

Abstract
Although there has been great progress in the treatment of human cancers, especially leukemias, many remain resistant to treatment. A major current focus is the development of so-called epigenetic drugs. Epigenetic states are stable enough to persist through multiple cell divisions, but by their very nature are reversible and thus are amenable to therapeutic manipulation. Exciting work in this area has produced a new breed of highly specific small molecules designed to inhibit epigenetic proteins, some of which have entered clinical trials. The current and future development of epigenetic drugs is greatly aided by highly detailed information about normal and aberrant epigenetic changes at the molecular level. In this review we focus on a class of aggressive acute leukemias caused by mutations in the Mixed Lineage Leukemia (MLL) gene. We provide an overview of how detailed molecular analysis of MLL leukemias has provided several early-stage epigenetic drugs and propose that further study of MLL leukemogenesis may continue to provide molecular details that potentially have a wider range of applications in human cancers.
Keywords: acetylation, chromatin modifications, epigenetic, fusion proteins, histone, leukemia, methylation, MLL, small molecule inhibitors, therapy, transcription elongation
Abbreviations
- Ac
acetylation
- ALL
acute lymphoid leukemia
- AML
acute myeloid leukemia
- BRD4
bromodomain containing protein 4
- CBP
Creb binding protein
- DOT1L
Disruptor of telomeric silencing 1-Like
- HSC
haematopoietic stem cell
- HOX
homeobox
- KAT
lysine acetyltransferase
- KMT
lysine methyltransferase
- Me
methylation
- MLL
mixed lineage leukemia
- MOF
Males absent On the First
- P
phosphorylation
- Ub
ubiquitination
- PAF1C
polymerase associated factor 1 complex
- PHD
plant homeodomain
The Importance of Epigenetics in Human Disease
Genome-wide sequencing data have revealed mutations in a large number of proteins that control epigenetic states, suggesting that epigenetic changes are a key driving force in human disease.1 Epigenetics is generally defined as heritable changes in gene expression that do not alter the underlying DNA sequence. Epigenetic states are stable enough to persist through the cell cycle, but are also reversible and can respond to changes in the cellular environment. On the molecular level, epigenetic information is controlled by DNA cytosine modifications (e.g., methylation, hydroxylation, formylation, and carboxylation), the expression of noncoding RNAs, and by the covalent modification of histone proteins and their variants.1,2
Histone proteins are the core constituents of the protein/DNA complex termed chromatin. The basic subunit of chromatin is the nucleosome consisting of DNA wrapped around a core of 4 canonical histones (H2A, H2B, H3, and H4).2 A fifth histone, H1, binds to the linker DNA between nucleosomes and is thought to contribute to higher order chromatin structure.2 Further adding to the complexity of this basic structure is the existence of multiple histone variants, many of which control different aspects of gene regulation and are occasionally mutated in some human diseases.1,2
Histone proteins can be covalently modified at specific amino acid residues with “marks” such as phosphorylation (P), acetylation (Ac), methylation (Me, which can be added as mono [1], di [2], or tri [3] methylation), ubiquitination (Ub), and many others.3 Many of these histone marks function as docking sites for specific effector proteins and can be used to demarcate different functional regions of the genome.3 For example, H3 lysine 4 monomethylation (H3K4Me1) is generally considered to be a mark of enhancers whereas H3K4Me3 is found at promoters that are either “poised” or active, and H3K79Me2/3 and H3K36Me3 are both found in actively elongating genes.3 Many proteins that control gene expression and are implicated in human disease are involved in “writing” (i.e., adding modifications), “erasing” (i.e., removing modifications), or “reading” (i.e., binding to) histone modifications.4
The wide range of possible epigenetic states makes analysis of epigenetic changes in human patients challenging, as there may be a great deal of variation from patient to patient or even between different cancer cells within the same patient.5 In this review, we argue that leukemias caused by mutations in the Mixed Lineage Leukemia (MLL) gene provide a good model system for analyzing epigenetic mechanisms in cancer. Work on MLL has not only provided novel epigenetic drugs for MLL leukemias, but we believe that it also has the capability of providing insight into epigenetic disease mechanisms in general. In this review, we start with an overview of wild-type MLL activity because this is an essential component of understanding MLL leukemogenesis. As we will discuss, the function of the wild-type protein also has very specific implications for therapy.
Wild-Type MLL in Development and Hematopoiesis
Chromosome 11q23 is a long-recognized common breakpoint site for chromosome translocations in a subset of highly aggressive acute leukemias. Cloning of the breakpoint revealed the presence of the Mixed Lineage Leukemia (MLL or MLL1) gene, a homolog of the Drosophila gene trithorax (trx).6,7 The trx protein is a member of the trithorax group (TrxG) of proteins, important regulators that are required to maintain gene activation throughout development.8 TrxG protein activity is balanced by the maintenance of gene repression as mediated by the polycomb group (PcG) of proteins.8 These early results provided key insight into potential mechanisms of MLL leukemogenesis because it was known that TrxG and PcG proteins control gene expression through epigenetic mechanisms.
Similar to trx in Drosophila, the MLL protein is required for the proper anterior-posterior axis patterning of the developing embryo.9,10 MLL is also required for the maintenance of stem cells and their progenitors during neurogenesis11 and hematopoiesis.12,13 The role of MLL in normal hematopoiesis has been most clearly studied using 2 different inducible knockout systems in which loss of MLL causes a failure of haematopoietic stem cell (HSC) self renewal.12,13 One possible complicating factor in interpreting these phenotypes is that although each model targets different exons of the Mll gene, they all have the potential of producing either long13 or short10,12 MLL peptides that contain the N-terminal high-affinity binding sites for the MENIN and LEDGF proteins (see below and Fig. 1). The MLL N terminus can act as a dominant negative in zebrafish development,14 potentially as a result of sequestration of MENIN and/or LEDGF. If the MLL N terminus can also function as a dominant negative in mammalian systems, this could complicate interpretation of the phenotypes of the above Mll knockout models.
Figure 1.
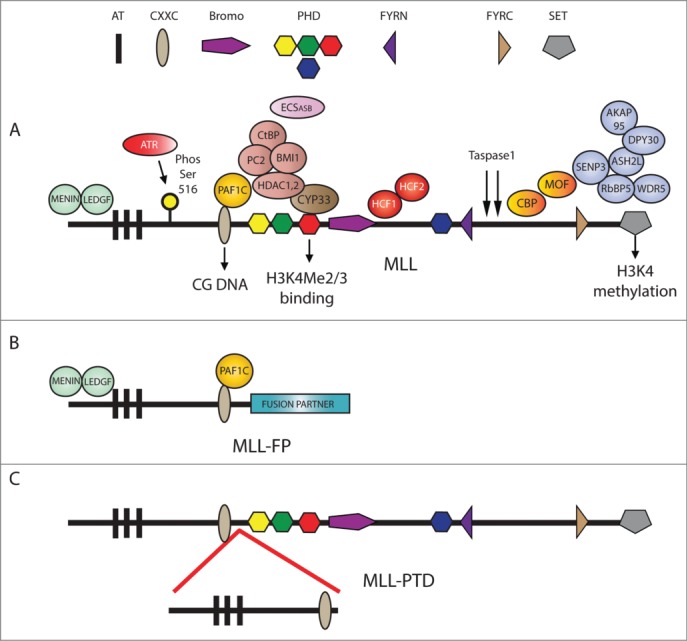
Structure of the MLL protein. (A) Important protein domains and interactions. MLL possesses 3 AT hooks for binding to AT-rich DNA, a CXXC domain for binding to unmethylated CpG islands, 4 plant homeodomain (PHD) fingers (the third PHD binds to H3K4Me2/3 and CYP33 on the opposite surface), an atypical bromodomain (Bromo), FYRN and FYRC domains, and a C-terminal SET domain that methylates histone H3 on lysine 4. Wild-type MLL is cleaved by Taspase 1 to yield 2 fragments: MLL-N and MLL-C. MLL-N can directly interact with different proteins/complexes, including MENIN, LEDGF, the PAF1 complex (PAF1C), CYP33, PC2, HDAC1, HCF1, and HCF2, and can indirectly bind to BMI-1 and CtBP. The PHD fingers may also interact directly with the ECSASB complex. MLL-N is directly phosphorylated by the ATR protein at serine 516. MLL-C can interact with CBP and MOF. The SET domain interacts directly with WDR5 and RBBP5. Interactions with SENP3, DPY30, and AKAP95 are all indirect or partially characterized. (B) Representation of MLL fusion proteins. MLL-FPs retain the N terminus of the wild type protein and lose the C terminus. The breakpoint lies in the region between the CXXC domain and the PHD fingers. (C) Representation of MLL partial tandem duplication. MLL-PTDs duplicate the N terminus of the wild-type protein, which contains the MENIN/LEDGF interaction region, the AT hooks, and the CXXC domain.
