rodriguez-Vita J, Bancroft TA, Eichmann A, Simons M, Kyriakides TR, Morales-Ruiz M, Sessa WC, et al. Endothelial Akt1 mediates angiogenesis by phosphorylating multiple angiogenic substrates. Proc Natl Acad Sci U S A 2014, 111:12865-70; PMID:25136137; http://dx.doi.org 10.1073/pnas.1408472111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gambardella L, Hemberger M, Hughes B, Zudaire E, Andrews S, Vermeren S. PI3K signaling through the dual GTPase-activating protein ARAP3 is essential for developmental angiogenesis. Sci Signal 2010; 3:ra76; PMID:20978237; http://dx.doi.org 10.1126/scisignal.2001026 [DOI] [PubMed] [Google Scholar]
- 35. Hamada K, Sasaki T, Koni PA, Natsui M, Kishimoto H, Sasaki J, Yajima N, Horie Y, Hasegawa G, Naito M, et al. The PTEN/PI3K pathway governs normal vascular development and tumor angiogenesis. Genes Dev 2005; 19:2054-65; PMID:16107612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yuan TL, Choi HS, Matsui A, Benes C, Lifshits E, Luo J, Frangioni JV, Cantley LC. Class 1A PI3K regulates vessel integrity during development and tumorigenesis. Proc Natl Acad Sci U S A 2008; 105:9739-44; PMID:18621722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nicoli S, Knyphausen CP, Zhu LJ, Lakshmanan A, Lawson ND. miR-221 is required for endothelial tip cell behaviors during vascular development. Dev Cell 2012; 22:418-29; PMID:22340502; http://dx.doi.org/ 10.1016/j.devcel.2012.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fish JE, Santoro MM, Morton SU, Yu S, Yeh RF, Wythe JD, Ivey KN, Bruneau BG, Stainier DY, Srivastava D. miR-126 regulates angiogenic signaling and vascular integrity. Dev Cell 2008; 15:272-84; PMID:18694566; http://dx.doi.org/ 10.1016/j.devcel.2008.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Luo J, Field SJ, Lee JY, Engelman JA, Cantley LC. The p85 regulatory subunit of phosphoinositide 3-kinase down-regulates IRS-1 signaling via the formation of a sequestration complex. J Cell Biol 2005; 170, 455-64; PMID:16043515; http://dx.doi.org/ 10.1083/jcb.200503088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ueki K, Fruman DA, Brachmann SM, Tseng YH, Cantley LC, Kahn CR. Molecular balance between the regulatory and catalytic subunits of phosphoinositide 3-kinase regulates cell signaling and survival. Mol Cell Biol 2002; 22:965-77; PMID:11784871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jain RK, Carmeliet P. SnapShot: tumor angiogenesis. Cell 2012; 149:1408-1408 e1; PMID:22682256; http://dx.doi.org/ 10.1016/j.cell.2012.05.025 [DOI] [PubMed] [Google Scholar]
- 42. Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med 2011; 17:1359-70; PMID:22064426; http://dx.doi.org/ 10.1038/nm.2537 [DOI] [PubMed] [Google Scholar]
- 43. Madeddu P, Kraenkel N, Barcelos LS, Siragusa M, Campagnolo P, Oikawa A, Caporali A, Herman A, Azzolino O, Barberis L, et al. Phosphoinositide 3-kinase gamma gene knockout impairs postischemic neovascularization and endothelial progenitor cell functions. Arterioscler Thromb Vasc Biol 2008; 28:68-76; PMID:17962628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nishishita T, Lin P. Angiopoietin 1, PDGF-B, and TGF-beta gene regulation in endothelial cell and smooth muscle cell interaction. J Cell Biochem 2004; 91:584-93; PMID:14755687; http://dx.doi.org/ 10.1002/jcb.10718 [DOI] [PubMed] [Google Scholar]
- 45. Pukac L, Huangpu J, Karnovsky MJ. Platelet-derived growth factor-BB, insulin-like growth factor-I, and phorbol ester activate different signaling pathways for stimulation of vascular smooth muscle cell migration. Exp Cell Res 1998; 242:548-60; PMID:9683541; http://dx.doi.org/ 10.1006/excr.1998.4138 [DOI] [PubMed] [Google Scholar]
