

Abstract
Numerous studies have shown that normal cells often respond to the activation of oncogenes by undergoing reactive oxygen species-dependent induction of senescence. Here, we discuss our recent publication identifying protein tyrosine phosphatase PTP1B as an important redox-controlled checkpoint for senescence downstream of oncogenic RAS.
Keywords: argonaute, cancer, gene silencing, microRNAs, oncogene, phosphatase, PTP1B, p21, reactive oxygen species, senescence
Abbreviations
- AGO2
argonaute 2
- CDK
cyclin-dependent kinase
- miRNA
micro RNA
- p16
cyclin-dependent kinase inhibitor 2A
- p21
cyclin-dependent kinase inhibitor 1
- p53
tumor protein 53
- PTP
protein tyrosine phosphatase
- ROS
reactive oxygen species
- RB
retinoblastoma protein
It is now widely believed that cellular senescence, first described in the 1960s by Hayflick and Moorhead as an in vitro replicative phenomenon, plays a wide range of roles in vivo. Cellular senescence is a stable form of cell cycle arrest that is implicated in pathological responses controlling cancer progression, wound healing, and aging, and in physiological responses occurring in embryonic development.1 In normal cells, the activation of RAS (e.g., H-RASV12)2 and several other oncogenes triggers cellular senescence, a crucial barrier against cancer progression. p53 (TP53), p21 (p21WAF1/CIP1, cyclin-dependent kinase inhibitor 1), p16 (p16Ink4A, cyclin-dependent kinase inhibitor 2A), RB (retinoblastoma protein), increased formation of heterochromatic foci, and increased β-galactosidase activity (indicative of increased autophagocytotic activity) have been studied extensively in this context, and are used as markers of oncogene-induced and other types of senescence. However, despite the fact that a significant body of evidence supports an important role for reactive oxygen species (ROS) in senescence, surprisingly little is known about the targets of ROS and how redox signaling is integrated in the cell cycle arrest-mediated oncogenic response.
The concept that ROS may be involved in cancer dates from the 1950s; however, the important role of ROS in oncogene-induced senescence was first revealed in the 1990s when studies from the Ames, Aaronson, and Finkel groups demonstrated that treatment with antioxidants delays or prevents senescence.1 Using oncogenic RAS-transformed human diploid fibroblasts, the Finkel group clearly established that scavenging ROS (e.g., hydrogen peroxide) with N-acetylcysteine or placing the transformed cells in a low-oxygen environment hindered development of the senescent phenotype.3 The exact mechanism of ROS-induced senescence is unknown. It is, however, known that ROS can activate the DNA damage response, the p38 mitogen-activated protein kinase pathway, and Seladin,4 and that all of these responses contribute to p53 stabilization and activation.5
At the center of redox signaling are oxidoreductive chemical reactions involving low and moderate levels of ROS and reductants that are able to modify proteins reversibly. These structural alterations allow acute or sustained ROS production to be coupled to alterations in cell function, in contrast to oxidative stress which irreversibly damages DNA, lipids, and proteins. Attention has been drawn to protein tyrosine phosphatases (PTPs) as ROS targets because of the signature motif of this family. The architecture of the PTP catalytic center creates an environment that lowers the pKa of the catalytic cysteine residue. The side chain of the conserved cysteine residue is thus in a negatively charged thiolate form, making it a good nucleophile for phosphorylated substrates and also particularly sensitive to cellular oxidants.6 Oxidation of the catalytic cysteine residue of specific members of the PTP family abrogates their activity and transiently facilitates phosphorylation-dependent signaling.