The Molecular Activity of Wild Type MLL
The MLL gene encodes a large protein that contains many important functional domains (Fig. 1A). Taspase1 proteolytically cleaves the full-length MLL protein into MLL-N (∼300 kDa) and MLL-C (∼180 kDa) fragments (Fig. 1A), and this cleavage is essential for in vivo activity of MLL.15 Although the MLL-N and MLL-C portions of the MLL complex are in theory independent from each other, biochemical purifications suggest that they tightly associate through FYRN and FYRC domain interactions.16,17
MLL binds directly to important gene targets during development and is required for their activation.9,11,16,18,19 The best-studied MLL and trx gene targets are the clustered Homeobox (or HOX) genes. Similar to mice with trx mutations, Mll mutant mice initially display normal Hox gene expression patterns and it is only as development proceeds that gene activation patterns break down.9 This suggests that MLL is not required for the initiation of gene expression, but is instead necessary for the maintenance of gene expression patterns through cell division. This observation is supported by the demonstration that MLL binds directly to gene promoters throughout mitosis and is required for the rapid induction of transcriptional activation at specific gene targets after mitotic exit.20
How Does MLL Mediate Transcriptional Activation?
MLL interacts with a large and varied range of proteins (see Fig. 1A for an overview). Not all of these protein interactions have been equally well characterized, but one is left with the impression that the major function of the MLL protein is to act as a scaffold for the assembly of different protein complexes, perhaps in a highly gene-specific or context-dependent manner.
Starting at the N terminus of MLL, the MENIN and LEDGF proteins interact with MLL in a trimeric complex.21,22 It was initially suggested that the MENIN/LEDGF interaction functions to stabilize binding of MLL to chromatin.21,23 Some support for this idea came from the fact that MENIN knockouts show reduced binding of MLL to gene targets,23,24 but more recent work suggests that a minimal MLL recruitment domain that lacks the MENIN and LEDGF interaction domains can still bind to some MLL target genes.25 Importantly, wild-type MLL and MENIN appear to have both overlapping and distinct functions in hematopoiesis, but MENIN is not absolutely required for HSC maintenance or normal hematopoiesis.26 Although the molecular function of MENIN and LEDGF in the context of MLL activity remains to be fully elucidated, these proteins are generally implicated in gene activation.
The CXXC domain of MLL binds to unmethylated CG-rich DNA.27 This does not appear to stabilize binding of the wild type MLL protein,25 but it may instead prevent gene repression by protecting loci from DNA methylation.28 The region containing the CXXC domain also interacts with the polymerase associated factor 1 complex (PAF1C).25,29 PAF1C can promote transcription elongation through chromatin templates30 and may function in part by specifically recruiting wild-type MLL to some active genes.25
MLL is thought to keep genes active primarily through the H3K4 methyltransferase (KMT) activity of its C-terminal SET domain18,19 and by recruiting the histone lysine acetyltransferases Males absent On the First (MOF) and Creb Binding Protein (CBP).16,31 The KMT and lysine acetyltransferase (KAT) activities of MLL both have important therapeutic implications and it is worth discussing them in greater detail.
MLL and H3K4 Methylation
Based on SET domain homology, MLL belongs to a family of 6 mammalian H3K4 methyltransferases that includes MLL, MLL2, MLL3, MLL4, and SET domain containing protein (SET1) A and B.32 The overall protein architecture of MLL2 is the most highly similar to MLL and it likely that these 2 proteins are related through a gene duplication event.32 As an aside, we would like to note that there is often confusion about the nomenclature of MLL2 versus MLL4 (which is also referred to as MLL2, and was originally called ALR) and we discuss this in some detail in a previous review.33 In the current review we will be using MLL2 to refer to the gene on human chromosome 19 (gene ID 9757) as this is the most common usage in the MLL literature.33
The MLL SET domain requires the activity of a core complex of proteins (WRAD,Fig. 1A) that includes WDR5 (WD repeat-containing protein 5), RBBP5 (retinoblastoma binding protein 5), ASH2L (absent, small, or homeotic-like (Drosophila) ash2), and DPY30 for full H3K4Me3 activity,34,35 and can also be further stimulated by the component AKAP95.36 RBBP5 may also interact directly with SENP3 and its activity is modulated by de-SUMOylation.37 Despite their similar SET domain structure, MLL family members are important for the regulation of both overlapping and unique sets of genes32,38 and display different intrinsic H3K4Me activity. For example, MLL is able to mono-, di-, and tri- methylate K4, whereas MLL3 appears to be primarily a monomethylase in vitro.39 Also, although WRAD interactions are common to the entire MLL family, the MLL-WDR5 interaction is critical for MLL SET domain activity but appears to be dispensable for the in vitro activity of other MLL family members.40
In mammals, H3K4Me3 can function as a docking site for different reader molecules, including the TAF3 protein.32 TAF3 directly binds to H3K4Me3 and can promote increased transcription by stabilizing formation of the RNA polymerase II (RNA pol II) pre-initiation complex.41 Interestingly, the third plant homeodomain (PHD) finger of MLL binds directly to H3K4Me3 (Fig. 1A) and there is some evidence that this interaction functions to stabilize binding of MLL to its gene targets.25
The actual importance of MLL SET domain H3K4me activity varies with the system under study. In Drosophila, a trx SET domain point mutation (trxZ11) displays a similar Hox gene mutant phenotype as trx gene knockouts.42 This suggests that trx SET domain function is an essential component of trx-mediated Hox gene regulation in vivo. Early work using cell culture systems suggested that MLL SET domain H3K4Me3 activity was not required for global H3K4me3 levels,18 but was essential for MLL-mediated activation of HOX genes.18,19 Consistent with this, knockdown of different core components of WRAD disrupt HOX gene activation as well as H3K4Me3 levels.34 However, mice with an MLL SET domain deletion generally develop normally and display only a slight defect in H3K4Me1 levels with no alteration in H3K4Me3.43 This could partly be due to redundancy with other mammalian MLL family members such as MLL2,38 but this result contrasts rather strongly with the more drastic phenotype observed in Mll−/- knockout mice.9,10
There are several other lines of evidence indicating that MLL H3K4Me activity may not be essential for many aspects of MLL-mediated gene regulation. First, in neural development, MLL is required for activation of the Dlx2 gene but the main effect (likely indirect) is through demethylation of H3K27Me3 rather than through H3K4Me3.11 Second, loss of MLL disrupts reactivation of target genes after mitotic exit without an associated loss of H3K4Me3.20 Finally, in a more recent analysis of the role of MLL in hematopoiesis, Mishra and others have shown that MLL SET domain knockouts have no effect on normal hematopoiesis and there is no change in gene expression or any change in H3K4 methylation at any target genes.44 Interestingly, they also report that, even in MLL knockout models, although there is a reduction in gene expression of MLL targets there is no associated change in H3K4Me1 or H3K4Me3.44 Together, these results suggest that in several biologic systems there is redundancy for maintaining H3K4Me at many MLL target genes, but MLL may have other important functions in maintaining gene activation. This has important therapeutic implications as discussed below.