- 46. Cospedal R, Abedi H, Zachary I. Platelet-derived growth factor-BB PDGF-BB) regulation of migration and focal adhesion kinase phosphorylation in rabbit aortic vascular smooth muscle cells: roles of phosphatidylinositol 3-kinase and mitogen-activated protein kinases. Cardiovasc Res 1999; 41:708-21; PMID:10435043; http://dx.doi.org/ 10.1016/S0008-6363(98)00232-6 [DOI] [PubMed] [Google Scholar]
- 47. Guillermet-Guibert J, Bjorklof K, Salpekar A, Gonella C, Ramadani F, Bilancio A, Meek S, Smith AJ, Okkenhaug K, Vanhaesebroeck B. The p110beta isoform of phosphoinositide 3-kinase signals downstream of G protein-coupled receptors and is functionally redundant with p110gamma. Proc Natl Acad Sci U S A 2008; 105:8292-7; PMID:18544649; http://dx.doi.org/ 10.1073/pnas.0707761105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Soler A, Serra H, Pearce W, Angulo A, Guillermet-Guibert J, Friedman LS, Viñals F, Gerhardt H, Casanovas O, Graupera M, et al. Inhibition of the p110alpha isoform of PI 3-kinase stimulates nonfunctional tumor angiogenesis. J Exp Med 2013; 210:1937-45; PMID:24043760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cooke V G, LeBleu VS, Keskin D, Khan Z, O'Connell JT, Teng Y, Duncan MB, Xie L, Maeda G, Vong S, et al. Pericyte depletion results in hypoxia-associated epithelial-to-mesenchymal transition and metastasis mediated by met signaling pathway. Cancer Cell 21:66-81 2012; PMID:22264789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xian X, Håkansson J, Ståhlberg A, Lindblom P, Betsholtz C, Gerhardt H, Semb H. Pericytes limit tumor cell metastasis. J Clin Invest 2006; 116:642-51; PMID:16470244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jiang BH, Zheng JZ, Aoki M, Vogt PK. Phosphatidylinositol 3-kinase signaling mediates angiogenesis and expression of vascular endothelial growth factor in endothelial cells. Proc Natl Acad Sci U S A 2000; 97:1749-53; PMID:10677529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Skinner HD, Zheng JZ, Fang J, Agani F, Jiang BH. Vascular endothelial growth factor transcriptional activation is mediated by hypoxia-inducible factor 1alpha, HDM2, and p70S6K1 in response to phosphatidylinositol 3-kinase/AKT signaling. J Biol Chem 2004; 279:45643-51; PMID:15337760 [DOI] [PubMed] [Google Scholar]
- 53. Fang J, Ding M, Yang L, Liu L, Jiang BH. PI3K/PTEN/AKT signaling regulates prostate tumor angiogenesis. Cell Signal 2007; 19, 2487-97; PMID:17826033; http://dx.doi.org/10.1016/j.cellsig.2007.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xia C, Meng Q, Cao Z, Shi X, Jiang BH. Regulation of angiogenesis and tumor growth by p110 alpha and AKT1 via VEGF expression. J Cell Physiol 2006; 209:56-66; PMID:16775835; http://dx.doi.org/ 10.1002/jcp.20707 [DOI] [PubMed] [Google Scholar]
- 55. Hu L, Hofmann J, Jaffe RB. Phosphatidylinositol 3-kinase mediates angiogenesis and vascular permeability associated with ovarian carcinoma. Clin Cancer Res 2005; 11:8208-12; PMID:16299254; http://dx.doi.org/ 10.1158/1078-0432.CCR-05-0206 [DOI] [PubMed] [Google Scholar]
- 56. Mazure NM, Chen EY, Laderoute KR, Giaccia AJ. Induction of vascular endothelial growth factor by hypoxia is modulated by a phosphatidylinositol 3-kinase/Akt signaling pathway in Ha-ras-transformed cells through a hypoxia inducible factor-1 transcriptional element. Blood 1997; 90:3322-31; PMID:9345014 [PubMed] [Google Scholar]
- 57. Saharinen P, Eklund L, Pulkki K, Bono P, Alitalo K. VEGF and angiopoietin signaling in tumor angiogenesis and metastasis. Trends Mol Med 2011; 17:347-62; PMID:21481637; http://dx.doi.org/ 10.1016/j.molmed.2011.01.015 [DOI] [PubMed] [Google Scholar]