While testing whether certain PTPs were inactivated by ROS in the original model of oncogene-induced senescence established by the Lowe group,2 we found that reversible oxidation of the protein tyrosine phosphatase PTP1B (encoded by the PTPN1 gene) was necessary for RAS-induced senescence.7 We identified PTP1B using a cysteinyl-labeling assay in which reversible oxidation of the low-pKa cysteine residue of PTPs was converted to a modification by biotin.8 To our surprise, even though other PTPs were shown to be inactivated in the cysteinyl-labeling assay, the prevention of senescence observed by the Finkel group3 (i.e., by treating H-RASV12-transformed fibroblasts with N-acetylcysteine), could be offset by specifically inhibiting PTP1B using pharmacological inhibitors. We then identified argonaute 2 (AGO2), a member of the RNA-induced silencing complex, as a major substrate of PTP1B in RAS-mediated senescence and showed that tyrosine 393 of AGO2 was specifically dephosphorylated by PTP1B. We further examined the effect of PTP1B-regulated phosphorylation of AGO2 tyrosine 393 on the association of AGO2 with DICER (an endoribonuclease encoded by the DICER1 gene). These experiements confirmed that phosphorylation of AGO2 tyrosine 393 compromised its association with DICER, as previously observed by others in hypoxic conditions,9 and showed that overexpressing PTP1B (thus compensating for the inactivated pool of PTP1B in H-RASV12-expressing fibroblasts) could prevent this dissociation. The report from Shen et al.9 shows that AGO2 tyrosine 393 phosphorylation alters the maturation of microRNAs (miRNAs) in hypoxic conditions; however, in our model AGO2 tyrosine 393 phosphorylation prevented miRNA loading altogether. In human diploid fibroblasts expressing H-RASV12, phospho-rylation of AGO2 tyrosine 393 prevents it from interacting with miR-106b, miR-20a, and miR-20b, among other abundant miRNAs, and from silencing p21 mRNA (Fig. 1). This strongly supports the notion that inhibition of overall miRNA silencing as a consequence of reversible oxidation of PTP1B can prevent the action of oncogenic miRNAs and preserve a senescence program. Several other mRNAs were not regulated in our model; however, given that p21 expression inhibits cell cycle progression by inhibiting CDK (cyclin-dependent kinase)-cyclin complexes as well as by contributing to ROS accumulation,10 its contribution to establishing the senescent phenotype is likely to be critical. Although previous studies have demonstrated that PTP1B is a critical regulator of metabolism and can act as a tumor suppressor or as an oncogene, our data demonstrate that PTP1B is also a regulator of senescence. Therefore, since the biological purpose of senescence appears to be to eliminate damaged cells, developing an inhibitor for PTP1B could provide an interesting avenue for senescence-inducing therapy to block oncogenic RAS-mediated tumor progression.
Figure 1.
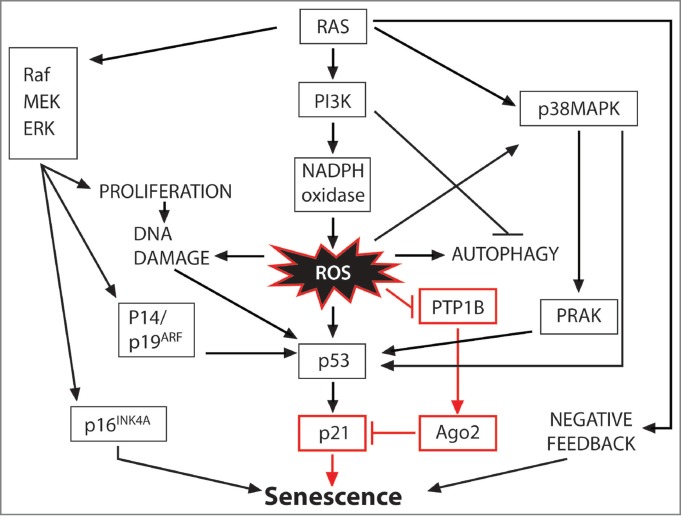
Molecular mechanisms of oncogenic RAS-induced senescence. The numerous pathways of oncogenic RAS-induced senescence converge on common effectors, such as p53 and p16, to execute the senescence response. On one hand, oncogenic RAS activates the ERK MAPK pathway and promotes the transcriptional upregulation of p16 and senescence. Several pathways downstream of oncogenic RAS have been shown to activate p53. In addition to increased p16 transcription, the ERK MAPK pathway can lead to transcriptional upregulation of p14/19ARF and activation of the DNA damage response, both shown to activate p53. Similarly, expression of oncogenic RAS