Histone Acetylation
Both MOF and CBP are lysine acetyltransferases, an activity that is generally associated with gene activation. CBP can acetylate H3 at lysine 27, but is also capable of acetylating a wide range of other histone residues,45 whereas MOF activity is more specific and appears to focus on the H4 lysine 16 (H4K16) residue.46 Both MOF and CBP interact directly with the MLL protein (Fig. 1A).16,31 MLL-mediated gene activation strongly correlates with increases in H4K16Ac and histone acetylation in general.16,44,47 Interestingly, although Mishra et al. saw no changes in H3K4Me3 in MLL knockouts in hematopoiesis they did notice a decrease in gene expression that was strongly associated with loss of MOF binding and H4K16 acetylation.44 This led them to suggest that the major function of MLL is to recruit MOF to gene targets where it can acetylate H4K16 and promote gene activation.44
MLL and Gene Repression
Other known activities of MLL include a possible role in gene repression as mediated by an interaction with cyclophilin 33 (CyP33), and a repression complex containing the histone deacetylase HDAC1 and the PcG proteins CtBP, HPC2, and BMI-1.48,49 There is not sufficient space here for a detailed discussion of the possible implications of these interactions, but structural studies have raised the interesting possibility that CyP33 could function as a regulatory switch by altering the structure of MLL and enabling recruitment of the repressive complex.50 Thus it is possible that this interaction functions as a way of mediating MLL activity and changing it from an activator to a repressor. The PHD fingers have also been shown to interact with the ECSASB E3 ubiquitin ligase complex, which is thought to control MLL activity through ubiquitin-mediated degradation of MLL.51
MLL and the S Phase Cell Cycle Checkpoint
Work from Liu and colleagues has also shown that MLL H3K4Me3 activity has a role in delaying S-phase progression.52 ATR phosphorylates MLL at serine 516 (Fig. 1A), preventing the SCFSkp2- and APCCdc20-mediated degradation of MLL. This results in increased H3K4Me3 at replication origins and inhibits CDC45 binding, which results in a delay of DNA replication and productive DNA checkpoint repair. Interestingly, MLL mutations in leukemia inhibit this pathway, potentially causing increased genome instability in MLL leukemias.52
Other MLL Functional Interactions
A region proximal to the bromodomain contains a high-affinity interaction site for the host cell factor C1 and C2 (HCF1 and HCF2) proteins.53 HCF1 and 2 have been implicated in the recruitment of MLL family complexes to some gene targets,54 but knockdown of HCF1 or HCF2 does not appear to have any effect on MLL activation of HOX genes.53
MLL Mutations in Leukemia
MLL gene mutations are associated with acute myeloid leukemia (AML) and acute lymphoid leukemia (ALL) in both children and adults.55 In AML, MLL mutations can be found in as many as 10% of adult and 18% of childhood cases,56,57 whereas MLL mutations are responsible for 8% of childhood and 10% of adult ALL cases.57,58 Common MLL mutations include chromosome translocations that fuse the MLL gene with partner genes to create novel fusion proteins (MLL-FPs, Fig. 1B). Partial tandem duplications (MLL-PTDs, Fig. 1C) of the N terminal portion of the MLL gene are also fairly common mutations in adult AML.55 For the MLL-FPs, over 79 different fusion partner genes have been identified, but 4 partners (AF9, ELL, AF10, and AF6) account for most MLL rearrangements in AML and 3 partners (AF4, AF9, and ENL) account for the majority of ALL rearrangements.55
In most cases, the reciprocal fusion (FP-MLL) is not expressed, but t(4;11)(q21;q23) translocations, referred to as t(4;11), represent a special case in that 50–80% of patients express the AF4-MLL fusion as well as the MLL-AF4 fusion.59,60 Important work in mouse model systems suggests that AF4-MLL, but not MLL-AF4, is sufficient to initiate leukemogenesis.61 Mouse models and molecular data also suggest that these 2 protein complexes may cooperate,61,62 but a complete understanding of this interplay has yet to be fully elucidated.
In general, the prognosis of patients carrying MLL mutations is quite poor.63 However, a more detailed analysis of patients with MLL-FP has suggested that they actually display a range of different prognostic outcomes. For example, MLL-AF9 produces an intermediate prognosis in AML63 but a poor prognosis in infant ALL.64 MLL-ENL is associated with a good prognosis in t-ALL,57 whereas t(4;11) and MLL-AF6 are both associated with very poor prognoses.57,63,65 This review will focus primarily on the activity of MLL-FPs.
Cooperating Mutations in MLL-FP Leukemias
MLL-FP leukemias in humans have very few cooperating mutations56,60,66 and retroviral transduction models produce rapid leukemias in mice.67 This suggests that MLL-FPs alone might be sufficient to drive leukemogenesis. However, knock-in mouse models have a much longer latency68 and it has also been shown that 30–50% of MLL-FP patients harbor a RAS mutation.69 Other more rare events such as Fms-like tyrosine kinase 3 (FLT-3) mutations are present in approximately 3% of MLL cases.70 Recent data have also shown that, although rare at diagnosis, copy number abnormalities associated with infant MLL-AF4 are present at relapse.71 It is thus possible that MLL-FPs alone are sufficient for initiating leukemogenesis, but that the presence of additional mutations such as RAS or FLT3 might contribute to tumor development and to the rise of more aggressive clones. That said, relative to other acute leukemias, MLL-FP leukemias present a somewhat simple genetic landscape,56,60,66 probably because most changes are on the epigenetic level.
The Molecular Activity of MLL Fusion Proteins (MLL-FPs)
We provide a comprehensive discussion of MLL-FP complexes in a recent review33 and will only give a brief overview of their proposed function here (for a summary see Fig. 2). MLL-FPs are thought to function by binding to and inappropriately activating a small set of key target genes.33,62 Okuda and colleagues have shown that only 3 domains are necessary for MLL-FP–mediated transformation: the MEN/LEDGF interaction domain(s), the CXXC domain, and the fusion partner itself.72 It was initially suggested that PAF1C, MENIN, and LEDGF are important for the recruitment and stable binding of MLL-FPs at target genes,21,25,29 but this model needs to be more rigorously tested. Irrespective of the specific functional mechanism, it has been shown that disruption of these interactions is sufficient for disrupting MLL-FP–mediated leukemogenesis.21,25,29
Figure 2.