- 58. Okkenhaug K. Signaling by the phosphoinositide 3-kinase family in immune cells. Annu Rev Immunol 2013; 31:675-704; PMID:23330955; http://dx.doi.org/ 10.1146/annurev-immunol-032712-095946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Murillo MM, Zelenay S, Nye E, Castellano E, Lassailly F, Stamp G, Downward J. RAS interaction with PI3K p110alpha is required for tumor-induced angiogenesis. J Clin Invest 2014; 124:3601-11; PMID:25003191; http://dx.doi.org/ 10.1172/JCI74134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol 2013; 10:143-53; PMID:23400000; http://dx.doi.org/ 10.1038/nrclinonc.2013.10 [DOI] [PubMed] [Google Scholar]
- 61. Schnell CR, Stauffer F, Allegrini PR, O'Reilly T, McSheehy PM, Dartois C, Stumm M, Cozens R, Littlewood-Evans A, García-Echeverría C, et al. Effects of the dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 on the tumor vasculature: implications for clinical imaging. Cancer Res 2008; 68:6598-607; PMID:18701483; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-1044 [DOI] [PubMed] [Google Scholar]
- 62. Maira SM, Pecchi S, Huang A, Burger M, Knapp M, Sterker D, Schnell C, Guthy D, Nagel T, Wiesmann M, et al. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol Cancer Ther 2012; 11:317-28; PMID:22188813; http://dx.doi.org/ 10.1158/1535-7163.MCT-11-0474 [DOI] [PubMed] [Google Scholar]
- 63. Qayum N, Muschel RJ, Im JH, Balathasan L, Koch CJ, Patel S, McKenna WG, Bernhard EJ. Tumor vascular changes mediated by inhibition of oncogenic signaling. Cancer Res 2009; 69:6347-54; PMID:19622766; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-0657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Qayum N, Im J, Stratford MR, Bernhard EJ, McKenna WG, Muschel RJ. Modulation of the tumor microvasculature by phosphoinositide-3 kinase inhibition increases doxorubicin delivery in vivo. Clin Cancer Res 2012; 18:161-9; PMID:22065081; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-1413 [DOI] [PubMed] [Google Scholar]
- 65. Fokas E, Yoshimura M, Prevo R, Higgins G, Hackl W, Maira SM, Bernhard EJ, McKenna WG, Muschel RJ. NVP-BEZ235 and NVP-BGT226, dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitors, enhance tumor and endothelial cell radiosensitivity. Radiat Oncol 2012; 7:48; PMID:22452803; http://dx.doi.org/ 10.1186/1748-717X-7-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Xu S, Li S, Guo Z, Luo J, Ellis MJ, Ma CX. Combined targeting of mTOR and AKT is an effective strategy for basal-like breast cancer in patient-derived xenograft models. Mol Cancer Ther 2013; 12:1665-75; PMID:23689832; http://dx.doi.org/ 10.1158/1535-7163.MCT-13-0159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Juvekar A, Burga LN, Hu H, Lunsford EP, Ibrahim YH, Balmañà J, Rajendran A, Papa A, Spencer K, Lyssiotis CA, et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov 2012; 2:1048-63; PMID:22915751; http://dx.doi.org/ 10.1158/2159-8290.CD-11-0336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Fokas E, Im JH, Hill S, Yameen S, Stratford M, Beech J, Hackl W, Maira SM, Bernhard EJ, McKenna WG, et al. Dual inhibition of the PI3K/mTOR pathway increases tumor radiosensitivity by normalizing tumor vasculature. Cancer Res 2012; 72:239-48; PMID:22108822 [DOI] [PubMed] [Google Scholar]
- 69. Kong D, Okamura M, Yoshimi H, Yamori T. Antiangiogenic effect of ZSTK474, a novel phosphatidylinositol 3-kinase inhibitor. Eur J Cancer 2009; 45, 857-65; PMID:19144509; http://dx.doi.org/ 10.1016/j.ejca.2008.12.007 [DOI] [PubMed] [Google Scholar]