activates the p38 MAPK pathway and PRAK, which has been shown to phosphorylate and activate p53. Importantly, RAS also leads to activation of a NADPH oxidase and the generation of ROS in a PI3K-dependent mechanism. ROS contribute to the stabilization and activation of the tumor suppressor p53, which in turn leads to the transcription of p21, a mediator of p53-induced cell-cycle arrest. Reversible oxidation and inactivation of PTP1B prevents it from keeping AGO2 in an active, microRNA-loaded state. Hence, ROS-mediated inhibition of PTP1B and AGO2 allows the transcription of p21 and the onset of senescence. (Modified from DeNicola and Tuveson5). AGO2, argonaute 2; ERK, extracellular-signal regulated kinase; MAPK, mitogen-activated protein kinase; PI3K, phosphoinositide 3-kinase; PRAK, p38 regulated/activated kinase; PTP1B, protein tyrosine phosphatase 1B; ROS, reactive oxygen species.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental Material may be downloaded here: publisher's website
References
- 1. Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 2014; 15:482-96; PMID:24954210; http://dx.doi.org/ 10.1038/nrm3823 [DOI] [PubMed] [Google Scholar]
- 2. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997; 88:593-602; PMID:9054499; http://dx.doi.org/ 10.1016/S0092-8674(00)81902-9 [DOI] [PubMed] [Google Scholar]
- 3. Lee AC, Fenster BE, Ito H, Takeda K, Bae NS, Hirai T, Yu ZX, Ferrans VJ, Howard BH, Finkel T, Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J Biol Chem 1999; 274:7936-40; PMID:10075689; http://dx.doi.org/ 10.1074/jbc.274.12.7936 [DOI] [PubMed] [Google Scholar]
- 4. Wu C, Miloslavskaya I, Demontis S, Maestro R, Galaktionov K. Regulation of cellular response to oncogenic and oxidative stress by Seladin-1. Nature 2004; 432:640-5; PMID:15577914; http://dx.doi.org/ 10.1038/nature03173 [DOI] [PubMed] [Google Scholar]
- 5. DeNicola GM, Tuveson DA. RAS in cellular transformation and senescence. Eur J Cancer 2009; 45:211-6; PMID:19775620; http://dx.doi.org/ 10.1016/S0959-8049(09)70036-X [DOI] [PubMed] [Google Scholar]
- 6. Tonks NK. Protein tyrosine phosphatases–from housekeeping enzymes to master regulators of signal transduction. FEBS J 2013; 280:346-78; PMID:23176256; http://dx.doi.org/ 10.1111/febs.12077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang M, Haase A, Huang FK, Coulis G, Rivera KD, Dickinson BC, Chang CJ, Pappin D, Neubert TA, Hannon GJ, et al. Dephosphorylation of tyrosine 393 in argonaute 2 by protein tyrosine phosphatase 1B regulates gene silencing in oncogenic RAS-induced senescence. Mol Cell 2014; 55:782-90; PMID:25175024; http://dx.doi.org/ 10.1016/j.molcel.2014.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boivin B, Zhang S, Arbiser JL, Zhang ZY, Tonks NK. A modified cysteinyl-labeling assay reveals reversible oxidation of protein tyrosine phosphatases in angiomyolipoma cells. Proc Natl Acad Sci USA 2008; 105:9959-64; PMID:18632564; http://dx.doi.org/ 10.1073/pnas.0804336105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shen J, Xia W, Khotskaya YB, Huo L, Nakanishi K, Lim SO, Du Y, Wang Y, Chang WC, Chen CH, et al. EGFR modulates microRNA maturation in response to hypoxia through phosphorylation of AGO2. Nature 2013; 497:383-7; PMID:23636329; http://dx.doi.org/ 10.1038/nature12080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Macip S, Igarashi M, Fang L, Chen A, Pan ZQ, Lee SW, Aaronson SA. Inhibition of p21- mediated ROS accumulation can rescue p21-induced senescence. EMBO J 2002; 21:2180-8; PMID:11980715; http://dx.doi.org/ 10.1093/emboj/21.9.2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.