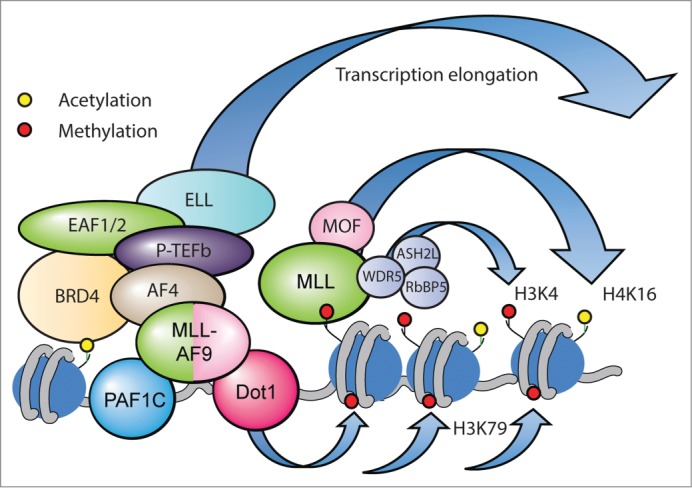
MLL and MLL-FP complexes bound to a gene target. The most common MLL-FPs, such as MLL-AF9, are members of a large super-elongation complex (SEC) that includes the most common MLL fusion partners (AF4, AF9, AF10, ENL), as well as the H3K79 methyltransferase DOT1L, the RNA pol II pause release complex P-TEFb, the elongation factors ELL and EAF, and the PAF1 complex (PAF1C). BRD4 also interacts with this complex. The wild-type MLL complex promotes both H3K4Me and H4K16Ac. The current model proposes that MLL-FPs recruit components of these complexes and then activate RNA polymerase II that is paused at the proximal promoter to promote productive transcription elongation.
The most common MLL fusion partners (AF4, AF9, ENL, AF10, and ELL) are all members of a large transcription elongation complex that has been alternatively called the ENL-associated protein complex (or EAP),73 the AF4 family/ENL/P-TEFb complex (or AEP),74 and the super elongation complex (or SEC).75 This complex also contains the H3K79 methyltransferase Disruptor of Telomeric Silencing 1-Like (DOT1L) protein.76 A high level of H3K79Me2/3 at gene targets is dependent on MLL-FP binding77 and DOT1L activity is necessary for MLL-FP leukemia growth.78 The functional model that has been proposed is that MLL-FPs recruit components of these complexes and then activate RNA polymerase II that is paused at the proximal promoter to promote productive transcription elongation.
Interestingly, a link between the bromodomain containing protein 4 (BRD4, an acetylated histone mark reader), MLL-FPs, PAF1C, and the SEC complex has also been identified.79,80 Disruption of BRD4 through RNAi or treatment with specific inhibitors disrupts MLL-FP leukemic growth both in vivo and in vitro.79,80 According to one model, BRD4 recognizes and binds acetylated histones and then recruits PAF1C, MLL-FPs, and the remaining transcription elongation components to a subset of important target genes such as BCL2 and MYC.79 Treatment with BRD4 inhibitors prevents BRD4 binding to acetyl residues on H3 and H4, leading to displacement of the other complexes from chromatin and transcriptional inhibition.79 However, this model cannot be completely correct as many MLL-FP target genes are unaffected by the loss of BRD4 binding.
MLL-AF6, another common MLL fusion protein, does not interact with any of these transcription elongation complexes74 but instead appears to function through dimerization81 and the aberrant activation of RAS signaling.82 Interestingly, MLL-AF6 is still dependent on DOT1L H3K79Me2/3 activity,83 potentially because of indirect mechanisms of gene activation. Dimerization of the N terminus of MLL appears to be a sufficient mechanism for transformation,84,85 and thus could potentially be a common mechanism among the approximately 70 MLL-FPs that are not part of the above complexes.
Taken together, a simple unifying model does not seem to explain how all of these MLL-FPs cause leukemia. A more detailed analysis of normal transcription elongation and the role of H3K79me2/3 will provide a better understanding of the leukemogenic process. However, the discovery that most MLL leukemias require the activity of specific proteins that includes MENIN, LEDGF, PAF1, BRD4, and DOT1L has been an important step in the rational design of therapeutic inhibitors.
Targeting the Activity of the MLL-FP Complex
Although MLL-FPs combine the activity of 2 separate complexes, from a biochemical perspective the functional activity may not have changed. That is, MLL-FPs may inappropriately activate gene expression through recruitment of a transcription elongation complex, but transcription elongation is required for normal gene activation. Thus, one of the initial concerns was that anything that inhibited MLL-FP complex activity could also be toxic to normal cells. Recent work has suggested that inhibiting transcription elongation in MLL leukemias with improved inhibition of CDK9 (a component of the P-TEFb complex) shows promise in disrupting the growth of MLL as well as potentially other acute leukemias.86 The toxicity of these inhibitors needs to be established, but clinical trials with the CDK9 inhibitor flavopiridol suggest that they may be well tolerated by patients.86
MLL-FP leukemias are also dependent on H3K79 methylation mediated by DOT1L.78 DOT1L is the only known H3K79 methyltransferase and experiments with Dot1L knockout mice have already shown that its inhibition mainly affects MLL-AF9 target genes.78 Recently, 3 different compounds against DOT1L have been developed.78 All 3 compete with S-adenosyl methionine, which is needed for the methyltransferase activity. Treatment of leukemia cells with these inhibitors decreases H3K79Me and downregulates important MLL-FP target genes, such as HOXA9 and MEIS1, leading to cellular apoptosis.78 It has been shown that at least one of these compounds (EPZ-5676) causes tumor regression in a rat xenograft model with no signs of toxicity.87 On the basis of these promising results, DOT1L inhibitors are now currently undergoing early stage clinical trials.78
As already discussed above, MENIN apparently does not contribute to normal MLL activity in hematopoiesis26 but is essential for MLL-FP–mediated leukemogenesis.88 This makes disruption of the MENIN-MLL interaction an appealing target for therapy. Exciting work performed over a long period of time has produced the first of what may potentially be a new breed of MLL-FP inhibitors: a small molecule that disrupts the MLL–MENIN protein–protein interaction and disrupts the growth of MLL leukemias.89
Along a similar line, dominant negative peptides designed to disrupt the MLL-FP–PAF190 and the ML-FP–LEDGF interactions91 have allowed proof-of-principal studies showing that targeting other MLL-FP complex components could provide additional therapeutic avenues.
A completely different way of targeting MLL-FP activity has been recently revealed by work showing that MLL-AF4 leukemias are highly sensitive to proteasome inhibitors.92 The suggested mechanism is that proteasome inhibition increases MLL-AF4 protein levels, causing increased expression of cell cycle inhibitors and increased sensitivity to apoptosis.92 Although the molecular underpinnings of this mechanism appear to be less precise than targeting a specific activity of the MLL-FP complex, this is still a very interesting observation that is potentially very promising.
Cooperation Between Wild-Type MLL and MLL-FP Activity
It was originally observed that wild-type MLL and MLL-FPs co-localize at important gene targets.77 This led to the suggestion that both wild-type MLL and MLL-FPs cooperate somehow in the promotion of leukemogenesis.77 Although there are rare patient samples that have a deletion of MLL and only express the MLL-FP, in the large majority of patients the wild-type MLL gene is retained. The possibility that wild-type MLL contributes to leukemogenesis was formally tested by Thiel et al., who elegantly showed that loss of the wild-type MLL protein disrupts the growth of MLL-AF9 leukemias in vivo.24
A possible mechanism for this cooperation comes from the observation that binding of MLL-AF9 to target genes such as HOXA9 is partially dependent on MLL-mediated gene activation.25 This has led to the suggestion that gene activation itself provides an “open” chromatin conformation that allows an MLL-FP to bind to a gene target (Fig. 2). The important question then becomes: what aspect of MLL mediated gene activation is the most important for MLL-FP activity and can it be targeted?