- 70. Mayer IA, Abramson VG, Isakoff SJ, Forero A, Balko JM, Kuba MG, Sanders ME, Yap JT, Van den Abbeele AD, Li Y, et al. Stand up to cancer phase Ib study of pan-phosphoinositide-3-kinase inhibitor buparlisib with letrozole in estrogen receptor-positive/human epidermal growth factor receptor 2-negative metastatic breast cancer. J Clin Oncol 2014; 32:1202-9; PMID:24663045; http://dx.doi.org/ 10.1200/JCO.2013.54.0518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bendell JC, Rodon J, Burris HA, de Jonge M, Verweij J, Birle D, Demanse D, De Buck SS, Ru QC, Peters M, et al. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol 2012; 30:282-90; PMID:22162589; http://dx.doi.org/ 10.1200/JCO.2011.36.1360 [DOI] [PubMed] [Google Scholar]
- 72. Jia S, Roberts TM, Zhao JJ. Should individual PI3 kinase isoforms be targeted in cancer? Curr Opin Cell Biol 2009; 21:199-208; PMID:19200708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ramadani F, Bolland DJ, Garcon F, Emery JL, Vanhaesebroeck B, Corcoran AE, Okkenhaug K. The PI3K isoforms p110alpha and p110delta are essential for pre-B cell receptor signaling and B cell development. Sci Signal 2010; 3(134):ra60; PMID:20699475; http://dx.doi.org/ 10.1126/scisignal.2001104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. So L, Yea SS, Oak JS, Lu M, Manmadhan A, Ke QH, Janes MR, Kessler LV, Kucharski JM, Li LS, et al. Selective inhibition of phosphoinositide 3-kinase p110alpha preserves lymphocyte function. J Biol Chem 2013; 288:5718-31; PMID:23275335; http://dx.doi.org/ 10.1074/jbc.M112.379446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Guba M, von Breitenbuch P, Steinbauer M, Koehl G, Flegel S, Hornung M, Bruns CJ, Zuelke C, Farkas S, Anthuber M, et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nat Med 2002; 8:128-35; PMID:11821896; http://dx.doi.org/ 10.1038/nm0202-128 [DOI] [PubMed] [Google Scholar]
- 76. Manegold PC, Paringer C, Kulka U, Krimmel K, Eichhorn ME, Wilkowski R, Jauch KW, Guba M, Bruns CJ.l. Antiangiogenic therapy with mammalian target of rapamycin inhibitor RAD001 (Everolimus) increases radiosensitivity in solid cancer. Clin Cancer Res 2008; 14:892-900; PMID:18245553; http://dx.doi.org/ 10.1158/1078-0432.CCR-07-0955 [DOI] [PubMed] [Google Scholar]
- 77. Vinals F, Chambard JC, Pouyssegur J. p70 S6 kinase-mediated protein synthesis is a critical step for vascular endothelial cell proliferation. J Biol Chem 1999; 274:26776-82; PMID:10480882; http://dx.doi.org/ 10.1074/jbc.274.38.26776 [DOI] [PubMed] [Google Scholar]
- 78. Julien LA, Carriere A, Moreau J, Roux PP. mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol Cell Biol 2010; 30, 908-21; PMID:19995915; http://dx.doi.org/ 10.1128/MCB.00601-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Falcon BL, Barr S, Gokhale PC, Chou J, Fogarty J, Depeille P, Miglarese M, Epstein DM, McDonald DM. Reduced VEGF production, angiogenesis, and vascular regrowth contribute to the antitumor properties of dual mTORC1/mTORC2 inhibitors. Cancer Res 2011; 71:1573-83; PMID:21363918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tibolla G, Piñeiro R, Chiozzotto D, Mavrommati I, Wheeler AP, Norata GD, Catapano AL, Maffucci T, Falasca M. Class II phosphoinositide 3-kinases contribute to endothelial cells morphogenesis. PLoS One 2013; 8, e53808; PMID:23320105; http://dx.doi.org/ 10.1371/journal.pone.0053808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Yoshioka K, Yoshida K, Cui H, Wakayama T, Takuwa N, Okamoto Y, Du W, Qi X, Asanuma K, Sugihara K, et al. Endothelial PI3K-C2alpha, a class II PI3K, has an essential role in angiogenesis and vascular barrier function. Nat Med 18, 1560-9 2012; PMID:22983395; http://dx.doi.org/ 10.1038/nm.2928 [DOI] [PubMed] [Google Scholar]
- 82. Biswas K, Yoshioka K, Asanuma K, Okamoto Y, Takuwa N, Sasaki T, Takuwa Y. Essential role of class II phosphatidylinositol-3-kinase-C2alpha in sphingosine 1-phosphate receptor-1-mediated signaling and migration in endothelial cells. J Biol Chem 2013; 288:2325-39; PMID:23192342 [DOI] [PMC free article] [PubMed] [Google Scholar]