Targeting Wild-Type MLL in MLL-FP Leukemias
Mishra et al. have shown that the wild-type MLL SET domain is dispensable for MLL-AF9–mediated leukemogenesis in vivo,44 although it is important to note that they used a retroviral transduction system that can be prone to overexpression artifacts. Conversely, work by Cao et al. suggests that an inhibitor that specifically blocks the methyltransferase activity of MLL leads to growth inhibition of MLL-AF9 blasts as a result of cell cycle arrest, apoptosis, and differentiation.40 RNA-seq analysis indicates that cells treated with the inhibitor exhibit the same changes in gene expression as those carrying an MLL deletion.40 Despite this apparent overlap between the inhibitor and wild-type MLL function, the inhibitor itself actually targets the WRAD component WDR5. WDR5 is more globally required for H3K4Me3 than MLL alone18,34 therefore it remains formally possible that the inhibitor is disrupting some non-MLL function of WDR5. However, another possibility could simply be that MLL-FP leukemias are sensitive to slight changes in H3K4Me3 levels. It is also possible that leukemia cells are more sensitive to slight variations in the stable binding of transcription promoting complexes. A slight decrease in H3K4Me3 at important gene targets could result in reduced binding of wild-type MLL and/or reduced binding of TAF3 that could result in destabilized protein complex formation and reduced transcription initiation in addition to elongation. Since MLL H3K4Me3 activity may be dispensable for MLL function in hematopoiesis,44 this raises the exciting possibility that an MLL H3K4Me3 inhibitor might disrupt leukemogenesis without having a strong effect on normal cells.
Common Epigenetic Pathways in Cancer
Although all the work described in this review is specific to MLL leukemias, BRD4 in particular has been found to have a much wider role in promoting leukemogenesis, and BRD4 inhibitors are now in Phase I clinical trials as potential inhibitors of all cancers that overexpress MYC.4 BRD4 is not commonly mutated or overexpressed in acute leukemias and would not have been identified as a potential therapeutic target in conventional genomic studies. This underscores the importance of studying rare leukemias such as MLL because these studies may reveal information about more general key pathways. It remains to be seen whether inhibitors of DOT1L, MLL-WDR5, or MENIN have the same potential for a broader scope of efficacy.
MLL family proteins are also mutated in other hematologic malignancies and some solid tumors.1 As the ability to develop inhibitors to a wider range of different proteins increases, the range of potential therapeutic targets could expand to include important downstream target genes or other components of these protein complexes. Understanding more about how MLL and associated proteins function may highlight important regulatory interactions that could be exploited in the context of different cancers. Thus, MLL leukemias may continue to be an important source of new information about epigenetic pathways in cancer.
Funding Statement
TAM and EB are supported by funding from the Medical Research Council (MRC) UK.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1. Timp W, Feinberg AP. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat Rev Cancer 2013; 13:497-510; PMID:23760024; http://dx.doi.org/ 10.1038/nrc3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Luger K, Dechassa ML, Tremethick DJ. New insights into nucleosome and chromatin structure: an ordered state or a disordered affair? Nat Rev Mol Cell Biol 2012; 13:436-47; PMID:22722606; http://dx.doi.org/ 10.1038/nrm3382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet 2011; 12:7-18; PMID:21116306; http://dx.doi.org/ 10.1038/nrg2905 [DOI] [PubMed] [Google Scholar]
- 4. Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell 2012; 150:12-27; PMID:22770212; http://dx.doi.org/ 10.1016/j.cell.2012.06.013 [DOI] [PubMed] [Google Scholar]
- 5. Greaves M, Maley CC. Clonal evolution in cancer. Nature 2012; 481:306-13; PMID:22258609; http://dx.doi.org/ 10.1038/nature10762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gu Y, Nakamura T, Alder H, Prasad R, Canaani O, Cimino G, Croce CM, Canaani E. The t(4;11) chromosome translocation of human acute leukemias fuses the ALL-1 gene, related to Drosophila trithorax, to the AF-4 gene. Cell 1992; 71:701-8; PMID:1423625 [DOI] [PubMed] [Google Scholar]
- 7. Tkachuk DC, Kohler S, Cleary ML. Involvement of a homolog of Drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell 1992; 71:691-700; PMID:1423624 [DOI] [PubMed] [Google Scholar]
- 8. Schuettengruber B, Martinez AM, Iovino N, Cavalli G. Trithorax group proteins: switching genes on and keeping them active. Nat Rev Mol Cell Biol 2011; 12:799-814; PMID:22108599; http://dx.doi.org/ 10.1038/nrm3230 [DOI] [PubMed] [Google Scholar]
- 9. Yu BD, Hanson RD, Hess JL, Horning SE, Korsmeyer SJ. MLL, a mammalian trithorax-group gene, functions as a transcriptional maintenance factor in morphogenesis. Proc Natl Acad Sci U S A 1998; 95:10632-6; PMID:9724755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yu BD, Hess JL, Horning SE, Brown GA, Korsmeyer SJ. Altered Hox expression and segmental identity in Mll-mutant mice. Nature 1995; 378:505-8; PMID:7477409 [DOI] [PubMed] [Google Scholar]
- 11. Lim DA, Huang YC, Swigut T, Mirick AL, Garcia-Verdugo JM, Wysocka J, Ernst P, Alvarez-Buylla A. Chromatin remodelling factor Mll1 is essential for neurogenesis from postnatal neural stem cells. Nature 2009; 458:529-33; PMID:19212323; http://dx.doi.org/ 10.1038/nature07726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jude CD, Climer L, Xu D, Artinger E, Fisher JK, Ernst P. Unique and independent roles for MLL in adult hematopoietic stem cells and progenitors. Cell Stem Cell 2007; 1:324-37; PMID:18371366; http://dx.doi.org/ 10.1016/j.stem.2007.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McMahon KA, Hiew SY, Hadjur S, Veiga-Fernandes H, Menzel U, Price AJ, Kioussis D, Williams O, Brady HJ. Mll has a critical role in fetal and adult hematopoietic stem cell self-renewal. Cell Stem Cell 2007; 1:338-45; PMID:18371367; http://dx.doi.org/ 10.1016/j.stem.2007.07.002 [DOI] [PubMed] [Google Scholar]
- 14. Wan X, Hu B, Liu JX, Feng X, Xiao W. Zebrafish mll gene is essential for hematopoiesis. J Biol Chem 2011; 286:33345-57; PMID:21784840; http://dx.doi.org/ 10.1074/jbc.M111.253252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hsieh JJ, Cheng EH, Korsmeyer SJ. Taspase1: a threonine aspartase required for cleavage of MLL and proper HOX gene expression. Cell 2003; 115:293-303; PMID:14636557 [DOI] [PubMed] [Google Scholar]
- 16. Dou Y, Milne TA, Tackett AJ, Smith ER, Fukuda A, Wysocka J, Allis CD, Chait BT, Hess JL, Roeder RG. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell 2005; 121:873-85; PMID:15960975 [DOI] [PubMed] [Google Scholar]
- 17. Hsieh JJ, Ernst P, Erdjument-Bromage H, Tempst P, Korsmeyer SJ. Proteolytic cleavage of MLL generates a complex of N- and C-terminal fragments that confers protein stability and subnuclear localization. Mol Cell Biol 2003; 23:186-94; PMID:12482972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, Hess JL. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell 2002; 10:1107-17; PMID:12453418 [DOI] [PubMed] [Google Scholar]
- 19. Nakamura T, Mori T, Tada S, Krajewski W, Rozovskaia T, Wassell R, Dubois G, Mazo A, Croce CM, Canaani E. ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell 2002; 10:1119-28; PMID:12453419 [DOI] [PubMed] [Google Scholar]
- 20. Blobel GA, Kadauke S, Wang E, Lau AW, Zuber J, Chou MM, Vakoc CR. A reconfigured pattern of MLL occupancy within mitotic chromatin promotes rapid transcriptional reactivation following mitotic exit. Mol Cell 2009; 36:970-83; PMID:20064463; http://dx.doi.org/ 10.1016/j.molcel.2009.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yokoyama A, Cleary ML. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell 2008; 14:36-46; PMID:18598942; http://dx.doi.org/ 10.1016/j.ccr.2008.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huang J, Gurung B, Wan B, Matkar S, Veniaminova NA, Wan K, Merchant JL, Hua X, Lei M. The same pocket in menin binds both MLL and JUND but has opposite effects on transcription. Nature 2012; 482:542-6; PMID:22327296; http://dx.doi.org/ 10.1038/nature10806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Milne TA, Hughes CM, Lloyd R, Yang Z, Rozenblatt-Rosen O, Dou Y, Schnepp RW, Krankel C, Livolsi VA, Gibbs D, et al. . Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc Natl Acad Sci U S A 2005; 102:749-54; PMID:15640349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thiel AT, Blessington P, Zou T, Feather D, Wu X, Yan J, Zhang H, Liu Z, Ernst P, Koretzky GA, et al. . MLL-AF9-induced leukemogenesis requires coexpression of the wild-type Mll allele. Cancer Cell 2010; 17:148-59; PMID:20159607; http://dx.doi.org/ 10.1016/j.ccr.2009.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Milne TA, Kim J, Wang GG, Stadler SC, Basrur V, Whitcomb SJ, Wang Z, Ruthenburg AJ, Elenitoba-Johnson KS, Roeder RG, et al. . Multiple interactions recruit MLL1 and MLL1 fusion proteins to the HOXA9 locus in leukemogenesis. Mol Cell 2010; 38:853-63; PMID:20541448; http://dx.doi.org/ 10.1016/j.molcel.2010.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li BE, Gan T, Meyerson M, Rabbitts TH, Ernst P. Distinct pathways regulated by menin and by MLL1 in hematopoietic stem cells and developing B cells. Blood 2013; 122:2039-46; PMID:23908472; http://dx.doi.org/ 10.1182/blood-2013-03-486647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Birke M, Schreiner S, Garcia-Cuellar MP, Mahr K, Titgemeyer F, Slany RK. The MT domain of the proto-oncoprotein MLL binds to CpG-containing DNA and discriminates against methylation. Nucleic Acids Res 2002; 30:958-65; PMID:11842107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Erfurth FE, Popovic R, Grembecka J, Cierpicki T, Theisler C, Xia ZB, Stuart T, Diaz MO, Bushweller JH, Zeleznik-Le NJ. MLL protects CpG clusters from methylation within the Hoxa9 gene, maintaining transcript expression. Proc Natl Acad Sci U S A 2008; 105:7517-22; PMID:18483194; http://dx.doi.org/ 10.1073/pnas.0800090105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Muntean AG, Tan J, Sitwala K, Huang Y, Bronstein J, Connelly JA, Basrur V, Elenitoba-Johnson KS, Hess JL. The PAF complex synergizes with MLL fusion proteins at HOX loci to promote leukemogenesis. Cancer Cell 2010; 17:609-21; PMID:20541477; http://dx.doi.org/ 10.1016/j.ccr.2010.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim J, Guermah M, Roeder RG. The human PAF1 complex acts in chromatin transcription elongation both independently and cooperatively with SII/TFIIS. Cell 2010; 140:491-503; PMID:20178742; http://dx.doi.org/ 10.1016/j.cell.2009.12.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ernst P, Wang J, Huang M, Goodman RH, Korsmeyer SJ. MLL and CREB bind cooperatively to the nuclear coactivator CREB-binding protein. Mol Cell Biol 2001; 21:2249-58; PMID:11259575; http://dx.doi.org/ 10.1128/MCB.21.7.2249-2258.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell 2007; 25:15-30; PMID:17218268; http://dx.doi.org/ 10.1016/j.molcel.2006.12.014 [DOI] [PubMed] [Google Scholar]
- 33. Ballabio E, Milne TA. Molecular and Epigenetic Mechanisms of MLL in Human Leukemogenesis. Cancers 2012; 4:904-44; PMID:24213472; http://dx.doi.org/ 10.3390/cancers4030904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, Verdine GL, Allis CD, Roeder RG. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol 2006; 13:713-9; PMID:16878130; http://dx.doi.org/ 10.1038/nsmb1128 [DOI] [PubMed] [Google Scholar]
- 35. Patel A, Dharmarajan V, Vought VE, Cosgrove MS. On the mechanism of multiple lysine methylation by the human mixed lineage leukemia protein-1 (MLL1) core complex. J Biol Chem 2009; 284(36):24242-56; PMID:19556245; http://dx.doi.org/ 10.1074/jbc.M109.014498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jiang H, Lu X, Shimada M, Dou Y, Tang Z, Roeder RG. Regulation of transcription by the MLL2 complex and MLL complex-associated AKAP95. Nat Struct Mol Biol 2013; 20:1156-63; PMID:23995757; http://dx.doi.org/ 10.1038/nsmb.2656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nayak A, Viale-Bouroncle S, Morsczeck C, Muller S. The SUMO-Specific Isopeptidase SENP3 Regulates MLL1/MLL2 Methyltransferase Complexes and Controls Osteogenic Differentiation. Mol Cell 2014; 55:47-58; PMID:24930734; http://dx.doi.org/ 10.1016/j.molcel.2014.05.011 [DOI] [PubMed] [Google Scholar]
- 38. Denissov S, Hofemeister H, Marks H, Kranz A, Ciotta G, Singh S, Anastassiadis K, Stunnenberg HG, Stewart AF. Mll2 is required for H3K4 trimethylation on bivalent promoters in embryonic stem cells, whereas Mll1 is redundant. Development 2014; 141:526-37; PMID:24423662; http://dx.doi.org/ 10.1242/dev.102681 [DOI] [PubMed] [Google Scholar]
- 39. Wu L, Lee SY, Zhou B, Nguyen UT, Muir TW, Tan S, Dou Y. ASH2L regulates ubiquitylation signaling to MLL: trans-regulation of H3 K4 methylation in higher eukaryotes. Mol Cell 2013; 49:1108-20; PMID:23453805; http://dx.doi.org/ 10.1016/j.molcel.2013.01.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cao F, Townsend, Karatas H, Xu J, Li L, Lee S, Liu L, Chen Y, Ouillette P, Zhu J, et al. . Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol Cell 2014; 53:247-61; PMID:24389101; http://dx.doi.org/ 10.1016/j.molcel.2013.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lauberth SM, Nakayama T, Wu X, Ferris AL, Tang Z, Hughes SH, Roeder RG. H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell 2013; 152:1021-36; PMID:23452851; http://dx.doi.org/ 10.1016/j.cell.2013.01.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Breen TR. Mutant alleles of the Drosophila trithorax gene produce common and unusual homeotic and other developmental phenotypes. Genetics 1999; 152:319-44; PMID:10224264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Terranova R, Agherbi H, Boned A, Meresse S, Djabali M. Histone and DNA methylation defects at Hox genes in mice expressing a SET domain-truncated form of Mll. Proc Natl Acad Sci U S A 2006; 103:6629-34; PMID:16618927; http://dx.doi.org/ 10.1073/pnas.0507425103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mishra BP, Zaffuto KM, Artinger EL, Org T, Mikkola HK, Cheng C, Djabali M, Ernst P. The Histone Methyltransferase Activity of MLL1 Is Dispensable for Hematopoiesis and Leukemogenesis. Cell Rep 2014; 7:1239-47; PMID:24813891; http://dx.doi.org/ 10.1016/j.celrep.2014.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McManus KJ, Hendzel MJ. Quantitative analysis of CBP- and P300-induced histone acetylations in vivo using native chromatin. Mol Cell Biol 2003; 23:7611-27; PMID:14560007; http://dx.doi.org/ 10.1128/MCB.23.21.7611-7627.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Smith ER, Pannuti A, Gu W, Steurnagel A, Cook RG, Allis CD, Lucchesi JC. The drosophila MSL complex acetylates histone H4 at lysine 16, a chromatin modification linked to dosage compensation. Mol Cell Biol 2000; 20:312-8; PMID:10594033; http://dx.doi.org/ 10.1128/MCB.20.1.312-318.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Milne TA, Dou Y, Martin ME, Brock HW, Roeder RG, Hess JL. MLL associates specifically with a subset of transcriptionally active target genes. Proc Natl Acad Sci U S A 2005; 102:14765-70; PMID:16199523; http://dx.doi.org/ 10.1073/pnas.0503630102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fair K, Anderson M, Bulanova E, Mi H, Tropschug M, Diaz MO. Protein interactions of the MLL PHD fingers modulate MLL target gene regulation in human cells. Mol Cell Biol 2001; 21:3589-97; PMID:11313484; http://dx.doi.org/ 10.1128/MCB.21.10.3589-3597.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xia ZB, Anderson M, Diaz MO, Zeleznik-Le NJ. MLL repression domain interacts with histone deacetylases, the polycomb group proteins HPC2 and BMI-1, and the corepressor C-terminal-binding protein. Proc Natl Acad Sci U S A 2003; 100:8342-7; PMID:12829790; http://dx.doi.org/ 10.1073/pnas.1436338100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang Z, Song J, Milne TA, Wang GG, Li H, Allis CD, Patel DJ. Pro isomerization in MLL1 PHD3-bromo cassette connects H3K4me readout to CyP33 and HDAC-mediated repression. Cell 2010; 141:1183-94; PMID:20541251; http://dx.doi.org/ 10.1016/j.cell.2010.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang J, Muntean AG, Hess JL. ECSASB2 mediates MLL degradation during hematopoietic differentiation. Blood 2012; 119:1151-61; PMID:22174154; http://dx.doi.org/ 10.1182/blood-2011-06-362079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liu H, Takeda S, Kumar R, Westergard TD, Brown EJ, Pandita TK, Cheng EH, Hsieh JJ. Phosphorylation of MLL by ATR is required for execution of mammalian S-phase checkpoint. Nature 2010; 467:343-6; PMID:20818375; http://dx.doi.org/ 10.1038/nature09350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yokoyama A, Wang Z, Wysocka J, Sanyal M, Aufiero DJ, Kitabayashi I, Herr W, Cleary ML. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol 2004; 24:5639-49; PMID:15199122; http://dx.doi.org/ 10.1128/MCB.24.13.5639-5649.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tyagi S, Chabes AL, Wysocka J, Herr W. E2F activation of S phase promoters via association with HCF-1 and the MLL family of histone H3K4 methyltransferases. Mol Cell 2007; 27:107-19; PMID:17612494; http://dx.doi.org/ 10.1016/j.molcel.2007.05.030 [DOI] [PubMed] [Google Scholar]
- 55. Meyer C, Hofmann J, Burmeister T, Groger D, Park TS, Emerenciano M, Pombo de Oliveira M, Renneville A, Villarese P, Macintyre E, et al. . The MLL recombinome of acute leukemias in 2013. Leukemia 2013; 27(11):2165-76; PMID:23628958; http://dx.doi.org/ 10.1038/leu.2013.135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson AG, Hoadley K, Triche TJ, Laird PW, Baty JD, et al. . The cancer genome atlas research network: genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013; 368:2059-74; PMID:23634996; http://dx.doi.org/ 10.1056/NEJMoa1301689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pui CH, Carroll WL, Meshinchi S, Arceci RJ. Biology, risk stratification, and therapy of pediatric acute leukemias: an update. J Clin Oncol 2011; 29:551-65; PMID:21220611; http://dx.doi.org/ 10.1200/JCO.2010.30.7405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Downing JR, Wilson RK, Zhang J, Mardis ER, Pui CH, Ding L, Ley TJ, Evans WE. The pediatric cancer genome project. Nat Genet 2012; 44:619-22; PMID:22641210; http://dx.doi.org/ 10.1038/ng.2287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kowarz E, Burmeister T, Lo Nigro L, Jansen MW, Delabesse E, Klingebiel T, Dingermann T, Meyer C, Marschalek R. Complex MLL rearrangements in t(4;11) leukemia patients with absent AF4.MLL fusion allele. Leukemia 2007; 21:1232-8; PMID:17410185 [DOI] [PubMed] [Google Scholar]
- 60. Dobbins SE, Sherborne AL, Ma YP, Bardini M, Biondi A, Cazzaniga G, Lloyd A, Chubb D, Greaves MF, Houlston RS. The silent mutational landscape of infant MLL-AF4 pro-B acute lymphoblastic leukemia. Genes Chromosomes Cancer 2013; 52:954-60; PMID:23893660; http://dx.doi.org/ 10.1002/gcc.22090 [DOI] [PubMed] [Google Scholar]
- 61. Bursen A, Schwabe K, Ruster B, Henschler R, Ruthardt M, Dingermann T, Marschalek R. The AF4.MLL fusion protein is capable of inducing ALL in mice without requirement of MLL.AF4. Blood 2010; 115:3570-9; PMID:20194896; http://dx.doi.org/ 10.1182/blood-2009-06-229542 [DOI] [PubMed] [Google Scholar]
- 62. Wilkinson AC, Ballabio E, Geng H, North P, Tapia M, Kerry J, Biswas D, Roeder RG, Allis CD, Melnick A, et al. . RUNX1 Is a Key Target in t(4;11) Leukemias that Contributes to Gene Activation through an AF4-MLL Complex Interaction. Cell Rep 2013; 3:116-27; PMID:23352661; http://dx.doi.org/ 10.1016/j.celrep.2012.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Grimwade D, Hills RK, Moorman AV, Walker H, Chatters S, Goldstone AH, Wheatley K, Harrison CJ, Burnett AK, National Cancer Research Institute Adult Leukaemia Working G. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 2010; 116:354-65; PMID:20385793; http://dx.doi.org/ 10.1182/blood-2009-11-254441 [DOI] [PubMed] [Google Scholar]
- 64. Pieters R, Schrappe M, De Lorenzo P, Hann I, De Rossi G, Felice M, Hovi L, LeBlanc T, Szczepanski T, Ferster A, et al. . A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet 2007; 370:240-50; PMID:17658395; http://dx.doi.org/ 10.1016/S0140-6736(07)61126-X [DOI] [PubMed] [Google Scholar]
- 65. Balgobind BV, Raimondi SC, Harbott J, Zimmermann M, Alonzo TA, Auvrignon A, Beverloo HB, Chang M, Creutzig U, Dworzak MN, et al. . Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: results of an international retrospective study. Blood 2009; 114:2489-96; PMID:19528532; http://dx.doi.org/ 10.1182/blood-2009-04-215152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kihara R, Nagata Y, Kiyoi H, Kato T, Yamamoto E, Suzuki K, Chen F, Asou N, Ohtake S, Miyawaki S, et al. . Comprehensive analysis of genetic alterations and their prognostic impacts in adult acute myeloid leukemia patients. Leukemia 2014; 28(8):1586-95; PMID:24487413; http://dx.doi.org/ 10.1038/leu.2014.55 [DOI] [PubMed] [Google Scholar]
- 67. Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z, Zon LI, Armstrong SA. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science 2010; 327:1650-3; PMID:20339075; http://dx.doi.org/ 10.1126/science.1186624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Corral J, Lavenir I, Impey H, Warren AJ, Forster A, Larson TA, Bell S, McKenzie AN, King G, Rabbitts TH. An Mll-AF9 fusion gene made by homologous recombination causes acute leukemia in chimeric mice: a method to create fusion oncogenes. Cell 1996; 85:853-61; PMID:8681380; http://dx.doi.org/ 10.1016/S0092-8674(00)81269-6 [DOI] [PubMed] [Google Scholar]
- 69. Liang DC, Shih LY, Fu JF, Li HY, Wang HI, Hung IJ, Yang CP, Jaing TH, Chen SH, Liu HC. K-Ras mutations and N-Ras mutations in childhood acute leukemias with or without mixed-lineage leukemia gene rearrangements. Cancer 2006; 106:950-6; PMID:16404744; http://dx.doi.org/ 10.1002/cncr.21687 [DOI] [PubMed] [Google Scholar]
- 70. Armstrong SA, Mabon ME, Silverman LB, Li A, Gribben JG, Fox EA, Sallan SE, Korsmeyer SJ. FLT3 mutations in childhood acute lymphoblastic leukemia. Blood 2004; 103:3544-6; PMID:14670924; http://dx.doi.org/ 10.1182/blood-2003-07-2441 [DOI] [PubMed] [Google Scholar]
- 71. Bardini M, Woll PS, Corral L, Luc S, Wittmann L, Ma Z, Lo Nigro L, Basso G, Biondi A, Cazzaniga G, et al. . Clonal variegation and dynamic competition of leukemia-initiating cells in infant acute lymphoblastic leukemia with MLL rearrangement. Leukemia 2014; PMID:24798483; http://dx.doi.org/ 10.1038/leu.2014.154. [DOI] [PubMed] [Google Scholar]
- 72. Okuda H, Kawaguchi M, Kanai A, Matsui H, Kawamura T, Inaba T, Kitabayashi I, Yokoyama A. MLL fusion proteins link transcriptional coactivators to previously active CpG-rich promoters. Nucleic Acids Res 2014; 42:4241-56; PMID:24465000; http://dx.doi.org/ 10.1093/nar/gkt1394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mueller D, Bach C, Zeisig D, Garcia-Cuellar MP, Monroe S, Sreekumar A, Zhou R, Nesvizhskii A, Chinnaiyan A, Hess JL, et al. . A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood 2007; 110:4445-54; PMID:17855633; http://dx.doi.org/ 10.1182/blood-2007-05-090514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yokoyama A, Lin M, Naresh A, Kitabayashi I, Cleary ML. A higher-order complex containing AF4 and ENL family proteins with P-TEFb facilitates oncogenic and physiologic MLL-dependent transcription. Cancer Cell 2010; 17:198-212; PMID:20153263; http://dx.doi.org/ 10.1016/j.ccr.2009.12.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lin C, Smith ER, Takahashi H, Lai KC, Martin-Brown S, Florens L, Washburn MP, Conaway JW, Conaway RC, Shilatifard A. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell 2010; 37:429-37; PMID:20159561; http://dx.doi.org/ 10.1016/j.molcel.2010.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Okada Y, Feng Q, Lin Y, Jiang Q, Li Y, Coffield VM, Su L, Xu G, Zhang Y. hDOT1L links histone methylation to leukemogenesis. Cell 2005; 121:167-78; PMID:15851025; http://dx.doi.org/ 10.1016/j.cell.2005.02.020 [DOI] [PubMed] [Google Scholar]
- 77. Milne TA, Martin ME, Brock HW, Slany RK, Hess JL. Leukemogenic MLL fusion proteins bind across a broad region of the Hox a9 locus, promoting transcription and multiple histone modifications. Cancer Res 2005; 65:11367-74; PMID:16357144 [DOI] [PubMed] [Google Scholar]
- 78. Neff T, Armstrong SA. Recent progress toward epigenetic therapies: the example of mixed lineage leukemia. Blood 2013; 121:4847-53; PMID:23649466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C, Savitski MM, et al. . Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011; 478:529-33; PMID:21964340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, et al. . RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011; 478:524-8; PMID:21814200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Liedtke M, Ayton PM, Somervaille TC, Smith KS, Cleary ML. Self-association mediated by the Ras association 1 domain of AF6 activates the oncogenic potential of MLL-AF6. Blood 2010; 116:63-70; PMID:20395419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Manara E, Baron E, Tregnago C, Aveic S, Bisio V, Bresolin S, Masetti R, Locatelli F, Basso G, Pigazzi M. MLL-AF6 fusion oncogene sequesters AF6 into the nucleus to trigger RAS activation in myeloid leukemia. Blood 2014; 124(2):263-72; PMID:24695851 [DOI] [PubMed] [Google Scholar]
- 83. Deshpande AJ, Chen L, Fazio M, Sinha AU, Bernt KM, Banka D, Dias S, Chang J, Olhava EJ, Daigle SR, et al. . Leukemic transformation by the MLL-AF6 fusion oncogene requires the H3K79 methyltransferase Dot1l. Blood 2013; 121(13):2533-41; PMID:23361907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Martin ME, Milne TA, Bloyer S, Galoian K, Shen W, Gibbs D, Brock HW, Slany R, Hess JL. Dimerization of MLL fusion proteins immortalizes hematopoietic cells. Cancer Cell 2003; 4:197-207; PMID:14522254 [DOI] [PubMed] [Google Scholar]
- 85. So CW, Lin M, Ayton PM, Chen EH, Cleary ML. Dimerization contributes to oncogenic activation of MLL chimeras in acute leukemias. Cancer Cell 2003; 4:99-110; PMID:12957285 [DOI] [PubMed] [Google Scholar]
- 86. Garcia-Cuellar MP, Fuller E, Mathner E, Breitinger C, Hetzner K, Zeitlmann L, Borkhardt A, Slany RK. Efficacy of cyclin-dependent-kinase 9 inhibitors in a murine model of mixed-lineage leukemia. Leukemia 2014; 28:1427-35; PMID:24445865 [DOI] [PubMed] [Google Scholar]
- 87. Daigle SR, Olhava EJ, Therkelsen CA, Basavapathruni A, Jin L, Boriack-Sjodin PA, Allain CJ, Klaus CR, Raimondi A, Scott MP, et al. . Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013; 122:1017-25; PMID:23801631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Yokoyama A, Somervaille TC, Smith KS, Rozenblatt-Rosen O, Meyerson M, Cleary ML. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell 2005; 123:207-18; PMID:16239140 [DOI] [PubMed] [Google Scholar]
- 89. Grembecka J, He S, Shi A, Purohit T, Muntean AG, Sorenson RJ, Showalter HD, Murai MJ, Belcher AM, Hartley T, et al. . Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat Chem Biol 2012; 8:277-84; PMID:22286128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Muntean AG, Chen W, Jones M, Granowicz EM, Maillard I, Hess JL. MLL fusion protein-driven AML is selectively inhibited by targeted disruption of the MLL-PAFc interaction. Blood 2013; 122:1914-22; PMID:23900238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Mereau H, De Rijck J, Cermakova K, Kutz A, Juge S, Demeulemeester J, Gijsbers R, Christ F, Debyser Z, Schwaller J. Impairing MLL-fusion gene-mediated transformation by dissecting critical interactions with the lens epithelium-derived growth factor (LEDGF/p75). Leukemia 2013; 27:1245-53; PMID:23318960 [DOI] [PubMed] [Google Scholar]
- 92. Liu H, Westergard TD, Cashen A, Piwnica-Worms DR, Kunkle L, Vij R, Pham CG, DiPersio J, Cheng EH, Hsieh JJ. Proteasome inhibitors evoke latent tumor suppression programs in pro-B MLL leukemias through MLL-AF4. Cancer Cell 2014; 25:530-42; PMID:24735925; http://dx.doi.org/ 10.1016/j.ccr.2014.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]